

Abstract
Background
Alzheimer's disease (AD) co‐pathology is common in dementia with Lewy bodies and is associated with increased decline. Plasma pTau181 is a blood‐based biomarker that can detect AD co‐pathology.
Objectives
We investigated whether pTau181 was associated with cognitive decline in mild cognitive impairment with Lewy bodies (MCI‐LB) and MCI with AD (MCI‐AD).
Methods
We assessed plasma pTau181 using a single‐molecule array (Simoa) immunoassay at baseline and follow‐up in a longitudinal cohort of MCI‐LB, MCI‐AD, and controls.
Results
One hundred forty‐six subjects (56 probable MCI‐LB, 22 possible MCI‐LB, 44 MCI‐AD, and 24 controls) were reviewed for up to 5.7 years. Probable MCI‐LB had significantly higher pTau181 (22.2% mean increase) compared with controls and significantly lower (24.4% mean decrease) levels compared with MCI‐AD. Receiver operating characteristic analyses of pTau181 in discriminating probable MCI‐LB from controls showed an area under the curve (AUC) of 0.68 (83% specificity, 57% sensitivity); for discriminating MCI‐AD from healthy controls, AUC was 0.8 (83.3% specificity, 72.7% sensitivity). pTau181 concentration was less useful in discriminating between probable MCI‐LB and MCI‐AD: AUC of 0.64 (71.4% specificity, 52.3% sensitivity).
There was an association between pTau181 and cognitive decline in MCI‐AD but not in MCI‐LB. In a subset with repeat samples there was a nonsignificant 3% increase per follow‐up year in plasma pTau181. The rate of change in pTau181 was not significantly different in different diagnostic subgroups.
Conclusions
pTau181 was not associated with an increased decline assessed using either baseline or repeat pTau181. pTau181 partially discriminated probable MCI‐LB from controls and MCI‐AD from controls but was not useful in distinguishing probable MCI‐LB from MCI‐AD. © 2022 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: biomarkers, plasma, pTau181, mild cognitive impairment, dementia with Lewy bodies
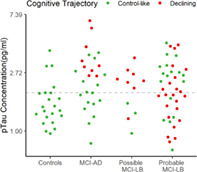
Elevated plasma pTau181 is specifically associated with a declining cognitive trajectory in mild cognitive impairment due to Alzheimer's disease. However, this is not the case in probable mild cognitive impairment with Lewy bodies: whereas many who decline have elevated pTau181 concentrations, many also decline who do not.
Alzheimer's disease (AD) and dementia with Lewy bodies (DLB) are the two most common neurodegenerative causes of dementia, between them accounting for 79% of cases in a UK brain bank study. 1 Both these diseases have a mild cognitive impairment (MCI) prodrome, and we have reported that MCI with Lewy bodies (MCI‐LB) has a faster decline and increased conversion to dementia compared with MCI‐AD, 2 , 3 which is consistent with evidence that at the dementia stage people with DLB have more rapid decline 4 and decreased survival times 5 compared with AD.
A large UK brain bank study with prospective clinical review reported that additional neurodegenerative co‐pathologies were associated with a 20‐fold increase in conversion to dementia, 1 and another report from a large prospective autopsy series found additionally that LB disease was a potent contributor at the person‐specific level to such accelerated decline. 6 Reduced Aβ42 in cerebrospinal fluid (CSF), a marker of AD, is predictive of faster decline in DLB. 7 Other work has reported that such multiple pathologies are already present at the MCI stage,8 making it plausible that the presence of AD pathologies in MCI‐LB patients would accelerate decline.
Recently, blood‐based biomarkers for identifying AD have been reported, in particular plasma measures of phosphorylated tau. 9 Phosphorylated tau at threonine 181 of the human tau protein (pTau181) has shown utility in detecting AD, 10 with performance comparable to CSF biomarkers and positron emission tomography (PET) tau 11 and validated against autopsy assessment of AD pathology. 11 , 12 Elevated pTau181 measures may track not only tau pathology but also early amyloid deposition. 13 pTau181 also detects preclinical AD and predicts both longitudinal decline and conversion from MCI to AD dementia. 10 , 14 , 15
Less is known about the use of these plasma biomarkers in DLB. One study of 35 people with Lewy body dementia (30 DLB and 5 PDD) found that pTau181 appears to be a useful marker of AD co‐pathology, because it correlated with CSF measures of AD and PET tau and predicted PET tau status. 16 No studies appear to have reported on MCI‐LB. A key issue is whether such biomarkers can facilitate the identification of the common AD co‐pathology in DLB/MCI‐LB and thus help predict individuals at risk of having a more rapid decline. pTau181 may be able to identify cases with coexisting AD 16 and enable an assessment of their longitudinal progression. We therefore conducted a prospective longitudinal study in MCI‐LB and MCI‐AD in which we measured pTau181 at baseline and conducted annual follow‐up reviews. We hypothesized that those with probable MCI‐LB who had baseline levels of pTau181 consistent with the presence of AD co‐pathology would have a more rapid decline and pTau181 would not differentiate between probable MCI‐LB and MCI‐AD but would show accuracy in differentiating probable MCI‐LB from controls. We also report on a subset of participants who had repeat pTau181 measures and on those with possible MCI‐LB.
