

Abstract
Objectives
To present the very long‐term follow up of patients with cobalamin A (cblA) deficiency.
Methods
A retrospective case series of adult (>16 years) patients with molecular or enzymatic diagnosis of cblA deficiency.
Results
We included 23 patients (mean age: 27 ± 7.6 years; mean follow‐up: 24.9 ± 7.6 years). Disease onset was mostly pediatric (78% < 1 year, median = 4 months) with acute neurologic deterioration (65%). Eight patients presented with chronic symptoms, and one had an adult‐onset mild cblA deficiency. Most of the patients (61%) were initially classified as vitamin B12‐unresponsive methylmalonic aciduria (MMA); in vitro B12 responsiveness was subsequently found in all the tested patients (n = 13). Initial management consisted of protein restriction (57%), B12 (17%), or both (26%). The main long‐term problems were intellectual disability (39%) and renal failure (30%). However, 56.5% of the patients were living independently. Intellectual disability was equally distributed among the initial treatment groups, while renal failure (moderate and beginning at the age of 38 years) was present in only one out of seven patients initially treated with B12.
Conclusions
We provide a detailed picture of the long‐term outcome of a series of adult cblA patients, mostly diagnosed before the enzymatic and molecular era. We confirm that about 35% of the patients do not present acutely, underlining the importance of measuring MMA in any case of unexplained chronic renal failure, intellectual disability, or growth delay. In addition, we describe a patient with a milder adult‐onset form. Early B12 supplementation seems to protect from severe renal insufficiency.
Keywords: Adult, cobalamin A deficiency, kidney disease, late‐onset, mental retardation, Methylmalonic aciduria, vitamin B12
Synopsis.
CblA deficiency can present as a chronic disease (35% in this series) underlining the importance of measuring MMA in any case of unexplained chronic renal failure, intellectual disability, or growth delay. Less severe adult‐onset forms are possible even if extremely rare. Early B12 supplementation seems to partially protect from severe renal insufficiency and should be given to every patient with cblA deficiency.
1. INTRODUCTION
Cobalamin A deficiency (MIM#251100) is a rare genetic cause of isolated methylmalonic aciduria due to mutations in the MMAA gene (MIM*607481). MMAA is a GTPase which exerts a role of chaperone of the methylmalonyl‐CoA mutase (MCM) by preventing the oxidation of its cofactor adenosylcobalamin (AdoCbl) during the catalytic cycles and by promoting the replacement of the inactivated cofactor by a new unoxidized cofactor. 1 , 2 , 3 , 4
Other causes of isolated methylmalonic acid (MMA) accumulation are deficiency of the MCM apoenzyme (MMUT gene), 5 responsible for the AdoCbl‐dependent conversion of L‐methylmalonyl‐CoA to succinyl‐CoA, defects in its cofactor AdoCbl (including cblA disease but also cblB (MMAB gene) and cblD disease (MMADHC gene)), and deficiency of the enzyme methylmalonyl‐CoA epimerase. 6 , 7 , 8
Individuals affected with cblA typically present with severe disease in infancy or early childhood and are prone to potentially life threatening metabolic decompensation with severe acidosis. 9 Although initial papers presented series including cblA deficient subjects and patients with the entire spectrum of isolated MMA 9 , 10 , 11 , 12 , 13 , 14 or organic acidurias, 15 it soon became clear that cblA had a unique evolution and that precise characterization of patients with MMA had important diagnostic, therapeutic and prognostic implications. Notably, patients with cblA deficiency are reported to have a slightly later disease onset (infantile, generally in the first months of life, instead of neonatal), to be responsive to B12 treatment and to have a more attenuated phenotype than cblB and MMUT patients. 16 Detailed data are available concerning the molecular and biological characterization of cblA patients 10 , 17 , 18 , 19 and their early outcome, 9 but more clinical data is needed on the long‐term outcome, management and possible complications in these patients. 12 , 20
We present here detailed clinical data and very long‐term outcome on 23 adult patients with cblA deficiency.
2. METHODS
We recruited adult (aged more than 16 years) patients with cblA deficiency who were being followed up at the Charles Dent Metabolic Unit, UCLH in London (seven patients followed by EM and RL) and by members of the French Society for Inherited Metabolic Diseases (SFEIM) (seven patients followed in Lyon by AF and NGF; six patients followed at Necker Hospital, in Paris by AS, AB, and PDL; one patient followed in Montpellier by CM; one patient followed at Debré Hospital in Paris by MS; one patient followed in Marseille by AC and BC). We retrospectively collected clinical, dietary, radiological, genetic and laboratory parameters related to these patients. For dietary information, we considered as a strict low protein diet a natural protein intake equal to or less than 0.8 g/kg/day (the Recommended Dietary Allowance, RDA for adults); we considered as a mild protein‐restricted diet a natural protein intake between 0.8 g/kg/day and 1.2–1.5 g/kg/day, and this often corresponded to the protein intake of patients declaring a “self ‐restricted protein diet.” The patients included in the study consented to collection and sharing of their data for scientific purposes. The protocol was approved by the IRB of the University Hospital of Montpellier (IRB‐MTP_2022_01_202201028).
3. RESULTS
We identified a total of 28 adult English and French patients with cblA deficiency; however detailed clinical and laboratory parameters were only available for 23 patients from 19 families, and only these have been included in the study. Three English patients were previously included in a paper reporting adult cases with organic acidemia, 20 but neither enzyme activity nor genetic analyses were available at that time. Six French patients were reported in a paper on the follow‐up of B12‐unresponsive MMA 14 or in other papers.15, 21
Demographic and clinical data are presented in Table 1. The mean age at onset was 1.6 ± 5.3 years (median 4 months; range 2 days–25 years); the mean age at the diagnosis of isolated MMA was 4.7 ± 9.8 years (median 6 months; range 2 days–35 years), while the mean age at cblA diagnosis (with molecular and/or enzymatic confirmation) 22 was 16.4 ± 11.8 years (median 17.5 years; range 2 days–35 years). Therefore, in this series the confirmation of the diagnosis of cblA deficiency was obtained on average about 15 years after disease onset. The mean age at the last visit was 27 ± 7.6 years (range16–40 years), with a mean follow‐up of 24.9 ± 7.6 years (range 11–40 years).
TABLE 1.
