

Abstract
Coproporphyrin I (CPI) is an endogenous biomarker of organic anion transporting polypeptide 1B transporter (OATP1B). CPI plasma baseline was reported to increase with severity of chronic kidney disease (CKD). Further, ratio of CPI area under the plasma concentration‐time curve (AUCR) in the presence/absence of OATP1B inhibitor rifampin was higher in patients with CKD compared with healthy participants, in contrast to pitavastatin (a clinical OATP1B probe). This study investigated mechanism(s) contributing to altered CPI baseline in patients with CKD by extending a previously developed physiologically‐based pharmacokinetic (PBPK) model to this patient population. CKD‐related covariates were evaluated in a stepwise manner on CPI fraction unbound in plasma (fu,p ), OATP1B‐mediated hepatic uptake clearance (CLactive ), renal clearance (CLR ), and endogenous synthesis (ksyn ). The CPI model successfully recovered increased baseline and rifampin‐mediated AUCR in patients with CKD by accounting for the following disease‐related changes: 13% increase in fu,p , 29% and 39% decrease in CLactive in mild and moderate to severe CKD, respectively, decrease in CLR proportional to decline in glomerular filtration rate, and 27% decrease in ksyn in severe CKD. Almost complete decline in CPI renal elimination in severe CKD increased its fraction transported by OATP1B, rationalizing differences in the CPI–rifampin interaction observed between healthy participants and patients with CKD. In conclusion, mechanistic modeling performed here supports CKD‐related decrease in OATP1B function to inform prospective PBPK modeling of OATP1B‐mediated drug‐drug interaction in these patients. Monitoring of CPI allows detection of CKD–drug interaction risk for OATP1B drugs with combined hepatic and renal elimination which may be underestimated by extrapolating the interaction risk based on pitavastatin data in healthy participants.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Patients with chronic kidney disease (CKD) had increased plasma CPI baseline and rifampin OATP1B interaction relative to healthy participants. Pitavastatin AUC also increased in CKD, whereas its rifampin DDI was insensitive to the disease.
WHAT QUESTION DID THIS STUDY ADDRESS?
What is the mechanism of altered CPI PK or CPI‐drug interaction in CKD? How informative is monitoring of CPI in patients with CKD?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Increased CPI baseline and pitavastatin AUC in patients with CKD p were attributed to decreased OATP1B clearance. Almost complete decrease in CPI renal clearance (parallel elimination route) increased its sensitivity to OATP1B DDI (shift in fraction transported by OATP1B), whereas it had marginal impact on pitavastatin interaction due to its negligible renal elimination.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Use of probe drugs eliminated by hepatic route only may underestimate OATP1B DDI in CKD for drugs with combined hepatic‐renal elimination. CPI is a valuable tool to evaluate OATP1B‐mediated DDI risk for such drugs in both healthy participants and patients with CKD and to support PBPK modelling in this patient cohort.
Chronic kidney disease (CKD) can change pharmacokinetics (PK) of both renally and nonrenally eliminated drugs / endogenous substances due to altered physiological functions in the kidney and other organs. 1 , 2 , 3 , 4 Changes include decrease in glomerular filtration rate (GFR) and serum albumin, accumulation of uremic solutes, and proposed decline in expression/activity of metabolizing enzymes and transporters in the kidney and liver. 5 , 6 , 7 Analyses of clinical data have suggested CKD‐related effects on the activity of hepatic cytochrome P450 2D6 isozyme (CYP2D6) and organic anion transporting polypeptide transporter (OATP) 1B, and intestinal P‐glycoprotein (P‐gp), leading to altered PK of substrates of these proteins in patients with CKD. 5 , 6 , 8 , 9 , 10 In contrast, minimal or variable disease effect was reported for CYP1A2‐mediated, CYP2C9‐mediated, CYP2C19‐mediated, or CYP3A4‐mediated clearances. 5 , 6 , 8 Since altered PK could lead to unexpected adverse effects in patients, regulatory authorities require evaluation of the impact of CKD on the PK of new drug entities. 11 CKD may also alter the magnitude of drug–drug interactions (DDIs) if the disease changes exposure of perpetrators or alters the fraction of the victim drug eliminated by DDI‐related route. 7 , 12 , 13 , 14 Despite supporting statements in regulatory guidance documents around inclusion of special populations in clinical trials and consideration of DDI risk in such patients, 15 , 16 evidence of investigating DDI in CKD is limited 12 due to general exclusion of such patients from clinical trials. Therefore, understanding changes in the magnitude of DDI (AUC ratio (AUCR)) caused by CKD‐related physiological changes is an important consideration.
Endogenous biomarkers have increasingly been used for early evaluation of transporter‐mediated perpetrator DDI, with the aim to guide the need and design of subsequent dedicated DDI studies with clinical probes. In the case of OATP1B, the most established endogenous biomarker is coproporphyrin I (CPI), a by‐product of heme synthesis. CPI has high specificity for this transporter and is 85% and 15% eliminated via liver and kidney, respectively. 17 , 18 CPI plasma concentrations are sensitive to altered OATP1B activity caused by either OATP1B inhibitors (e.g., rifampin) or genetic polymorphism. 19 , 20 In contrast to DDI, use of biomarkers to evaluate potential changes in transporter function in disease populations is still scarce. Recent clinical study reported ~ 31% higher plasma exposure of CPI, as well as pitavastatin (an OATP1B clinical probe with minor renal elimination 21 , 22 ) in CKD compared with healthy individuals (Figure S1 ). 8 This observation is in agreement with increasing clinical evidence of higher exposure of OATP1B substrates in patients with severe CKD (e.g., SN‐38, repaglinide). 5 , 8 , 10 , 22 , 23 , 24 The reported magnitude of CPI–rifampin OATP1B‐mediated DDI (AUCRCPI) was ~ 56% higher in patients with severe CKD compared with healthy participants, while the AUCR for pitavastatin–rifampin DDI (AUCRPTV) in the same individuals was only marginally affected by the disease. 8 These apparently contradictory findings require mechanistic explanation to facilitate accurate translation of OATP1B‐mediated DDI data from healthy participants to patients with CKD.
This study aimed (i) to understand the effect of CKD on OATP1B activity, and (ii) to interpret the inconsistent disease effect on the magnitude of CPI–rifampin and pitavastatin–rifampin DDI in this population, to inform the necessity for clinical OATP1B DDI assessment in patients with CKD. These questions were addressed by extending the reported population physiologically‐based pharmacokinetic (PBPK) model for CPI 25 to the population with CKD. Combined with a rifampin CKD model, the CPI model was optimized to recover observed increases in biomarker plasma concentrations and AUCRCPI in patients with CKD by incorporating disease‐related physiological changes using a combination of top‐down and bottom‐up approaches (Figure 1 ). The CKD effects on fraction unbound in plasma (f u,p ) and hepatic uptake clearance (CL active ) were informed from in vitro data 8 and the pitavastatin CKD model developed using data from the same individuals. The CKD effects on renal clearance (CL R ) and synthesis rate (k syn ) (suggested by reported decrease in blood hemoglobin in CKD 26 ) were also investigated. Finally, mechanisms for the inconsistency in rifampin OATP1B‐mediated AUCR between CPI and pitavastatin in patients with CKD were investigated.
Figure 1.