Patients and Methods
Participants
As detailed previously, 17 , 18 , 19 medically stable patients aged 60 or above with a clinical diagnosis of MCI were recruited from local memory services in the northeast of England between April 2013 and September 2019 in two cohort studies, with the second having 123I‐metaiodobenzylguanidine (MIBG) cardiac scintigraphy in addition to dopaminergic 123I‐N‐fluoropropyl‐2β‐carbomethoxy‐3β‐(4‐iodophenyl) single‐photon emission computed tomography (FP‐CIT). Potential study participants reported the presence of either any core clinical feature of DLB (complex visual hallucinations, rapid eye movement sleep behavior disorder, cognitive fluctuations, or parkinsonism) or any supportive clinical feature found in DLB but not specific to this and so also common in AD (eg, depression, anxiety, and hyposmia). Exclusion criteria were dementia at screening, no objective cognitive impairment, or possible vascular or frontotemporal etiology (as evidenced by significant vascular or frontotemporal pathology on magnetic resonance imaging [MRI], strokes reported in clinical notes, or behavioral change consistent with a frontotemporal etiology). In addition, participants (healthy controls) with no evidence of cognitive impairment, parkinsonism, or other brain diseases and a normal structural MRI brain scan were recruited through the Join Dementia Research platform of volunteers and from friends or families of the patients. All identified participants provided written informed consent before detailed screening and medical review before final inclusion. The study obtained ethical approval from a UK Research Ethics Committee (NRES Committee North East—Newcastle and North Tyneside 2, reference: 12/NE/0290, 15/NE/0420). Written informed consent was obtained from all study participants in accordance with this approval. Participants from both cohorts were selected for inclusion when they had at least two observations available, with the exception of those who died.
Clinical Assessments and Imaging
After participants provided consent, they underwent a research‐level assessment involving a semistructured interview; clinical and neurocognitive assessment and neurological examination by a medical doctor (R.D., S.L., and P.C.D.); and imaging with FP‐CIT, MIBG (second cohort only), and structural neuroimaging 18 , 19 , 20 at baseline and then had longitudinal review at approximately annual follow‐ups.
The MDS Unified Parkinson's Disease Rating Scale—Motor Examination (UPDRS, Part III), Epworth Sleepiness Scale (ESS), North‐East Visual Hallucinations Inventory (NEVHI), and Geriatric Depression Scale (GDS) were administered to patients. The Instrumental Activities of Daily Living (IADL) scale, Neuropsychiatric Inventory (NPI), Mayo Sleep Questionnaire (MSQ), Clinician Assessment of Fluctuation (CAF), and Dementia Cognitive Fluctuation Scale (DCFS) were administered to informants. Clinical Dementia Rating (CDR) scale and Cumulative Illness Rating Scale for Geriatrics (CIRS‐G) were completed based on the clinical history and other research assessments. These scales were used to guide the clinical interview, but formalized cutoff scores were not used by the clinical panel—all available information was incorporated from the research notes into clinical judgments. A detailed neuropsychological evaluation was also carried out as reported previously, 17 which included the Addenbrooke's Cognitive Examination ‐ Revised (ACE‐R), a 100‐point cognitive screening test from which Mini‐Mental State Examination (MMSE) score was derived. FP‐CIT and imaging were provided to all participants as previously described. 18 , 19 FP‐CIT images were visually rated as normal or abnormal by a 5‐person panel of experienced image analysts, blind to clinical information. MIBG images were classified blind to clinical information as abnormal given a heart:mediastinum uptake ratio of <1.86 based on data from locally recruited healthy research controls. 21
Differential Diagnosis
As detailed previously 17 , 18 , 19 diagnoses were performed by a three‐person expert clinical panel (A.J.T., P.C.D., and J.‐P.T.), who independently reviewed research data and health service record and performed MCI diagnoses according to the National Institute on Aging—Alzheimer's Association (NIA‐AA) criteria. 22 This consensus panel method has previously been validated against autopsy and is recognized by regulatory authorities as the clinical gold standard. 23 The panel was unaware of pTau181 levels as assays were undertaken after diagnostic classification.
To determine MCI etiology, the presence or absence of core DLB symptoms was also rated by the panel, in accordance with the fourth consensus criteria for DLB. 24 Those with evidence (including on MRI) of vascular or frontotemporal etiologies, or parkinsonism pre‐dating cognitive impairment by more than 1 year, were excluded. In accordance with the research diagnostic criteria for MCI‐LB,25 a diagnosis of probable MCI‐LB was made if a patient had two or more core LB symptoms or one core symptom in addition to a positive FP‐CIT or MIBG scan. Patients were diagnosed with possible MCI‐LB when they had only one core symptom or one abnormal scan. MCI‐AD was diagnosed following the criteria of Albert et al 22 : subjective and objective cognitive decline consistent with AD was established, along with generally maintained independence of function in everyday life, and the absence of dementia and other causes was then excluded as mentioned earlier. These diagnoses were updated at each annual follow‐up, again without using information from pTau181 levels. When judged to meet NIA‐AA criteria for all‐cause dementia,26 participants received a diagnosis of dementia and ended involvement in the study. Diagnosis of all‐cause dementia was based on reported loss of independent function along with evidence of cognitive decline as determined by the panel. Core symptom presence and abnormal biomarkers were subsequently assessed as mentioned earlier, and their final dementia diagnosis was rated according to current consensus clinical criteria for DLB or AD. 24 , 26 MRI scans were clinically reviewed as part of the diagnostic process so that all with MCI‐AD had scans consistent with this diagnosis.
Sample Collection and Processing
Blood samples were obtained by venepuncture and collected in ethylenediaminetetraacetic acid tubes. They were centrifuged to isolate plasma, aliquoted, and stored at −70°C until further analyses. Plasma assays were conducted at the biomarker lab at the UK Dementia Research Institute, UCL, London. Plasma samples were thawed and centrifuged at 13,000g for 5 minutes at room temperature. Calibrators (neat) and samples (plasma: 1:4 dilution) were measured in duplicates. pTau181 was measured using the Simoa pTau181 Advantage kit on an HD‐X Analyzer (Quanterix, Billerica, MA). All samples were analyzed at the same time using the same batch of reagents. A four‐parameter logistic curve fit data reduction method was used to generate a calibration curve. Two control samples of known concentration of the protein of interest (high‐ctrl and low‐ctrl) were included as quality control. The mean coefficient of variation for duplicate samples run with the assay was 2.41%.
