

Abstract
Aim
To evaluate the ability of ladarixin (LDX, 400 mg twice‐daily for three cycles of 14 days on/14 days off), an inhibitor of the CXCR1/2 chemokine receptors, to maintain C‐peptide production in adult patients with newly diagnosed type 1 diabetes.
Materials and Methods
A double‐blind, randomized (2:1), placebo‐controlled study was conducted in 45 males and 31 females (aged 18‐46 years) within 100 days of the first insulin administration. The primary endpoint was the area under the curve (AUC) for C‐peptide in response to a 2‐hour mixed meal tolerance test (AUC[0‐120 min]) at week 13 ± 1. Secondary endpoints included C‐peptide AUC(15‐120 min), HbA1c, daily insulin requirement, severe hypoglycaemic events (SHE), the proportion of subjects achieving HbA1c less than 7.0% without SHE and maintaining a residual beta cell function. Follow‐up assessments were scheduled at weeks 13 ± 1, 26 ± 2 and 52 ± 2.
Results
In total, 26/26 (100%, placebo) and 49/50 (98%, LDX) patients completed week 13. The mean change from baseline to week 13 in C‐peptide AUC(0‐120 min) was −0.144 ± 0.449 nmol/L with placebo and 0.003 ± .322 nmol/L with LDX. The difference was not significant (0.149 nmol/L, 95% CI −0.04 to 0.33; P = .122). At week 26, the proportion of patients with HbA1c less than 7.0% without SHE was transiently higher in the LDX group (81% vs. 54%, P = .024). Otherwise, no significant secondary endpoint differences were noted. Transient metabolic benefit was seen at week 26 in favour of the LDX group in the prespecified subpopulation with fasting C‐peptide less than the median value at screening.
Conclusions
In newly diagnosed patients with type 1 diabetes, short‐term LDX treatment had no appreciable effect on preserving residual beta cell function.
Keywords: beta cell function, phase I‐II study, randomized trial, type 1 diabetes
1. INTRODUCTION
Type 1 diabetes is an immune‐mediated chronic disease resulting in progressive failure of pancreatic beta cells. Despite important improvements in diabetes care in recent decades, type 1 diabetes results in short‐ 1 and long‐term complications and is one of the leading causes of cardiovascular diseases, end‐stage renal disease, blindness and amputations. 2 Despite more than 2 decades of efforts and dozens of clinical trials with a variety of immune and non‐immune interventions, only six immunotherapies mainly targeting the adaptive lymphocyte‐mediated attack of beta cells have been shown to preserve insulin secretion in stage 3 type 1 diabetes (teplizumab, 3 otelixizumab, 4 rituximab, 5 abatacept, 6 low‐dose antithymocyte globulin 7 and alefacept 8 ) and teplizumab have been shown to delay the onset of stage 2 disease. 9 Type 1 diabetes is generally depicted as a beta cell‐specific T cell‐mediated autoimmune disease, with an associated non‐beta cell–specific inflammatory component. 10 Not surprisingly, some randomized controlled trials targeting innate immune mediators (such as tumour necrosis factor alpha [TNFα], interleukin [IL]‐1 and IL‐6R) have been conducted. 11 , 12 , 13 Neutrophils were proposed as relevant players in the pathogenesis of type 1 diabetes. 14 Pancreas‐infiltrating neutrophils were observed at the level of very small blood vessels in the exocrine pancreas of multiorgan donors with type 1 diabetes (both at onset and at later stages of the disease), but not in those of multiorgan non‐diabetic donors or donors with type 2 diabetes. 15 A tissue‐specific pathogenic role of these pancreas‐infiltrating neutrophils is suggested by their ability to extrude neutrophil extracellular traps. 16 Moreover, a mild but significant and reproducible peripheral neutropenia both precedes and parallels the onset of type 1 diabetes. 7 Blood neutrophils in type 1 diabetes revealed a unique molecular signature that is distinguished by an overabundance of interferon (IFN)‐associated genes; despite being healthy, said signature is already present in type 1 diabetes‐autoantibody‐negative at‐risk subjects. 16 The role of neutrophils in the pathogenesis of type 1 diabetes has also emerged as pivotal in non‐obese diabetic (NOD) mice. Diana et al. showed that neutrophils, lymphocytes B‐1a and plasmacytoid dendritic cells are involved in the initiation of the diabetogenic T cell response and autoimmune diabetes development. 17 Moreover, chemokine ligand 8 (CXCL8), commonly called IL‐8, appears to be an important mediator in the progression of type 1 diabetes, modulating neutrophil trafficking and recruitment through specific CXCR1 and CXCR2 receptors. 18 Indeed, we showed that the inhibition of the neutrophil recruitment by ladarixin (LDX), an allosteric inhibitor of the IL‐8 receptors CXCR1/CXCR2, 19 could prevent and revert the hyperglycaemia in NOD mice. This evidence provided the basis for this phase 2 safety and efficacy study of LDX in newly diagnosed type 1 diabetes patients, testing the ability of the drug to preserve beta cell function and delay further disease progression.