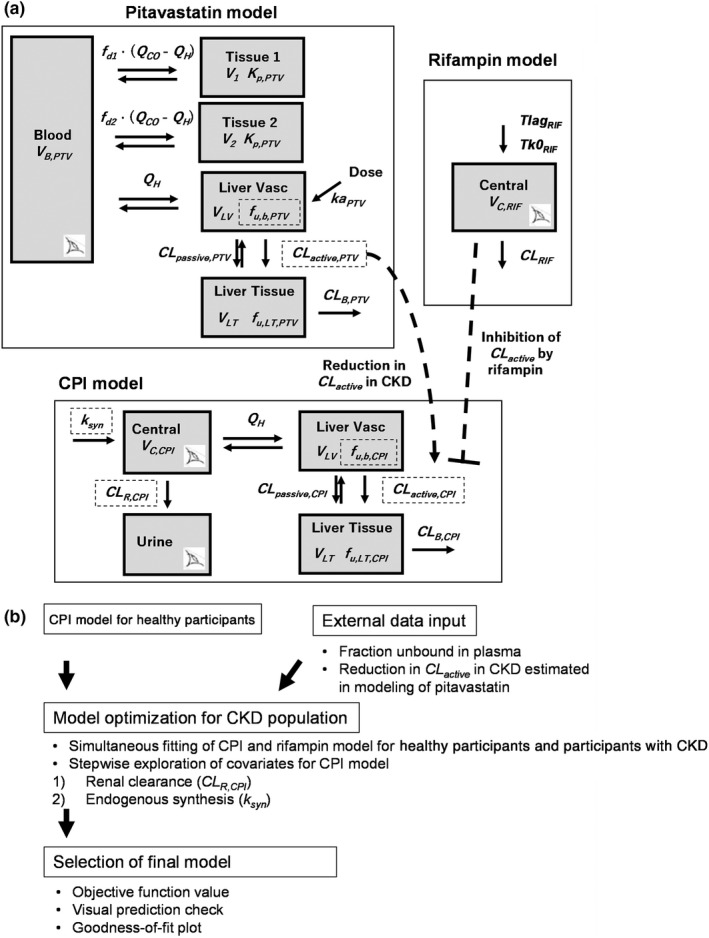
Strategy of CPI model development for the population with CKD. (a) Structure of models for coproporphyrin I (CPI), pitavastatin (PTV), and rifampin (RIF). Eye symbols represent observed compartments. Dashed squares represent parameters for which CKD effects were evaluated. (b) Workflow of CPI model development for the population with chronic kidney disease (CKD). CL active , hepatic uptake clearance (unbound); CL B , biliary clearance; CL passive , hepatic passive clearance (unbound); CL R , renal clearance; CL RIF , clearance of rifampin; f d1 and f d2 , fractions of Q CO for tissue compartment 1 and 2, respectively; f u,b , fraction unbound in the blood; f u,LT , fraction unbound in liver tissue; ka, first‐order absorption rate constant; Ki, total rifampin inhibition constant for CL active of CPI; Kp ; tissue partition coefficient; Q CO ; cardiac blood flow; Q H , hepatic blood flow; Tk0 RIF , duration of zero‐order absorption; Tlag RIF , lag time; V 1 and V 2 , volumes of tissue compartment 1 and 2, respectively; V B , volume of blood compartment; V C , volume of central compartment; V LT , volume of liver tissue; V LV , volume of liver vascular.
METHODS
Clinical data
Reported clinical data from 32 individuals with normal renal function or mild to end‐stage CKD 8 were used in this study. Patients with CKD were categorized based on estimated glomerular filtration rate (eGFR) 27 as: healthy: >90 mL/min/1.73 m2; mild CKD: 60–89 mL/min/1.73 m2; moderate CKD: 30–59 mL/min/1.73 m2; and severe CKD: 15–29 mL/min/1.73 m2. Patients with end‐stage CKD (<15 mL/min/1.73 m2) were excluded from the analysis. Each group included 6–7 individuals, and there was no clear bias in demographics such as ethnicity, sex, or age among groups. 8 The clinical study consisted of two occasions: a cocktail of microdose of probe drugs for multiple transporters (e.g., pitavastatin (OATP1B), rosuvastatin (OATP1B, BCRP), and dabigatran etexilate (intestinal P‐gp) among others 8 ) was administered orally on both occasions; a single dose of 600 mg rifampin was coadministered orally during the second occasion. Among the probes, pitavastatin was selected for the current analysis as the most selective OATP1B clinical probe. Plasma (CPI and pitavastatin) and urine data (CPI only) pre‐ and post‐administration of probe drugs were available for CPI and pitavastatin modeling. Noncompartment analysis was initially performed to identify potential covariates. PK parameters considered were mean plasma CPI concentration without rifampin (Cbase,CPI), pitavastatin AUC without rifampin (AUCPTV,control), AUCRCPI, AUCRPTV, CL R , and net secretory renal excretion clearance (CL R,sec,net , Eq. 1) of CPI.
| (1) |
where individual CL R,sec,net was calculated based on CPI f u,p in patient i (f u,p,i , Eq. 2) 28 :
| (2) |
where [P] i and [P] HV represent individual plasma albumin concentration and a mean value in healthy participants measured in this study (4.39 g/dL, Table S1 ), respectively; f u,p,HV represents CPI f u,p in the healthy population (0.069). 25
Development of PK models for pitavastatin, rifampin, and CPI in population with CKD
Population PK models for pitavastatin, rifampin, and CPI (Figure 1 ) were developed in Monolix 2019R2 (Lixoft, France) to recover PK in both healthy populations and populations with CKD. The models include both between‐participant variability (BPV) of model parameters log‐normally distributed and residual variability based on a combined proportional and additive error model. 29 Inclusion of each covariate was evaluated based on the objective function value (−2 times log‐likelihood) at a significance level of P < 0.05. The models were also evaluated using goodness‐of‐fit plots and visual predictive checks.
Pitavastatin CKD model
A semimechanistic multicompartment PK model 30 was developed to describe bi‐exponential decline of pitavastatin in plasma. The model described hepatic active and passive permeability of pitavastatin between the liver blood and tissue compartments mechanistically, as implemented in our previous models 25 , 31 (Appendix S1). Blood flow rates and volumes of compartments were calculated based on individual's body weight (BW). Passive hepatic intrinsic clearance and fraction unbound in the liver blood and tissue were obtained in vitro 8 , 32 (Tables S1 and S2 ). Hepatic uptake of pitavastatin (CL active,PTV ) was assumed to be the rate‐determining step in its hepatic disposition. Minor routes (e.g., metabolism) were not considered, based on previous studies. 21 , 22 , 33
CKD‐related changes in pitavastatin f u,p and CL active,PTV were included, whereas any possible CKD‐related changes in pitavastatin disposition (e.g., absorption) were not considered due to limited information for model parameterization. CL active,PTV in CKD category Gx (CL active,PTV,Gx , G2: mild CKD; and G34: lumped moderate to severe CKD) was based on the population values for healthy participants (CL active,PTV,HV ) and its fractional change in CKD relative to healthy (COV CLactive,Gx ) (Eq. 3). e η represents BPV, where eta is assumed distributed N(0,OMEGA). Pooling of moderate to severe CKD was based on apparent nonlinear increase in AUCPTV,control with severity of CKD, as suggested previously 8 (Appendix S1).
| (3) |
In vitro studies showed that hepatic non‐OATP1B transporters such as sodium taurocholate cotransporting polypeptide (NTCP) mediated 2–29% of pitavastatin hepatic uptake. 34 , 35 Therefore, CL active,PTV was assumed to represent the combined OATP1B‐mediated (CL OATP1B,PTV ) and non‐OATP1B‐mediated clearances (CL non‐OATP1B,PTV ), where proportion of each route in the healthy population was 82% and 18%, respectively, based on pitavastatin–rifampin interaction data in healthy participants used in this analysis (Appendix S1). Change in CL active,PTV by CKD assumed the same degree of disease effect on CL OATP1B,PTV and CL non‐OATP1B,PTV , supported by studies on the decline in NTCP expression/activity by CKD‐derived uremic solutes. 36 An alternative scenario, which assumed no CKD effect on CL non‐OATP1B,PTV , was also evaluated (Appendix S1). The model parameters including CL active,PTV,HV and COV CLactive,Gx were optimized by fitting the model simultaneously to pitavastatin plasma concentrations (without rifampin) from all groups.
Rifampin CKD model
A one‐compartment PK model with zero‐order absorption described rifampin plasma concentration in all groups (Appendix S1). Covariates for rifampin parameters were evaluated as described above.