Statistical Analyses
All analyses were conducted in R statistical software. Group differences in continuous baseline demographic and clinical data were assessed using exact 2‐tailed permutation tests and Fisher's exact tests for categorical data. Age‐adjusted differences in plasma pTau181 concentrations were assessed with a log‐normal linear model, with a visual inspection of residual and fitted values to assess whether model assumptions were met. Receiver operating characteristic (ROC) curves were estimated using the pROC package, with best cutoffs identified by Youden's index.
Associations between plasma pTau181 and baseline cognitive function were assessed with a linear model controlling for age and education; pTau181 concentrations were included as a log‐transformed predictor interacting with a diagnostic group.
To assess whether probable MCI‐LB with an AD‐like plasma pTau181 concentration (and thus likely significant AD co‐pathology) would decline at a faster rate, we estimated a latent class mixed model with the lcmm package for R software to identify latent trajectories of cognitive function, incorporating the repeated ACE‐R measures from this cohort. To account for the possibility that individuals enter the study at different stages of decline, cognitive trajectories were centered on each individual's last observation. Onset of dementia was a cause for exclusion from future follow‐up, so this method meant that those who developed dementia could be more directly compared, as could those who died before dementia was observed. Age‐ and education‐adjusted trajectories in ACE‐R performance were estimated, and heterogeneous groups were considered, with these allowed to differ in their intercept (function at last observation, dementia onset, or death) and time slope (rate of decline up to the intercept). The model with the most appropriate number of latent trajectories was identified based on it showing an improved fit over the null homogeneous model, with the stipulation that any additional latent class identified must not be too small to be generalizable (eg, classifying only a single participant's trajectory) or conceptually indistinct from those already identified (eg, separating one clearly defined class into two poorly defined classes with similar trajectories). Healthy controls were included in this comparison for further context on the expected rates of change in locally recruited healthy older adults. Participants were assigned to the group trajectory most similar to their own individual trajectory (the latent class to which they had the highest posterior probability of membership). Those with an uncertain trajectory that could reasonably fit with more than one class (a posterior classification probability of <0.80) were considered as not clearly classified and excluded from the following primary analyses.
Longitudinal pTau181 trajectories were assessed with age‐adjusted homogeneous log‐linear mixed models without the estimation of latent trajectories due to smaller numbers.
Data Sharing
The data for this study are available with permission from the authors at Dementias Platform UK.
Results
One hundred forty‐six subjects (56 probable MCI‐LB, 22 possible MCI‐LB, 44 MCI‐AD, and 24 controls) met the entry criteria, and the findings are summarized in Table 1. The longest follow‐up was for 5.67 years, mean 1.92 (standard deviation 1.36) years. The results show the probable MCI‐LB and MCI‐AD groups were matched in age with the control group (Z = −0.99, P = 0.332) and with each other on cognition (Z = −0.62, P = 0.544), CDR (Z = −1.33, P = 0.234), education (Z = −1.53, P = 0.131), and overall health rated by the CIRS‐G (Z = 1.53, P = 0.131), but as expected the probable MCI‐LB had more men (P < 0.001), greater overall neuropsychiatric symptomatology (Z = 3.20, P = 0.001) and resultant carer distress (Z = 3.99, P < 0.001) reported in the NPI, a higher UPDRS score (Z = 2.72, P = 0.006), greater loss of daily function (lower IADL score: Z = −2.48, P = 0.014), and were receiving more cholinesterase inhibitors (P = 0.001). Further consistent with their diagnosis, probable MCI‐LB also had greater visual hallucination symptom scores on the NEVHI (Z = 3.14, P = 0.001), fluctuation symptom severity rated by the CAF (Z = 3.38, P < 0.001) and DCFS (Z = 3.50, P < 0.001), depressive symptoms rated by the GDS (Z = 2.20, P = 0.028), daytime sleepiness rated by the ESS (Z = 4.94, P < 0.001), and sleep disturbance reported by the MSQ (W = −2.22, P = 0.026).
TABLE 1.
Baseline characteristics for each diagnostic group
| Control (N = 24) | MCI‐AD (N = 44) | Probable MCI‐LB (N = 56) | Possible MCI‐LB (N = 22) | |
|---|---|---|---|---|
| Age at baseline | ||||
| Mean (SD) | 73.9 (7.76) | 76.8 (7.20) | 75.4 (6.89) | 73.1 (7.18) |
| Sex | ||||
| Female | 7 (29.2%) | 28 (63.6%) | 12 (21.4%) | 12 (54.5%) |
| Male | 17 (70.8%) | 16 (36.4%) | 44 (78.6%) | 10 (45.5%) |
| Years in education | ||||
| Median [min, max] | 13 [10, 24] | 11 [9, 20] | 11 [7, 18] | 10 [9, 22] |
| Instrumental Activities of Daily Living | ||||
| Median [min, max] | – | 8 [2, 8] | 7 [2, 8] | 7 [3, 8] |
| ACE‐R total score | ||||
| Mean (SD) | 92.6 (4.33) | 81.8 (9.79) | 80.6 (9.76) | 74.4 (12.0) |
| ACE‐R MMSE score | ||||
| Median [min, max] | 29 [27, 30] | 27 [22, 30] | 27 [20, 30] | 25 [20, 30] |
| CDR_Total | ||||
| Median [min, max] | 0 [0, 0] | 0.5 [0, 0.5] | 0.5 [0, 0.5] | 0.5 [0.5, 0.5] |
| UPDRS‐III total score | ||||
| Median [min, max] | 4.50 [0, 16] | 16 [0, 62] | 21.5 [1, 53] | 15 [1, 40] |
| Receiving cholinesterase inhibitors | ||||
| Yes | 0 (0%) | 8 (18.2%) | 28 (50.0%) | 4 (18.2%) |
| pTau concentration (pg/mL) | ||||
| Mean (SD) | 1.65 (0.759) | 2.88 (1.30) | 2.25 (0.996) | 2.44 (1.17) |
Abbreviations: SD, standard deviation; MCI, mild cognitive impairment; AD, Alzheimer's disease; LB, Lewy bodies; CDR, Clinical Dementia Rating; MMSE, Mini‐Mental State Examination; UPDRS, Unified Parkinson's Disease Rating Scale.