Adult cblA defective patients: demographic, clinical, and molecular data
| N/Family | Year of birth/sex | Onset Age | Dia MMA age | Dia cblA age | FU, y | Presenting symptoms | Initial diagnosis | Mutation | Consanguinity | Propionate incorporation (fibroblasts; variable methods used); °°ref range for proprionate incorporation: 35–60 nmol/mg/16 h 31 | MCM activity (+ AdoCbl) 32 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ‐OHCbl | + OHCbl | |||||||||||
| a , e 1/Lond01 | 1980/M | 3 m | 6 m | NA | 40 | Acute encephalopathy | MMA B12‐U | c.433C > T p.Arg145* c.433C > T p.Arg145* | NA | NA | NA | NA |
| 2/Lond02 | 1994/M | 2 d | 2 y | 16 | 26 | Acute encephalopathy | MMA B12‐U | c.433C > T p.Arg145* c.434G > A p.Arg145Gln | No | NA | NA | NA |
| e 3/Lond04 | 1993/M | 5 m | 11 m | NA | 27 | Acute encephalopathy | MMA B12‐R | c.433C > T p.Arg145* c.586C > T p.Arg196* | No | NA | NA | NA |
| 4/Lond05 | 1984/F | 25 y | 30 y | 30 y | 11 | Muscle pain, chronic fatigue, headache | MMA B12‐R | c.791 T > C p.Leu264Pro c.‐66 + 2 T > C p.? | No | 1.61 nmol/mg prot/16 h | 11.7 nmol/mg prot/16 h | N |
| 5/Lond05 | 1981/M | 10 m | 35 y | 35 y | 38 | Psychomotor delay (after measles) | MMA B12‐R |
c.791 T > C p.Leu264Pro c.‐66 + 2 T > C p.? |
No | NA | NA | NA |
| a , e 6/Lond06 | 1987/F | 6 m | 7 m | 19 y | 33 | Acute encephalopathy | MMA B12‐U | c.728C > T p.Thr243Ile c.728C > T p.Thr243Ile | No | NA | NA | NA |
| e 7/Lond07 | 1989/F | 3 m | 6 m | 21 y | 31 | Acute encephalopathy | MMA B12‐U | NA | NA | 0.90 nmol/mg prot/16 h | 4.7 nmol/mg prot/16 h | N |
| b , c 8/Neck01 | 1990/M | 8 m | 8 m | 19 y | 28 | Acute encephalopathy | MMA B12‐U | c.828A > T p.Lys276Asn c.828A > T p.Lys276Asn | Yes | 6.8 nmol/mg prot/16 h °° | 16.9 nmol/mg prot/16 h | N |
| c 9/Neck02 | 1984/M | 2 y | 14 y | 29 y | 31 | Acute encephalopathy (2 y); Growth delay and CKD (14y) | MMA B12‐U |
(c.31 + 1_c.32–1)_(c.733 + 1_c.734‐1)del (c.31 + 1_c.32–1)_(c.733 + 1_c.734‐1)del |
NA | 3.5 nmol/mg prot/16 h °° | 20.1 nmol/mg prot/16 h | N |
| c , d 10/Neck02 | 1979/F | NA | 18 y | 34 y | NA | Early CKD | MMA B12‐R |
(c.31 + 1_c.32–1)_(c.733 + 1_c.734‐1)del (c.31 + 1_c.32–1)_(c.733 + 1_c.734‐1)del |
NA | 3.9 nmol/mg prot/16 h °° | 40.3 nmol/mg prot/16 h | N |
| b , c 11/Neck03 | 1992/M | 14 m | 2 y | NA | 23 | Acute encephalopathy | MMA B12‐U | c.593_596delCTGA p.Thr198Sfs*6 c.595_599delGAGTT p.E199IfsX14 | No | 3.2 nmol/mg prot/16 h °° | 17.1 nmol/mg prot/16 h | N |
| b , c 12/Neck04 | 1992/F | 3 m | 3 m | NA | 26 | Psychomotor delay and hypotonia | MMA B12‐U |
c.433C > T p.Arg145* not found |
No | 4.7 nmol/mg prot/16 h °° | 35.1 nmol/mg prot/16 h | N |
| b , c 13/Neck05 | 2002/M | 3 m | 6 m | 12 y | 16 | Weight stagnation, hypotonia, subacute encephalopathy | MMA B12‐U |
c.64C > 7 p.Arg22* c.64C > 7 p.Arg22* |
Yes | 4.7 nmol/mg prot/16 h °° | 10.6 nmol/mg prot/16 h | N |
| 14/Lyo01 | 1985/F | NA | 45 d | 2 y | 32 | Weight stagnation | MMA B12‐R | c.586C > T p.Ala196* c.586C > T p.Ala196* | Yes | 6.3 nmol/mg prot/16 h °° | 10.3 nmol/mg prot/16 h | N |
| 15/Lyo02 | 1985/F | 4 m | 5 m | NA | 32 | Acute encephalopathy | MMA B12‐U | c.586C > T p.Ala196* c.586C > T p.Ala196* | Yes | 4.6 nmol/mg prot/16 h °° | 9.7 nmol/mg prot/16 h | N |
| 16/Lyo03 | 2002/F | 2 d | 2 d | 2 d | 16 | Acute encephalopathy | MMA B12‐R |
c.455delC p.Pro152Leufs9 c.455delC p.Pro152Leufs*9 |
Yes | NA | NA | NA |
| 17/Lyo03 | 1994/F | 2 d | 4 d | 2 y | 22 | Acute encephalopathy | MMA B12‐R |
c.455delC p.Pro152Leufs*9 c.455delC p.Pro152Leufs*9 |
Yes | NA | NA | NA |
| 18/Lyo02 | 1981/M | 9 d | 12 d | NA | 20 | Acute encephalopathy | MMA B12‐U |
c.586C > T p.Ala196* c.586C > T p.Ala196* |
Yes | NA | NA | NA |
| 19/Lyo04 | 1995/F | 8 m | 9 m | 12 y | 22 | Acute encephalopathy | MMA B12‐U | c.1162C > T p.Gln388* c.1162C > T p.Gln388* | Yes | 19 nmol/mg prot/16 h °° | 34.9 nmol/mg prot/16 h | N |
| 20/Lyo05 | 1998/M | 4 m | 4 m | NA | 19 | Acute encephalopathy | MMA B12‐U |
c.521 T > G p.Leu174* c.521 T > G p.Leu174* |
Yes | 6.9 nmol/mg prot/16 h °° | 18.6 nmol/mg prot/16 h | N |
| e 21/Mars01 | 2002/M | 21 d | 1 m | 10 y | 16 | Acute encephalopathy | MMA B12‐U |
c.433C > T p.Arg145* c.589A > G p.Met197Val |
No | 5.3 nmol/mg prot/16 h °° | 10.1 nmol/mg prot/16 h | N |
| 22/Debr01 | 2000/M | 4 m | 4 m | 4 m | 18 | Acute encephalopathy | MMA B12‐R |
c.586C > T p.Ala196* c.586C > T p.Ala196* |
NA | NA | NA | NA |
| 23/ Mont01 | 1998/F | 1 m | 9 m | 22 y | 22 | Psychomotor delay; growth failure; subacute encephalopathy | MMA B12‐R |
c.616_619delGCAT p.Ala206Thrfs*11 c.433C > T p.Arg145* |
No | NA | NA | NA |
Abbreviations: AdoCbl, adenosylcobalamin; B12‐U, vitamin B12‐unrensposive; B12‐R, vitamin B12‐responsive; d, day; Dia, diagnosis; FU, follow up; m, month; MMA, methylmalonic aciduria; MCM, methylmalonil‐CoA mutase enzyme; OHCbl, hydroxoCobalamin; y, year.
previously reported. 20
previously reported. 14
previously reported. 15
previously reported. 21
included in the EIMD (European Registry and Network for Intoxication type Metabolic Diseases).
In most of the patients the symptom leading to diagnosis was acute neurologic deterioration (15/23, 65%). The other patients (8/23, 35%) presented with chronic problems: late onset chronic muscular pain, headache, and fatigue after a flu‐like episode (patient 5/Lon05); chronic growth delay and renal insufficiency (patient 9/Neck02); early onset chronic renal insufficiency in the context of a family history of cblA deficiency (patient 10/Neck02); early onset psychomotor retardation (patient 12/Neck04; patient 8/Lond05); early onset failure to thrive (patient 13/Lyo01); and early onset failure to thrive with psychomotor regression (patient 21/Neck05; patient 23/Mont01).