CPI CKD model
The CPI CKD model was based on the existing CPI population PBPK model for healthy participants, which incorporated renal excretion and mechanistic description of liver processes. 25 In the CPI CKD model, CL active,CPI was assumed to be mediated only by OATP1B. 37 The base model implemented (i) CKD‐related increase in f u,p using individuals' plasma albumin concentrations (Eq. 2), and (ii) CKD‐related decrease in CPI CL active (CL active,CPI ) estimated in the pitavastatin CKD model (COV CLactive,GX ; Eq. 3), assuming the same magnitude of CKD effect on both CL active,CPI and CL active,PTV . Validity of this assumption was confirmed by comparing Cbase,CPI, AUCPTV.control, and CL active,PTV estimated by post hoc analysis in the pitavastatin CKD model in respective patients.
Additionally, the CKD effects on CPI CL R (CL R,CPI ) was evaluated with an individual's eGFR (eGFR i ) as a covariate (Eq. 4):
| (4) |
where CL R ,CPI,HV and CL R,CPI ,i were CL R,CPI for a typical healthy participant with eGFR of 120 mL/min/1.73 m2 (eGFR HV ) and i th individual, respectively. COV CLR is the coefficient of the covariate effect. No effect of rifampin on CL R,CPI was assumed based on previous studies. 8 , 38 The CKD effect on k syn was assumed only for moderate to severe CKD based on the reported proportional decrease in blood hemoglobin and eGFR specific to these patients. 26 Individuals' k syn (k syn ,i ) was described as a function of eGFR i with a power coefficient (COV syn ) (Eq. 5):
| (5) |
where k syn , HV&G2 represents the population value for healthy and mild CKD groups and eGFR ref (60 mL/min/1.73 m2) represents a reference value for moderate to severe CKD groups. Liver volume, liver blood flow and hepatocellularity were assumed to be unaffected by CKD based on literature reports. 39 , 40 Biliary clearance of CPI (CL B,CPI ) was assumed to be the same in healthy participants and participants with CKD. 41 , 42 , 43
The rifampin CKD model was combined with the CPI CKD model to describe inhibition of CL active,CPI and recover CPI plasma and urine concentrations with and without rifampin. Because of the negligible change in rifampin f u,p by CKD (Table S1 ), inhibition of CL active,CPI by rifampin was linked to total plasma concentration of rifampin for all the groups, as described previously. 25 Inhibition of biliary transporter(s) by rifampin was not considered due to the lack of bile data for verification, as discussed previously. 17
The potential impact of a disease‐related change in model parameters on Cbase,CPI and AUCRCPI was evaluated by two approaches. The first approach was based on normalized sensitivity coefficients, as previously performed. 7 In the second approach, percent changes in Cbase,CPI and AUCRCPI in severe CKD relative to the healthy population were calculated for each potential CKD‐related change assuming it was the sole effect of severe CKD.
Evaluation of CKD effect on the magnitude of OATP1B‐mediated interactions
The mechanism of CKD‐related differences in the extent of rifampin interaction (AUCR) was investigated by calculating changes in the fraction transported by OATP1B (f OATP1B ) in CKD for hypothetical OATP1B substrates with different contributions of hepatic and renal elimination. The f OATP1B in CKD category Gx (f OATP1B,Gx ) was expressed based on fraction eliminated in urine in a healthy participant (f e,urine,HV ) and CKD‐derived decline in OATP1B‐mediated and renal clearances. The assumption was that any non‐OATP1B‐mediated hepatic clearance (f non‐OATP1B,HV ) declined to the same extent as OATP1B (Eq. 6, Appendix S1) and that passive uptake clearance is a minor contributor, as reported for CPI 25 and pitavastatin. 32 , 35 , 44
| (6) |
where the terms for hepatic uptake clearance (f OATP1B,HV and f non‐OATP1B,HV ) were reduced based on COV CLactive,GX estimated in the pitavastatin CKD model. Renal elimination (f e,urine,HV ) was reduced proportionally to a decrease in eGFR relative to the healthy population (eGFR HV ). CKD‐related change in the magnitude of OATP1B‐mediated interaction (AUCRGx/AUCRHV) was calculated from f OATP1B,Gx , assuming inhibition of only OATP1B‐mediated clearance by a rifampin‐equivalent OATP1B inhibitor (%inh of 90%) 45 (Eq. 7):
| (7) |
These calculations were performed for theoretical OATP1B substrates with different contribution of renal elimination (f e,urine,HV ranging from 0.01 to 0.5), including CPI‐equivalent (f e,urine,HV ; 0.15 (ref. 17)) and pitavastatin‐equivalent examples (f e,urine,HV ; 0.01 (refs. 21, 22)).
RESULTS
Estimated decrease in OATP1B activity in CKD based on modeling of pitavastatin data
The pitavastatin semimechanistic model adequately described pitavastatin plasma PK in patients with CKD (Figures S2 and S3 ). Model parameters were estimated with low relative standard error (RSE) of <40%, except BPV of ka PTV (Table 1). Inclusion of CKD effect on CL active,PTV significantly improved model fitting and enabled recovery of higher pitavastatin exposure in the population with CKD relative to the healthy population. Assuming the same degree of disease effect on both OATP1B and minor non‐OATP1B route resulted in the estimated decrease in CL active,PTV of 29% and 39% in mild and moderate to severe CKD groups, respectively, consistent with previous reports. 5 Alternative assumption of no disease effect on non‐OATP1B route resulted in slightly larger estimated disease effect on CL OATP1B,PTV : 37% and 50% decline in mild and moderate to severe CKD, respectively (Table S3 ).
Table 1.
Parameter estimates in models for PTV, RIF, and CPI in the healthy population and the population with CKD
| Drugs | Parameters (units) | Estimate (RSE) | ||
|---|---|---|---|---|
| Populationa | BPVb | |||
| PTV | System parameters | ka PTV (/h) | 0.727 (7) | 10.9 (73) |
| V 1_frac | 0.985 (1) | — | ||
| f d1 | 0.0564 (7) | 29.6 (26) | ||
| CL active,PTV,HV (L/h) | 8,136 (15) | 32.3 (20) | ||
| CL passive,PTV (L/h) | 134 FIXED | — | ||
| CL B,PTV (L/h) | 240 (34) | 47.9 (28) | ||
| Kp PTV | 3.46 (6) | 21.0 (28) | ||
| Covariates | COV CLactive,G2 | 0.29 (3) | — | |
| COV CLactive,G34 | 0.39 (7) | — | ||
| Residual unexplained variabilities | σ prop – PTV (%) | 27.1 (5) | — | |
| σ add – PTV (nM) | 0.00005 FIXED | — | ||
| RIF | System parameters | Tlag RIF (h) | 0.401 (17) | 76.4 (18) |
| Tk0 RIF,male (h) | 0.716 (12) | 34.9 (22) | ||
| V RIF (L) | 38.3 (6) | 26.5 (16) | ||
| CL RIF (L/h) | 6.35 (6) | 26.2 (15) | ||
| K i,total (μM) | 0.345 (6) | — | ||
| Covariates | COV Tk0,SEX | 0.639 (37) | — | |
| COV Vrif,BW | 1.01 (28) | — | ||
| COV CLrif,BW | 0.933 (29) | — | ||
| Corr V RIF and CL RIF | 0.668 (19) | — | ||
| Residual unexplained variabilities | σ prop – RIF (%) | 16.1 (8) | — | |
| σ add – RIF (μM) | 0.2 FIXED | — | ||
| CPI | System parameters | k syn,HV&mild (nmol/h) | 10.6 (11) | 13.4 (26) |
| CL B,CPI (L/h) | 7.23 (13) | — | ||
| CL R,CPI,HV (L/h) | 4.58 (12) | 33.4 (17) | ||
| V C,CPI (L) | 11 (17) | — | ||
| CL active,CPI (L/h) | 1,040 (22) | 28.9 (19) | ||
| CL passive,CPI (L/h) | 7.7 FIXED | — | ||
| Covariates | COV CLR | 1.05 (10) | — | |
| COV ksyn | 0.225 (10) | — | ||
| Residual unexplained variabilities | σ prop – CPI plasma (%) | 18.6 (4) | — | |
| σ add – CPI plasma (nM) | 0.0005 FIXED | — | ||
| σ prop – CPI urine (%) | 47.4 (6) | — | ||
| σ add – CPI urine (nMol) | 0.01 FIXED | — | ||
Parameter estimates in models for PTV (pitavastatin), RIF (rifampin), and CPI (coproporphyrin I) in the healthy population and the population with chronic kidney disease (CKD).