The lower daily function of MCI‐LB did not appear to be related to greater disease progression in this group but rather neuropsychiatric symptoms: there was a significant negative partial correlation between NPI and IADL scores (standardized β = −0.32, 95% confidence interval [CI]: −0.60 to −0.03) and no significant association between diagnosis and IADL ratings after adjusting for neuropsychiatric symptoms, age, and sex.
Comparison of pTau181 Levels
Because age is well recognized to be strongly associated with pTau181 levels, a log‐linear model adjusting for age compared the probable MCI‐LB subjects with controls and MCI‐AD (see Table 2). This analysis found that every 1‐year increase in age above the grand mean (75) was associated with an estimated 2.2% higher plasma pTau181 concentration. The probable MCI‐LB had significantly higher pTau181 compared with controls (who had, on average, 22.2% lower age‐adjusted pTau181 concentrations) and significantly lower levels compared with MCI‐AD (MCI‐AD 24.4% higher). The possible MCI‐LB group did not significantly differ from the probable MCI‐LB group, overlapping with both this and MCI‐AD (see unadjusted data in pTau181, Fig. 1A).
TABLE 2.
Estimated group differences from log‐linear model of plasma pTau181 at baseline, adjusting for age
| pTau181 concentration (pg/mL) | |||
|---|---|---|---|
| Predictors | Estimate* | 95% CI | P |
| Probable MCI‐LB (intercept—geometric mean) | 2.02 | 1.79 to 2.27 | <0.001 |
| Control vs. probable MCI‐LB % difference | −22% | −37% to −4% | 0.022 |
| MCI‐AD vs. probable MCI‐LB % difference | +24% | +4% to +49% | 0.016 |
| Possible MCI‐LB vs. probable MCI‐LB % difference | +14% | −9% to +42% | 0.256 |
| Age (mean centered) % difference per year | +2% | +1% to +3% | <0.001 |
| Observations | 146 | ||
Exponentiated estimates, expressed as % differences.
Bold p values denote effects significant at α < 0.05.
Abbreviations: MCI, mild cognitive impairment; LB, Lewy bodies; CI, confidence interval; AD, Alzheimer's disease.
FIG 1.
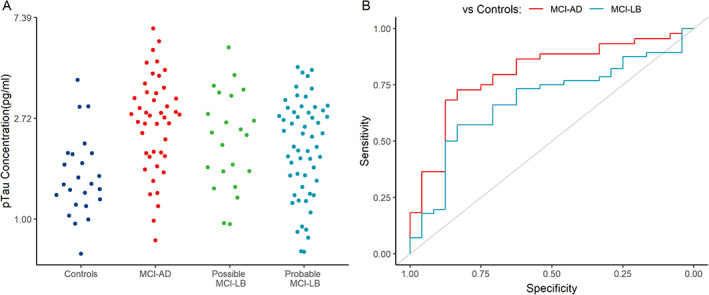
(A) Raw plasma pTau181 measurements in each diagnostic group at baseline; (B) ROC (receiver operating characteristic) curves for the differentiation of MCI‐AD and MCI‐LB from healthy controls. AD, Alzheimer's disease; LB, Lewy bodies; MCI, mild cognitive impairment. [Color figure can be viewed at wileyonlinelibrary.com]
Sensitivity and Specificity of pTau181
ROC analyses (see Fig. 1B) found that the area under the curve (AUC) of pTau181 in discriminating probable MCI‐LB from controls was 0.68; a threshold of 1.94 pg/mL provided the best discrimination of probable MCI‐LB from controls, with 83% specificity and 57% sensitivity. For discriminating MCI‐AD from healthy controls, the AUC was 0.8; the best cutoff threshold was 1.93 pg/mL, providing 83% specificity and 72% sensitivity. pTau181 concentration was less useful in discriminating between probable MCI‐LB and MCI‐AD, with an AUC of 0.64; a cutoff threshold of 2.79 pg/mL provided 71% specificity and 52% sensitivity.
Association between pTau181 and Baseline Cognitive Function
In MCI‐AD, higher plasma pTau181 was associated with lower baseline cognitive function, a nonlinear effect, where 1% higher plasma pTau181 was associated with 0.12% lower ACE‐R score (95% CI: −0.19% to −0.04%) controlling for age, education, and diagnostic group. This association was not observed in probable MCI‐LB (P = 0.211), possible MCI‐LB (P = 0.127), or healthy controls (P = 0.534).
Classifying Cognitive Decline by Baseline pTau181
Based on the best cutoff identified for MCI‐AD (1.93 pg/mL) in the ROC analysis, 32 (57.1%) probable MCI‐LB cases would be classified as also having AD co‐pathology, and 13 (59.1%) possible MCI‐LB cases would be similarly classified.