2. MATERIALS AND METHODS
2.1. Study design and patients
This phase 2 clinical trial was registered with ClinicalTrials.gov (NCT02814838) and conducted in compliance with all applicable regulatory requirements. This was a multicentre, randomized, double‐blind, parallel‐assignment study conducted at eight European Union centres (four in Italy, two in Germany and two in Belgium) in newly diagnosed type 1 diabetes patients. Because there were no data available to estimate the effect size of LDX in patients with type 1 diabetes, the sample size for this study was based on figures provided by Lachin et al., 20 considering an adult population (aged >18 years) and the log(x + 1)‐transformed C‐peptide area under the curve (AUC) from the mixed meal tolerance test (MMTT), initially selected by TrialNet as the appropriate transformation. With these assumptions, it was planned for 72 patients to be included in the trial, to provide 85% power to detect a 50% between‐group difference (α = .05, one‐sided) in the 2‐hour MMTT C‐peptide AUC(0‐120 min), assuming a 24% drop‐out rate. As a minimum, the inclusion criteria included: age 18 to 45 years, new‐onset (randomization within 100 days of the first insulin administration) type 1 diabetes confirmed by at least one positive diabetes‐related autoantibody (anti‐GAD [GADA], anti‐insulin [IAA], anti‐IA‐2 [IA‐2A] or anti‐ZnT8 [ZnT8A]), insulin requirement at some time and residual beta cell function as per peak stimulated (MMTT) C‐peptide level of more than 0.2 nmol/L. Exclusion criteria included: the patient taking premixed insulin or on an insulin pump, creatinine clearance less than 60 ml/min, alanine aminotransferase (ALT)/aspartate aminotransferase (AST) more than three times the upper limit of normal and total bilirubin of more than 3 mg/dl, hypoalbuminaemia (serum albumin <3 g/dl), the corrected QT interval by Fredericia (QTcF) of more than 470 ms, as well as other significant co‐morbid conditions or administration of concomitant medications that could have biased the efficacy outcome/readout.
2.2. Study treatment, randomization and masking
Patients received hard gelatine capsules of either LDX at a dose of 400 mg twice‐daily for three cycles of 14 days on/14 days off, or placebo (same schedule), according to their randomization number (Figure S1). LDX inhibits neutrophil (PMN) migration in vitro with a maximal inhibitory concentration (IC50) in the range of 1 ng/ml, as per preclinical data. Pharmacokinetics trials in humans have established that the 400‐mg dose provides an average steady state plasma concentration of the LDX unbound fraction of about 100 to 150 ng/ml. As a consequence, the 400‐mg dose was selected to ensure full inhibition of PMN migration. The two daily doses were administered orally in the morning and in the evening, 2 hours apart from breakfast and dinner, respectively. An independent statistician generated the master randomization list, balancing LDX and placebo in a 2:1 fashion within each centre. Individual treatment codes were provided as sealed envelopes to the investigators and sponsor pharmacovigilance for emergency/safety purposes. To maintain blindness, the appearance of the capsules, including packaging and labelling, did not allow the recognition of the actual treatment (either LDX or placebo).
2.3. Procedures and endpoints
Patients enrolled in this trial were admitted to intensive diabetes management, according to the American Diabetes Association recommendation, to ensure optimal glycaemic control. Insulin therapy was based on multiple daily insulin injections. Patients were instructed to self‐monitor (finger‐stick) their glucose values at least four times per day to allow insulin to be titrated up or down to the following targets: preprandial blood glucose of 70 to 130 mg/dl, postprandial blood glucose of less than 180 mg/dl and bedtime blood glucose of 110 to 150 mg/dl, consistent with an overall target of HbA1c less than 7%. Screening included evaluation of medical history and disease‐specific clinical information, including the date of first insulin administration and autoantibody status (at least one positive among GADA; IAA, if obtained within 10 days of insulin therapy; IA‐2A and ZnT8A), to confirm the diagnosis of type 1 diabetes. Baseline daily insulin requirement, HbA1c, C‐peptide and glucose from the MMTT were assessed within 3 weeks before randomization. Follow‐up assessments were scheduled at weeks 13 ± 1 (month 3), 26 ± 2 (month 6) and 52 ± 2 (month 12) from the beginning of treatment. The prespecified primary outcome was the AUC for the serum C‐peptide level during 2 hours (AUC[0‐120 min]) of an MMTT at weeks 13 ± 1. Secondary endpoints included MMTT C‐peptide increase above fasting values (AUC[15‐120 min]), HbA1c, daily insulin requirement, severe hypoglycaemic events (SHE), the proportion of subjects achieving an HbA1c of less than 7.0% without SHE and the proportion of patients maintaining a residual beta cell function (defined as at least one MMTT C‐peptide value ≥0.2 nmol/L). The incidence of treatment‐emergent adverse events (TEAEs), vital signs and standard laboratory variables (haematology and clinical chemistry) were specific safety endpoints.
2.4. Statistical analysis
Data are presented as mean ± standard deviation (SD) or median, according to their distribution. All the AUC analyses were based on actual rather than scheduled timings and were calculated using the trapezoidal rule. Analyses were performed according to the intention‐to‐treat (ITT) principle; all statistical tests were performed one‐sided with α = .05, unless otherwise specified. The AUC(0‐120 min) after the MMTT at week 13 ± 1 was transformed as log(x + 1) values; transformed AUC was analysed with an analysis of covariance (ANCOVA) model adjusting for sex, baseline age and baseline C‐peptide AUC(0‐120 min) and unpaired t test. The comparisons between treatment groups on log(x + 1)‐transformed AUC(0‐120 min), % change from baseline of AUC(0‐120 min), average daily insulin requirement and HbA1c value, were carried out using a mixed linear model with treatment group, visit and treatment by visit interaction as fixed factors of the model and patient as a random effect. Number and proportion along the 95% CI (Clopper–Pearson′s formula) of patients with HbA1c less than 7% and absence of SHE from the previous visit were calculated for each time point. The comparison between the two study treatment groups was performed by means of a Fisher′s exact text at each time point. Alternative approaches were explored, including subset analysis and AUC geometric mean ratios, as described in the sections below.