BPV, between‐participant variability; CL active or CL passive , hepatic active or passive uptake clearance (unbound) corrected for hepatocellularity (120 × 106 cells/g of liver); CL B , biliary clearance; CL R , renal clearance; CL RIF , clearance of rifampin; Corr V RIF and CL RIF , correlation between V RIF and CL RIF ; COV CLactive,Gx , a fractional change in CL active in CKD category Gx; COV CLR , power coefficient for CL R of CPI; COV CLrif,BW , continuous covariate (body weight) on CL RIF ; COV ksyn , power coefficient for k syn ; COV Tk0,SEX , categorical covariate (sex) on Tk0 RIF ; COV Vrif,BW , continuous covariate (body weight) on V RIF ; f d1, fractions of Q CO for tissue compartment 1; ka, first‐order absorption rate constant; K i,total , total rifampin inhibition constant for CL active of CPI; K p , tissue partition coefficient; k syn,HV&mild , rate of CPI synthesis in the healthy population and the population with mild CKD; Tk0 RIF,male , duration of zero‐order absorption for male; Tlag RIF , lag time of absorption; V 1_frac , a fraction of V 1 ; V c, volume of central compartment; V RIF , volume of distribution of rifampin; σ add , additive residual error; σ prop , proportional residual error; —, not applicable.
aThe population (fixed effect) parameters and relative standard errors (RSEs, %).
Estimated BPV (%) and its RSE (%).
Rifampin CKD model
The final rifampin model adequately described the observed plasma concentration data in all groups (Figures S11 and S12 ), with body weight and sex as covariates on total clearance (CL RIF ), volume of distribution (V RIF ), duration of zero‐order absorption, and correlation between CL RIF and V RIF (Table 1). Model parameters were estimated with RSE <40%. Use of eGFR as a covariate in the model did not improve model fitting significantly (data not shown), consistent with no trend between rifampin AUC and eGFR (Figure S13 ).
Physiologically‐based CPI CKD model
The base CPI model included CKD effects on f u,p and CL active,CPI . The CPI f u,p in severe CKD (0.078) was 13% higher than in healthy participants due to lower plasma albumin in this CKD group (Table S1 ). In contrast, measured albumin in patients with mild to moderate CKD was comparable to healthy individuals, resulting in similar CPI f u,p in those groups. Significant correlations between C base,CPI and estimated CL active,PTV (R = −0.61, P < 0.01) (Figure 2 ), as well as C base,CPI and AUCPTV,control (R = 0.76, P < 0.01), suggested that decreased OATP1B activity in CKD is contributing to increased exposure of CPI and pitavastatin in this patient population. It also rationalized implementation of CKD effect on CL active,CPI estimated by modeling of pitavastatin data in the CPI model.
Figure 2.
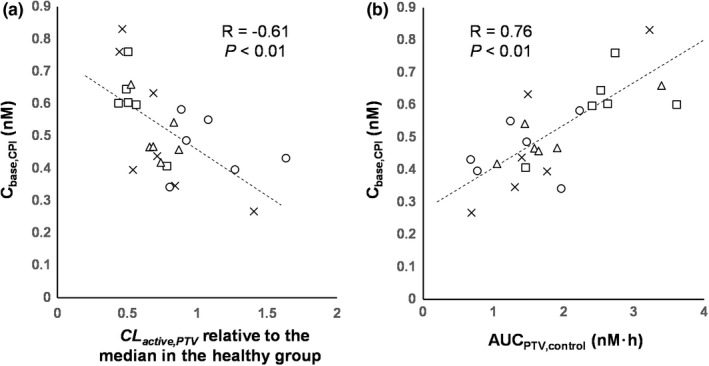
Correlation between baseline coproporphyrin I (CPI) exposure and pitavastatin PK parameters of healthy participants and participants with chronic kidney disease (CKD). Correlations between mean baseline concentration of CPI (Cbase,CPI) and (a) individuals' CL active,PTV estimated in the final pitavastatin CKD model (expressed as relative values to median of those in the healthy group) or (b) pitavastatin AUC without rifampin (AUCPTV,control). Marks represent individual participants, and symbols represent CKD groups: healthy (circle), mild CKD (triangle), moderate CKD (square), and severe CKD (cross). Dashed lines represent regression lines. AUC, area under the plasma concentration‐time curve; CL active,PTV , hepatic uptake clearance of pitavastatin; PK, pharmacokinetic.
Selection of the final CPI model was based on both disease‐related physiological changes and decrease in objective function value relative to the base model with CKD effects on f u,p and CL active,CPI . Implementation of the covariates on CL R,CPI and k syn was statistically significant, hence, the final model included CKD‐related effects on f u,p , CL active,CPI , CL R,CPI , and k syn . Fixed effects parameters were estimated with RSE <40% (Table 1). The final model adequately described the observed CPI data in both healthy and CKD groups (Figure 3 and Figure S14 ), including reduced urinary CPI elimination and increased Cbase,CPI. Additionally, the model also described well the increased magnitude of OATP1B interaction (AUCRCPI) in the population with CKD. Consistent with results from the previous CPI modeling work, 17 hepatic uptake was a major contributor to CPI elimination (88% in healthy population) and renal elimination accounted for the remaining component. CL R,CPI in CKD decreased proportionally to eGFR (COV CLR = 1.03), consistent with correlations between eGFR and CL R,CPI or net secretory renal excretion clearance of CPI (Figure S15 ). The estimated effect of CKD on CPI synthesis rate (COV ksyn ) was equivalent to 27% lower k syn in participants with severe CKD.
Figure 3.
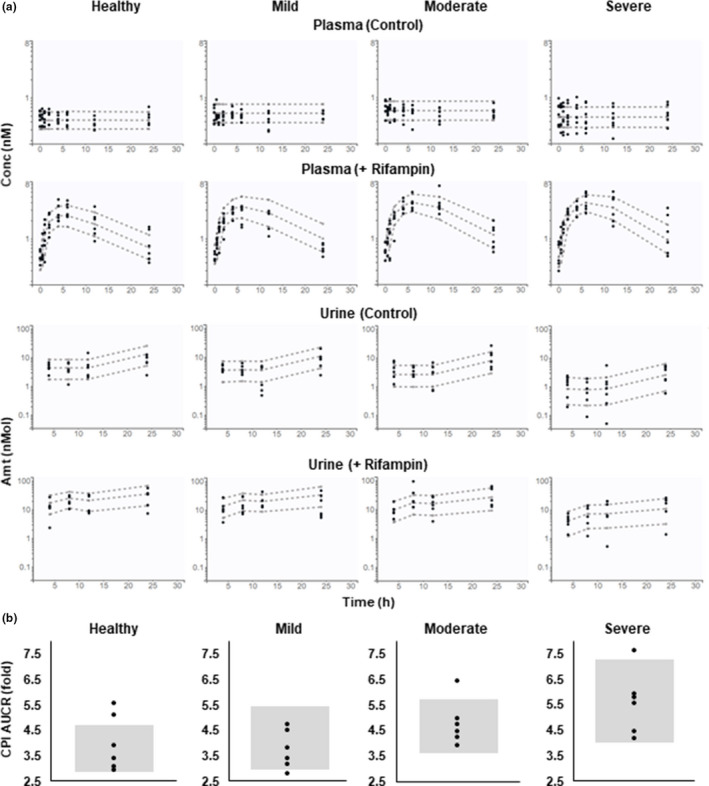
VPC (Visual predictive check) for coproporphyrin I (CPI) in plasma and urine, and CPI AUCR in healthy and chronic kidney disease (CKD) groups. (a) CPI in plasma and urine on two occasions with (+ Rifampin) or without rifampin treatment (Control) was simulated for each group. Circles, observed data; dashed lines, 10%, 50%, and 90% quantiles of the prediction (based on 5,000 simulated individuals in each group). (b) CPI AUCR (AUC with rifampin/AUC without rifampin) was simulated for each group. Circles and gray areas represent observed data and 10–90% quantiles of the prediction (based on 5,000 simulated individuals in each group), respectively. Amt, amount; AUC, area under the plasma concentration‐time curve; Conc, concentration.