The analysis of the molecular data (Table 1) showed the presence of 15 different variants: 5 missense mutations; 5 nonsense mutations; 3 frameshifts; 1 possible splice‐site defect (n = 2); and 1 deletion. Some mutations were recurrent, and the most frequent were two nonsense mutations: c.586C>T p.Ala196*, accounting for 9 alleles (21%), and c.433C>T p.Arg145*, accounting for 7 alleles (16%). Therefore, most of the patients (91%) had at least one severe mutation (nonsense, frameshift, splice ‐site) or a deletion, while only two patients harbored two missense mutations. Information about consanguinity was available for 18 out of 23 patients: 9 of them (50%) were issued from consanguineous parents.
Interestingly, before the molecular and enzymatic era, only 9/23 patients (39%) were clinically diagnosed as B12‐responsive MMA, while 14/23 patients (61%) were classified as B12‐unresponsive MMA. We have no details about how B12‐responsiveness was evaluated, but all the patients tested for propionate incorporation in cultured fibroblasts (n = 13) showed a variable increase in propionate incorporation after administration of B12, including 10 patients initially clinically classified has having vitamin B12‐unresponsive MMA; the functional in vitro test was sometimes only performed several years after disease onset and diagnosis.
Details of initial treatment are presented in Table 2. In the case of five patients, B12 was initially given in addition to protein‐restricted diet, but subsequently rapidly stopped after the clinical diagnosis of B12‐unresponsive MMA; therefore, we considered that these patients did not receive B12 treatment and had only protein‐restricted diet. As a whole, the initial treatment was quite variable but all the patients received either a protein restricted diet (12/23, 52%), B12 supplementation (3/23, 13%), or both (5/23, 22%). Additional medications included L‐ carnitine, neomycin and metronidazole. Three patients (13%) had an early‐onset disease but a late‐diagnosis, meaning that disease specific treatment was begun only many years after disease‐onset (this is the group considered as without treatment in Figure 1).
TABLE 2.
Adult cblA defective patients: initial and current treatments and methylmalonic acid levels
| N/Family | Year of birth/sex | Initial treatment (at diagnosis of MMA) | Current treatment (protein/B12) (year of onset if treatment is different from the initial one) | Current treatment: Other supplements | Current treatment: Other drugs | Metabolic decompensation after the first one | Initial/current MMA a. plasma (0–0.28 μmol/L |
|---|---|---|---|---|---|---|---|
| a , e 1/Lond01 | 1980/M | B12 IM | OHCbl IM 1 mg twice/w; no protein restriction. | B9, Ca++/vit D | aripiprazole, allopurinol | A few (childhood) | NA/293 |
| e2/Lond02 | 1994/M | Low protein; L‐Carnitine | Low Protein: 0.7–0.9 g/kg/d; CNCbl PO 2.5 mg/d (2010) | Ca++/vit D, L‐Carnitine, Phlexy‐vits | Carbamazepine; midazolam | No more | NA/56 |
| e3/LOnd04 | 1993/M | Low protein; B12 IM | OHCbl IM 1 mg/d; protein self ‐restriction: 1.2 g/Kg/d (2015) | No | No | No more | NA/106 |
| 4/Lond05 | 1984/F | B12 IM (late disease onset) | OHCbl IM 1 mg/d | L‐Carnitine | NA | Never | 11.6/8 |
| 5/Lond 05 | 1981/M | B12 IM (late diagnosis) | OHCbl IM 1 mg/d | NA | NA | Never | 41.3/21 |
| a , e 6/Lond06 | 1987/F | Low protein (mild) | OHCbl IM 1 mg/4w (2006); protein self‐restriction | Ca++/vit D, Flaxseed oil; cod liver oil; iron | No | No more | NA/21 |
| a , e 7/Lond07 | 1989/F | Low protein | OHCbl IM 1 mg/8w + CNCbl PO 5 mg/d (2009); protein self‐restriction | Ca++/vit D, L‐Carnitine, Phlexy‐vits, iron | No | A few; the last at the age of 14 | NA/63 |
| b , c 8/Neck01 | 1990/M | Low protein; B12 rapidly discontinued; L‐carnitine; Neomycin; metronidazole | OHCbl IM 1 mg/w (2009); low protein: 0.7 g/Kg/d | Ca++/vit D, L‐Carnitine, Phlexy‐vits | Metronidazole | No more | 500/101 |
| c9/Neck02 | 1984/M | Low protein; L‐carnitin (late‐diagnosis) | OHCbl IM 1 mg/w (2010); low protein: 0.5 g/Kg/d | B9, Ca++/vit D, L‐Carnitine, Vitaflo MMA/PA; iron | Anti‐AH | A few; the last at the age of 18 | NA/78 |
| c , d 10/Neck02 | 1979/F | B12 IM; low protein (late diagnosis) | B12 PO 1 mg/d; protein: 1 g/Kg/d | Ca++/vit D, L‐Carnitine, iron | Azathioprine; Tacrolimus | Never | NA/78 |
| b , c 11/Neck03 | 1992/M | Low protein; L‐carnitine | OHCbl IM 1 mg/w (2012); low protein: 0.5 g/Kg/d | Ca++/vit D, L‐Carnitine, Phlexyvit | No | No more | NA/46 |
| b , c 12/Neck04 | 1992/F | Low protein; metronidazole; L‐carnitine | OHCbl IM 1 mg/w (2013); protein: 0.9 g/Kg/d | Ca++/vit D, L‐Carnitine, Phlexyvit | No | A few; the last at the age of 10 | NA/30 |
| b , c 13/Neck05 | 2002/M | Low protein; L‐carnitine | CNCbl PO (2017); low protein: 0.7 g/Kg/d | Vit D, L‐Carnitine, Phlexyvit | NA | A few; the last at the age of 16 | NA/70 |
| 14/Lyo01 | 1985/F | Low protein: B12 IM; L‐carnitine | OHCbl IM 10 mg/w; animal protein self‐restriction | Ca++/vit D, L‐Carnitine, iron | No | Never | 8,3/14.4 |
| 15/Lyo02 | 1985/F | Low protein; B12 rapidly discontinued | CnCbl PO 1 mg/w (2011); protein restriction | Ca++/vit D, L‐Carnitine, iron, fruitivits; duocal | Darbepoietin alpha, Anti‐AH; metronidazole; Allopurinol; | A few; the last at the age of 26 | NA/857 |
| 16/Lyo03 | 2002/F | B12 IM; L‐carnitine | OHCbl IM 5 mg/w (2002); protein self‐ restriction | Ca++/vit D, L‐Carnitine | No | No more | 196/17.6 |
| 17/Lyo03 | 1994/F | B12 IM; low protein | OHCbl IM 5 mg/w (1994); protein restriction | Ca++/vit D, L‐Carnitine | Risperidone; Cyamemazine; Valproic Acid | No more | 1585/10.5 |
| 18/Lyo02 | 1981/M | Low protein; B12 discontinued; hemofiltration | Protein restriction: 1,2–1,5 g/kg/d | Ca++/vit D, L‐Carnitine | Allopurinol; anti‐AH; metronidazole | At least five; the last at 20 (hemofiltration) | NA/94 |
| 19/Lyo04 | 1995/F | Low protein; B12 IM rapidly discontinued (1998) | Low protein | L‐Carnitine, Phlexy‐vit | No | About six; the last at the age of 14 | 284/54 |
| 20/Lyo05 | 1998/M | Low protein; B12 IM rapidly discontinued (1998) | Low protein | Ca++/vit D, L‐Carnitine | Metronidazole; Methylphenidate. | No more | 107/60 |
| e 21/Mars01 | 2002/M | Low protein (strict) | OHCbl PO (2012) 4 mg/w; low protein (moderate) | L‐Carnitine, Phlexy‐vit; iron; MMA/PA express: | No | No more | NA/30.1 |
| 22/Debr01 | 2000/M | Low protein (mild); OHCbl PO (IM during acute decompensation); L‐carnitine | OHCbl PO 1 mg/d (2000); vegetarian diet | L‐Carnitine; iron; vit D | Metronidazole | Only one more | NA/176 |
| 23/Montp01 | 1998/F | B12 IM, O; low protein (mild) | OHCbl IM 5 mg twice/w and OHCbl PO 1 mg/d; protein: 2 g/kg/d mainly vegetarian | Folic acid; iron; Ca++/vit D | No | About 1/month; the last at the age of 14 | NA/18.1 |
Abbreviations: AH, arterial hypertension; CNCbl, cyanocobalamin; d, day; IM, intramuscular; MMA, methylmalonic aciduria; MMA a, methylmalonic acid; OHCbl, hydroxocobalamin; PO, oral; w, week.