2.5. Study approval
The protocol, protocol amendments and consent documents were approved by the appropriate ethics committees. All participants provided written, informed consent.
3. RESULTS
3.1. Patient disposition and baseline characteristics
The predefined ITT cohort included all 76 patients who underwent randomization and received at least one dose of study medication. Details of patient disposition and inclusion in analysis sets are shown in Figure 1. One patient of the 76 randomized did not complete the week 13 MMTT (because of early withdrawal of consent); therefore, 75 patients were included in the primary outcome analysis (49 on LDX, 26 on placebo). Seventy‐three of 75 patients completed the week 52 follow‐up (48 on LDX, 25 on placebo): one patient on placebo discontinued from the study because of consent withdrawal; one patient on LDX was lost to follow‐up. Demographic characteristics of the ITT patients are reported in Table 1. The mean exposure (% of scheduled total dose) to LDX was 97.7% ± 7.6%. This includes two patients with a study treatment compliance of less than 80%. The majority of patients were positive for two or more autoantibodies, with GADA being the most frequent, followed by ZnT8A. There were no notable differences between treatment groups with respect to demographic and baseline characteristics. Neutrophil count was comparable at screening in the two treatment groups (LDX 3.37 ± 1.21 109/L, placebo 3.25 ± 1.16 109/L) and remained as such at week 13 (treatment completion: LDX 3.49 ± 1.56 109/L, placebo 3.32 ± 1.23 109/L).
FIGURE 1.
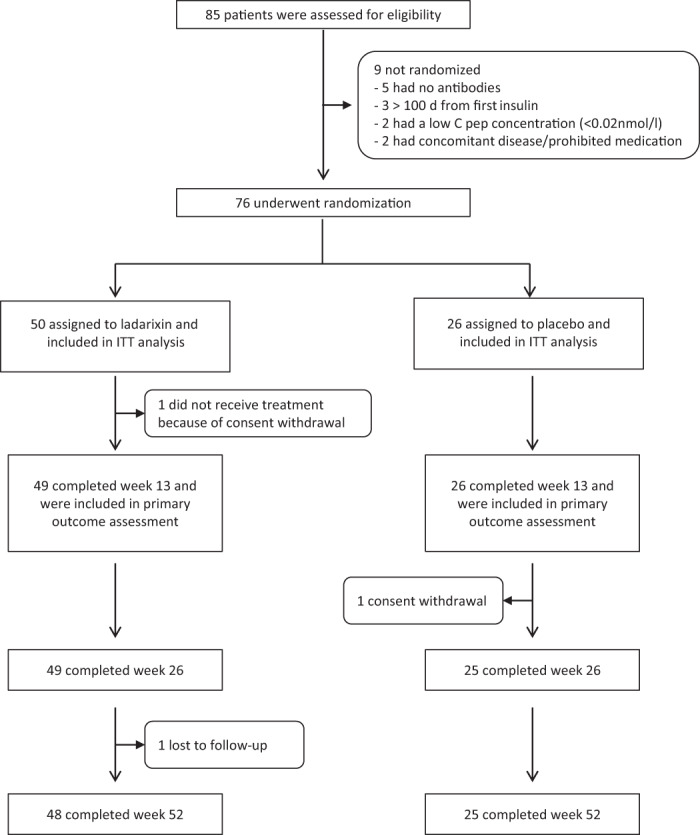
Enrolment, randomization and follow‐up of study participants. From August 2016 to May 2018, 85 new‐onset type 1 diabetes patients were assessed for eligibility and 76 were randomized. All randomized patients were included in the intention‐to‐treat (ITT) cohort
TABLE 1.
Characteristics of the study groups
| LDX (N = 50) | Placebo (N = 26) | |
|---|---|---|
| Age (y) | ||
| Mean | 27.6 ± 7.06 | 26.8 ± 6.35 |
| Median | 26 | 26.5 |
| Range | 18‐46 | 18‐38 |
| Male sex (N [%]) | 29 (58) | 16 (61.5) |
| Ethnic group (N [%]) | ||
| White/Caucasian | 49 (98) | 26 (100) |
| No. of autoantibodies (N [%]) | ||
| 1 | 7 (14) | 4 (15.4) |
| 2 | 19 (36) | 7 (26.9) |
| 3 | 13 (28) | 7 (30.8) |
| 4 | 11 (22) | 7 (26.9) |
| IAA+ | 21 (42) | 11 (42.3) |
| GADA | 47 (94) | 23 (88.5) |
| IA‐2A | 28 (56) | 17 (65.4) |
| ZnT8A | 32 (64) | 19 (73.1) |
| No. of days from first insulin to treatment | ||
| Median | 74 | 77 |
| Range a | 29‐104 | 40‐107 |
| Weight (kg) | 68.52 (47.2‐110.4) | 68.27 (44‐109.2) |
| BMI | 22.5 (18.2‐34.5) | 22.7 (18.8‐30.8) |
| White blood cells (cells/mm3) | 5.95 ± 1.54 | 5.77 ± 1.29 |
| Creatinine (μmol/L) | 71.8 ± 13.58 | 66.4 ± 10.91 |
| Creatinine clearance (ml/min) b | 127.3 ± 31.55 | 137.1 ± 35.34 |
| Fasting C‐peptide (nmol/L) | 0.218 ± 0.1087 | 0.225 ± 0.1416 |
| Peak stimulated C‐peptide (nmol/L) | 0.676 ± 0.2708 | 0.675 ± 0.2882 |
| C‐peptide AUC(0‐120) (nmol/L) | 60.381 ± 24.9210 | 59.092 ± 26.243 |
| HbA1c (mmol/mol [%]) | 60 (7.60 ± 1.62) | 50 (7.50 ± 1.37) |
| HbA1c ≥ 7% (N [%]) | 28 (56) | 15 (57.7) |
| Insulin requirement (U/kg/d) | 0.33 ± 0.192 | 0.33 ± 0.198 |
Note. All are means ± SD, unless otherwise specified.