Calculation of normalized sensitivity coefficients showed that Cbase,CPI was sensitive to CKD‐related changes in k syn , CL activeCPI , f u,p,CPI , blood to plasma ratio, and less sensitive to CL R,CPI (Figure 4 a ). The same tendency was seen when percent changes in each CPI parameter in severe CKD were calculated (Figure 4 b ). Reduced CL active,CPI and k syn (−39% and −27% in severe CKD, respectively) prominently increased and decreased Cbase,CPI, whereas the impact of f u,p,CPI and CL R,CPI on Cbase,CPI was less evident. In contrast to CPI baseline, differences in AUCRCPI in patients with CKD relative to healthy participants were predominantly attributed to prominent disease effect on parallel elimination route (~ 90% decrease in CL R,CPI in severe CKD) relative to CL active,CPI . Both k syn and f u,p,CPI had no effect on AUCRCPI.
Figure 4.
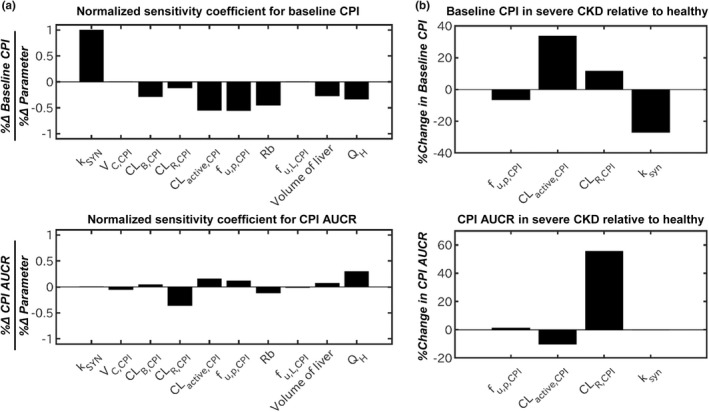
Parameter sensitivity analysis for baseline plasma concentration of coproporphyrin I (CPI) and AUCR for CPI–rifampin interaction. (a) Normalized sensitivity coefficient of CPI model parameters for baseline CPI or CPI AUCR (AUC with rifampin/AUC without rifampin) calculated based on the developed CPI model for a typical healthy participant. (b) Percent change in baseline CPI or CPI AUCR in severe CKD relative to healthy population calculated based on the magnitude of CKD‐related physiological changes estimated in the CPI CKD model. Volume of liver represents volume of the whole liver including both V LV and V LT . AUC, area under the plasma concentration‐time curve; CKD, chronic kidney disease; CL active,CPI , hepatic uptake clearance; CL B,CPI , biliary clearance; CL R,CPI , renal clearance; f u,p,CPI , fraction unbound in plasma; f u,L,CPI , fraction unbound in liver; k syn , endogenous synthesis rate; Q H , hepatic blood flow; Rb, blood to plasma ratio; V C,CPI , volume of central compartment; V LT , volume of liver tissue; V LV , volume of liver vascular.
Interpretation of CKD‐related difference in the magnitude of OATP1B‐mediated interactions
The mechanism(s) contributing to potential differences in OATP1B AUCR in the population with CKD were investigated for a range of hypothetical OATP1B probes with different contribution of renal elimination (f e, urine,HV ). For a CPI‐equivalent drug (f e,urine,HV ; 0.15), decline in CL R increased f OATP1B of 0.85 in the healthy population to 0.97 in severe CKD, regardless of disease‐related changes in CL active (Figure 5 a ). This shift in f OATP1B by CKD resulted in predicted 1.8‐fold higher AUCR in severe CKD relative to healthy participants (Figure 5 c ), in good agreement with the observed CPI data (1.6‐fold, Figure S1 ). However, these trends were not apparent for pitavastatin‐equivalent drug with a negligible renal elimination (f e,urine,HV ; 0.01), resulting in no increase in AUCR in severe CKD relative to the healthy population (Figure 5 b,d ), in line with comparable pitavastatin–rifampin DDI reported in healthy patients and patients with CKD.
Figure 5.
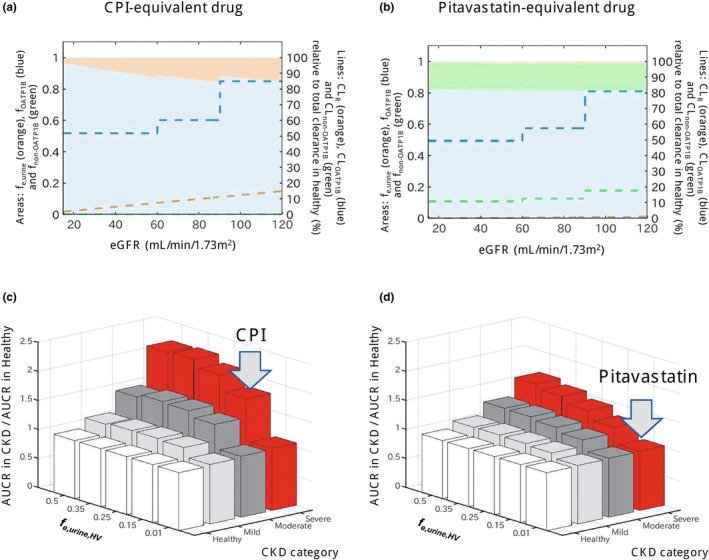
Effect of chronic kidney disease (CKD) on OATP1B‐mediated interactions for drugs with different contributions of hepatic and renal elimination. (a,b) Change in fraction transported of CPI‐equivalent and pitavastatin‐equivalent drugs by OATP1B (f OATP1B , blue area), non‐OATP1B hepatic uptake (f non‐OATP1B , green area), and renal elimination (f e,urine , orange area) derived from CKD‐derived decline in each route. Dashed lines represent clearances via each elimination route (CL OATP1B , CL non‐OATP1B , and CL R ) in different stages of CKD, expressed as relative values to a total clearance in healthy population (label on the right axis). The CPI‐equivalent drug has f e,urine in the healthy population (f e,urine,HV ) of 0.15 and hepatic uptake via OATP1B only (f OATP1B,HV of 0.85). The pitavastatin‐equivalent drug has minimal renal elimination (f e,urine,HV of 0.01) and hepatic uptake via OATP1B (f OATP1B of 0.812, 82% of nonrenal clearance) and non‐OATP1B route (f non‐OATP1B,HV of 0.178, 18% of nonrenal clearance). Simulations were performed assuming that both OATP1B and non‐OATP1B routes contributing to CL active decline to the same extent in CKD and that decrease in CL R is proportional to decline in eGFR (healthy: CL active 100% and CL R 100%; mild CKD: CL active 71% and CL R 75%; moderate CKD: CL active 61% and CL R 50%; severe CKD: CL active 61% and CL R 13%). (c,d) Ratio of AUCR (with/without OATP1B inhibitor) in the population with CKD relative to the healthy population calculated for hypothetical OATP1B drugs with f e,urine,HV ranging from 0.01 to 0.5 and different proportion of non‐OATP1B route to total hepatic uptake clearance (c: none, d: 18%); all assumptions as highlighted above. Gray arrows indicate drugs equivalent to CPI and pitavastatin. Simulations illustrate that presence of non‐OATP1B‐mediated hepatic clearance (assumed to decline in the same manner as OATP1B in CKD) decreases the difference in OATP1B AUCR between CKD and healthy, as the CKD‐derived shift in fraction transported is then not solely attributed to OATP1B. AUCR, ratio of area under the plasma concentration‐time curve; CL active , hepatic uptake clearance; CL R , renal clearance; CPI, coproporphyrin I; eGFR, estimated glomerular filtration rate; OATP1B, organic anion transporting polypeptide 1B.