previously reported. 20
previously reported. 14
previously reported. 15
previously reported. 21
included in the EIMD (European Registry and Network for Intoxication type Metabolic Disease).
FIGURE 1.
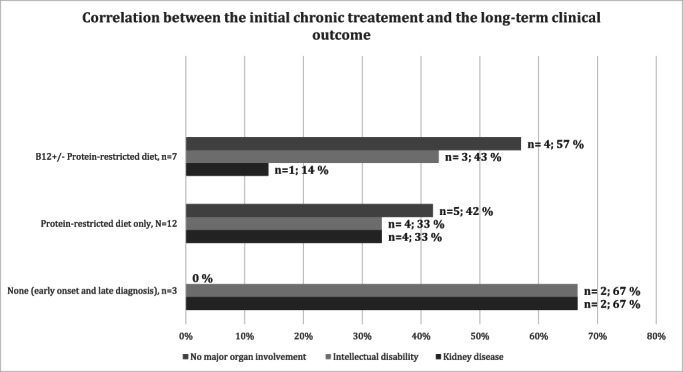
Correlation between the initial chronic treatment and the long‐term clinical outcome. The patients who received B12 (± protein restricted diet) are compared with the patients who received only a protein restricted diet or no treatment at all (patient with early onset and late diagnosis). Total n = 22/23: Patient #4 Lond 05 was not included because of adult‐onset disease). The patients without treatment had the worst outcome; intellectual disability was found in about 30%–40% of the patients, independently of the initial treatment modality (B12, protein restriction); instead, only one case (14%) of moderate and late‐onset renal failure was found in patients treated early with B12 as compared to 33% with chronic kidney disease in the group who did not initially receive B12 supplementation
Data about current treatment (Table 2) shows that most of the patients are taking B12 and a protein restricted or protein self‐limited diet (n = 16/23, 70%); only four patients (17%) are on B12 only; and three patients (13%) are on a protein‐restricted diet only. In total, 87% of the patients currently take B12 compared to 43% who were started on B12 at diagnosis. Initially, B12 was given mainly by intramuscular injection but at the last treatment one patient (10/Neck02) switched to oral B12 and MMA remained stable after that (data not shown); one patient (22/Debr 01) had since the beginning chronic oral B12 but IM B12 was administered in case of acute decompensation; four patients (2/Lond02; 13/Neck05; 15/Lyo02; 21/Mars01) did not initially have B12 and when B12 was introduced, at an adult age, an oral route of administration was preferred; three patients (2/Lond02; 7/Lond07; 23/Montp01) took both IM and oral B12. Dose and frequency of B12 administration varied greatly among patients (details of initial and on‐going treatments and supplementations are provided in Table 2).
Available baseline and post‐treatment values of plasma MMA are reported in Table 2 and showed great variability. The evolution of the disease was also very variable with subsequent episodes of metabolic decompensation in 10 patients (44%) (in general, less than 10 episodes lifelong and mostly during childhood/adolescence) and no more decompensations after the initial presentation in nine patients (39%); four patients (17%) had a sub‐acute presentation and never experienced an acute decompensation.
Social and clinical outcomes are presented in Table 3. Looking at educational status (data not available for one patient), only three patients had university degrees; nine patients were in mainstream education without support; four patients were in mainstream education with support; six patients required special school due to learning difficulties or autistic spectrum features. Thirteen patients (56.5%) are completely self‐caring and 10 (43.5%) need variable degrees of support, from residential care to living with family. Ten patients (45%) are working, two of them in sheltered work; five patients (23%) are students; seven patients (32%) are not employed (information not available for one patient).
TABLE 3.