Abbreviations: AUC, area under the curve; BMI, body mass index; GADA, anti‐GAD; IAA, anti‐insulin; IA‐2A, anti‐IA‐2, LDX, ladarixin; ZnT8A, Zinc Transporter 8 antibody.
One patient in each treatment group was randomized slightly after 100 days from the first insulin injection (day 103 and day 106 in the LDX e placebo group, respectively); exemption was granted because of patients being already committed to study participation. Such a delay was not considered to impact trial outcome.
Cockcroft–Gault formula.
3.2. Efficacy outcomes
MMTT‐stimulated C‐peptide AUC(0‐120 min) adjusted for age, sex and baseline C‐peptide value was similar between the groups at 13 weeks (LDX 4.03 nmol/L, 95% CI 3.89‐4.16; and placebo 3.87 nmol/L, 95% CI 3.54‐4.15; Figure 2). The difference was not significant (mean 0.14 nmol/L, 95% CI −0.14 to 0.42; P = .122 ANCOVA; t test, two‐sided P = .33). Specifically, the results of the linear mixed model for the AUC(0‐120 min) throughout the study showed statistically significant effects over time (P < .0001), while the factor treatment (P = .6928) and the interaction treatment by visit (P = .0993) were not statistically significant. Results on the primary outcome were not impacted by including in the analysis the time from first insulin to treatment (P = .2035, ANCOVA), even if the time itself was statistically significant at the .05 level (P = .0497, ANCOVA).
FIGURE 2.
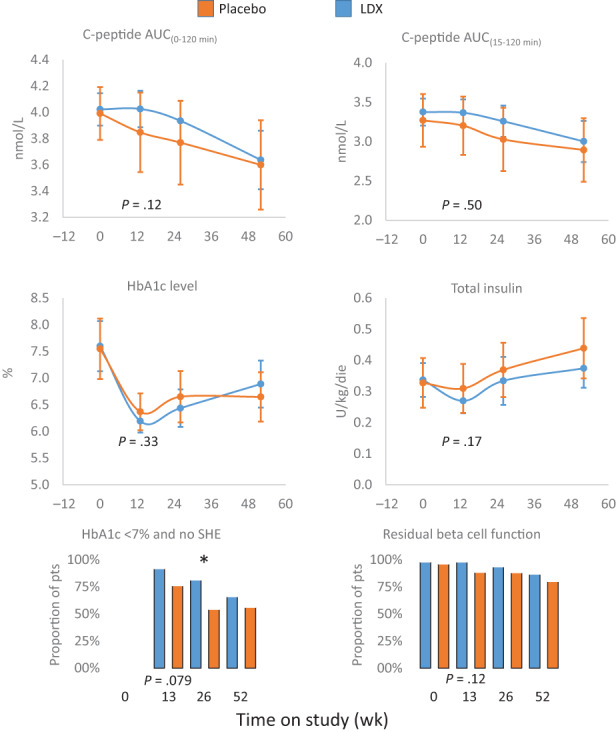
Trial primary and secondary outcomes. Effects of ladarixin (LDX) on 2‐hour area under the curve (AUC) of C‐peptide AUC(0‐120 min), C‐peptide AUC(15‐120 min) above fasting value, HbA1c level, insulin dose, proportion of patients with HbA1c less than 7% and absence of episodes of severe hypoglycaemia (SHE) and proportion of patients maintaining a residual beta cell function (defined as at least one MMTT C‐peptide value ≥0.2 nmol/L). Means (95% CI) or proportions for each treatment group are reported over time. The analysis of covariance model adjusted for age, sex, baseline value and treatment assignment or Fisher′s exact test for categorical independent variables were used to compare the two groups. All P values referring to week 13 are reported in full. MMTT, mixed meal tolerance test. * P < .05
Similarly, the adjusted mean difference between LDX and placebo for C‐peptide AUC(15‐120 min) was not statistically significant throughout the study. The mean (±SD) insulin requirement at screening was 0.325 (±0.1923) IU/kg/day for the LDX group and decreased at week 13 (−0.067 [±0.1774] IU/kg/day); however, an increasing trend was seen at week 26 (−0.011 [±0.2625] IU/kg/day) and week 52 (0.025 [±0.2507] IU/kg/day). A similar profile was seen in the placebo group. The linear mixed model of daily insulin requirement throughout the study showed a statistically significant effect over time (P < .0001), while the factor treatment (P = .3668) and the interaction treatment by visit (P = .7121) were not statistically significant. The adjusted HbA1c mean differences between LDX and placebo were not statistically significant throughout the study. A maximum decrease in HbA1c level was seen at week 13 compared with week 26 and week 52 in both treatment groups. The results of the linear mixed model on HbA1c showed a statistically significant effect over time (P < .0001), while the factor treatment (P = .8988) and the interaction treatment by visit (P = .4588) were not statistically significant. The proportion of patients with HbA1c less than 7% in the absence of SHE is reported as a composite endpoint in Figure 2. The overall mean cumulative SHE/patient occurring from randomization was 0.1 in the LDX group (two patients) and 0.1 in the placebo group (one patient). The comparison between treatment groups for the proportion of patients with HbA1c less than 7% and absence of SHE was statistically significant at week 26 (P = .0248) in favour of the LDX group (LDX = 39 patients [81.3%] vs. placebo = 13 patients [50%]) and a trend was also evident at week 13 (P = .0779). The results at week 26 were also confirmed by a logistic regression model, which included time elapsed from first insulin injection (P = .0087). The proportion of patients maintaining a residual beta cell function throughout the study is also presented in Figure 2. At week 52, 78% of patients maintained a residual beta cell function in the LDX group compared with 76.9% in the placebo group. The comparison between treatment groups was not statistically significant throughout the study.