The heterogeneous mixed model with best model fit and well‐defined classes identified a three‐trajectory model:
Class 1: an estimated −12.7 (95% CI: −16.7 to −8.6) points lost on the ACE‐R per year.
Class 2: an estimated −2.2 (95% CI: −3.2 to −1.1) points lost on the ACE‐R per year.
Class 3: an estimated −0.7 (95% CI: −1.4 to 0.1) points lost on the ACE‐R per year.
Final classifications are present in Table 3. Subject‐specific unadjusted trajectories and estimated class trajectories adjusted for covariates are shown in Figure 2A. Figure 2B shows baseline pTau181 concentrations for each group, stratified by their subsequent cognitive trajectory. The rapid‐declining group was excluded from further analysis due to its small size: of the four in this group, three (two MCI‐AD and one probable MCI‐LB) had pTau181 concentrations above the MCI‐AD threshold, and one, from the possible MCI‐LB group, did not. The probable MCI‐LB group contains decliners spread fairly evenly throughout, and consistent with this there was no association between pTau181 and the overall rate of cognitive decline on ACE‐R (age and education adjusted) (estimate = −1.23 more points lost per year, 95% CI: −3.20 to 0.75, P = 0.224). In contrast, in the MCI‐AD group all decliners had higher levels of pTau181, and there was a faster decline per year overall in the ACE‐R (estimate: −3.22, CI: −6.38 to −0.05, P = 0.046).
TABLE 3.
N (%) of each diagnostic group allocated to each trajectory class (posterior classification probabilities of at least 0.8)
| Class 1 (rapid decline) | Class 2 (intermediate decline) | Class 3 (slow/stable) | |
|---|---|---|---|
| Control | 0 (0%) | 0 (0%) | 22 (100%) |
| MCI‐AD | 2 (7.1%) | 9 (32.1%) | 17 (60.7%) |
| Possible MCI‐LB | 1 (8.3%) | 8 (66.7%) | 3 (25%) |
| Probable MCI‐LB | 1 (2.5%) | 22 (55%) | 17 (42.5%) |
Abbreviations: MCI, mild cognitive impairment; AD, Alzheimer's disease; LB, Lewy bodies.
FIG 2.
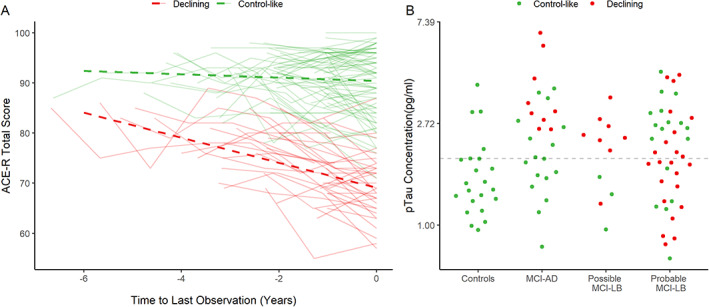
Latent trajectories of cognitive function, centered on the last observation: (A) with adjusted estimated trajectories for each class and (B) raw pTau181 concentrations for each diagnostic group, stratified by latent trajectory; dashed line indicates a cutoff of 1.929 pg/mL pTau181. [Color figure can be viewed at wileyonlinelibrary.com]
A sensitivity analysis was conducted with a less‐stringent trajectory classification cutoff (posterior probability of <0.5 for exclusion) to provide a larger sample size for comparison (n = 145): this did not meaningfully change the results, with elevated pTau181 being predictive of cognitive decline in MCI‐AD but not probable MCI‐LB.
Longitudinal Change in Plasma pTau181
A subset of the cohort (37 MCI participants with 85 measurements in total) provided at least one repeated plasma sample at first‐ or second‐year follow‐up (see Fig. S1): all but 1 were MCI patients, so the control was excluded from further analysis. Yearly change in plasma pTau181 was assessed with a log‐linear mixed model with random slopes, adjusting for baseline age (see Table S1). There was a nonsignificant 3% increase per follow‐up year in plasma pTau181 across MCI, in addition to the significant 3% increase per year associated with age. Although individual pTau181 trajectories varied (see Fig. S1), any systematic follow‐up change in pTau181 concentration was limited and largely indistinguishable from broader age‐associated changes. In further exploratory analyses, the rate of change in pTau181 was not significantly different in the diagnostic subgroups or in those with a declining cognitive trajectory (weighted by posterior classification probability). However, these analyses are limited by the number of individuals with repeat plasma pTau181 measurements available and likely lack statistical power to assess such group × time interactions.
Discussion
People with DLB have a worse prognosis than people with AD, with evidence of more rapid decline at both MCI 2 , 3 and dementia 4 , 5 stages. One of the reasons for this is considered to be the high frequency of AD co‐pathology in DLB, the presence of more than one brain pathology in dementia being associated with more rapid decline,1 and in DLB, previous work has reported this specifically for AD co‐pathology. 16 , 27 Because pTau181 has recently been established as a valid biomarker for AD pathology 11 , 12 and is a marker for AD co‐pathology in DLB, 16 we hypothesized that increased levels of pTau181 in people with MCI‐LB, the cognitive prodrome of DLB, would be associated with a more rapid decline. However, in our longitudinal study we did not find such an association, nor in a subgroup with longitudinally repeated pTau181 did we find any association between cognitive decline in MCI‐LB and these repeat pTau181 levels.
