

Abstract
There is growing evidence that active tubular secretory clearance (CLs) may not decline proportionally with the glomerular filtration rate (GFR) in chronic kidney disease (CKD), leading to the overestimation of renal clearance (CLr) when using solely GFR to approximate disease effect on renal elimination. The clinical pharmacokinetic data of 33 renally secreted OAT1/3 substrates were collated to investigate the impact of mild, moderate, and severe CKD on CLr, tubular secretion and protein binding (f u,p). The f u,p of the collated substrates ranged from 0.0026 to 1.0 in healthy populations; observed CKD‐related increase in the f u,p (up to 2.7‐fold) of 8 highly bound substrates (f u,p ≤ 0.2) was accounted for in the analysis. Use of prediction equation based on disease‐related changes in albumin resulted in underprediction of the CKD‐related increase in f u,p of highly bound substrates, highlighting the necessity to measure protein binding in severe CKD. The critical analysis of clinical data for 33 OAT1/3 probes established that decrease in OAT1/3 activity proportional to the changes in GFR was insufficient to recapitulate effects of severe CKD on unbound tubular secretion clearance. OAT1/3‐mediated CLs was estimated to decline by an additional 50% relative to the GFR decline in severe CKD, whereas change in active secretion in mild and moderate CKD was proportional to GFR. Consideration of this additional 50% decline in OAT1/3‐mediated CLs is recommended for physiologically‐based pharmacokinetic models and dose adjustment of OAT1/3 substrates in severe CKD, especially for substrates with high contribution of the active secretion to CLr.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Recent studies have shown that active tubular secretory of renally cleared drugs may not decline proportionally with glomerular filtration rate (GFR) in chronic kidney disease (CKD), leading to potential overestimation of renal clearance when using solely GFR to approximate disease effect on renal elimination. However, this has not been evaluated for highly protein‐bound drugs.
WHAT QUESTION DID THIS STUDY ADDRESS?
Does the activity of OAT1/3 decrease proportionally to GFR in CKD? How do CKD‐associated changes to plasma protein binding affect this evaluation, in particular for highly bound drugs?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Critical analysis of 33 renally excreted OAT1/3 probes established that decrease in OAT1/3‐mediated active secretion exceeded the decline in GFR by ~ 50% in severe CKD.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Analysis refines virtual CKD populations in physiologically‐based pharmacokinetic models to increase the confidence in prospective quantitative predictions of pharmacokinetics in severe CKD, aiding the dose adjustment recommendations for OAT1/3 substrates in CKD populations in the absence of clinical data.
Chronic kidney disease (CKD) is a growing public health problem, affecting ~ 10% of the global population, 1 with significant consequences on pharmacokinetics and drug dosing. 2 Glomerular filtration rate (GFR) is commonly used as a surrogate marker to estimate CKD progression and measure the residual kidney function. 3 Among the various methods available to estimate/measure GFR, use of serum creatinine with one of several equations to obtain estimated GFR (eGFR) remains the most clinically applied method. 4 Likewise, the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) both recommend eGFR or creatinine clearance (CLcr) to guide drug dosing in these patients. 5 , 6 However, glomerular filtration is only one of three mechanisms contributing to the renal excretion of drugs. Despite this, drug dosing guidance is often based on the assumption that in CKD, the total renal excretion clearance (CLr) of a drug declines proportionally with GFR, built on the intact nephron hypothesis (INH). 7 , 8 INH assumes that any loss of glomerular/tubular function in a nephron represents a simultaneous and proportional loss of function to the entire nephron, thus, tubular secretion and re‐absorption should decrease proportionally to GFR in CKD.
However, there is growing evidence that the active tubular secretion of drugs, mediated by various renal transporters, such as organic anion transporters (OATs) and organic cation transporter 2 (OCT2), does not decline proportionally with GFR. 9 , 10 , 11 OAT1 and OAT3 are membrane‐bound solute carrier transporters predominantly expressed on the basolateral membrane of the renal proximal tubule and are responsible for the intracellular uptake of organic anions, including acidic drugs and endogenous compounds. 12 The active renal secretion clearance (CLs) of OAT1/3 drugs has been proposed to decline faster than GFR with progressively severe CKD, with the likelihood of overestimating the CLr when using GFR to approximate CKD effect on this parameter. 9 , 13 However, this disease effect has not been studied with highly bound OAT1/3 drugs and clinically observed changes to protein binding during CKD were mostly unaccounted for in prior analyses. 13
Changes to the protein binding of drugs during CKD are well‐established and are typically observed with acidic drugs that are primarily bound to albumin in plasma. 14 As a consequence, tubular secretion and protein binding (f u,p) of acidic drugs increases with disease progression, 15 whereas basic drugs tend to show an opposite trend due to their propensity to bind to α1‐acid glycoprotein, which increases during CKD. 15 Despite the multifactorial reasons behind the disease‐related changes in protein binding, f u,p values in CKD are often predicted by accounting solely for changes in plasma protein concentrations. 16 , 17 This prediction method was initially applied to estimate protein binding in infants 18 and its use in predicting f u,p in CKD has not been validated with a large dataset of drugs. Moreover, binding is assumed to occur with one protein (albumin or α1‐acid glycoprotein) whereas many drugs bind to both proteins to some extent.
In recent years, there has been an acceleration of physiologically‐based pharmacokinetic (PBPK) modeling in both commercial and academic applications, 19 but only 4% of PBPK modeling submissions to the FDA in 2018–2019 were for renal impairment. 20 Despite recent promising examples, 21 , 22 , 23 PBPK modeling of CKD‐related modulation of active secretion is still challenging, in contrast to PBPK modeling of metabolized drugs. 24 Lack of quantitative proteomic measurements of renal transporter expression in CKD populations contributes to this inability to recapitulate pharmacokinetics in CKD‐PBPK and the use of these models in a prospective manner. Therefore, there is a need for further understanding into the mechanistic changes that occur during CKD and its impact on transporter‐mediated renal secretion to build confidence in CKD‐PBPK models.
This study aimed to investigate the impact of CKD on plasma protein binding and active renal secretion of renally excreted OAT1/3 substrates using published clinical pharmacokinetic data in different disease stages. The work has built upon previous analysis into the appropriateness of the INH 9 , 13 and analyzed changes in active secretion for a wider range of OAT1/3 substrates, including highly protein bound compounds. Furthermore, the possible influence of CKD modifications to plasma protein binding on the OAT1/3‐mediated renal secretion was explored for highly bound substrates, together with CKD‐mediated changes in transporter function. The accuracy of predicting the change in f u,p based solely on the decrease in albumin levels during CKD was evaluated against the clinically observed data.