Adult cblA defective patients: social status, educational, cognitive, and clinical outcomes
| N/Family | Psycho‐motor development | Studies/special need | Work | Housing | H, cm | W, kg | BMI | Neurol. examination | CRF (GFR ml/min/1.73sqm) | Hyperuricaemia | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a , e 1/Lond01 | N. Autistic tracts, Behavioral problems | Special school (autism) | No remunerate activity | Dependent. Parents | 184 | 90 | 26.7 | UL tremor. Mild left UL bradykinesia | Moderate (GFR = 47) | Yes | No |
| 2 e /Lond02 | Delayed; autistic tracts | NA | Not working | Dependent. Parents | 152 | 51 | 22.1 | N; epilepsy | No (GFR > 90) | No | Osteoporosis |
| 3 e /Lond04 | N | College degree | Sheltered work | Partially independent. Parents | 190 | 94 | 26 | Walking difficulties. Dystonia |
No (GFR > 90) |
No | No |
| 4/Lond05 | N | University degree/no help | Remunerate working | Autonomous. Married | NA | 52 | NA | N | No (GFR > 90) | No | No |
| 5/Lond05 | Delayed (mild) | At mainstream school up until the age of 11; special school afterward | Part‐time kitchen porter | Dependent. Parents | 166 | 75 | 27 | Bradykinesia; incoordination |
No (GFR > 90) |
NA | No |
| a , e 6/Lond06 | N | High school with support | Teacher | Autonomous | 153 | 64 | 27.1 | N | No (GFR = 75) | No | Osteopenia |
| a , e 7/Lond07 | Delayed (moderate to severe) | Mainstream school/ help | Not working | Dependent; social and family help | 157 | 71 | 28.5 | N | No (GFR > 90) | No | Vit D, iron, and B12 deficiency; osteopenia |
| b , c 8/Neck01 | N | NA/no help | Mechanic | Autonomous | 158 | 56 | 22.4 | N | Moderate (GFR = 45) | No | Osteoporosis |
| 9 c /Neck02 | Delayed (mild) | Special school | Cook | Live in “CAT”; dependent | 166 | 54 | 19.6 | N | Severe (GFR = 23) | Yes | NA |
| c , d 10/Neck02 | N | N | Nurse | Autonomous | 160 | 52 | 20.3 | N | Severe (kidney transplant in 2011) | No | Osteopenia |
| b , c 11/Neck03 | N | Sale professional certificate/ no help | Interim work | Autonomous | 174 | 66 | 21.8 | NA | Moderate (GFR = 53) | Yes | NA |
| b , c 12/Neck04 | N | Professional certificate/no help | NA | Autonomous | 160 | 49 | 19.1 | N | No (GFR > 90) | No | NA |
| b , c 13/Neck05 | N | Mainstream school (study certificate)/no help | Student | Autonomous. Parents | 175 | 59 | 19.3 | N |
No (GFR > 90) |
No | No |
| 14/Lyo01 | N | NA/ No help | Waitress | Autonomous. Married | 163 | 71 | 26.8 | N |
No (GFR > 90) |
No | NA |
| 15/Lyo02 | N | Mainstream school until 16 y | Not working | Autonomous. Parents | 162 | 56 | 21.1 | N | Severe | Yes | Anorexia; VH due to AH |
| 16/Lyo03 | N | Mainstream school/No help | Student | Autonomous. Parents | 161 | 53 | 20.3 | N |
No (GFR > 90) |
No | NA |
| 17/Lyo03 | Delayed (severe) | Special school | Not working | Institution. Dependent. | 165 | 65 | 23.9 | N; epilepsy |
No (GFR > 90) |
No | Psychomotor agitation |
| 18/Lyo02 | Delayed (mild) | Special school | Not working | Institution. Dependent. | 170 | 38 | 13 | Ataxia; epilepsy | Severe | Yes | Severe bilateral optic atrophy (13 y). VH due to AH (11 y). Pubertal delay, depression, anorexia. Died in 2001 |
| 19/Lyo04 | N | University degree/no help | Student | Autonomous. Alone | 174 | 68 | 22.2 | N |
No (GFR > 90) |
No | NA |
| 20/Lyo05 | Delayed (moderate) | Special school | Sheltered work | Partially dependent. Parents | 160 | 47 | 18.4 | N |
No (GFR > 90) |
No | VH |
| ¨21/Mars01 | N | Mainstream school/ no help | Student | Autonomous; Parents | 168 | 46 | 15.9 | N |
No (GFR > 90) |
No | No |
| 22/Debr01 | N | High school/ no help | Student | Autonomous; Parents | 164 | 58 | 21.4 | N |
No (GFR > 90) |
NA | No |
| 23/Montp01 | Delayed (mild) | At mainstream school with extra help | NA | Dependent. Parents. | 152 | 44 | 19 | Mild upper limb dystonia; bradykinesia; incoordination |
No (GFR > 90) |
NA | NA |
Abbreviations: AH, arterial hypertension; BMI, body mass Index; CAP, professional certificate; CAT, centre for aide in working; CRF, chronic renal failure; GFR, glomerular filtration rate; H, height; N, normal; NA, not available/assessed; UL, upper limb; VH, ventricular hypertrophy; vit D, vitamin D; w, weight.
previously reported. 20
previously reported. 14
previously reported. 15
previously reported. 21
included in the EIMD (European Registry and Network for Intoxication type Metabolic Disease).
Three female patients had children of their own. Patient 14/Lyo01 had three children (at the age of 24, 26, and 28 years): she continued B12 at the same regimen and she gave birth to eutrophic babies after full term pregnancies without renal insufficiency. Patient 10/Neck02 had two pregnancies (at the age of 35 and 38 years) and gave birth to two healthy babies (born at 36 and 35 weeks of gestation): during the first pregnancy she had IM OHCbl, and the pregnancy was complicated by a pre‐eclampsia state; the switch from IM to PO OHCbl was done between the two pregnancies; she continued oral OHCbl during the second pregnancy. Patient 07/Lond07 had three pregnancies: at 20 years she gave birth to a healthy girl, born following an early elective cesarean for intrauterine growth retardation (33 weeks of gestation; 1530 g; Apgar score 9/9/10), with apparently no further concerns about development; she subsequently gave birth to a healthy boy (38 weeks of gestation; 3008 g; spontaneous vaginal delivery) and to a premature girl (35 weeks of gestation; 1087 g; induced delivery and cesarean section). The general nutrition of patient 07/Lond07 has always been reported as of poor quality, however, her renal function remained normal during pregnancies and the B12 regimen was maintained without changes; during her second pregnancy she developed gestational diabetes which remained then as type 2 diabetes. Each pregnancy was closely monitored and recommendations were provided for delivery and the puerperium. 23 , 24 , 25
As regards to long‐term neurological and clinical outcomes (Table 3), the main problems were intellectual disability and/or chronic kidney disease (globally 13/23, 57% of the patients); 10 patients (43%) have no major organ involvement.
A total of 14/23 patients (61%) had normal cognitive function, while 9/23 patients (39%) had intellectual disability. Neurological examination was normal in 17/23 patients (74%): only five patients had mild extrapyramidal features, such as bradykinesia or involuntary movements, and in at least one patient (patient 1/Lon01) this was probably secondary to long standing neuroleptic treatments; one patient had ataxia; three patients had seizures. Eleven patients had at least one brain imaging procedure performed (cerebral MRI in nine; CT scan in two). For the most part, these were normal or showed non‐specific findings, such as white matter abnormalities or mild atrophy. Two patients showed basal ganglia signal changes, but these did not correlate with their neurological examination, which was normal, or with cognitive function, normal as well. Except for patient 18/Lyo02, all the patients are still alive.
Chronic renal failure (CRF), defined as a glomerular filtration rate less than 60 ml/min/1.73 m2, was present in 7/23 patients (30%) and was moderate in three and severe in four cases (one requiring renal transplantation). Hyperuricemia was present in 5/23 patients (22%), three of whom were treated with allopurinol. One patient presented with symptomatic marked bilateral optic atrophy at the age of 15, associated with severe renal insufficiency and intellectual disability.
Bone densitometry was performed in 9 patients and was abnormal in 5 of them with osteoporosis (n = 2) or osteopenia (n = 3). The results of a cardiac evaluation with echocardiography were rarely available (5/23) and showed cardiac hypertrophy only in two siblings who also suffered from arterial hypertension and severe renal insufficiency (family Lyo02).
When we tried to correlate the initial treatment with the clinical outcome (Figure 1) we found that a better final outcome was found in patients early treated with B12 supplementation (alone or plus protein restriction) (n = 7: four patients with no major organ involvement; mental retardation in three patients; only one patient with mild and late onset kidney disease). Patients initially treated only with protein restriction have a worse final outcome (n = 12; 5 with no major organ involvement; intellectual disability in four patients; chronic renal failure in four patients). Patients with early onset and late diagnosis, considered as having had no initial treatment, have the worst outcome, as expected (n = 3; all have major organ involvement with mental retardation in two patients and kidney disease also in two patients). Importantly, only one (14%) of the seven patients early treated with B12 supplementation (alone or plus protein restriction) developed moderate late ‐onset chronic renal failure, while the percentage of intellectual disability was comparable and independent of the initial treatment (B12±protein restriction, or protein restriction only) (Figure 1).