DISCUSSION
A recent clinical study reported increased baseline exposure of CPI and pitavastatin in patients with CKD, 8 consistent with previous studies with other OATP1B substrates. 5 , 8 , 10 , 22 , 23 , 24 Those patients with CKD had higher rifampin‐associated AUCR for CPI, but not for pitavastatin. Using population PBPK modeling, the current study investigated the complex interplay of multiple CKD‐related physiological changes to understand mechanism(s) of disease‐related increase in CPI baseline and its utility as a biomarker for evaluation of OATP1B function and DDIs in the population with CKD. Additionally, modeling was used to provide mechanistic interpretation for inconsistency in magnitude of OATP1B‐mediated interaction of CPI and pitavastatin in patients with CKD, with the aim to inform the necessity of prospective clinical OATP1B DDI studies in the population with CKD.
Interpretation of altered PK of CPI in CKD based on PBPK modeling
The CPI CKD model was developed by optimizing covariate structure of the existing population PBPK model for CPI 25 considering physiological changes in CKD; the final model included the disease effects on f u,p , CL R,CPI , CL active,CPI , and k syn . Because of the high extent of CPI plasma protein binding in healthy participants (f u,p of 0.069 (ref. 25)), CKD‐related changes in plasma albumin were relevant for interpretation of clinical findings. In the absence of measured CPI f u,p in CKD, this parameter was predicted based on f u,p in healthy participants and measured plasma albumin in patients with CKD 28 ; this approach was considered appropriate as CPI is predominantly bound to albumin. 46 Prominent decrease in plasma albumin in severe CKD resulted in 13% higher predicted CPI f u,p compared with healthy participants, in agreement with trends seen in measured binding data for pitavastatin (Table S1 , 23% increase in severe CKD).
The noncompartment analysis indicated that CL R,sec,net accounted for 78% of CL R,CPI in the healthy population, implying the presence of tubular renal secretion. Glomerular filtration and tubular secretion were lumped as a single parameter in the CPI model (CL R,CPI ) and assumed to decrease in proportion to eGFR in CKD. The latter assumption was supported by the linear relationship between eGFR and CL R,CPI (Figure S15 ). The mechanism of tubular renal secretion of CPI is unknown, but multidrug resistance‐associated protein(s) (MRP(s)) located on the apical membrane could be involved. 37
Because of the correlation between CL active,CPI and k syn , direct estimation of CKD effects on these parameters in the CPI CKD model resulted in parameter identifiability issue. Therefore, the CKD effect on OATP1B‐mediated uptake in the CPI model was informed by independent modeling of pitavastatin data (a well‐characterized clinical OATP1B probe 47 ) in the same individuals. This approach resolved the identifiability issue and enabled separation of the disease effect on CL active from other confounding factors. A good correlation between an individual's Cbase,CPI and AUCPTV,control or CL active,PTV estimated with the pitavastatin CKD model (Figure 2 ) also rationalized this approach. Estimated decrease in OATP1B‐mediated CL active was 29% and 39% in mild and moderate to severe CKD, respectively, consistent with a previous meta‐analysis of OATP1B activity in patients with CKD (~ 50% decline). 5 The possibility of reduced CPI synthesis in CKD was based on a strong relationship between CPI and hemoglobin in blood 48 and proportional decrease in blood hemoglobin and eGFR in moderate to severe CKD. 26 Estimated 27% lower k syn in severe CKD was consistent with ~ 20% lower blood hemoglobin in the same CKD group.
Estimated disease effect on OATP1B activity was conditional upon assumed fixed contributions of non‐OATP1B (18% of CL active ) or passive diffusion clearance (1.6% of hepatic uptake) in the pitavastatin model, both of which were in agreement with previous studies. 32 , 34 , 35 , 44 The pitavastatin CKD model assumed the same degree of disease effect on both OATP1B‐mediated and non‐OATP1B‐mediated hepatic uptake of pitavastatin. The alternative model (assuming no disease effect on the non‐OATP1B route) showed comparable goodness‐of‐fit plots, but a slightly larger estimated disease effect on CL OATP1B (37% and 50% decline in mild and moderate to severe CKD) (Table S3 ). Similarly, this assumption estimated more pronounced disease effect on k syn in the CPI model (36% decline in severe CKD) considering correlation between CL active and k syn , highlighting the necessity for sensitivity analysis on these parameters when used in further analyses.
Parameter sensitivity analysis showed that CKD effects on CL active,CPI and CL R,CPI affect prominently Cbase,CPI and AUCRCPI, respectively. Although the clinical DDI study 8 attributed higher Cbase,CPI in participants with CKD solely to decreased renal elimination, the modeling revealed the importance of changes in CL active,CPI (Figure 4 b ). Additionally, CKD‐related changes in f u,p,CPI (specific to severe CKD) and k syn decreased Cbase,CPI, which might partially contribute to observed nonlinear increase in Cbase,CPI in severe CKD and larger variability (Figure S1 ). CKD‐related decline in CL active,CPI (39% lower in severe CKD relative to healthy) was expected to decrease magnitude of OATP1B‐mediated DDI in these patients, analogous to our previous study where 42% lower CL active,CPI in Asian‐Indian participants relative to White participants resulted in up to 22% lower rifampin OATP1B‐mediated interaction. 25 However, more prominent disease effect on CPI parallel elimination route (CL R,CPI ) than on the OATP1B resulted in f OATP1B shift and apparent increase in AUCRCPI in the population with CKD. CKD‐related changes in k syn and f u,p,CPI had marginal effect on AUCRCPI, as these did not affect f OATP1B . CKD effects on CPI biliary clearance and blood to plasma ratio were not considered despite moderate sensitivity of CPI PK to these parameters. The CKD effect on CPI biliary clearance was inconclusive due to the lack of information in human and inconsistency in rat data. 41 , 42 , 43 Implementation of lower blood to plasma ratio, attributed to decreased hematocrit because of decreased production of red blood cells in CKD, 49 had marginal impact on results (data not shown) and therefore was not included in the final model.
Utility of CPI as an endogenous biomarker of OATP1B activity and corresponding interaction risk in CKD
Good correlation between individual Cbase,CPI and AUCPTV,control values (Figure 2 b ) supported the assumption that decreased OATP1B activity in CKD was the common underlying mechanism for increased exposure of pitavastatin and CPI in this population. These observations are in agreement with previously reported correlations between AUCs of OATP1B probe drugs and CPI in a healthy population. 50 This correlation between individual plasma CPI and pitavastatin AUC in patients with CKD provides a possibility to enable individualized therapeutic dosing of statins based on an individual's OATP1B activity informed by CPI levels.