These findings do not appear to be due to pTau181 not being a valid biomarker of AD co‐pathology because not only, as mentioned earlier, have other studies indicated that it is (and is associated with faster cognitive decline in probable DLB 28 ), but in the MCI‐AD group there was a relationship between baseline higher levels of pTau181 and increased cognitive decline. Rather we suggest our findings are due to the heterogeneity within MCI‐LB (and DLB). LB pathology is recognized as being a potent cause of cognitive decline, 6 and the more widespread this pathology is, especially when involving neocortical areas, the greater the decline. 29 , 30 Within our probable MCI‐LB group there are patients with all four core clinical features (and some of these with abnormal [positive] biomarkers) and others with only one (plus a positive biomarker), indicating a diversity of spread of LB pathology. Similarly some of the probable MCI‐LB are more cognitively impaired than others (albeit within the narrow MCI range), again supporting the presence of variable amounts of brain pathology within the group. Thus, we suggest that within the probable MCI‐LB group those with low levels of pTau181 but a more rapid cognitive decline (red dots below the dotted line in Fig. 2B) have a greater burden of LB pathology, and it is this rather than AD co‐pathology that is driving their decline. In other probable MCI‐LB patients with high pTau181 and more rapid decline, AD co‐pathology is likely a greater contributor to this decline. This interpretation requires future direct testing with biomarkers for LB disease, which can quantify the overall brain burden of LB disease. The current diagnostic biomarkers (dopaminergic imaging, MIBG cardiac scintigraphy, and polysomnography) are not able to do this. An alternative, and compatible, interpretation is that other brain co‐pathologies are present in our MCI‐LB patients, which are driving their decline. Because we endeavored to exclude frontotemporal dementia and cerebrovascular disease, it seems less likely, though not impossible, that these pathologies are relevant here, but other co‐pathologies, especially TDP‐43, which is recognized to be a cause of accelerated decline in dementia, 31 may be relevant. Unfortunately, this interpretation is not testable at the MCI stage currently, though as we are following our patients to autopsy we will eventually be able to explore this possibility.
Our other findings were as hypothesized and consistent with the wider literature. We found that pTau181 is not useful in differentiating probable MCI‐LB from MCI‐AD, which we interpret as related to the high burden of AD co‐pathology in DLB/MCI‐LB, an interpretation supported by many of our probable (and possible) MCI‐LB patients having high (AD‐like) pTau181 levels consistent with the presence of such co‐pathology (see Figs. 1A and 2B). For the same reasons pTau181 was significantly higher in probable MCI‐LB than in controls and differentiated reasonably well between these groups, so that finding increased pTau181 in someone with MCI supports the presence of neurodegeneration (AD) even if they may also have LB disease. As expected pTau181 differentiated very well between MCI‐AD and controls. The possible MCI‐LB group was not distinguishable by pTau181 from either MCI‐AD or probable MCI‐LB, having substantial overlap with both these groups (see Fig. 1A), consistent with previous evidence for the higher degree of heterogeneity 32 , 33 in this diagnostically less‐certain group. Interestingly, pTau181 levels were high in a large proportion of this group (Fig. 1A) and in almost all of those who declined more rapidly (Fig. 2B), again supporting a role for AD co‐pathology in driving decline. In such patients this larger burden of AD co‐pathology, indicated by higher pTau181, might also have obscured the full extent of LB disease, thereby preventing them being recognized as probable MCI‐LB. Such diagnostic overshadowing by AD, and especially tau, pathology has been reported previously. 34 , 35
Our study is a well‐characterized and relatively large cohort of probable MCI‐LB and MCI‐AD with detailed clinical and cognitive assessments and both structural and radionuclide imaging biomarkers. It also benefits from using consensus panel diagnosis and longitudinal reviews and reevaluation of the diagnoses. Although using autopsy diagnosis may be regarded as the gold standard, this is not realistic for MCI studies, and our use of consensus clinical panel diagnosis is the standard recognized by regulatory authorities and has been validated against autopsy. 23 Evaluation of pTau181 used the established and highly accurate Simoa methodology. A limitation we have acknowledged before is that patients were recruited from memory services based on the possible presence of symptoms characteristic of LB disease identified in these services, such as core clinical diagnostic features or supportive features such as depression, anxiety, postural hypotension, and falls. Although this was necessary to ensure a high proportion of MCI‐LB in the study sample, it does mean those diagnosed with MCI‐AD may not be entirely representative of all AD in such services, tending to have more symptoms consistent with LB disease, although the rigorous clinical and biomarker research assessments would likely exclude any with good clinical evidence of LB disease. It remains possible that these MCI‐AD cases may have an unrecognized non‐AD etiology or LB disease without clearly manifest clinical symptoms, a limitation of this prospective sample. We also acknowledge the absence of amyloid CSF and PET biomarkers as a limitation of this study.
As a single‐site study incorporating detailed and longitudinal assessment, the overall sample size available for this cohort was small: this is especially important when considering the heterogeneity within groups of probable and, in particular, possible MCI‐LB. The sample size of these is therefore a limitation of this study and may obscure any real associations between or within MCI subgroups. It is also possible that individuals may have entered the study at different stages of disease severity; although we endeavored to account for this when assessing cognitive decline, how this relates to baseline plasma biomarkers is unclear.
In conclusion, although pTau181 did appear to identify AD co‐pathology in probable MCI‐LB, this was not associated with the hypothesized increased decline assessed using either baseline or repeat pTau181 levels. This is likely due to heterogeneity in the burden of LB pathology in these patients, which might be determined with larger cohorts. pTau181 did discriminate probable MCI‐LB from controls and MCI‐AD from controls but was not useful in distinguishing probable MCI‐LB from MCI‐AD.