3.1. Patient 4/Lond05: a patient with adult‐onset cblA deficiency and mild disease
This is a 36‐year‐old lady who presented at the age of 25 years because of non‐specific chronic fatigue, muscle pain, exercise intolerance, continuous headaches, and nausea after a flu‐like illness. These symptoms had a major impact on her day‐to‐day life, leading her to plan a career change; they could relapse after mild health problems such as upper respiratory tract infection. Neurological examination was completely normal. She was investigated by rheumatologists, hematologists, and neurologists and diagnosed variously as having Lyme disease, fibromyalgia, chronic fatigue symptoms and, eventually, an undefined neuro‐immunological condition. She had been treated with antibiotics, antidepressants, and intravenous immunoglobulins, without any persistent amelioration. All laboratory parameters were normal except for an isolate increased in MMA at 11.6 μmol/L (nv < 0.28), decrease of B12 at 129 pg/ml (nv > 190 pg/ml) with normal plasma homocysteine, MCV, hemoglobin, folate, and renal function. After treatment with B12 up to 1 mg IM daily her chronic symptoms completely disappeared; plasma B12 level normalized although MMA remained elevated (8 μmol/L).
Enzymatic studies of her fibroblasts showed a deficit in propionate incorporation (1.61 nmol/mg prot/16 h vs. 6.08 nmol/mg prot/16 h in the experimental control; range in controls 3.5–24.4) with a clear increase after B12 supplementation (11.7 nmol/mg prot/16 h vs. 7.79 nmol/mg prot/16 h in the experimental control; range in controls 4.33–28.9), and normal MCM activity (measured with excess of AdoCbl), indicating a B12‐responsive defect of AdoCbl synthesis. Molecular analysis showed compound heterozygous mutations in the MMAA gene; no other mutation could be found in the other genes known to be responsible for increased MMA (ABCD4/cblJ, ACSF3/CMAMMA, CD320, CUBN, CBLIF, HCFC1/cblX, LMBRD1/cblF, MCEE/methylmalonyl‐CoA epimerase deficiency, MLYCD/MA, MMAB/cblB, MMACHC/cblC, MMADH/cblD, MMUT/MMA mut, SUCLA2, TCN2); the gene SUCLG1 was non tested.
This patient has a 35‐year‐old brother (patient 5/Lond05) suffering from mild intellectual disability of unknown origin. He was born after a normal pregnancy. His early milestones were normal until an episode of measles at the age of 10 months. After that, his mother noticed delay in his speech and mild developmental delay. He attended a mainstream school up until the age of 11 years and was then oriented towards a special school because of learning difficulties. He never experienced acute decompensation and had never been on any regular medication. After his sister was found with increased MMA he also was tested and displayed increased MMA (41.3 μmol/L) with normal homocysteine, B12, and folate. Treatment with B12 by IM injection every 2 weeks was prescribed. Neurological examination, at age 35, showed a global bradykinesia and incoordination. He requires supported living and is currently working as a part‐time kitchen porter. He did not develop renal, hematological, ophthalmological or hepatic complications.
The segregation was confirmed in the family: the proband's brother was also compound heterozygous for two mutations in the MMAA gene; the parents of these two siblings were asymptomatic and had normal MMA levels and each of them carried one of the two mutations in the MMAA gene.
3.2. Patient 18/Lyo02: A patient with an early onset and severe cblA deficiency disease
This patient presented at 9 days of life with a severe acute decompensation with ketoacidosis and hyperammonia (up to 500 micromol/l) requiring hemofiltration; despite following treatment with low protein and B12 he subsequently developed many severe decompensations. B12 was subsequently stopped because he was considered as unresponsive to this treatment. He developed intellectual disability, ataxia, epilepsy, and renal insufficiency (since the age of 11 years) with arterial hypertension, cardiac left hypertrophy and cardiac rhythm anomalies (atrioventricular block, long QT at the age of 15 years). Optic atrophy was found since the age of 13 years. This patient was issued from consanguineous parents and molecular analysis found the variant c.586C>T p.Ala196* at the homozygous state in the MMAA gene, confirming the diagnosis of cblA disease; enzymatic studies on fibroblast were not available. A renal biopsy was performed at the age of 14 years, and showed tubulo‐interstitial nephropathy with glomerulosclerosis compatible with MMA. This patient died at the age of 20 years during an acute decompensation of MMA. Unfortunately, this is a quite old case (more than 20 years ago) and no more clinical information is available.
4. DISCUSSION
We presented here the long‐term follow up (mean 24.9 ± 7.6 years; range 11–40 years) of a series of 23 adult patients with cblA disease. Our series confirm that this is predominantly a pediatric, early onset‐disease with a median age at onset of 4 months and with 85.7% (18/21) of the patients presenting before the age of 1 year. However, rare cases of adult‐onset disease are possible. Indeed, we present here a woman with cblA deficiency (confirmed by moderately increased plasma MMA, fully compatible enzymatic activity results, compound heterozygous mutations in the MMAA gene) and adult‐onset disease, presenting at the age of 25 years with chronic fatigue, muscular pain, nausea and headache beginning after a flu‐like illness and relapsing after infections. Her brother had also a globally mild presentation, even if much more similar to that previously reported (early‐onset chronic intellectual disability, beginning after a viral infection with no more decompensation and no kidney disease). This data suggests the existence of rare adult‐onset and mild presentations of cblA deficiency, with intrafamilial variability.
Most patients presented with an acute decompensation rapidly leading to the diagnosis of isolated MMA. However, we describe here four patients with an early‐onset chronic presentation (renal insufficiency; weight stagnation; intellectual disability) and no acute decompensation, for whom diagnosis had been considerably delayed. This underlines the occurrence of chronic presentation and the importance of considering MMA in any case of unexplained renal disease, intellectual disability or growth delay. 26
This data also suggests that cblA is a disease potentially eligible for newborn ‐screening, since most of the patient (19/23, 83%) had a disease onset after day 9 of life; however, 17% of the patients still had an earlier disease onset. Newborn‐ screening applied to cblA deficiency could significantly reduce time until diagnosis 27 and be useful in preventing decompensation with acute encephalopathy and in avoiding late‐diagnosis in the case of misdiagnosed chronic symptoms. However, cblA deficiency is in most cases a less severe disease than other forms of methylmalonic aciduria and the real impact of early treatment on neurological complications of the disease need to be further evaluated.
On the basis of the molecular data, we confirmed that for the MMAA gene the proportion of nonsense mutations, frameshift mutations, possible splice‐site defect or deletions was much higher than that of missense mutations, with some recurrent mutations (c.586C>T p.Ala196*; c.433C>T p.Arg145*). 17 We did not find any correlation between molecular and phenotypic findings. The mutation c.433C>T p.Arg145* accounted for 16% of the alleles and was less frequent than in other European series (19% 17 ; 43% 19 ) perhaps because in our series many patients (43%) were not of European descendent.