METHODS
Collation of clinical dataset of OAT substrates
A stepwise approach using four inclusion criteria (A–D) was applied to shortlist OAT1/3 substrates that were renally secreted and clinically impacted by CKD (Figure 1 ). Relevant data were collated from the PubMed electronic database (https://pubmed.ncbi.nlm.nih.gov, accessed between November 2020 and September 2021) and the Clinical Pharmacology and Biopharmaceutics Review uploaded on the Drugs@FDA database (https://www.accessdata.fda.gov/scripts/cder/daf/, accessed between November 2020 and September 2021). Criterion (A) shortlisted substrates with in vitro evidence of OAT1/3 activity as identified from published databases of renal transporter substrates 25 , 26 or in vivo evidence based on presence of a clinically significant drug–drug interaction (DDI) with an OAT1/3 inhibitor (area under the plasma concentration‐time curve ratio (AUCR) ≥ 1.25). Substrates with the fraction excreted unchanged in the urine equal or greater than 0.3 (f e,urine ≥0.3) 5 were considered (B). Active secretion defined as the ratio of CLr to filtration clearance (CLf) in healthy subjects (R nf,Healthy; Eqs. 1, 2) was criterion (C), where substrates with R nf,Healthy >1.5 were considered to have significant net active renal secretion and minimal re‐absorption, and were shortlisted. 9 , 13
| (1) |
where
| (2) |
Finally, only OAT1/3 substrates with clinically measured CLr and GFR in both healthy and CKD populations were considered (D). CKD stages were defined following the Kidney Disease Outcomes Quality Initiative (KDOQI) 2002 clinical practice guidelines: stage 1/healthy (GFR ≥ 90 mL/min/1.73 m2), stage 2/mild (GFR = 60–89 mL/min/1.73 m2), stage 3/moderate (GFR = 30–59 mL/min/1.73 m2), and stage 4/severe (GFR = 15–29 mL/min/1.73 m2). CKD subjects undergoing dialysis or on non‐dialysis days were excluded. Subjects with GFR < 15 mL/min but not receiving dialysis treatment were included in the severe CKD group. No distinction was made between the various methods to obtain eGFR or measured GFR (mGFR), although mGFR was preferred if available. In addition, AUC and f u,p in healthy and each CKD stage were collated, together with drug related properties (e.g., LogP and pKa) to investigate potential associations using Spearman correlation analysis (Supplementary Material).
Figure 1.

A flowchart describing the methodology used to shortlist the OAT1/3 substrates in this study. Only renally excreted and renally secreted OAT1/3 substrates with clinical pharmacokinetic data in CKD populations were shortlisted. In vitro evidence of OAT1/3 activity was accepted if greater intracellular uptake of a substrate with an OAT1/3 overexpressing cellular systems vs. the wildtype/control system or upon inhibition of OAT1/3 with probenecid were found. Evidence of in vivo OAT1/3 activity was a clinically relevant OAT‐mediated DDI with > 1.25‐fold increase in AUC in the presence of a US Food and Drug Administration recommended clinical inhibitor of OAT1/3. 48 In vitro inhibition of OAT1/3 by the shortlisted substrates were not considered as evidence of OAT1/3 activity. Due to species differences in OAT1/3 activity and protein expression, 49 in vitro studies using systems expressing mice Oat1/3 were excluded. The number of OAT1/3 substrates (N) that passed each inclusion criterion is shown. AUC, area under the plasma concentration‐time curve; AUCRDDI, area under the plasma concentration‐time curve ratio drug‐drug interaction/AUCNo‐DDI; CLr, renal clearance; CKD, chronic kidney disease; DDI, drug–drug interaction; f e,urine, fraction excreted unchanged in urine; GFR, glomerular filtration rate; HCTZ, hydrochlorothiazide; OAT1/3, organic anion transporters 1/3; PK, pharmacokinetic; R nf, ratio of renal clearance to filtration clearance in healthy populations.
Examining the impact of CKD on protein binding
The f u,p‐Severe‐CKD was predicted using Eq. 3, 16 assuming that human serum albumin (HSA) is the primary drug‐binding protein in blood and that there are no changes to the protein binding affinity or capacity of HSA during CKD. Predicted f u,p‐Severe‐CKD were compared with the collated clinically reported data to evaluate predictive performance of Eq. 3 for the CKD population.
| (3) |
A 1.25‐fold error acceptance criteria were used to compare the predicted f u,p‐Severe‐CKD against the observed values, and the overall accuracy of Eq. 3 was assessed using the geometric mean fold error (GMFE, Eq. 4).
| (4) |
The maximum predicted fold change in f u,p in severe CKD was approximated using Eq. 5 (Derivation in Supplementary Material):
| (5) |
where [HSA]Healthy and [HSA]CKD‐Stage represent the average concentration of HSA in healthy and severe CKD populations, respectively. The [HSA]Healthy (43.4 g/L) was obtained with the midpoint of the concentration found in healthy men (44.9 g/L) and women (41.8 g/L). 16 , 17 The [HSA]Severe‐CKD of 36.3 g/L is based on the mean concentration in men (37.6 g/L) and women (35.0 g/L) in severe CKD.
Evaluating the CKD effect on active tubular secretion
To evaluate the appropriateness of the INH, clinical evidence of the decline in active secretion compared with decline in GFR during CKD was evaluated using the collated clinical studies in our OAT1/3 database. CLs of the shortlisted substrates in mild, moderate, and severe CKD were calculated using Eqs. 6 and 7:
| (6) |
| (7) |
The reported mean, geometric mean or median GFR and CLr of each corresponding CKD stage were used in Eqs. 6 and 7. If average GFR was not reported, either the midpoint of the GFR range used to recruit subjects for each CKD stage was considered or 120 mL/min was assumed for healthy subjects. When necessary, the CLr and GFR of individual subjects were digitized using WebPlotDigitizer (version 4.4; Pacifica, CA). Subsequent descriptive statistical analyses were performed using Microsoft Excel (Microsoft, Redmond, WA). Because f u,p has been observed to vary depending on severity of CKD, 16 measured f u,p in the corresponding CKD stages were used when reported. For low to moderately bound OAT1/3 substrates (f u,p‐Healthy >0.2), the f u,p in CKD was assumed to be equal to f u,p‐Healthy when measurements of f u,p in the various CKD stages were not available. Negligible passive re‐absorption along the renal tubule was assumed when calculating CLs.