Predefined subgroup analyses were performed on the efficacy endpoints according to age class (<25 and ≥25 years), fasting C‐peptide (pre‐MMTT) (< median value and ≥ median value) and the number (from one to four) of positive autoantibodies at screening/diagnosis (Figure 3). In patients with fasting C‐peptide at screening of less than 0.205 nmol/L (median value), the difference in C‐peptide AUC(15‐120 min) between LDX (n = 26) and placebo (n = 11) reached statistical significance at week 26 (LDX = 3.22 ± 0.55 vs. placebo = 2.53 ± 1.18, adjusted mean differences 0.6304 [95% CI 0.061‐1.199]; P = .031) (Figure 4). Accordingly, the proportion of patients with HbA1c less than 7% in the absence of SHE was significantly higher at week 26 for patients receiving LDX compared with placebo (LDX = 88.5% vs. placebo = 36.4%; P = .0074). Moreover, clear trends were evident in favour of LDX for HbA1c at week 26 (LDX = 6.3% [95% CI 6.1‐6.5] vs. placebo = 7.01% [95% CI 5.8‐8.1], P = .053) and for the proportion of patients maintaining a residual beta cell function at week 13 (LDX = 100.0% vs. placebo = 81.8%, P = .0826) and week 26 (LDX = 95.8% vs. placebo = 70.0%, P = .0666) (Figure 4). No statistically significant changes were observed between the two treatments in the other subgroups, with the exception of the proportion of patients maintaining a residual beta cell function at week 13 in patients aged 25 years or older, which was higher in the LDX than in the placebo group (LDX = 100% vs. placebo = 81.3%, P = .0345).
FIGURE 3.
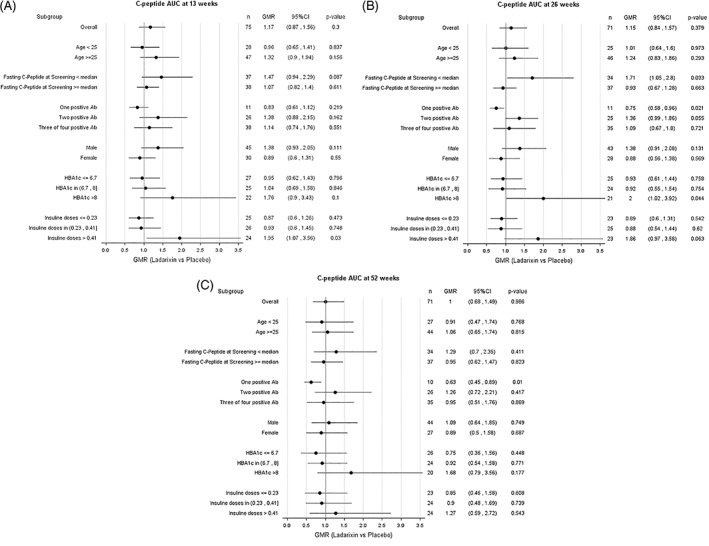
A‐C, Subgroup plot of ratios for the effect of treatment on mean AUC C‐peptide at 13 ± 1 (month 3), 26 ± 2 (month 6) and 52 ± 2 (month 12) weeks from the beginning of treatment. Ratio of geometric means for ladarixin (LDX) versus placebo, with 95% confidence intervals, within subgroups of patients as defined at the baseline. When adjusted for multiple subgroup analyses, there was no significant heterogeneity (test of treatment by subgroup interaction) among subgroups. When considering the subgroup with fasting C‐peptide less than the median value (0.205 nmol/L), the 71% improvement seen with LDX versus control at 26 weeks was nominally significant (P = .033, not adjusted for multiple tests), while it was not significant in the other subgroups or at weeks 13 and 52. Ab, antibody; AUC, area under the curve; GMR, Geometric Mean Ratio
FIGURE 4.