A key finding of this study is that the magnitude of the OATP1B interaction (AUCR) may increase in patients with CKD relative to the healthy population due to increased f OATP1B caused by abolished renal clearance as a parallel elimination route. Our analysis showed that the extent of this CKD‐related increase in AUCR depended on the severity of CKD and f e,urine,HV of OATP1B probes (Figure 5 ). Monitoring of CPI as a probe with high selectivity to OATP1B and combined hepatic and renal elimination provides valuable information on such CKD‐related difference/increase in OATP1B AUCR. The current modeling showed that CKD‐mediated decrease in CPI renal clearance (~ 90% lower in severe CKD) increased its f OATP1B , compensating for the disease effect on OATP1B (39% lower activity in severe CKD). For OATP1B probes with negligible renal elimination (low f e,urine,HV as pitavastatin), difference in OATP1B interaction between healthy population and patients with CKD was marginal, indicating that OATP1B DDI data for such drugs obtained in healthy patients are informative also for patients with CKD. Additional considerations are selectivity of probes for OATP1B and understanding of any confounding effects of CKD‐related change(s) on non‐OATP1B pathway(s) (hepatic and nonhepatic). Rosuvastatin showed marginal CKD‐related increase in rifampin OATP1B interaction relative to a healthy population 8 despite its combined hepatic‐renal elimination of rosuvastatin (f e,urine,HV of 0.28 (ref. 5)). In contrast to CPI, the role of intestinal transporters (BCRP, P‐gp, and to some extent OATP2B1) and disease‐related changes in these processes is an additional consideration. Partial contribution of non‐OATP1B‐mediated hepatic uptake (28% via NTCP 34 ) and proposed decrease in activity of intestinal P‐gp (likely also for BCRP) reported in patients with CKD 8 may have contributed to large variability in rosuvastatin PK and confounded interpretation of the OATP1B DDI data for dual OATP1B and P‐gp/BCRP substrates. The small number of OATP1B substrates evaluated highlights the necessity for further clinical DDI studies in the population with CKD to confirm these findings.
In conclusion, mechanistic analysis using population PBPK modeling attributed increased CPI baseline and pitavastatin exposure in patients with CKD to disease‐related decrease in OATP1B activity (in addition to changes in plasma protein binding and renal excretion). The current modeling work provides invaluable information on altered OATP1B in CKD for the refinement of corresponding PBPK models and improvement of prospective prediction of OATP1B‐mediated DDI risk in these patients. Mechanistic modeling of complex disease–drug interaction data revealed a shift in fraction of CPI eliminated by OATP1B in CKD attributed to decline in renal elimination, resulting in increased sensitivity to rifampin inhibition in severe CKD. In contrast, such change was not apparent for pitavastatin due to its negligible renal elimination. The current analysis highlights that use of a clinical probe eliminated solely by hepatic route (e.g., pitavastatin) may underestimate the extent of OATP1B DDI in CKD of drugs with combined hepatic and renal elimination. To that end, partial contribution of renal excretion to elimination of CPI reinforces its value as a clinical tool for evaluation of OATP1B‐mediated DDI risk in both healthy patients and patients with CKD. Monitoring of this endogenous biomarker also provides data in patients with CKD critical for validation of PBPK modeling, bridging the translation of interaction risk from the healthy population to the population with CKD.
FUNDING
H.T. was financially supported by a fellowship grant from Asahi Kasei Pharma Corporation.
CONFLICT OF INTEREST
The other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
H.T., D.S., X.C., K.L.Y., K.O., and A.G. wrote the manuscript. H.T., D.S., X.C., K.L.Y., K.O., and A.G. designed the research. H.T. performed the research. H.T. analyzed the data.
Supporting information
Appendix S1
Appendix S2
References
- 1. Evers, R. et al. Disease‐associated changes in drug transporters may impact the pharmacokinetics and/or toxicity of drugs: a white paper from the International Transporter Consortium. Clin. Pharmacol. Ther. 104, 900–915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lea‐Henry, T.N. , Carland, J.E. , Stocker, S.L. , Sevastos, J. & Roberts, D.M. Clinical pharmacokinetics in kidney disease: fundamental principles. Clin. J. Am. Soc. Nephrol. 13, 1085–1095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeung, C.K. et al. Organ impairment‐drug‐drug interaction database: a tool for evaluating the impact of renal or hepatic impairment and pharmacologic inhibition on the systemic exposure of drugs. CPT Pharmacometrics Syst. Pharmacol. 4, 489–494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miners, J.O. , Yang, X. , Knights, K.M. & Zhang, L. The role of the kidney in drug elimination: transport, metabolism, and the impact of kidney disease on drug clearance. Clin. Pharmacol. Ther. 102, 436–449 (2017). [DOI] [PubMed] [Google Scholar]
- 5. Tan, M.L. et al. Effect of chronic kidney disease on nonrenal elimination pathways: a systematic assessment of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP. Clin. Pharmacol. Ther. 103, 854–867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoshida, K. et al. Systematic and quantitative assessment of the effect of chronic kidney disease on CYP2D6 and CYP3A4/5. Clin. Pharmacol. Ther. 100, 75–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takita, H. , Scotcher, D. , Chinnadurai, R. , Kalra, P.A. & Galetin, A. Physiologically‐based pharmacokinetic modelling of creatinine‐drug interactions in the chronic kidney disease population. CPT Pharmacometrics Syst. Pharmacol. 9, 695–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tatosian, D.A. et al. A microdose cocktail to evaluate drug interactions in patients with renal impairment. Clin. Pharmacol. Ther. 109, 403–415 (2021). [DOI] [PubMed] [Google Scholar]
- 9. Tan, M.‐L. et al. Use of physiologically based pharmacokinetic modeling to evaluate the effect of chronic kidney disease on the disposition of hepatic CYP2C8 and OATP1B drug substrates. Clin. Pharmacol. Ther. 105, 719–729 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujita, K. et al. Decreased disposition of anticancer drugs predominantly eliminated via the liver in patients with renal failure. Curr. Drug Metab. 20, 361–376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. US Food and Drug Administration . Guidance for industry: pharmacokinetics in patients with impaired renal function — study design, data analysis, and impact on dosing and labeling <https://www.fda.gov/media/78573/download> (2020). Accessed April 10, 2022.
- 12. Storelli, F. , Samer, C. , Reny, J.L. , Desmeules, J. & Daali, Y. Complex drug–drug–gene–disease interactions involving cytochromes P450: systematic review of published case reports and clinical perspectives. Clin. Pharmacokinet. 57, 1267–1293 (2018). [DOI] [PubMed] [Google Scholar]
- 13. Zhao, P. , Zhang, L. & Huang, S.M. Complex drug interactions: significance and evaluation. In Enzyme‐ Transporter‐Based Drug‐Drug Interactions: Progress and Future Challenges (eds. Pang, K.S. , Rodrigues, A.D. & Peter, R.M. ) 667–692 (Springer, New York, 2010). [Google Scholar]
- 14. Galetin, A. , Zhang, L. , Rodrigues, A.D. & Huang, S.M. Drug‐drug interactions. In Atkinson's Principles of Clinical Pharmacology 4th edn., chapter 14 (eds. Huang, S.M. , Lertora, J.J.L. , Vicini, P. & Atkinson, J.A.J. ) 241–265 (Academic Press, Boston, MA, 2022). [Google Scholar]
- 15. European Medicines Agency . Guideline on the investigation of drug interactions <https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf>. Accessed April 10, 2022.
- 16. US Food and Drug Administration . Enhancing the diversity of clinical trial populations — eligibility criteria, enrollment practices, and trial designs <https://www.fda.gov/media/127712/download> (2020). Accessed April 10, 2022.