Author Roles
A.J.T.: drafting of the manuscript, formulation of research question, design of the study, interpretation of data, and review and critique of the manuscript; C.A.H.: data collection and analysis and drafting of the manuscript; A.H.: assay validation, data collection, and review and critique of the manuscript; S.B.: data collection and review and critique of the manuscript; R.D.: data collection and review and critique of the manuscript; S.L.: data collection and review and critique of the manuscript; N.B.: study administration, data collection, and review and critique of the manuscript; D.L.: data collection and review and critique of the manuscript; M.F.: data collection and review and critique of the manuscript; G.R.: data collection and review and critique of the manuscript; J.P.‐T.: design of the study, data collection, and review and critique of the manuscript; P.C.D.: design of the study, formulation of research question, data collection, and review and critique of the manuscript; H.Z.: assay development and validation, data collection, and review and critique of the manuscript; and J.O.: design of the study, formulation of research question, and review and critique of manuscript.
Full financial disclosures for the previous 12 months
A.J.T. has received funding from GE Healthcare for investigator‐led studies. C.A.H. has received a research grant from the NIHR Newcastle Biomedical Research Centre, unrelated to this work. J.O. was consultant for TauRx, Eisai, Novo Nordisk, Biogen, and GE Healthcare, and received grant support from Merck and Alliance Medical. H.Z. has served on scientific advisory boards and/or as consultant for AbbVie, Alector, Annexon, Eisai, Denali, Roche, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies, CogRx, and Red Abbey Labs; has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, and Biogen; and is cofounder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. P.C.D. has received honoraria for teaching from the Neurology Academy. J.‐P.T. received speaker fees from GE Healthcare and was consultant for Kyowa‐Kirin. He is on the scientific advisory panel of the Lewy Body Society, UK, and the Lewy Body Disease Association and is a grant panel member for Alzheimer's Research UK. He has received an educational grant from Sosei‐Heptares and has been supported by the Newcastle NIHR Biomedical Research Centre. All other authors declare no conflicts of interest.
Supporting information
Supplementary Figure S1. Estimated plasma pTau181 change over time in subset of MCI cases with follow‐up plasma sample, with subject‐specific trajectories.
Supplementary Table S1. Estimated plasma pTau181 change over time in subset of MCI cases with follow‐up plasma sample.
Acknowledgments
We acknowledge the support of Ms Helen Kain in the undertaking of this study and the NIHR Clinical Research Network North East and Cumbria for their support in recruiting participants.
Funding agencies: This work was supported by Alzheimer's Research UK (ARUK‐PG2015‐13) and by the NIHR Newcastle Biomedical Research Centre. GE Healthcare provided the FP‐CIT ligand for this investigator‐led study. H.Z. is a Wallenberg Scholar supported by grants from the Swedish Research Council (no. 2018‐02532); the European Research Council (no. 681712); the Swedish State Support for Clinical Research (no. ALFGBG‐720931); the Alzheimer Drug Discovery Foundation (ADDF), USA (no. 201809‐2016862); the AD Strategic Fund and the Alzheimer's Association (nos. ADSF‐21‐831376‐C, ADSF‐21‐831381‐C, and ADSF‐21‐831377‐C); the Olav Thon Foundation; the Erling‐Persson Family Foundation; Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (no. FO2019‐0228); the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement number 860197 (MIRIADE); and the UK Dementia Research Institute at UCL.
Data Availability Statement
The data for this study are available with permission from the authors at DPUK (dementias platforms UK)
References
- 1. McAleese KE, Colloby SJ, Thomas AJ, et al. Concomitant neurodegenerative pathologies contribute to the transition from mild cognitive impairment to dementia. Alzheimers Dement 2021;17:1121–1133. 10.1002/alz.12291 [DOI] [PubMed] [Google Scholar]
- 2. Hamilton CA, Matthews FE, Donaghy PC, et al. Progression to dementia in mild cognitive impairment with Lewy bodies or Alzheimer disease. Neurology 2021;96(22):e2685–e2693. 10.1212/wnl.0000000000012024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamilton CA, Matthews FE, Donaghy PC, et al. Prospective predictors of decline v. stability in mild cognitive impairment with Lewy bodies or Alzheimer's disease. Psychol Med 2021;51(15):2590–2598. 10.1017/s0033291720001130 [DOI] [PubMed] [Google Scholar]
- 4. Rongve A, Soennesyn H, Skogseth R, et al. Cognitive decline in dementia with Lewy bodies: a 5‐year prospective cohort study. BMJ Open 2016;6(2):e010357. 10.1136/bmjopen-2015-010357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Price A, Farooq R, Yuan JM, et al. Mortality in dementia with Lewy bodies compared with Alzheimer's dementia: a retrospective naturalistic cohort study. BMJ Open 2017;7(11):e017504. 10.1136/bmjopen-2017-017504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boyle PA, Yu L, Wilson RS, et al. Person‐specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018;83(1):74–83. 10.1002/ana.25123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abdelnour C, van Steenoven I, Londos E, et al. Alzheimer's disease cerebrospinal fluid biomarkers predict cognitive decline in Lewy body dementia. Mov Disord 2016;31(8):1203–1208. 10.1002/mds.26668 [DOI] [PubMed] [Google Scholar]
- 8. Abner EL, Kryscio RJ, Schmitt FA, et al. Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 2017;81(4):549–559. 10.1002/ana.24903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hansson O. Biomarkers for neurodegenerative diseases. Nat Med 2021;27(6):954–963. 10.1038/s41591-021-01382-x [DOI] [PubMed] [Google Scholar]