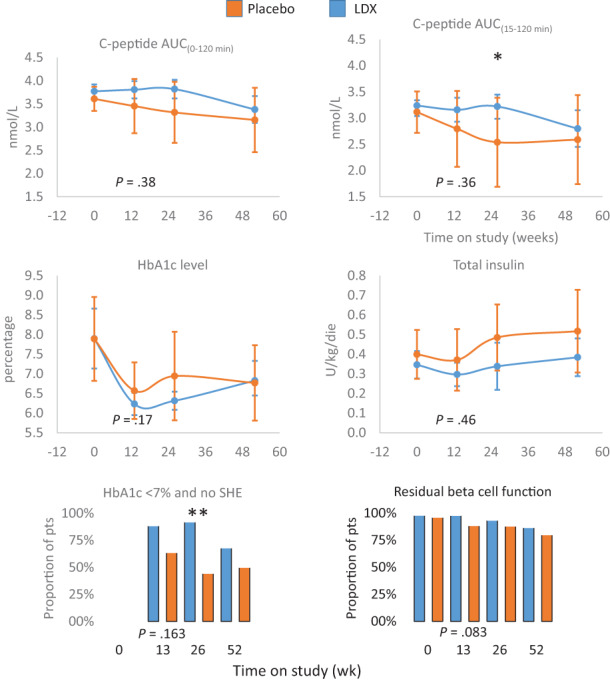
Primary and secondary outcomes in the predefined subgroup with fasting C‐peptide (pre‐MMTT) of less than 0.205 nmol/L (median value). The effects of ladarixin (LDX) on the 2‐hour area under the curve (AUC) of C‐peptide AUC(0‐120 min), C‐peptide AUC(15‐120 min) above fasting value, HbA1c level, insulin dose, proportion of patients with HbA1c less than 7% and absence of episodes of severe hypoglycaemia (SHE) and proportion of patients maintaining a residual beta cell function (defined as at least one MMTT C‐peptide value >0.2 nmol/L). Means (95% CI) or proportions for each treatment group are reported over time. The analysis of covariance model adjusted for age, sex, baseline value and treatment assignment or Fisher′s exact test for categorical independent variables were used to compare the two groups. All P values referring to week 13 are reported in full. MMTT, mixed meal tolerance test. * P < .05, ** P < .001
3.3. Adverse events and safety
Overall, a good safety profile was observed for LDX. It was safe and well tolerated and no clinically relevant safety observations were detected (Table S1). Specifically, no differences between treatment groups were observed for rates, severity and distribution of TEAEs or treatment‐emergent serious adverse events (TESAEs). Thirty‐seven patients (74.0%) in the LDX group and 21 patients (80.77%) in the placebo group reported at least one TEAE during study participation. The majority of the TEAEs reported in the study were considered mild in severity. The most common TEAEs presented by primary “System Organ Classes” (SOCs) were infections and infestations (LDX = 46.0% vs. placebo = 46.2%), followed by gastrointestinal disorders (LDX = 36.0% vs. placebo = 34.6%) and nervous system disorders (LDX = 34.0% vs. placebo = 26.9%). TEAEs were considered related to study treatment (adverse drug reactions [ADRs]) in 20 patients (40.0%) in the LDX group (52 ADRs) and eight patients (30.8%) in the placebo group (17 ADRs). ADRs occurring in 10% or more of patients included dyspepsia (LDX = 16% vs. placebo = 0%) and headache (LDX = 16% vs. placebo = 15.4%). A total of three patients in the LDX group and one patient in the placebo group reported TESAEs, none of which were considered related to the study treatment. TEAEs led to treatment discontinuation in one patient (2%) in the LDX group because of an increase in ALT/AST, and in one patient (3.8%) in the placebo group because of rash (Table S1). No patients died during the study. There were no clinically meaningful changes in mean values from screening to assessment time points for haematology and blood chemistry variables across the treatment groups. Analyses of vital signs did not reveal any clinically relevant differences across the treatment groups.
4. DISCUSSION
Our results showed that a short‐term transient inhibition of the IL‐8 receptors CXCR1/CXCR2 with the allosteric inhibitor LDX 21 did not consistently slow the decline in beta cell function in adults with recent‐onset type 1 diabetes. The use of LDX in type 1 diabetes was proposed on the basis of preclinical evidence, where transient blockade of CXCR1/CXCR2 was effective in preventing the inflammatory damage in the mouse model of multiple low‐dose streptozotocin injections and in preventing and reversing diabetes in NOD mice. 18 In addition to the well‐known limitations of the preclinical models in predicting the success of treatments at the onset of type 1 diabetes in humans, more than one hypothesis can explain the outcome difference between preclinical studies and this human trial. In mouse models, LDX consistently decreased the percentage of pancreas‐infiltrating polymorphonuclear cells and modulated the distribution of various leukocyte populations targeting CXCR2+ leukocyte subpopulations, which are known to be required for the initiation of beta cell disruptive insulitis in both mouse and human type 1 diabetes. 10 , 15 , 16 , 17 , 18 , 22 In support of this, the most extensive anti‐inflammatory changes and the highest efficacy of LDX were observed in 12‐week‐old NOD mice, a disease phase characterized by marked beta cell disruptive insulitis immediately preceding the onset of hyperglycaemia. In this trial, LDX treatment was limited to three cycles of 14 days on/14 days off, in compliance with regulatory requirements based on the preclinical safety data available at the time of trial submission. Consistently, considering that polymorphonuclear cells are short half‐life cells, we chose the end of treatment (13 weeks) as the time to evaluate the primary endpoint. This schedule was probably undersized to keep beta cell‐specific autoimmunity in humans under control. Supporting this hypothesis, in the preclinical model, 14 day of treatment rapidly reverted diabetes in 78% of animals; however, this did not insure any long‐term benefit on glucose levels because the disease recurred, and more quickly if glycaemia was higher at onset. More generally, the inability to identify the time in which the local inflammatory response is at its peak may have contributed to the negative result. The absence of validated biomarkers of islet inflammation may represent a problem for any clinical study targeting pancreatic inflammation in type 1 diabetes, limiting the ability to anticipate trial enrolment and treatment initiation versus the predisease onset phase. In line with this observation, other anti‐inflammatory strategies have been tested in type 1 diabetes patients at the onset of the disease. Findings from small pilot clinical trials suggest that inhibition of IL‐1, 23 TNF‐α 24 or IL‐6 signals might have a beneficial effect in type 1 diabetes, but the results were only partially confirmed in randomized phase 2 trials. 