- 17. Barnett, S. et al. Gaining mechanistic insight into coproporphyrin I as endogenous biomarker for OATP1B‐mediated drug–drug interactions using population pharmacokinetic modeling and simulation. Clin. Pharmacol. Ther. 104, 564–574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barnett, S. , Ogungbenro, K. , Ménochet, K. , Shen, H. , Humphreys, W.G. & Galetin, A. Comprehensive evaluation of the utility of 20 endogenous molecules as biomarkers of OATP1B inhibition compared with rosuvastatin and coproporphyrin I. J. Pharmacol. Exp. Ther. 368, 125–135 (2019). [DOI] [PubMed] [Google Scholar]
- 19. Mori, D. et al. Effect of OATP1B1 genotypes on plasma concentrations of endogenous OATP1B1 substrates and drugs, and their association in healthy volunteers. Drug Metab. Pharmacokinet. 34, 78–86 (2019). [DOI] [PubMed] [Google Scholar]
- 20. Yee, S.W. et al. Organic anion transporter polypeptide 1B1 polymorphism modulates the extent of drug–drug interaction and associated biomarker levels in healthy volunteers. Clin. Transl. Sci. 12, 388–399 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Livalo (pitavastatin) [highlights of prescribing information]. (Kowa Pharmaceuticals, Montgomery, AL, 2019). <https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/022363s015lbl.pdf>. Accessed April 10, 2022. [Google Scholar]
- 22. Morgan, R.E. , Campbell, S.E. , Yu, C.Y. , Sponseller, C.A. & Muster, H.A. Comparison of the safety, tolerability, and pharmacokinetic profile of a single oral dose of pitavastatin 4 mg in adult subjects with severe renal impairment not on hemodialysis versus healthy adult subjects. J. Cardiovasc. Pharmacol. 60, 42–48 (2012). [DOI] [PubMed] [Google Scholar]
- 23. Bergman, A. et al. Effect of hepatic organic anion‐transporting polypeptide 1B inhibition and chronic kidney disease on the pharmacokinetics of a liver‐targeted glucokinase activator: a model‐based evaluation. Clin. Pharmacol. Ther. 106, 792–802 (2019). [DOI] [PubMed] [Google Scholar]
- 24. Zhao, P. et al. Evaluation of exposure change of nonrenally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. J. Clin. Pharmacol. 52, 91S–108S (2012). [DOI] [PubMed] [Google Scholar]
- 25. Takita, H. et al. PBPK model of coproporphyrin I: evaluation of the impact of SLCO1B1 genotype, ethnicity, and sex on its inter‐individual variability. CPT Pharmacometrics Syst. Pharmacol. 10, 137–147 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Astor, B.C. , Muntner, P. , Levin, A. , Eustace, J.A. & Coresh, J. Association of kidney function with anemia: the third National Health and Nutrition Examination Survey (1988‐1994). Arch. Intern. Med. 162, 1401–1408 (2002). [DOI] [PubMed] [Google Scholar]
- 27. Levey, A.S. et al. A new equation to estimate glomerular filtration rate. Arch. Intern. Med. 150, 604–612 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rowland Yeo, K. , Aarabi, M. , Jamei, M. & Rostami‐Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 4, 261–274 (2011). [DOI] [PubMed] [Google Scholar]
- 29. Proost, J.H. Combined proportional and additive residual error models in population pharmacokinetic modelling. Eur. J. Pharm. Sci. 109, S78–S82 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Cao, Y. & Jusko, W.J. Applications of minimal physiologically‐based pharmacokinetic models. J. Pharmacokinet. Pharmacodyn. 39, 711–723 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gertz, M. , Tsamandouras, N. , Säll, C. , Houston, J.B. & Galetin, A. Reduced physiologically‐based pharmacokinetic model of repaglinide: impact of OATP1B1 and CYP2C8 genotype and source of in vitro data on the prediction of drug‐drug interaction risk. Pharm. Res. 31, 2367–2382 (2014). [DOI] [PubMed] [Google Scholar]
- 32. Ménochet, K. , Kenworthy, K.E. , Houston, J.B. & Galetin, A. Use of mechanistic modeling to assess interindividual variability and interspecies differences in active uptake in human and rat hepatocytes. Drug Metab. Dispos. 40, 1744–1756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elsby, R. , Hilgendorf, C. & Fenner, K. Understanding the critical disposition pathways of statins to assess drug‐drug interaction risk during drug development: it's not just about OATP1B1. Clin. Pharmacol. Ther. 92, 584–598 (2012). [DOI] [PubMed] [Google Scholar]
- 34. Bi, Y.A. et al. Quantitative assessment of the contribution of sodium‐dependent taurocholate co‐transporting polypeptide (NTCP) to the hepatic uptake of rosuvastatin, pitavastatin and fluvastatin. Biopharm. Drug Dispos. 34, 452–461 (2013). [DOI] [PubMed] [Google Scholar]
- 35. Mitra, P. , Weinheimer, S. , Michalewicz, M. & Taub, M.E. Prediction and quantification of hepatic transporter‐mediated uptake of pitavastatin utilizing a combination of the relative activity factor approach and mechanistic modeling. Drug Metab. Dispos. 46, 953–963 (2018). [DOI] [PubMed] [Google Scholar]
- 36. Weigand, K.M. et al. Uremic solutes modulate hepatic bile acid handling and induce mitochondrial toxicity. Toxicol. In Vitro 56, 52–61 (2019). [DOI] [PubMed] [Google Scholar]
- 37. Kunze, A. , Ediage, E.N. , Dillen, L. , Monshouwer, M. & Snoeys, J. Clinical investigation of coproporphyrins as sensitive biomarkers to predict mild to strong OATP1B‐mediated drug–drug interactions. Clin. Pharmacokinet. 57, 1559–1570 (2018). [DOI] [PubMed] [Google Scholar]
- 38. Lai, Y. et al. Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug‐drug interactions mediated by organic anion transporting polypeptide inhibition. J. Pharmacol. Exp. Ther. 358, 397–404 (2016). [DOI] [PubMed] [Google Scholar]
- 39. Leblanc, M. , Roy, L.F. , Villeneuve, J.P. , Malo, B. , Pomier‐Layrargues, G. & Legault, L. Liver blood flow in chronic hemodialysis patients. Nephron 73, 396–402 (1996). [DOI] [PubMed] [Google Scholar]
- 40. Blum, M. , Tchetchik, M. , Schujman, E. & Aviram, A. Liver enlargement in long‐term hemodialysis patients. Arch. Intern. Med. 140, 343–344 (1980). [PubMed] [Google Scholar]
- 41. Naud, J. , Michaud, J. , Leblond, F.A. , Lefrancois, S. , Bonnardeaux, A. & Pichette, V. Effects of chronic renal failure on liver drug transporters. Drug Metab. Dispos. 36, 124–128 (2008). [DOI] [PubMed] [Google Scholar]
- 42. Lu, H. & Klaassen, C. Gender differences in mRNA expression of ATP‐binding cassette efflux and bile acid transporters in kidney, liver, and intestine of 5/6 nephrectomized rats. Drug Metab. Dispos. 36, 16–23 (2008). [DOI] [PubMed] [Google Scholar]
- 43. Holzer, B. , Stieger, B. , Folkers, G. , Meier, P.J. & Fattinger, K. Differential regulation of basolateral and canalicular transporter expression in rat liver in chronic renal failure. Clin. Pharmacol. Ther. 77, P34(2005). [Google Scholar]
- 44. Varma, M.V. , Bi, Y.A. , Kimoto, E. & Lin, J. Quantitative prediction of transporter‐ and enzyme‐mediated clinical drug‐drug interactions of organic anion‐transporting polypeptide 1B1 substrates using a mechanistic net‐effect model. J. Pharmacol. Exp. Ther 351, 214–223 (2014). [DOI] [PubMed] [Google Scholar]
- 45. Kunze, A. , Poller, B. , Huwyler, J. & Camenisch, G. Application of the extended clearance concept classification system (ECCCS) to predict the victim drug‐drug interaction potential of statins. Drug Metab. Pers. Ther. 30, 175–188 (2015). [DOI] [PubMed] [Google Scholar]
- 46. Ding, Y. , Lin, B. & Huie, C.W. Binding studies of porphyrins to human serum albumin using affinity capillary electrophoresis. Electrophoresis 22, 2210–2216 (2001). [DOI] [PubMed] [Google Scholar]
- 47. Prueksaritanont, T. et al. Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suzuki, Y. et al. Simultaneous quantification of coproporphyrin‐I and 3‐carboxy‐4‐methyl‐5‐propyl‐2‐furanpropanoic acid in human plasma using ultra‐high performance liquid chromatography coupled to tandem mass spectrometry. J. Pharm. Biomed. Anal. 184, 113202 (2020). [DOI] [PubMed] [Google Scholar]
- 49. Hsu, C.Y. , Bates, D.W. , Kuperman, G.J. & Curhan, G.C. Relationship between hematocrit and renal function in men and women. Kidney Int. 59, 725–731 (2001). [DOI] [PubMed] [Google Scholar]
- 50. Mori, D. et al. Dose‐dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin. Pharmacol. Ther. 107, 1004–1013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2