The decline in GFR and CLs at different stages of CKD relative to healthy subjects (R GFR and R CLs, Eqs. 8, 9), and magnitude by which CLs deviates from the INH (F x) were determined using Eq. 10.
| (8) |
| (9) |
| (10) |
F x represents the additional decline in tubular secretion that was not accounted for by the INH. If INH is valid, CLs and GFR decrease proportionally with each other and F x = 1. In contrast, if the substrate does not obey the INH, F x would be larger or smaller than one. Using a sensitivity analysis, the impact of severe CKD on CLr for OAT‐secreted substrates was investigated for the INH and the non‐INH scenario for a hypothetical dataset of substrates with CLs ranging from 0.5 to 500 mL/min and f u,p ranging from 0.001 to 1.0 (for details see Supplementary Material). Sensitivity analysis was performed assuming that substrates are only renally eliminated.
RESULTS
Clinical CKD database of OAT1 and OAT3 substrates
A preliminary list of 73 OAT1/3 substrates was identified (criterion A). Among them, 20 did not meet our criteria for renal excretion (criterion B), 3 were not renally secreted (criterion C), and 17 were excluded as appropriate data in CKD populations were unavailable (criterion D). Final database included 33 OAT1/3 substrates that met our inclusion criteria, comprising of 32 drugs and one endogenous biomarker of OAT1/3, 4‐pyridoxic acid. 27 Complete list of OAT1/3 substrates together with pharmacokinetic properties is in Table S1 , the majority also had in vitro evidence of being an OAT1/3 substrate. The exceptions were ertapenem, cefuroxime, and cefmetazole, which were included based on clinically significant DDIs with probenecid (AUCR ≥1.25). In the database, f e,urine ranged from 0.30 (rosuvastatin) to 0.95 (acamprosate), with varied importance of active secretion (R nf,healthy from 1.60 (meropenem) to 27.55 (furosemide); Figure 2 a,b ). The protein binding ranged from 0.0026 (olmesartan) to 1.0 (acamprosate), with 6 OAT1/3 substrates with f u,p ≤ 0.1 (Figure 2 c ). The effect of CKD on systemic exposure differed across OAT1/3 substrates in the database, with the AUCR in severe CKD (AUCRSevere‐CKD) ranging from 1.64 (rivaroxaban) to 11.62 (acamprosate).
Figure 2.
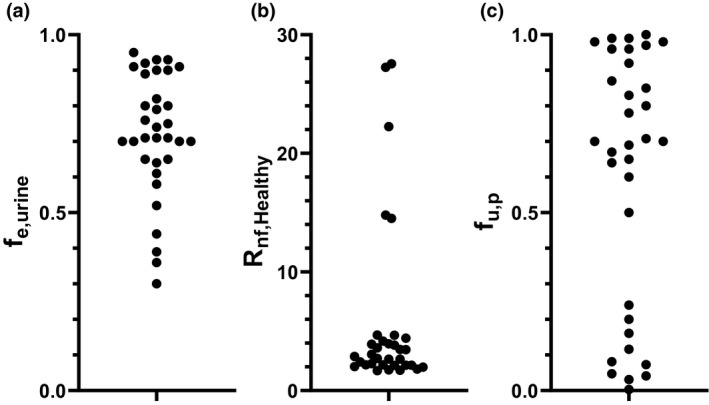
The distribution of (a) fraction excreted unchanged in the urine (f e,urine) (b) contribution of renal secretion in healthy population determined as the ratio of renal to filtration clearance (R nf,Healthy) and (c) measured fraction unbound in plasma (f u,p) of 33 OAT1/3 substrates in the CKD database. R nf,Healthy was calculated using Eq. 1. CKD, chronic kidney disease.
Impact of CKD on plasma protein binding
The literature analysis identified 9 published clinical measurements of f u,p‐Severe‐CKD for eight OAT1/3 substrates in our dataset (Figure 3 ). Two separate protein binding measurements in CKD for bumetanide were obtained from different clinical studies and analyzed. Although methotrexate was not included in the final shortlist due to the absence of CLr measurements in the published CKD clinical study, 28 it fulfilled all the other inclusion criteria and measurements of f u,p in CKD populations were collated. In general, highly protein bound OAT1/3 substrates (f u,p‐Healthy ≤ 0.2) in our database experienced a more pronounced increase in f u,p during severe CKD; for example, the highest fold change (f u,p‐Severe‐CKD/f u,p‐Healthy) was 2.71‐fold for bumetanide (f u,p‐Healthy = 0.0058 to 0.0304). Consideration of only CKD‐related changes in albumin concentration (Eq. 3) underpredicted the effect of disease when compared with the observed f u,p‐Severe‐CKD data (Figure 3 ). Maximum predicted fold change in f u,p in CKD was 1.20 (Figure 3 ), with 44% of datapoints within prediction limits (0.8 ≤ fold error ≤1.25) and a GMFE of 1.43. The most pronounced underprediction of f u,p‐Severe‐CKD was apparent for bumetanide and olmesartan, as predicted f u,p represented < 50% of the measured value (details in Figure S1 ).
Figure 3.
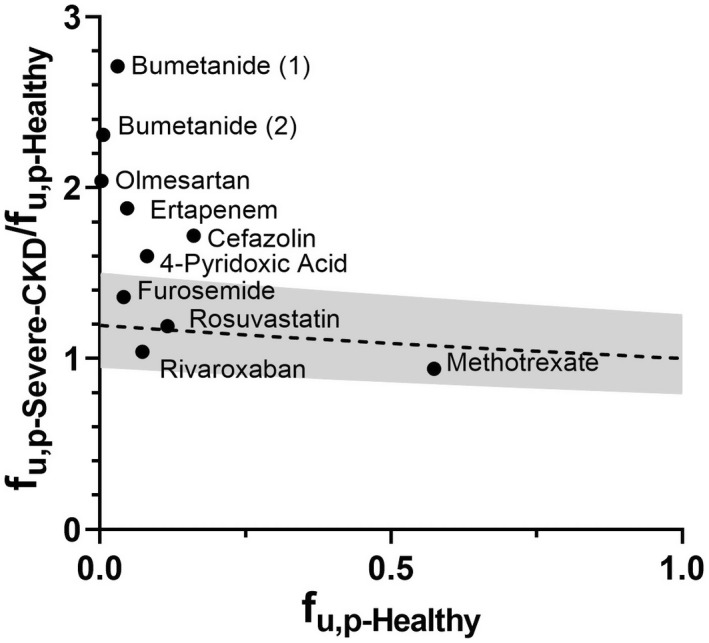
The comparison between fraction unbound in plasma (f u,p) in healthy populations (f u,p‐Healthy) and the predicted fold change of f u,p in severe CKD relative to the value in healthy populations (f u,p‐Severe‐CKD/f u,p‐Healthy, ‐‐‐) obtained using Eq. 3. The gray area bounds the 1.25‐fold error limit. The black solid circles (●) are the observed fold changes for OAT1/3 substrates with reported measurements of f u,p in severe CKD. CKD, chronic kidney disease.