12 , 25 , 26 On the other hand, six immunotherapies mainly targeting the adaptive lymphocyte‐mediated attack of beta cells have been shown to preserve insulin secretion in stage 3 type 1 diabetes (teplizumab, 3 otelixizumab, 4 rituximab, 5 abatacept, 6 low‐dose antithymocyte globulin 7 and alefacept 8 ) and teplizumab have been shown to delay the onset of stage 2 disease. 9 However, the transient nature of the efficacy observed or the associated side effects, or both, have until now prevented the marketing approval of these therapies. In our study, LDX showed an excellent tolerability profile and no safety issues emerged. While the complexity of type 1 diabetes and of its clinical management are clear, further research should not be discouraged in order to attempt to halt disease progression, possibly also considering combination therapies with agents presenting a favourable safety profile. In this context, an orally bioavailable small molecule such as the CXCR1/CXCR2 inhibitor LDX constitutes an opportunity for testing a second generation of disease‐modifying treatment in type 1 diabetes. 27 The study is underpowered to detect a true difference in the subpopulation analysis. Despite this, some transient metabolic benefits were seen in favour of the LDX group, in particular in patients with lower fasting C‐peptide and higher insulin requirement at screening. Further metabolic and immunological studies are ongoing and will allow to better understand the meaning of such findings, but the more pronounced positive effect observed in a population with lower fasting C‐peptide at screening that is also characterized by a severely impaired metabolic function, may suggest the existence of a clinical condition (either a population or a disease stage) responsive to IL‐8 inhibition. Nevertheless, together with the duration of treatment, the study has some other limitations that are consistent with phase 2 trials, like the small number of participants and the limited range of age within the studied population. Moreover, children were not included in the study, because of the early development phase, so the impact in younger ages that may have different progression characteristics is not known.
In conclusion, short‐term transient inhibition of the CXCL8 receptors CXCR1/CXCR2 with the allosteric inhibitor LDX did not consistently slow the decline in beta cell function in newly diagnosed type 1 diabetes patients. The lack of response to LDX in this study suggests that the role of IL‐8 and its receptors CXCR1/2 in type 1 diabetes is complex. In the future, therapeutic interventions targeting IL‐8 and its receptors CXCR1/2 may be the most beneficial, extending exposure to LDX or its use in combination with therapies that synergize with the IL‐8–driven pathways, which are the most important in the pathogenesis of type 1 diabetes.
AUTHOR CONTRIBUTIONS
LP: conceptualization, methodology, formal analysis and writing (original draft); BK: investigation, methodology, review and editing; PG: investigation, review and editing; TL: investigation, review and editing; EB: investigation, review and editing. LR: investigation, review and editing. PP: investigation, review and editing; FG: investigation, review and editing; EC: investigation, review and editing; LD: conceptualization and methodology; GG: formal analysis; PAR: writing review and editing; ARM: writing review and editing; FM: resources; and MA: resources. LP is the guarantor of this work and, as such, had full access to all the data presented in the study and takes responsibility for their integrity and for the accuracy of data analysis. The final version of the manuscript was read and approved by all the named authors.
CONFLICT OF INTEREST
LD, GG, PAR, ARM, FM and MA are employees of Dompé farmaceutici spa. LP served as a consultant for Dompé farmaceutici spa (Milan, Italy). BK, PG, TL, EB, LR, PP, FG and EC have no conflict of interest to declare
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14770.
Supporting information
TABLE S1 Safety results
FIGURE S1. Study design
ACKNOWLEDGEMENTS
This work was funded by Dompé farmaceutici spa (Milan, Italy). Each study centre received research support and compensation from Dompé farmaceutici spa (Milan, Italy) to conduct the study and collect data. No data outside those published in this manuscript and the material presented in the supporting information are available for sharing.