This series of patients shows that, before the molecular and enzymatic era, most of the cblA patients (61%) were classified as B12‐unresponsive MMA on the basis of a clinical and biochemical evaluation. Unfortunately, details about how B12 responsiveness was evaluated were not available. In contrast, in a recent large series of younger patients with cblA deficiency (mean age: 11.9 years) B12 responsiveness was reported in 27 out of 28 patients. 16 Indeed, testing with vitamin B12 has never been standardized and responsiveness criteria poorly determined, although recommendations have been proposed. 22 Moreover, in vitro enzymatic B12 responsiveness was subsequently found in all the tested patients (n = 13) of our series. This data suggests that the initial evaluation was probably not reliable and that in newly diagnosed patients with isolated methylmalonic aciduria careful enzymatic evaluation of B12 responsiveness and molecular analysis are required. This has important therapeutic consequences because in some patients with cblA deficiency B12 supplementation was discontinued because of presumed unresponsiveness.
In our series the initial treatment was quite variable but almost all the patients (with the exception of three patients with early‐onset disease and late diagnosis) received at least a protein restricted diet or B12 supplementation, or both. After the molecular confirmation of the cblA defect (on average about 15 years after disease onset), B12 supplementation was administered in most of the patients (from 43% at initial treatment to 87% at the final visit) and protein‐restricted diets were maintained but often relaxed. 16
IM B12 was preferred to oral B12, probably because it is more readily available and more indicate for children in case of relapsing acute decompensations. However, the use of oral vs IM B12 in adults is still a matter of debate: adult patients seem to be less prone to acute decompensation and in some patients oral B12 is well tolerated without increase of MMA; future prospective studies will better answer to these questions.
No clear correlation was found between initial MMA levels and disease severity or clinical outcome. The follow‐up showed that the occurrence of episodes of decompensation was also very variable and in general decompensation occurred more frequently in childhood than in adulthood.
We showed that intellectual disability and renal failure are the major clinical consequences of a cblA deficiency and that they are present in 39% and 30% of the patients, respectively. In the long‐term, patients with cblA deficiency can also present osteoporosis, likely related to low‐protein diet, and hypertension and cardiac hypertrophy, likely related to chronic kidney disease. The long‐term clinical outcome was very variable and seemed to be unrelated to the severity of the first decompensation. Moreover, we confirm that cblA deficiency has a better prognosis than that of patients with MMA due to mutation of MCM enzyme, 16 since 43% of the patients had normal cognition and no major organ involvement, neurological examination was normal in most of the patients (74%), and eventually 56.5% are completely autonomous. However, we also presented the case of a patient (18/Lyo02) with a particularly severe disease including neurological, cognitive, and renal involvement who died during a severe decompensation at the age of 20 years. This patient also developed optic atrophy, a rare but possible complication of cblA disease. 28 , 29
Importantly, our data suggests that, although protein restriction, B12, and energy supply are essential in the emergency management and prevention of acute decompensations, no clear‐cut correlation exist between the initial long‐term treatments and the final cognitive outcome, since intellectual disability was found in about 30%–40% of the patients, independently of the initial treatment modality (B12, protein restriction, or both). However, we cannot exclude that late diagnosis and treatment still contributed to the global high percentage of intellectual disability in this series. The high percentage of patients with intellectual disability was not linked to the co‐occurrence of consanguinity, since intellectual disability was rather equally present in the patients with and without consanguinity (33% and 44%, respectively). Instead, only one case (14%) of moderate and late‐onset renal failure was found in patients treated early with B12 as compared to 33% with chronic kidney disease in the group who did not initially receive B12 supplementation, suggesting that B12 treatment, despite being administered at variable doses and frequency, is very important in preventing or at least delaying renal dysfunction. Indeed, in a recent series in which B12 was administered to 78% of the patients, chronic kidney disease was reported only in 9% of cblA patients. 16
This is also in line with the pathophysiological hypotheses suggesting that chronic kidney disease is more directly linked to MMA toxicity, while brain involvement seems to be more complex and related partially to acute decompensation and energy deficit, and partially to de novo synthesis and trapping of toxic metabolites in brain tissue. 26 , 30
The present series is valuable because it provides a detailed picture of the very long‐term outcome of a series of adult patients with methylmalonic aciduria due to cblA deficiency, mostly diagnosed before the enzymatic and molecular era. It has the limitations associated with retrospective and incomplete data collection and the number of patients was not enough to perform statistical analysis when comparing the different treatment groups; it is also possible that some patients with a more severe clinical presentation could have died before reaching adult age and therefore have not been included in this series. However, we were able to draw important conclusion about cblA deficiency. We confirmed that about 35% of patients with cblA disease can have a chronic presentation, with no acute decompensation, underlining the importance of considering MMA in any case of unexplained chronic renal failure, mental retardation and growth delay. Moreover, we showed that milder adult‐onset onset forms can exist and possibly remain under diagnosed. Importantly, we showed that early diagnosis, based on precise molecular and enzymatic studies, is essential to properly take care of these patients and we provided further evidence about the importance of B12 supplementation in preventing chronic kidney disease; in contrast, the occurrence of intellectual disability seemed to develop partially independently of the initial long‐term treatment, although the optimal management and prevention of acute decompensations remains essential. Our data therefore suggests that every patient affected by cblA disease should be treated with vitamin B12. Future prospective studies will be of course essential to definitively answer to the questions about appropriate treatment and outcome.
FUNDING INFORMATION
CM was supported from a grant from the University Hospital of Montpellier.
CONFLICTS OF INTEREST
A Fouilhoux, JF Benoist, P De Lonlay, N Guffon‐Fouilhoux, A Brassier, A Cano, B Chabrol, A Pennisi, M Schiff, C Acquaviva, E Murphy, A Servais and R Lachmann declare that they have no conflict of interest. CM has received travel and congress support from Biomarin and honoraria for participating to a workshop from Roche. Conflicts of interest with the content of this work are not perceived.
ETHICS STATEMENT
All authors were compliant and followed the ethical guidelines, according to the requirements of the JIMD. The patients included in the study agreed to collection and sharing of their data for scientific purposes.
ACKNOWLEDGMENTS
We would like to thank all the clinicians, geneticists, and biologists who shared data about their patients. We thank Pr Gueant and his laboratory in Nancy, France, for performing molecular analysis in patient 23# and also the laboratory of the Metabolic Division of the University Hospital of Zurich, for performing enzymatic analysis in some of the English patients. We would also like to thank patients for their trust in our work and care.
Marelli C, Fouilhoux A, Benoist J‐F, et al. Very long‐term outcomes in 23 patients with cblA type methylmalonic acidemia. J Inherit Metab Dis. 2022;45(5):937‐951. doi: 10.1002/jimd.12525
Communicating Editor: Saadet Mercimek‐Andrews
Contributor Information
Cecilia Marelli, Email: c-marelli@chu-montpellier.fr.
Robin Lachmann, Email: r.lachmann@nhs.net.
DATA AVAILABILITY STATEMENT
Data archiving is not mandated but data will be made available on reasonable request.