Estimation of the effect of CKD on OAT1/3‐mediated renal secretion
In total, 38 CKD clinical studies for the 33 shortlisted OAT1/3 substrates were analyzed. Several drugs (famotidine, penciclovir, bumetanide, furosemide, and cefotaxime) had repeated clinical studies in CKD populations. The assumption of no disease‐related change in f u,p for OAT1/3 substrates with low to moderate plasma protein binding (Figure 2 c, f u,p‐Healthy >0.5) and with no reported data was reasonable, as their f u,p was not expected to vary significantly during CKD (Figure 3 ). CKD‐associated change in net renal secretion (R CLs) was compared against the decline in GFR (100 − R GFR) to evaluate the impact of disease progression (Figure 4 a ). In the mild to moderate stage of CKD, R CLs were scattered evenly around the line of unity, in overall agreement with the INH, illustrated also by estimated F x of 1.13 ± 0.53 and 0.84 ± 0.36, respectively. However, in severe CKD, active secretion declined faster than GFR, resulting in F x of 0.50 ± 0.29 (median = 0.44; Figure 4 b ). Only the R CLs of four drugs, olmesartan, rivaroxaban, sitagliptin, and adefovir, were above or on the line of unity in severe CKD. Conversely, famotidine, hydrochlorothiazide, piperacillin, meropenem, and avibactam exhibited an almost complete deterioration of active secretion in severe CKD (R CLs <2%) despite functioning residual glomerular filtration.
Figure 4.
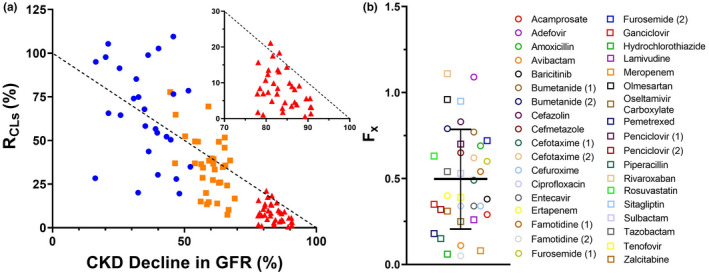
(a) Relationship between R
CLs (%) and the disease‐related % decline in GFR across mild ( ), moderate (
), moderate ( ), and severe (
), and severe ( ) chronic kidney disease (CKD) with the inset graph highlighting the severe CKD data only. R
CLs represents the decline in secretion clearance (CLs) at different stages of CKD relative to healthy subjects (). Each colored symbol represents an individual data point from 33 OAT1/3 substrates in the CKD database. The intact nephron hypothesis assumption where GFR declines proportionally to CLs is represented by the line of unity (‐‐‐). (b) The magnitude of deviation in R
CLs from the intact nephron hypothesis in severe CKD was estimated by calculating the F
x ratio (F
x = R
CLs/R
GFR, R
GFR = ). The black solid line ( – ) and error bars represent the estimated mean F
x and SD of 0.50 ± 0.29. This estimate was derived from an analysis of 38 CKD clinical studies for 33 OAT1/3 substrates shown in the figure with the corresponding names in the side figure legend. Data for 4‐pyridoxic acid were not reported in severe CKD, while ceftizoxime provided a negative F
x estimate in severe CKD hence both were excluded from the analysis of severe CKD group. GFR, glomerular filtration rate.
) chronic kidney disease (CKD) with the inset graph highlighting the severe CKD data only. R
CLs represents the decline in secretion clearance (CLs) at different stages of CKD relative to healthy subjects (). Each colored symbol represents an individual data point from 33 OAT1/3 substrates in the CKD database. The intact nephron hypothesis assumption where GFR declines proportionally to CLs is represented by the line of unity (‐‐‐). (b) The magnitude of deviation in R
CLs from the intact nephron hypothesis in severe CKD was estimated by calculating the F
x ratio (F
x = R
CLs/R
GFR, R
GFR = ). The black solid line ( – ) and error bars represent the estimated mean F
x and SD of 0.50 ± 0.29. This estimate was derived from an analysis of 38 CKD clinical studies for 33 OAT1/3 substrates shown in the figure with the corresponding names in the side figure legend. Data for 4‐pyridoxic acid were not reported in severe CKD, while ceftizoxime provided a negative F
x estimate in severe CKD hence both were excluded from the analysis of severe CKD group. GFR, glomerular filtration rate.
Majority (88%) of the OAT1/3 substrates in our database had an estimated F x <0.8 at severe CKD. The F x for all the analyzed OAT1/3 substrates and their rank‐order at every CKD stage is in Figure S2 and Table S2 . If the observed change in f u,p during CKD was not considered in our analysis (i.e., if f u,p‐Severe‐CKD was assumed to be equal to f u,p‐Healthy for all substrates), then estimated decrease in OAT1/3 activity would have been less prominent (mean F x in severe CKD of 0.53). The importance of including highly protein‐bound OAT1/3 substrates (f u,p‐Healthy ≤0.2) in this analysis was reinforced by the F x calculated for a subset of the substrates with f u,p‐Healthy >0.2, which resulted in a mean and median Fx value of 0.42 and 0.35, respectively.