Piemonti L, Keymeulen B, Gillard P, et al. Ladarixin, an inhibitor of the interleukin‐8 receptors CXCR1 and CXCR2, in new‐onset type 1 diabetes: A multicentre, randomized, double‐blind, placebo‐controlled trial. Diabetes Obes Metab. 2022;24(9):1840‐1849. doi: 10.1111/dom.14770
Funding information This work was funded by Dompé farmaceutici spa (Milan, Italy).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Sauder KA, Stafford JM, Mayer‐Davis EJ, et al. Co‐occurrence of early diabetes‐related complications in adolescents and young adults with type 1 diabetes: an observational cohort study. Lancet Child Adolesc Health. 2019;3(1):35‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perkins BA, Lovblom LE, Lanctot SO, Lamb K, Cherney DZI. Discoveries from the study of longstanding type 1 diabetes. Diabetologia. 2021;64(6):1189‐1200. [DOI] [PubMed] [Google Scholar]
- 3. Herold KC, Gitelman SE, Willi SM, et al. Teplizumab treatment may improve C‐peptide responses in participants with type 1 diabetes after the new‐onset period: a randomised controlled trial. Diabetologia. 2013;56(2):391‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3‐antibody therapy in new‐onset type 1 diabetes. N Engl J Med. 2005;352(25):2598‐2608. [DOI] [PubMed] [Google Scholar]
- 5. Pescovitz MD, Greenbaum CJ, Krause‐Steinrauf H, et al. Rituximab, B‐lymphocyte depletion, and preservation of beta‐cell function. N Engl J Med. 2009;361(22):2143‐2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orban T, Bundy B, Becker DJ, et al. Co‐stimulation modulation with abatacept in patients with recent‐onset type 1 diabetes: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2011;378(9789):412‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haller MJ, Long SA, Blanchfield JL, et al. Low‐dose anti‐thymocyte globulin preserves C‐peptide, reduces HbA1c, and increases regulatory to conventional T‐cell ratios in new‐onset type 1 diabetes: two‐year clinical trial data. Diabetes. 2019;68(6):1267‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rigby MR, DiMeglio LA, Rendell MS, et al. Targeting of memory T cells with alefacept in new‐onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013;1(4):284‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herold KC, Bundy BN, Long SA, et al. An anti‐CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. 2019;381(7):603‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Citro A, Dugnani E, Campo F, Piemonti L. Innate immunity mediated inflammation and beta cell function: neighbors or enemies? Front Endocrinol. 2020;11:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mastrandrea L, Yu J, Behrens T, et al. Etanercept treatment in children with new‐onset type 1 diabetes: pilot randomized, placebo‐controlled, double‐blind study. Diabetes Care. 2009;32(7):1244‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moran A, Bundy B, Becker DJ, et al. Interleukin‐1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double‐blind, placebo‐controlled trials. Lancet. 2013;381(9881):1905‐1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hundhausen C, Roth A, Whalen E, et al. Enhanced T cell responses to IL‐6 in type 1 diabetes are associated with early clinical disease and increased IL‐6 receptor expression. Sci Transl Med. 2016;8(356):356ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang J, Xiao Y, Xu A, Zhou Z. Neutrophils in type 1 diabetes. J Diabetes Investig. 2016;7(5):652‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Valle A, Giamporcaro GM, Scavini M, et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes. 2013;62(6):2072‐2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vecchio F, Buono NL, Stabilini A, et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight. 2018;3(18):e122146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Diana J, Simoni Y, Furio L, et al. Crosstalk between neutrophils, B‐1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. 2013;19(1):65‐73. [DOI] [PubMed] [Google Scholar]
- 18. Citro A, Valle A, Cantarelli E, et al. CXCR1/2 inhibition blocks and reverses type 1 diabetes in mice. Diabetes. 2015;64(4):1329‐1340. [DOI] [PubMed] [Google Scholar]
- 19. Bertini R, Barcelos L, Beccari A, et al. Receptor binding mode and pharmacological characterization of a potent and selective dual CXCR1/CXCR2 non‐competitive allosteric inhibitor. Br J Pharmacol. 2012;165(2):436‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lachin JM, McGee PL, Greenbaum CJ, et al. Sample size requirements for studies of treatment effects on beta‐cell function in newly diagnosed type 1 diabetes. PLoS One. 2011;6(11):e26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Allegretti M, Cesta MC, Garin A, Proudfoot AE. Current status of chemokine receptor inhibitors in development. Immunol Lett. 2012;145(1‐2):68‐78. [DOI] [PubMed] [Google Scholar]
- 22. Niu S, Bian Z, Tremblay A, et al. Broad infiltration of macrophages leads to a proinflammatory state in streptozotocin‐induced hyperglycemic mice. J Immunol. 2016;197(8):3293‐3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sumpter KM, Adhikari S, Grishman EK, White PC. Preliminary studies related to anti‐interleukin‐1β therapy in children with newly diagnosed type 1 diabetes. Pediatr Diabetes. 2011;12(7):656‐667. [DOI] [PubMed] [Google Scholar]
- 24. Peters MJ. Etanercept treatment in children with new‐onset type 1 diabetes: pilot randomized, placebo‐controlled, double‐blind study: response to Mastrandrea et al. Diabetes Care. 2009;32(12):e153 author reply e154. [DOI] [PubMed] [Google Scholar]
- 25. Quattrin T, Haller MJ, Steck AK, et al. Golimumab and beta‐cell function in youth with new‐onset type 1 diabetes. N Engl J Med. 2020;383(21):2007‐2017. [DOI] [PubMed] [Google Scholar]
- 26. Greenbaum CJ, Serti E, Lambert K, et al. IL‐6 receptor blockade does not slow β cell loss in new‐onset type 1 diabetes. JCI Insight. 2021;6(21):e150074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. von Herrath M, Bain SC, Bode B, et al. Anti‐interleukin‐21 antibody and liraglutide for the preservation of β‐cell function in adults with recent‐onset type 1 diabetes: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2021;9(4):212‐224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Safety results
FIGURE S1. Study design
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.