- 10. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol 2020;19(5):422–433. 10.1016/s1474-4422(20)30071-5 [DOI] [PubMed] [Google Scholar]
- 11. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med 2020;26(3):379–386. 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 12. Lantero Rodriguez J, Karikari TK, Suárez‐Calvet M, et al. Plasma p‐tau181 accurately predicts Alzheimer's disease pathology at least 8 years prior to post‐mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol 2020;140(3):267–278. 10.1007/s00401-020-02195-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suárez‐Calvet M, Karikari TK, Ashton NJ, et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer's continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med 2020;12(12):e12921. 10.15252/emmm.202012921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karikari TK, Benedet AL, Ashton NJ, et al. Diagnostic performance and prediction of clinical progression of plasma phospho‐tau181 in the Alzheimer's disease neuroimaging initiative. Mol Psychiatry 2021;26(2):429–442. 10.1038/s41380-020-00923-z [DOI] [PubMed] [Google Scholar]
- 15. Palmqvist S, Tideman P, Cullen N, et al. Prediction of future Alzheimer's disease dementia using plasma phospho‐tau combined with other accessible measures. Nat Med 2021;27(6):1034–1042. 10.1038/s41591-021-01348-z [DOI] [PubMed] [Google Scholar]
- 16. Hall S, Janelidze S, Londos E, et al. Plasma Phospho‐tau identifies Alzheimer's co‐pathology in patients with Lewy body disease. Mov Disord 2021;36(3):767–771. 10.1002/mds.28370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Donaghy PC, Ciafone J, Durcan R, et al. Mild cognitive impairment with Lewy bodies: neuropsychiatric supportive symptoms and cognitive profile. Psychol Med 2020;1–9. 10.1017/s0033291720002901 [DOI] [PubMed] [Google Scholar]
- 18. Roberts G, Donaghy PC, Lloyd J, et al. Accuracy of dopaminergic imaging as a biomarker for mild cognitive impairment with Lewy bodies. Br J Psychiatry 2021;218(5):276–282. 10.1192/bjp.2020.234 [DOI] [PubMed] [Google Scholar]
- 19. Roberts G, Durcan Mrcpi R, Donaghy PC, et al. Accuracy of cardiac innervation scintigraphy for mild cognitive impairment with Lewy bodies. Neurology 2021;96:e2801–e2811. 10.1212/wnl.0000000000012060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Firbank MJ, Durcan R, O'Brien JT, et al. Hippocampal and insula volume in mild cognitive impairment with Lewy bodies. Parkinsonism Relat Disord 2021;86:27–33. 10.1016/j.parkreldis.2021.03.011 [DOI] [PubMed] [Google Scholar]
- 21. Roberts G, Lloyd JJ, Kane JPM, et al. Cardiac (123)I‐MIBG normal uptake values are population‐specific: results from a cohort of controls over 60 years of age. J Nucl Cardiol 2019;28:1692–1701. 10.1007/s12350-019-01887-6 [DOI] [PubMed] [Google Scholar]
- 22. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7(3):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKeith I, O'Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I‐FP‐CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol 2007;6(4):305–313. [DOI] [PubMed] [Google Scholar]
- 24. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 2017;89(1):88–100. 10.1212/WNL.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McKeith IG, Ferman TJ, Thomas AJ, et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 2020;94(17):743–755. 10.1212/wnl.0000000000009323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKhann GM, Knopman D, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Donaghy PC, Firbank MJ, Thomas AJ, et al. Amyloid imaging and longitudinal clinical progression in dementia with Lewy bodies. Am J Geriatr Psychiatry 2020;28(5):573–577. 10.1016/j.jagp.2019.12.009 [DOI] [PubMed] [Google Scholar]
- 28. Gonzalez MC, Ashton NJ, Gomes BF, et al. Association of Plasma p‐tau181 and p‐tau231 concentrations with cognitive decline in patients with probable dementia with Lewy bodies. JAMA Neurol 2022;79(1):32–37. 10.1001/jamaneurol.2021.4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruffmann C, Calboli FC, Bravi I, et al. Cortical Lewy bodies and Aβ burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol Appl Neurobiol 2016;42(5):436–450. 10.1111/nan.12294 [DOI] [PubMed] [Google Scholar]
- 30. Schneider JA, Arvanitakis Z, Yu L, et al. Cognitive impairment, decline and fluctuations in older community‐dwelling subjects with Lewy bodies. Brain 2012;135(Pt 10):3005–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boyle PA, Yang J, Yu L, et al. Varied effects of age‐related neuropathologies on the trajectory of late life cognitive decline. Brain 2017;140(3):804–812. 10.1093/brain/aww341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Brien JT, McKeith IG, Walker Z, et al. Diagnostic accuracy of 123I‐FP‐CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatry 2009;194(1):34–39. [DOI] [PubMed] [Google Scholar]
- 33. Walker Z, Moreno E, Thomas A, et al. Clinical usefulness of dopamine transporter SPECT imaging with 123I‐FP‐CIT in patients with possible dementia with Lewy bodies: randomised study. Br J Psychiatry 2015;206(2):145–152. [DOI] [PubMed] [Google Scholar]
- 34. Ferman TJ, Aoki N, Boeve BF, et al. Subtypes of dementia with Lewy bodies are associated with α‐synuclein and tau distribution. Neurology 2020;95(2):e155–e165. 10.1212/wnl.0000000000009763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tiraboschi P, Attems J, Thomas A, et al. Clinicians' ability to diagnose dementia with Lewy bodies is not affected by beta‐amyloid load. Neurology 2015;84(5):496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Estimated plasma pTau181 change over time in subset of MCI cases with follow‐up plasma sample, with subject‐specific trajectories.
Supplementary Table S1. Estimated plasma pTau181 change over time in subset of MCI cases with follow‐up plasma sample.
Data Availability Statement
The data for this study are available with permission from the authors at DPUK (dementias platforms UK)