Sensitivity analysis
Simulations were performed to understand the impact of disease‐related changes in OAT1/3‐mediated CLs and f u,p on the overall decrease in CLr from healthy subjects (GFR = 120 ml/min) to a patient with severe CKD (GFR = 30 mL/min). In the INH scenario, the decline in CLr remained constant for all OAT1/3 substrates, reflecting the extent of GFR decline in severe CKD (R CLr = R GFR = 4). It was unaffected by protein binding or magnitude of secretion clearance in healthy. In the non‐INH scenario, the additional disease effect on OAT1/3 in severe CKD was implemented (Fx = 0.5 applied on the secretion clearance), resulting in a predicted R CLr that varied from 4 to 8 (Figure 5 a ). Sensitivity analysis showed more pronounced impact of CKD on CLr for OAT1/3 substrates with a larger CLs in healthy subjects. In contrast, extent of change in CLr was less pronounced for substrates with higher f u,p (Figure 5 a ), consistent with the correlation analysis of the clinical data (Figure S3 A). The contribution of active secretion to CLr was found to be a key determinant in predicting disease effect on CLr (Figure 5 b ). An OAT1/3 substrate with a larger contribution of secretion to total CLr is expected to be significantly affected in CKD (larger R CLr), regardless of the absolute value of CLs. The impact of CKD‐induced changes in f u,p, in addition to disease effect on transporter activity was simulated, assuming a two‐fold increase in f u,p in severe CKD for highly bound substrates (f u,p ≤ 0.2). The analysis highlighted lower predicted change in the overall CLr (Figure S4 ) compared with the scenario where f u,p remains unchanged in severe CKD (Figure 5 b ).
Figure 5.
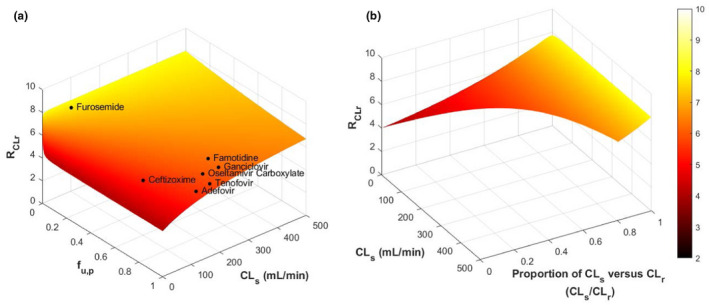
The 3D surface plots comparing the projected renal clearance (CLr) ratio of organic anion transporters 1/3 (OAT1/3) substrates in healthy vs. chronic kidney disease (CKD) subjects using Eqs. S5 and S6. Ratio of CLr in healthy vs. severe CKD was calculated as R CLr = CLr,Healthy/CLr,Severe‐CKD. Representative GFR values of 120 mL/min and 30 mL/min were used in the simulations for healthy and severe CKD populations, respectively. CKD‐related effect on OAT1/3 secretion clearance was simulated by assuming its decline beyond the intact nephron hypothesis (F x ratio of 0.5). (a) Predicted RCLr for a range of the secretion clearance (CLs = 0.5 to 500 mL/min) and fraction unbound in plasma (f u,p = 0.001 to 1.0) values. Clinically relevant OAT1/3 probes and in vitro substrates are shown on the surface of the 3D plot. (b) Predicted RCLr plotted against the proportion of active secretion against CLr (CLs/CLr) and CLs. The magnitude of CLs will determine the range of CLs/CLr that a certain drug may have. GFR, glomerular filtration rate.
DISCUSSION
The current study evaluated the appropriateness of the INH assumption to describe CKD‐related changes in OAT1/3‐mediated active secretion. A comprehensive database of 33 renally excreted OAT1/3 substrates was collated, together with retrospective analysis of the decline in GFR and CLs in mild to severe disease stages. Seven drugs in the database are FDA recommended probes either for OAT1/3‐mediated clinical DDI evaluation (adefovir, ceftizoxime, famotidine, furosemide, ganciclovir, and oseltamivir carboxylate) or as in vitro OAT1/3 substrates (adefovir and tenofovir). 29 The database also included 4‐pyridoxic acid, proposed endogenous biomarker of OAT1/3‐mediated DDI. 27 , 30 Considering complexities of CKD, this evaluation of disease‐related changes in activity of OAT1/3 included substrates with representative range in plasma protein binding (f u,p = 0.0026–1.0), extent of renal secretion and impact of CKD on plasma exposure (AUCRSevere‐CKD = 1.64–11.62). Furthermore, analysis into the disease impact on active secretion accounted for observed CKD‐associated changes in protein binding.
Clinical measurements of protein binding in CKD were available for only 8 of 33 (24%) of OAT1/3 substrates, highlighting a need for protein binding measurements for highly bound compounds, recognized also by regulatory guidance for renal impairment studies. 5 , 31 The analysis showed significant increase in f u,p of highly bound substrates in severe CKD (up to 2.7‐fold). The exception was rivaroxaban where minimal changes (3.7% increase at severe CKD) were reported despite its extensive binding mainly to albumin (f u,p‐Healthy = 0.073). 32 , 33 The small sample size (n = 8) and large experimental variability in the measurement of rivaroxaban f u,p‐Severe‐CKD (% coefficient of variation (%CV) = 23.2%) may be contributing factors. 32 A larger sample size in renal impairment clinical studies is needed to address the variability in the measured f u,p in this population. An additional confounding factor to consider is potential dual binding of drugs to albumin and α1‐acid glycoprotein, which would have an opposite effect on the f u,p‐Severe‐CKD. Expansion of the current dataset and inclusion of measurements of f u,p‐Severe‐CKD of nonrenally secreted drugs is needed to fully elucidate the effect of CKD on plasma protein binding. Our study demonstrated a likely underprediction of f u,p‐Severe‐CKD of highly bound OAT1/3 substrates based solely on disease‐related changes in albumin serum concentrations. Although prediction of f u,p‐Severe‐CKD was more accurate for OAT1/3 substrates with low plasma protein binding, their f u,p is not likely to change significantly during CKD anyway. Post‐translational guanidinylation in albumin that occurs in CKD reduces its binding capacity 34 and the accumulation of albumin‐bound uremic solutes might lead to the displacement of drug albumin binding. 35 Therefore, the inability to predict disease‐related alterations in f u,p‐Severe‐CKD (Figure 3 ) is likely due to the multifactorial changes that occur in CKD, unaccounted for in Eq. 3. Further refinement of the predictive equation and consideration of a possible nonlinear relationship between fold change in f u,p in CKD and f u,p‐healthy is required.