REFERENCES
- 1. Takahashi‐Iñiguez T, González‐Noriega A, Michalak C, Flores ME. Human MMAA induces the release of inactive cofactor and restores methylmalonyl‐CoA mutase activity through their complex formation. Biochimie. 2017;142:191‐196. [DOI] [PubMed] [Google Scholar]
- 2. Lofgren M, Padovani D, Koutmos M, Banerjee R. A switch III motif relays signaling between a B12 enzyme and its G‐protein chaperone. Nat Chem Biol. 2013;9:535‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Froese DS, Kochan G, Muniz JRC, et al. Structures of the human GTPase MMAA and vitamin B12‐dependent Methylmalonyl‐CoA mutase and insight into their complex formation. J Biol Chem. 2010;285:38204‐38213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Banerjee R, Gouda H, Pillay S. Redox‐linked coordination chemistry directs vitamin B 12 trafficking. Acc Chem Res. 2021;54:2003‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jansen R, Ledley FD. Heterozygous mutations at the Mut locus in fibroblasts with mut0 methylmalonic acidemia identified by polymerase‐chain‐reaction cDNA cloning. Am J Hum Genet. 1990;47:808‐814. [PMC free article] [PubMed] [Google Scholar]
- 6. Dobson CM, Wai T, Leclerc D, et al. Identification of the gene responsible for the cblA complementation group of vitamin B12‐responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc Natl Acad Sci U S A. 2002;99:15554‐15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dobson CM, Wai T, Leclerc D, et al. Identification of the gene responsible for the cblB complementation group of vitamin B12‐dependent methylmalonic aciduria. Hum Mol Genet. 2002;11:3361‐3369. [DOI] [PubMed] [Google Scholar]
- 8. Coelho D, Suormala T, Stucki M, et al. Gene identification for the cblD defect of vitamin B12 metabolism. N Engl J Med. 2008;358:1454‐1464. [DOI] [PubMed] [Google Scholar]
- 9. Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med. 1983;308:857‐861. [DOI] [PubMed] [Google Scholar]
- 10. Martínez MA, Rincón A, Desviat LR, Merinero B, Ugarte M, Pérez B. Genetic analysis of three genes causing isolated methylmalonic acidemia: identification of 21 novel allelic variants. Mol Genet Metab. 2005;84:317‐325. [DOI] [PubMed] [Google Scholar]
- 11. Vatanavicharn N, Champattanachai V, Liammongkolkul S, et al. Clinical and molecular findings in Thai patients with isolated methylmalonic acidemia. Mol Genet Metab. 2012;106:424‐429. [DOI] [PubMed] [Google Scholar]
- 12. Merinero B, Pérez B, Pérez‐Cerdá C, et al. Methylmalonic acidaemia: examination of genotype and biochemical data in 32 patients belonging to Mut, cblA or cblB complementation group. J Inherit Metab Dis. 2008;31:55‐66. [DOI] [PubMed] [Google Scholar]
- 13. Yang X, Sakamoto O, Matsubara Y, et al. Mutation analysis of the MMAA and MMAB genes in Japanese patients with vitamin B(12)‐responsive methylmalonic acidemia: identification of a prevalent MMAA mutation. Mol Genet Metab. 2004;82:329‐333. [DOI] [PubMed] [Google Scholar]
- 14. Cosson MA, Benoist JF, Touati G, et al. Long‐term outcome in methylmalonic aciduria: a series of 30 French patients. Mol Genet Metab. 2009;97:172‐178. [DOI] [PubMed] [Google Scholar]
- 15. Nizon M, Ottolenghi C, Valayannopoulos V, et al. Long‐term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J Rare Dis. 2013;8:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hörster F, Tuncel AT, Gleich F, et al. Delineating the clinical spectrum of isolated methylmalonic acidurias: cblA and Mut. J Inherit Metab Dis. 2021;44:193‐214. [DOI] [PubMed] [Google Scholar]
- 17. Plessl T, Bürer C, Lutz S, Yue WW, Baumgartner MR, Froese DS. Protein destabilization and loss of protein‐protein interaction are fundamental mechanisms in cblA‐type methylmalonic aciduria. Hum Mutat. 2017;38:988‐1001. [DOI] [PubMed] [Google Scholar]
- 18. Dempsey‐Nunez L, Illson ML, Kent J, et al. High resolution melting analysis of the MMAA gene in patients with cblA and in those with undiagnosed methylmalonic aciduria. Mol Genet Metab. 2012;107:363‐367. [DOI] [PubMed] [Google Scholar]
- 19. Lerner‐Ellis JP, Dobson CM, Wai T, et al. Mutations in the MMAA gene in patients with the cblA disorder of vitamin B12 metabolism. Hum Mutat. 2004;24:509‐516. [DOI] [PubMed] [Google Scholar]
- 20. Martín‐Hernández E, Lee PJ, Micciche A, Grunewald S, Lachmann RH. Long‐term needs of adult patients with organic acidaemias: outcome and prognostic factors. J Inherit Metab Dis. 2009;32:523‐533. [DOI] [PubMed] [Google Scholar]
- 21. Brassier A, Krug P, Lacaille F, et al. Long‐term outcome of methylmalonic aciduria after kidney, liver, or combined liver‐kidney transplantation: the French experience. J Inherit Metab Dis. 2020;43:234‐243. [DOI] [PubMed] [Google Scholar]
- 22. Fowler B, Leonard JV, Baumgartner MR. Causes of and diagnostic approach to methylmalonic acidurias. J Inherit Metab Dis. 2008;31:350‐360. [DOI] [PubMed] [Google Scholar]
- 23. Langendonk JG, Roos JCP, Angus L, et al. A series of pregnancies in women with inherited metabolic disease. J Inherit Metab Dis. 2012;35:419‐424. [DOI] [PubMed] [Google Scholar]
- 24. Murphy E. Pregnancy in women with inherited metabolic disease. Obstet Med. 2015;8:61‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raval DB, Merideth M, Sloan JL, et al. Methylmalonic acidemia (MMA) in pregnancy: a case series and literature review. J Inherit Metab Dis. 2015;38:839‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haarmann A, Mayr M, Kölker S, et al. Renal involvement in a patient with cobalamin a type (cblA) methylmalonic aciduria: a 42‐year follow‐up. Mol Genet Metab. 2013;110:472‐476. [DOI] [PubMed] [Google Scholar]
- 27. Heringer J, Valayannopoulos V, Lund AM, et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J Inherit Metab Dis. 2016;39:341‐353. [DOI] [PubMed] [Google Scholar]
- 28. Ku CA, Ng JK, Karr DJ, et al. Spectrum of ocular manifestations in cobalamin C and cobalamin a types of methylmalonic acidemia. Ophthalmic Genet. 2016;37:404‐414. [DOI] [PubMed] [Google Scholar]
- 29. Valayannopoulos V, Hubert L, Benoist JF, et al. Multiple OXPHOS deficiency in the liver of a patient with CblA methylmalonic aciduria sensitive to vitamin B(12). J Inherit Metab Dis. 2009;32:159‐162. [DOI] [PubMed] [Google Scholar]
- 30. Morath MA, Okun JG, Müller IB, et al. Neurodegeneration and chronic renal failure in methylmalonic aciduria‐‐a pathophysiological approach. J Inherit Metab Dis. 2008;31:35‐43. [DOI] [PubMed] [Google Scholar]
- 31. Willard HF, Rosenberg LE. Inherited deficiencies of human methylmalonyl CaA mutase activity: reduced affinity of mutant apoenzyme for adenosylcobalamin. Biochem Biophys Res Commun. 1977;78:927‐934. [DOI] [PubMed] [Google Scholar]
- 32. Kolhouse JF, Utley C, Allen RH. Isolation and characterization of methylmalonyl‐CoA mutase from human placenta. J Biol Chem. 1980;255:2708‐2712. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data archiving is not mandated but data will be made available on reasonable request.