Our analysis revealed that in severe CKD, OAT1/3‐mediated CLs deteriorated much faster than GFR and hence reliance on the INH assumption would lead to an overestimation of CLs by ~ 50% (F x = 0.50). This adds on to the growing body of evidence that the INH might not be appropriate for renal transporters. 9 , 10 , 11 , 13 Previous analysis of 18 low to moderately protein‐bound OAT1/3 drugs estimated a median reduction of 59% in the CLs beyond the INH in severe CKD. 13 , 36 A comparable decline in the CLint of OAT2 was implemented in a PBPK model of creatinine developed for the CKD population. 11 The current analysis suggests that the underlying mechanism causing deterioration in OAT1/3‐mediated active secretion manifests clinically in severe disease. CLs declined proportionally to GFR in mild stage of CKD and showed only small additional decline of 16% in moderate CKD. Accumulation of uremic solutes is associated with decreased hepatic metabolism and hepatic/renal transport during CKD, 37 , 38 including in vitro evidence of OAT1/3 inhibition. 13 Because plasma concentrations of uremic solutes have been shown to increase as CKD progresses, 39 the critical inhibitory concentration might be reached only in severe CKD. In addition, suggested decreased expression of renal transporters, 22 , 40 decreased albumin levels, and post‐translational modifications to albumin, 16 , 34 coupled with the proposed protein‐mediated uptake transport 41 , 42 may all contribute to the deviation from the INH, but more evidence is needed to ascertain their involvement in modifying OAT1/3 function during CKD. Further work is needed to determine if this Fx ratio could be applied to other populations, such as pediatric CKD populations where age‐dependent expression of OAT1/3 43 complicates the interpretation of the disease effect. Moreover, these trends may not apply to other transporters, as suggested for OCT2 and MATE 2. 11 , 44 In principle, the method used here can be applied for the analysis of the CKD impact on other renal transporters, but the availability of clinical data, relevance of transporter to renal secretion, and overlapping substrate specificity between different renal transporters must be considered.
There were several uncertainties in our analysis, which we considered and controlled for where possible. First, there is a large experimental variability in the clinical measurements of CLr and GFR as evident for baricitinib, rivaroxaban, and sulbactam (%CV > 40%). Such variability can be due to the small sample size (n = 6–8) and large interindividual variability in patients with severe CKD that often have different comorbidities, disease etiology, and demographics. Second, a subset of our database are also substrates for other renal transporters (Table S1 ). For example, 2 of the outlying drugs in our dataset with F x >1 in severe CKD were adefovir (multidrug resistance‐associated protein 4) and rivaroxaban (P‐glycoprotein). 25 , 45 , 46 The activity of these transporters may change in a different manner to OAT1 and/or GFR, thus confounding our analysis. Nevertheless, > 70% of our shortlisted OAT1/3 substrates had both in vitro and clinical evidence of OAT1/3 activity. Last, the use of creatinine‐based methods to obtain CLcr or eGFR in most of the clinical studies introduces uncertainty into the measurements GFR. Creatinine CLr is a combination of glomerular filtration and active secretion via multiple renal transporters. 47 Thus, using serum creatinine to estimate GFR may also be confounded by changes to its active renal secretion during CKD or DDI with concomitant medication, leading to an inaccurate GFR estimate. 11
Sensitivity analysis was performed to assess the impact of disproportional decline in OAT1/3 activity on CLr of renally cleared compounds with varying f u,p (0.001 to 1.0) and CLs (0.5 to 500 mL/min). In the INH scenario where various renal processes decline proportionally to each other, predicted decline in CLr was a constant value (= GFRhealthy/GFRSevere‐CKD) regardless of the f u,p or CLs. However, assuming additional decline of 50% in OAT1/3 (non‐INH scenario) in severe CKD, disease‐related changes to CLr depended on the magnitude of f u,p and CLs in healthy populations. The CKD‐mediated decrease in CLr ranged from 4 in the INH scenario to an upper limit of 8 for highly bound OAT1/3 substrates in the non‐INH scenario, illustrating a 2‐fold difference in the predicted RCLr that would be unaccounted for in the INH scenario. The analysis also highlighted relevance of the percent of contribution of active secretion to total CLr as a factor that might be sufficient to anticipate the extent of decrease in CLr in CKD. Our step‐wise approach and recommendations for consideration of additional decline in OAT1/3‐mediated active secretion in PBPK models of severe CKD populations is summarized as a decision tree (Figure 6 ). When secretion clearance forms majority of the CLr (CLs/CLr > 50%), an additional 1.25‐fold change in CLr relative to the extent of decline in GFR (INH) is expected. In contrast, a CLs/CLr < 50% results in < 1.25‐fold deviation in CLr from the INH. This analysis is in agreement with recent ganciclovir PBPK modeling where a modest difference in CLr between the INH and non‐INH assumed changes in OATs was observed and rationalized by relatively low contribution of secretion clearance. 23
Figure 6.
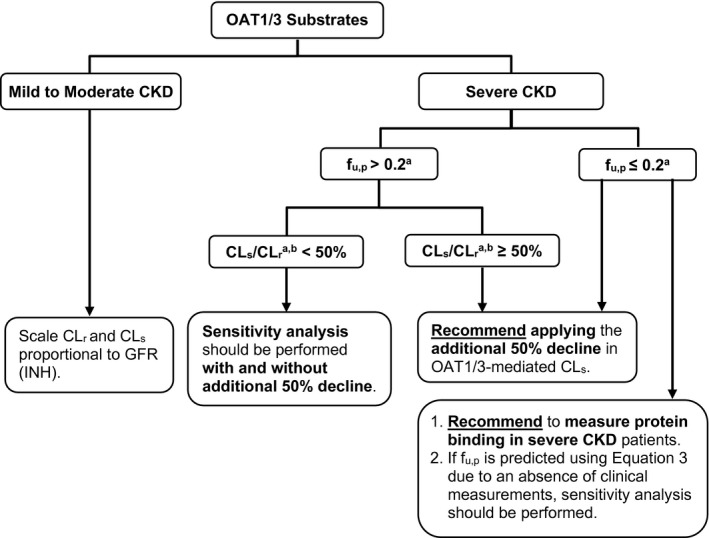
Proposed recommendations for physiologically‐based pharmacokinetic models of renally excreted OAT1/3 substrates in CKD populations. aPharmacokinetic properties in healthy population; b% contribution of active secretion clearance (CLs) to total renal clearance (CLr). CKD, chronic kidney disease; f u,p, fraction unbound in plasma; GFR, glomerular filtration rate; INH, intact nephron hypothesis; OAT1/3, organic anion transporters 1/3.
In conclusion, our study assessed the decline in OAT1/3‐mediated active secretion by establishing a database of 33 drugs/endogenous probes with various degree of renal excretion and protein binding and retrospectively analyzing the collated clinical data in mild, moderate, and severe CKD. The current analysis estimated 50% decline in OAT1/3 activity beyond the disease‐related changes in GFR, highlighting the inadequacy of INH assumptions in describing changes to tubular secretion via these transporters during severe CKD. Depending on the CKD severity, extent of protein binding and the contribution of active secretion to total CLr, application of 50% decline in OAT1/3 activity in addition to proportional decline in GFR is recommended in PBPK models of CKD populations (see decision tree in Figure 6 ). Moreover, this study emphasizes the critical need to measure the f u,p of highly bound drugs in CKD studies. Given the observed underprediction of protein‐binding changes in severe CKD, predicting the f u,p of highly bound transporter substrates based solely on changes to albumin concentrations should be avoided. Understanding the mechanisms behind the CKD‐associated changes to active tubular secretion and protein binding is critical when estimating the CLr of transporter substrates in CKD and for increasing confidence in CKD‐PBPK modeling of transporter‐mediated renal elimination.
FUNDING
S.P.F.T. was supported by a PhD scholarship from the Agency for Science, Technology, and Research (A*STAR), Singapore (BM/NDR/19/003).
CONFLICT OF INTEREST
A.R.‐H. is an employee of Certara UK Limited. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
S.P.F.T., D.S., A.R.‐H., and A.G. wrote the manuscript. S.P.F.T., D.S., and A.G. designed the research. S.P.F.T. performed the research. S.P.F.T., D.S., A.R.‐H., and A.G. analyzed the data. S.P.F.T., D.S., A.R.‐H., and A.G. contributed new reagents/analytical tools.
Supplementary information accompanies this paper on the Clinical Pharmacology & Therapeutics website (www.cpt-journal.com).
Supporting information
Appendix S1 Supporting information
References
- 1. Bikbov, B. et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395, 709–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lea‐Henry, T.N. , Carland, J.E. , Stocker, S.L. , Sevastos, J. & Roberts, D.M. Clinical pharmacokinetics in kidney disease: fundamental principles. Clin. J. Am. Soc. Nephrol. 13, 1085–1095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matzke, G.R. et al. Drug dosing consideration in patients with acute and chronic kidney disease—a clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 80, 1122–1137 (2011). [DOI] [PubMed] [Google Scholar]
- 4. Porrini, E. et al. Estimated GFR: time for a critical appraisal. Nat. Rev. Nephrol. 15, 177–190 (2019). [DOI] [PubMed] [Google Scholar]
- 5. U.S. Food and Drug Administration . Guidance for industry: pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing. <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pharmacokinetics-patients-impaired-renal-function-study-design-data-analysis-and-impact-dosing-and> (2020).
- 6. European Medicines Agency . Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. <https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf> (2015).
- 7. Bricker, N.S. On the meaning of the intact nephron hypothesis. Am. J. Med. 46, 1–11 (1969). [DOI] [PubMed] [Google Scholar]
- 8. Verbeeck, R.K. & Musuamba, F.T. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol 65, 757–773 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Chapron, A. , Shen, D.D. , Kestenbaum, B.R. , Robinson‐Cohen, C. , Himmelfarb, J. & Yeung, C.K. Does secretory clearance follow glomerular filtration rate in chronic kidney diseases? Reconsidering the intact nephron hypothesis. Clin. Transl. Sci. 10, 395–403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pradhan, S. , Duffull, S.B. , Walker, R.J. & Wright, D.F.B. The intact nephron hypothesis as a model for renal drug handling. Eur. J. Clin. Pharmacol. 75, 147–156 (2019). [DOI] [PubMed] [Google Scholar]
- 11. Takita, H. , Scotcher, D. , Chinnadurai, R. , Kalra, P.A. & Galetin, A. Physiologically‐based pharmacokinetic modelling of creatinine‐drug interactions in the chronic kidney disease population. CPT Pharmacometrics Syst. Pharmacol. 9, 695–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giacomini, K.M. et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsueh, C.H. et al. Identification and quantitative assessment of uremic solutes as inhibitors of renal organic anion transporters, OAT1 and OAT3. Mol. Pharm. 13, 3130–3140 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Reidenberg, M.M. & Drayer, D.E. Alteration of drug‐protein binding in renal disease. Clin. Pharmacokinet. 9(Suppl 1), 18–26 (1984). [DOI] [PubMed] [Google Scholar]
- 15. Vanholder, R. , Van Landschoot, N. , De Smet, R. , Schoots, A. & Ringoir, S. Drug protein binding in chronic renal failure: evaluation of nine drugs. Kidney Int 33, 996–1004 (1988). [DOI] [PubMed] [Google Scholar]
- 16. Rowland Yeo, K. , Aarabi, M. , Jamei, M. & Rostami‐Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 4, 261–274 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Tan, M.L. et al. Effect of chronic kidney disease on nonrenal elimination pathways: a systematic assessment of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP. Clin. Pharmacol. Ther. 103, 854–867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McNamara, P.J. & Alcorn, J. Protein binding predictions in infants. AAPS PharmSci 4, E4 (2002), 19, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. El‐Khateeb, E. , Burkhill, S. , Murby, S. , Amirat, H. , Rostami‐Hodjegan, A. & Ahmad, A. Physiological‐based pharmacokinetic modeling trends in pharmaceutical drug development over the last 20‐years; in‐depth analysis of applications, organizations, and platforms. Biopharm. Drug Dispos. 42, 107–117 (2021). [DOI] [PubMed] [Google Scholar]
- 20. Zhang, X. et al. Application of PBPK modeling and simulation for regulatory decision making and its impact on US prescribing information: an update on the 2018–2019 Submissions to the US FDA's Office of Clinical Pharmacology. J. Clin. Pharmacol. 60(Suppl 1), S160–S178 (2020). [DOI] [PubMed] [Google Scholar]
- 21. Yee, K.L. et al. Evaluation of model‐based prediction of pharmacokinetics in the renal impairment population. J. Clin. Pharmacol. 58, 364–376 (2018). [DOI] [PubMed] [Google Scholar]
- 22. Scotcher, D. , Jones, C.R. , Galetin, A. & Rostami‐Hodjegan, A. Delineating the role of various factors in renal disposition of digoxin through application of physiologically based kidney model to renal impairment populations. J. Pharmacol. Exp. Ther. 360, 484–495 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scotcher, D. & Galetin, A. PBPK simulation‐based evaluation of ganciclovir crystalluria risk factors: effect of renal impairment, old age, and low fluid intake. AAPS J 24, 13(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heimbach, T. et al. Physiologically‐based pharmacokinetic modeling in renal and hepatic impairment populations: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 110, 297–310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morrissey, K.M. , Stocker, S.L. , Wittwer, M.B. , Xu, L. & Giacomini, K.M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 53, 503–529 (2013). [DOI] [PubMed] [Google Scholar]
- 26. Ivanyuk, A. , Livio, F. , Biollaz, J. & Buclin, T. Renal drug transporters and drug interactions. Clin. Pharmacokinet. 56, 825–892 (2017). [DOI] [PubMed] [Google Scholar]
- 27. Willemin, M.E. et al. Clinical investigation on endogenous biomarkers to predict strong OAT‐mediated drug‐drug interactions. Clin. Pharmacokinet. 60, 1187–1199 (2021). [DOI] [PubMed] [Google Scholar]
- 28. Bressolle, F. , Bologna, C. , Kinowski, J.M. , Sany, J. & Combe, B. Effects of moderate renal insufficiency on pharmacokinetics of methotrexate in rheumatoid arthritis patients. Ann. Rheum. Dis. 57, 110–113 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. U.S. Food and Drug Administration . drug development and drug interactions: table of substrates, inhibitors and inducers. <https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers> (2019).
- 30. Ahmad, A. et al. Population pharmacokinetic modeling and simulation to support qualification of pyridoxic acid as endogenous biomarker of OAT1/3 renal transporters. CPT Pharmacometrics Syst. Pharmacol. 10, 467–477 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sahre, M.D. et al. Evaluating patients with impaired renal function during drug development: highlights from the 2019 US FDA Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting. Clin. Pharmacol. Ther. 110, 285–288 (2021). [DOI] [PubMed] [Google Scholar]
- 32. Kubitza, D. et al. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct Factor Xa inhibitor. Br. J. Clin. Pharmacol. 70, 703–712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mueck, W. , Stampfuss, J. , Kubitza, D. & Becka, M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin. Pharmacokinet. 53, 1–16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rueth, M. et al. Guanidinylations of albumin decreased binding capacity of hydrophobic metabolites. Acta Physiol. (Oxf) 215, 13–23 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Klammt, S. et al. Albumin‐binding capacity (ABiC) is reduced in patients with chronic kidney disease along with an accumulation of protein‐bound uraemic toxins. Nephrol. Dial Transplant. 27, 2377–2383 (2012). [DOI] [PubMed] [Google Scholar]
- 36. Hsueh, C.H. , Hsu, V. , Zhao, P. , Zhang, L. , Giacomini, K.M. & Huang, S.M. PBPK modeling of the effect of reduced kidney function on the pharmacokinetics of drugs excreted renally by organic anion transporters. Clin. Pharmacol. Ther. 103, 485–492 (2018). [DOI] [PubMed] [Google Scholar]
- 37. Tan, M.L. et al. Use of physiologically based pharmacokinetic modeling to evaluate the effect of chronic kidney disease on the disposition of hepatic CYP2C8 and OATP1B drug substrates. Clin. Pharmacol. Ther. 105, 719–729 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tatosian, D.A. et al. A microdose cocktail to evaluate drug interactions in patients with renal impairment. Clin. Pharmacol. Ther. 109, 403–415 (2021). [DOI] [PubMed] [Google Scholar]
- 39. Chen, Y. et al. Kidney clearance of secretory solutes is associated with progression of CKD: the CRIC study. J. Am. Soc. Nephrol. 31, 817–827 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Follman, K.E. & Morris, M.E. Prediction of the effects of renal impairment on clearance for organic cation drugs that undergo renal secretion: a simulation‐based study. Drug Metab. Dispos. 46, 758–769 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Francis, L.J. , Houston, J.B. & Hallifax, D. Impact of plasma protein binding in drug clearance prediction: a data base analysis of published studies and implications for in vitro‐in vivo extrapolation. Drug Metab. Dispos. 49, 188–201 (2021). [DOI] [PubMed] [Google Scholar]
- 42. van der Made, T.K. et al. Quantitative translation of microfluidic transporter in vitro data to in vivo reveals impaired albumin‐facilitated indoxyl sulfate secretion in chronic kidney disease. Mol. Pharm. 16, 4551–4562 (2019). [DOI] [PubMed] [Google Scholar]
- 43. Cheung, K.W.K. et al. A comprehensive analysis of ontogeny of renal drug transporters: mRNA analyses, quantitative proteomics, and localization. Clin. Pharmacol. Ther. 106, 1083–1092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheung, K.W.K. et al. The effect of uremic solutes on the organic cation transporter 2. J. Pharm. Sci. 106, 2551–2557 (2017). [DOI] [PubMed] [Google Scholar]
- 45. Gnoth, M.J. , Buetehorn, U. , Muenster, U. , Schwarz, T. & Sandmann, S. In vitro and in vivo P‐glycoprotein transport characteristics of rivaroxaban. J. Pharmacol. Exp. Ther. 338, 372–380 (2011). [DOI] [PubMed] [Google Scholar]
- 46. Imaoka, T. , Kusuhara, H. , Adachi, M. , Schuetz, J.D. , Takeuchi, K. & Sugiyama, Y. Functional involvement of multidrug resistance‐associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol. Pharmacol. 71, 619–627 (2007). [DOI] [PubMed] [Google Scholar]
- 47. Scotcher, D. et al. A novel physiologically based model of creatinine renal disposition to integrate current knowledge of systems parameters and clinical observations. CPT Pharmacometrics Syst. Pharmacol. 9, 310–321 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. U.S. Food and Drug Administration . Guidance for industry: in vitro drug interaction studies ‐ cytochrome P450 enzyme and transporter‐mediated drug interactions (2020).
- 49. Basit, A. , Radi, Z. , Vaidya, V.S. , Karasu, M. & Prasad, B. Kidney cortical transporter expression across species using quantitative proteomics. Drug Metab. Dispos. 47, 802–808 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting information