

Abstract
Insufficient oxygen supply (hypoxia) during fetal development leads to cardiac remodeling and a predisposition to cardiovascular disease in later life. Previous work has shown hypoxia causes oxidative stress in the fetal heart and alters the activity and expression of mitochondrial proteins in a sex‐dependent manner. However, the functional effects of these modifications on mitochondrial respiration remain unknown. Furthermore, while maternal antioxidant treatments are emerging as a promising new strategy to protect the hypoxic fetus, whether these treatments convey similar protection to cardiac mitochondria in the male or female fetus has not been investigated. Therefore, using an established rat model, we measured the sex‐dependent effects of gestational hypoxia and maternal melatonin treatment on fetal cardiac mitochondrial respiration, reactive oxygen species (ROS) production, and lipid peroxidation. Pregnant Wistar rats were subjected to normoxia or hypoxia (13% oxygen) during gestational days (GDs) 6–20 (term ~22 days) with or without melatonin treatment (5 µg/ml in maternal drinking water). On GD 20, mitochondrial aerobic respiration and H2O2 production were measured in fetal heart tissue, together with lipid peroxidation and citrate synthase (CS) activity. Gestational hypoxia reduced maternal body weight gain (p < .01) and increased placental weight (p < .05) but had no effect on fetal weight or litter size. Cardiac mitochondria from male but not female fetuses of hypoxic pregnancy had reduced respiratory capacity at Complex II (CII) (p < .05), and an increase in H2O2 production/O2 consumption (p < .05) without any changes in lipid peroxidation. CS activity was also unchanged in both sexes. Despite maternal melatonin treatment increasing maternal and fetal plasma melatonin concentration (p < .001), melatonin treatment had no effect on any of the mitochondrial parameters investigated. To conclude, we show that gestational hypoxia leads to ROS generation from the mitochondrial electron transport chain and affects fetal cardiac mitochondrial respiration in a sex‐dependent manner. We also show that maternal melatonin treatment had no effect on these relationships, which has implications for the development of future therapies for hypoxic pregnancies.
Keywords: fetal, heart, hypoxia, melatonin, metabolism, mitochondria, ROS
1. INTRODUCTION
The growth and survival of the developing fetus is critically dependent on optimal maternal health. Fluctuations in the intrauterine environment can be detrimental to the fetus and can permanently alter the functional capacity of vital organs. 1 In particular, insufficient fetal oxygen supply (hypoxia) is a common pregnancy complication that occurs in a wide range of conditions, including pre‐eclampsia, placental insufficiency, and high‐altitude pregnancies. 1 In the short term, hypoxia causes a redistribution of fetal blood flow to hypoxia‐sensitive organs, such as the fetal heart. 2 , 3 However, in the longer term, gestational hypoxia can overwhelm this circulatory protection, and lead to impaired cardiac development, 4 , 5 , 6 thickening of the aortic wall, 7 and reduced cardiac output in the fetus. 8 Offspring of hypoxic pregnancies display cardiac abnormalities in adulthood and appear to be sensitized to ischemia/reperfusion injury. 7 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 These findings support epidemiological evidence from human pregnancies, which show hypoxia during development can increase the risk of cardiac dysfunction in offspring. 18 , 19 , 20 , 21 , 22 , 23 Combined, evidence derived from human clinical studies and from experimental preclinical models strongly supports that fetal hypoxia is an independent risk factor for offspring cardiovascular disease. Understanding the factors that drive cardiac dysfunction in hypoxic pregnancies provides an exciting opportunity to develop therapeutics that prevent abnormal developmental programming of cardiovascular disease.
Cellular dysfunction in hypoxia mainly occurs because oxygen is vital for the production of ATP by oxidative phosphorylation (OXPHOS). Briefly, electrons derived from carbohydrates, fats, and proteins are transferred to the mitochondrial electron transport chain, where they move through a series of complexes and finally bind to oxygen at Complex IV (CIV). The movement of electrons provides the energy for CI, CIII, and CIV to pump protons against their electrochemical gradient and establish a proton‐motive force that drives ATP production through complex V (the F1F0 ATP‐synthase). 24 Although most electrons are transported to CIV, a small proportion slip from the chain and bind directly to molecular oxygen to produce superoxide. 25 When oxygen becomes limiting, the electron transport chain is inhibited and electron slip becomes more common, leading to reduced ATP production and the overproduction of reactive oxygen species (ROS). 26 Consequently, prolonged periods of hypoxia are associated with energy depletion and oxidative stress, which can ultimately lead to cell death. Nevertheless, studies on adult mammals have shown mitochondria are plastic organelles that are able to adapt and remodel in hypoxic environments. For example, acclimatization to high‐altitude hypoxia is commonly associated with a reduction in mitochondrial content, a downregulation of enzymes involved in OXPHOS and an upregulation of anaerobic pathways. 27 , 28 While the strategy is thought to limit ROS production and oxidative stress, these modifications will reduce ATP production, which can have adverse consequences for cellular survival. 27 Although these processes are relatively well‐studied in the adult mammalian heart, far less is known about the adaptive capacity of fetal mitochondria. It is important to fill this knowledge gap, because adaptive responses during early life can often be permanent, leading to life‐long changes in morphology, physiology, and behavior. 29
During normal fetal development, a metabolic shift from anaerobic to aerobic respiration occurs during mid‐late gestation and the heart relies on OXPHOS for the majority of ATP production. 30 Therefore, hypoxia is expected to significantly impact ATP production and ROS generation, particularly during the latter stages of fetal development. Indeed, work from our laboratory 31 and others 32 , 33 has shown that adult offspring from hypoxic pregnancies have sex‐dependent alterations in mitochondrial respiration, protein expression, enzymatic activity, and ROS production, suggesting that hypoxia programs mitochondrial pathways during fetal development. In support of this notion, previous work in guinea pigs has shown that late gestational hypoxia reduces protein expression of CI, CIII, and CV in male and female fetal hearts, as well as a reduction in CIV activity in the male fetal heart. 34 Similar to adult mammals, this response may represent a compensatory adjustment to limit mitochondrial ROS production by reducing respiration rates. However, the reduction in CI protein expression was not associated with lower enzymatic activity, and another study using the same model showed evidence of lipid peroxidation, suggesting that the overproduction of ROS persists despite these adaptations. 35 To better understand the functional effects of fetal hypoxia on fetal cardiac mitochondria and their adaptive significance, it is important to measure cardiac mitochondrial respiration and ROS production.
In addition to studying the mechanisms that drive fetal cardiac dysfunction in hypoxia, it is essential to develop an effective therapeutic strategy to prevent cardiovascular disease in adulthood. In this respect, maternal antioxidant treatment is emerging as a promising new approach. Several studies have shown that antioxidants can prevent fetal cardiac remodeling and dysfunction in adulthood, such as maternal treatment with vitamin C, allopurinol, statins, MitoQ, and melatonin. 9 , 11 , 36 , 37 , 38 , 39 , 40 , 41 Antioxidant protection on the offspring of hypoxic pregnancy manifests in several ways, including enhanced antioxidant capacity, improvements in transplacental oxygenation, diminished endothelial dysfunction, and normalization of arterial blood pressure. 38 Interestingly, the protective effects are enhanced if mitochondria‐targeted antioxidants, such as MitoQ, are used, 39 suggesting ROS production from the mitochondria may play a significant role in programming cardiac dysfunction. Despite the promise of antioxidants in treating hypoxic pregnancies, very little is known about the effects of these compounds on fetal mitochondrial function and ROS production, even in healthy pregnancy. In addition, no study has investigated the sex‐dependent impact of developmental hypoxia on fetal cardiac mitochondrial respiration, nor the impact of maternal antioxidant treatment. This information is essential to assess the suitability of antioxidants as therapeutic agents in complicated pregnancies.
Therefore, the first objective of the present study was to determine the sex‐dependent effects of developmental hypoxia on fetal cardiac mitochondrial respiratory capacity, ROS production, and lipid peroxidation. The second objective was to assess the suitability of maternal melatonin treatment as a therapeutic intervention for fetal hypoxia. We have chosen melatonin as our treatment because it is already safely consumed by people to avoid jetlag, 42 it can be transported across the mitochondrial membrane and it is also synthesized in the matrix, 43 so it is considered a mitochondria‐targeted antioxidant. 44 , 45 , 46 Furthermore, melatonin acts at multiple levels in the mitochondria by upregulating antioxidant enzymes, increasing the activity of respiratory complexes, and directly scavenging free radicals. 45 , 47 , 48 Importantly, recent studies have shown that treatment of hypoxic pregnancies with melatonin prevented all alterations in cardiovascular structure and function in adult offspring. 11 Melatonin is already being used in clinical trials for complicated pregnancies, 49 , 50 , 51 and can readily cross the placenta. 11 However, nothing is known about the effects of melatonin on fetal cardiac mitochondrial function.
2. MATERIALS AND METHODS
2.1. Animals
All procedures were carried out in accordance with The Animals (Scientific Procedures) Act, 1986. The ARRIVE guidelines were followed for reporting the use of animals in scientific experiments. 52 Local ethical approval was granted by The University of Manchester Animal Welfare Ethical and Review Board. Female Wistar rats (250–275 g) were purchased from Charles River, and maintained at the University of Manchester. All animals were allowed to acclimatize for at least 1 week upon arrival before use. Animals were group‐housed in individually ventilated cages under standard conditions, with a regular 12:12 h light/dark cycle. Access to food (standard rat chow, Envigo) and water was provided ad libitum.
2.2. Hypoxia model
Following acclimatization, dams were time‐mated overnight with Wistar males, with the presence of a copulatory plug the following morning used to confirm pregnancy. The date the plug was found was taken as the GD 0. On GD 6, pregnant rats were randomly assigned into four different groups: normoxic control (NC, n = 9), normoxic melatonin (NM, n = 11), hypoxic control (HC, n = 11), and hypoxic melatonin (HM, n = 8), and from this point onwards were individually housed. For hypoxic exposure, rats were transferred to an environmental chamber (Coy O2 In Vivo Glove Box, Coy Laboratory Products) at GD 6 where they were subjected to 13 ± 0.1% oxygen (O2). For antioxidant treatment, the drinking water of NM and HM animals was supplemented with 5 µg/ml melatonin (Sigma Aldrich) dissolved in the minimum required volume of ethanol (final concentration of ethanol, 0.03%). An equal concentration of ethanol was added to NC and HC drinking water as vehicle control. The dose of melatonin was chosen as it is comparable with the maximal effective dose recommended for overcoming jet lag in humans. 42 Melatonin was made up fresh every other day and water bottles were covered to prevent light‐induced breakdown. Maternal food and water intake were regularly measured throughout pregnancy, and maternal body weight was measured before and at the end of the exposure. On GD 20, dams were killed by cervical dislocation, decapitated, and maternal trunk blood was collected. The uterus was externalized, and fetuses and associated placentae were individually weighed and recorded. Fetal trunk blood was collected and pooled per litter. Maternal and fetal blood was collected in heparinized tubes and centrifuged at 5000 rpm for 5 min, aliquoted, and frozen at −80°C for subsequent analysis of plasma concentration of melatonin. From each litter, two hearts were used for mitochondrial analysis in separate chambers (one heart per chamber) and two hearts were snap frozen in liquid nitrogen and stored at −80°C for future analysis. For each set of experiments, pups were used from the same position on the uterine horn to account for the potential differential exposure to hypoxia within the uterus. 12 Tail tips were collected and stored at −20°C for sex determination by SRY genotyping. A schematic of the hypoxia model is shown in Figure 1.
Figure 1.
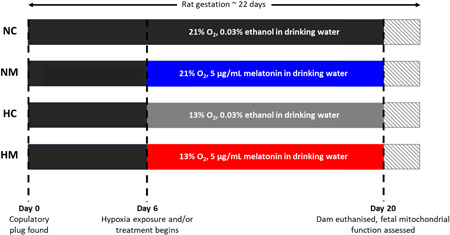
Timeline of hypoxia exposure and melatonin treatment during rat fetal development. Term rat gestation occurs around 22 days. HC, hypoxia control; HM, hypoxia melatonin; NC, normoxia control; NM, normoxia melatonin.
2.3. SRY genotyping
Tail tips were thawed and DNA extracted using RED Extract‐N‐Amp Tissue polymerase chain reaction (PCR) kit, according to the manufacturer's instructions. PCR was performed in tubes containing 0.5 μl of extracted DNA, and 19.5 μl of a master mixture (1× Red Taq DNA Polymerase Master Mix (VWR International)), 0.2 μM of each primer (Table 1). 53 PCR conditions were denaturation at 94°C for 4 min, followed by 36 cycles of amplification at 94°C for 1 min, 52°C for 1 min and 72°C for 1 min, and then extension at 72°C for 10 min with a thermocycler (C1000 Touch™; BioRad). For analysis of PCR products, each PCR mixture was subject to electrophoresis in a 2% agarose gel containing SafeView (Applied Biological Materials) for 40 min at 90 V. Gels were visualized using a ChemiDoc™ (MP; BioRad). The presence of an SRY band in addition to the positive control band for beta‐actin was used to confirm sex as male.
Table 1.
Primers for SRY genotyping
| Gene | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| Sry | 5′–TACAGCCTGAGGACATATTA–3′ | 5′–GCACTTTAACCCTTCGATGA–3′ |
| Actb | 5′–AGCCATGTACGTAGCCATCC–3′ | 5′–TGTGGTGGTGAAGCTGTAGC–3′ |
Note: Sequences for forward and reverse Sry and Actb primers. Actb was included as a control for DNA quality.
2.4. Melatonin assay
The concentration of maternal and fetal melatonin levels in plasma was determined in all samples within one assay/standard curve. Melatonin levels were determined using a colorimetric melatonin ELISA kit (code ‐ ab283259; Abcam). The sensitivity of the assay was 2.5 pg/ml.
2.5. Mitochondrial analysis
Hearts were excised from pups, homogenized in ice‐cold MIRO5 buffer (ethylene glycol‐bis(ß‐aminoethyl ether)‐N,N,N',N'‐tetraacetic aicd 0.5 mM, MgCl2 3 mM, K‐MES 60 mM, KH2PO4 10 mM, 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid 20 mM, sucrose 110 mM, and 0.1% bovine serum albumin (BSA)) and filtered through a nylon mesh (200 µm filter). Homogenate (60 µl) was quickly (within 10 min) injected into an Oroboros Oxygraph‐O2K (Oroboros Instruments) for simultaneous measurement of mitochondrial respiration and H2O2 production. A substrate–uncoupler–inhibitor–titration (SUIT) protocol was utilized (see details below) to examine oxygen consumption and ROS production at different electron transport chain complexes (Supporting information: Figure 1). Results were normalized to protein content (90 ± 3 µg), determined using the Bradford method, and corrected for the BSA content of the buffer (Bio‐Rad Laboratories). Remaining homogenate was frozen at −80°C for determination of CS activity.
2.6. Mitochondrial oxygen consumption and ROS production
Production of H2O2 was measured through the addition of Amplex® UltraRed (10 μM), 1 U/ml horseradish peroxidase (HRP), and 5 U/ml superoxide dismutase (SOD) to each chamber, before addition of homogenate. Amplex® UltraRed oxidizes in the presence of H2O2 to form resorufin, using HRP as a catalyst, while SOD converts extramitochondrial superoxide (O2−) to H2O2. Amplex® UltraRed was excited at 563 nm and emission measured at 587 nm. An H2O2 calibration was carried out once in each experiment by adding two additions of 40 µM H2O2 to achieve a chamber concentration of 0.1 and 0.2 µM. CI substrates pyruvate (6.25 mM), malate (2 mM), and glutamate (10 mM) were then added to each chamber, followed by the homogenate (90 ± 3 µg), to achieve LEAK respiratory state with CI substrates in the absence of adenylates (LEAKCI). Saturating ADP (5 mM) was then added to activate OXPHOS with CI substrates (OXPHOSCI). This was followed by succinate addition (10 mM), to measure the additive effects of CII substrates on OXPHOS (OXPHOSCI + CII). To assess maximum electron transfer capacity (ETCCI + II), mitochondria were uncoupled through the addition of carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP), which was titrated in steps to a final concentration of 0.75–3 µM, until additions failed to cause a subsequent increase in respiration. Following this, the CI inhibitor rotenone (0.15 μM) was added to assess ETCCII with CII substrates only. Next, CIII inhibitor antimycin A (12.5μM) was added to block the electron transport pathway so residual nonmitochondrial oxygen consumption (ROX) could be measured. To measure CIV activity, the electron donor N,N,N′,N′‐tetramethyl‐p‐phenylenediamine (TMPD, 0.5mM) was added, in combination with ascorbate (2 mM) to prevent autoxidation of TMPD. Finally, the CIV inhibitor sodium azide (50 mM) was added, to measure background nonmitochondrial oxygen consumption following the addition of TMPD.
2.7. Lipid peroxidation assay
Lipid peroxidation was assessed using the TBARS (thiobarbituric acid [TBA] reactive substance) assay, which measures the levels of malondialdehyde (MDA) in biological samples. Frozen hearts were briefly thawed and placed in ice‐cold 10 mM phosphate buffer (pH 7.2), containing 200 μM butylated hydroxytoluene to prevent homogenization‐induced artificial oxidative stress. Hearts were homogenized at 4°C and protein concentration was measured using the Bradford method. Homogenate was added to a TBA‐TCA‐HCl mix (0.375% TBA, 15% trichloroacetic acid [TCA], 0.25 N hydrochloric acid [HCl]) at a 1:2 ratio and incubated at 95°C for 30 min. Samples were then incubated on ice for 10 min, before the sample was added to 1‐butanol at a 2:1 ratio and centrifuged at 15 000g for 3min. The top layer containing the malondialdehyde‐thiobarbituric acid adduct was pipetted into a microplate in duplicate, and absorbance measured at 532 nm. MDA concentrations were determined by using 1,1,3,3‐tetraethoxypropane as a standard, normalized to protein concentration, and expressed as nmoles/mg protein.
2.8. CS activity
CS activity was measured using a spectrophotometric method. Frozen samples were thawed and diluted in CS assay buffer (100μM 5,5′‐dithiobis‐(2‐nitrobenzoic acid) [DTNB], 300 μM acetyl coenzyme‐A, 100 mM Tris, 0.1% Triton‐X‐100). Oxaloacetate (0.5 mM) was added, and absorbance was measured repeatedly for 10 min at 412 nm to determine V max.
2.9. Statistics
Fetal and placental weights were pooled to obtain litter averages. For maternal and fetal parameters, a two‐way analysis of variance (ANOVA) test was used with Tukey post‐hoc test (GraphPad Prism 8.1.2). For food and water intake, the two factors were the experimental group and GD. For maternal weight, fetal weight, placental weight, and relative placental weight, the two factors were treatment (hypoxic or normoxic) and drug (control or melatonin). For mitochondrial analysis, lipid peroxidation, and CS activity, multiple measurements were taken from the same litter. Therefore, linear mixed modeling (SPSS Statistics, IBM) was performed to account for the nested (clustered) design of the experiment, for example, observations from different pups from the same dam (with treatment [hypoxic or normoxic] and drug [control or melatonin] as cofactors). A Bonferroni post‐hoc test was used. A multivariate ANOVA (MANOVA) analysis was also performed on respiration and H2O2 production (treatment [hypoxic or normoxic] and drug [control or melatonin], and the interaction between treatment and drug). The significance of the interaction and the main effects were examined using the Wilks' Lambda statistic. For mitochondrial analysis, lipid peroxidation, and CS activity, data were log‐transformed to achieve normal distribution before statistical analysis. A p < .05 was considered significant and p < .1 was included on graphs and within the results section for clarity.
3. RESULTS
3.1. Fetal and maternal parameters
Hypoxia did not affect maternal food intake across the hypoxic incubation when compared to the NC group (Figure 2A). However, there was a reduction in maternal food intake at the start of the exposure in both hypoxic groups, compared to the NM group (interaction effect p = .027 [hypoxia control vs. normoxia melatonin], p = .024 [hypoxia melatonin vs. normoxia melatonin]), which may have reduced maternal weight gain at GD 20 (Figure 2C). However, maternal food intake recovered after 4 days of hypoxia (Figure 2A), and no difference was observed in water intake between the four groups (Figure 2B). While maternal body weight gain during GD 6–20 was lower in the hypoxic groups (main hypoxia effect p = .003), there were no differences in litter size or fetal body weight (Figures 2C,D and 3A,D). Hypoxia increased placental weight in both male and female fetuses, but this effect only reached significance in males (main hypoxia effect p = .026 [males], p = .055 [females]) (Figure 3B,E). Additionally, hypoxia decreased the ratio of fetal weight to placental weight, for both males and females (main hypoxia effect p = .001 and .003, respectively) (Figure 3C,F).
Figure 2.
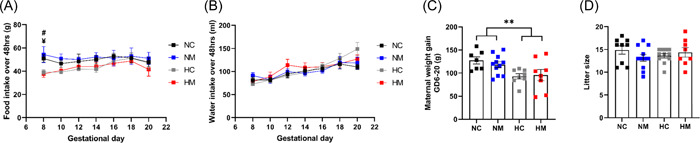
Maternal parameters for dams (gestational day [GD] 6–20). Food and water were monitored every 48 h in normoxic control, normoxic melatonin, hypoxic control, and hypoxic melatonin treated dams (A, B). Maternal weight was measured at GD 6 and 20, and the weights were used to calculate weight gain (C). Litter size was measured following euthanasia at GD 20 (D). n = 9 (NC), 11 (NM), 11 (HC), 8 (HM). For food intake, #Significant difference between NM and HC, ¥significant difference between NM and HM (p < .05). **p < .01 to represent significant effects of hypoxia. Significance was assessed using a two‐way ANOVA. Error bars show mean ± SEM. ANOVA, analysis of variance; GD, gestational day; HC, hypoxia control; HM, hypoxia melatonin; NC, normoxia control; NM, normoxia melatonin.
Figure 3.
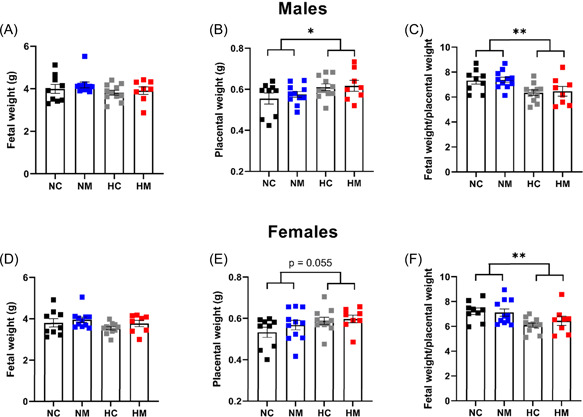
Fetal parameters at GD 20. Fetal weight and placental weight were measured for male fetuses (A–C) and female fetuses (D–F) at GD 20. Data points represent litter averages. n = 134 pups from 9 litters (NC), 145 pups from 11 litters (NM), 150 pups from 11 litters (HC), 115 and pups from 8 litters (HM). *p< .05, **p< .01 to represent significant effects of hypoxia. Significance was assessed using a two‐way ANOVA. Error bars show mean ± SEM. ANOVA, analysis of variance; GD, gestational day; HC, hypoxia control; HM, hypoxia melatonin; NC, normoxia control; NM, normoxia melatonin.
As expected, maternal treatment with melatonin increased the concentration of melatonin at GD 20 in both maternal and fetal plasma compared with untreated pregnancies (main melatonin effect p < .0001), and hypoxia did not affect fetal melatonin concentrations (Table 2). It should be noted that maternal melatonin concentrations were slightly higher in hypoxic dams compared to normoxic (interaction effect p = .081 [hypoxia melatonin vs. normoxia melatonin]). Nevertheless, maternal melatonin treatment had no effect on any of the measured fetal and maternal parameters (Figures 2 and 3).
Table 2.
Plasma melatonin concentrations
| NC | NM | HC | HM | |
|---|---|---|---|---|
| Maternal plamsa (melatonin) (pg/ml) | 80 ± 13.7 | 515 ± 106.5 *** | 92.9 ± 17.1 | 800.7 ± 118.1 *** |
| Fetal plasma (melatonin) (pg/ml) | 33.2 ± 4.5 | 308.1 ± 50.7 *** | 57.6 ± 11.8 | 326.9 ± 51.3 *** |
Note: Values are mean ± SEM for the concentration of melatonin in plasma collected on gestational day 20 from NC, NM, HC, and HM dams and fetuses. n = 9–10 per group.
Abbreviations: ANOVA, analysis of variance; NC, normoxic control; NM, normoxic melatonin; HC, hypoxic control; HM, hypoxic melatonin.
p < .0001 significant effect of melatonin (two‐way ANOVA).
3.2. Mitochondrial respiration and CS activity
Mitochondrial homogenate preparations were of high quality, as attested by high respiratory control ratios (9.5 ± 0.4) and OXPHOS coupling efficiency ratios (0.88 ± 0.01), with CI substrates (malate, pyruvate, and glutamate) (Figure 4G,H). Homogenates responded to SUIT chemicals as expected 54 (Supporting Information: Figure 1).
Figure 4.
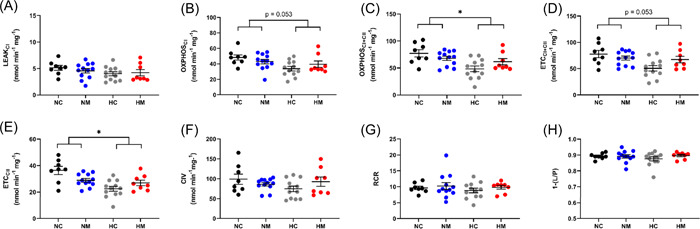
Developmental hypoxia reduces oxidative capacity in heart mitochondria from male fetuses at multiple points of the electron transport chain. Mitochondrial oxidative capacity was measured following a SUIT protocol. Each panel represents a different respiratory state. RCR values (G) and coupling efficiencies (H) were high for all groups, demonstrating mitochondrial homogenate preparations were of good quality. n = 8 (NC from 6 litters), 12 (NM from 7 litters), 12 (HC from 7 litters), 8 (HM from 6 litters). *p< .05 (effects of hypoxia). Significance was assessed using a linear mixed model (nested). Error bars show mean ± SEM. 1‐(L/P), coupling efficiency; CI, complex I; CII, complex II; CIV, complex IV; ETC, electron transport capacity; HC, hypoxia control; HM, hypoxia melatonin; NC, normoxia control; NM, normoxia melatonin; OXPHOS, oxidative phosphorylation; RCR, respiratory control ratio; SUIT, substrate–uncoupler–inhibitor–titration.
In heart homogenates from male fetuses, chronic hypoxia caused a decrease in mitochondrial oxygen consumption in the OXPHOXCI + CII and ETCCII states (main hypoxia effect p = .031 and .037, respectively) (Figure 4C,E). This effect was also evident in the OXPHOSCI and ETCCI + CII states, but did not reach significance (main hypoxia effect p = .053) (Figure 4B,D). All the other respiratory parameters in males (LEAK, CIV respiration, RCR, and OXPHOS‐coupling efficiency) were unaffected by hypoxia (Figure 4A,F,G,H). In contrast to males, there was no effect of hypoxia on any mitochondrial respiratory parameter in female fetuses (Figure 5), with all four groups showing comparable respiration across respiratory states. In both sexes, maternal melatonin treatment had no effects on mitochondrial oxygen consumption in normoxic or hypoxic pregnancies (Figures 4, 5). We also measured enzymatic activity of CS activity as a common marker for mitochondrial content and found no differences between any of the experimental groups in males or females (Figure 8A,C).
Figure 5.
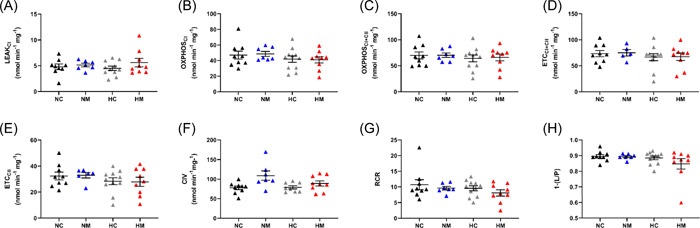
Developmental hypoxia does not change oxidative capacity in heart mitochondria from female fetuses. Mitochondrial oxidative capacity was measured following a SUIT (substrate‐uncoupler‐inhibitor‐titration) protocol. Each panel represents a different respiratory state. RCR values (G) and coupling efficiencies (H) were high for all groups, demonstrating mitochondrial homogenate preparations were of good quality. n = 9 (NC from 7 litters), 7 (NM from 5 litters), 11 (HC from 8 litters), 9 (HM from 7 litters). Significance was assessed using a linear mixed model (nested). Error bars show mean ± SEM. 1‐(L/P), coupling efficiency; CI, complex I; CII, complex II; CIV, complex IV; ETC, electron transport capacity; HC, hypoxia control; HM, hypoxia melatonin; NC, normoxia control; NM, normoxia melatonin; OXPHOS, oxidative phosphorylation; RCR, respiratory control ratio; SUIT, substrate–uncoupler–inhibitor–titration.
Figure 8.
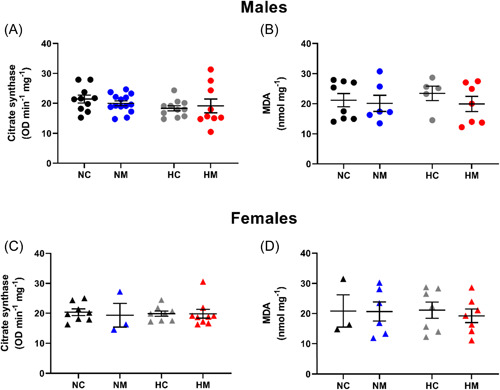
Effects of developmental hypoxia on citrate synthase activity and lipid peroxidation in fetal ventricles. Citrate synthase activity and MDA were measured in homogenized ventricular tissue from male (A, B) and female (C, D) fetuses. Circular symbols show males, triangular symbols show females. For MDA (males), n = 8 (NC from 6 litters), 6 (NM from 6 litters), 5 (HC from 4 litters), 7 (HM from 6 litters). For MDA (females), n = 3 (NC from 3 litters), 6 (NM from 5 litters), 7 (HC from 6 litters), 7 (HM from 6 litters). For citrate synthase (males), n = 10 (NC from 7 litters), 14 (NM from 7 litters), 11 (HC from 6 litters), 9 (HM from 6 litters). For citrate synthase (females), n = 8 (NC from 6 litters), 3 (NM from 3 litters), 8 (HC from 6 litters), 9 (HM from 7 litters). Significance was assessed using a linear mixed model (nested). Error bars show mean ± SEM. HC, hypoxia control; HM, hypoxia melatonin; MDA, malondialdehyde; NC, normoxia control; NM, normoxia melatonin; OD, optical density.
3.3. Mitochondrial ROS production and oxidative stress
As mitochondrial H2O2 production is dependent on O2 consumption, we expressed H2O2 production normalized to O2 consumption and found that H2O2 production/O2 consumption was increased in the hypoxic male cohort (Figure 6). This effect was apparent in the OXPHOSI + II, ETCI + II, and ETCCII states, but only reached significance in the ETCCII state (main hypoxia effect p = .027) (Figure 6C–E). There were no differences in basal mitochondrial H2O2 production when normalized to protein in any of the male experimental groups (Supporting Information: Figure 2). Similar to males, there were no differences in basal mitochondrial H2O2 production normalized to protein in any of the female experimental groups (Supporting Information: Figure 3), and there were also no differences when H2O2 production was normalized to O2 consumption (Figure 7). To determine whether changes in male H2O2 production/O2 consumption led to oxidative stress, we measured lipid peroxidation (assessed with the TBARS assay) and found no differences between normoxic or hypoxic groups in either sex (Figure 8B,D). Finally, melatonin treatment had no effect on H2O2 production, H2O2 production/O2 consumption or lipid peroxidation in any of the groups studied (Figures 6, 7, 8). The MANOVA analysis agreed with the above findings, showing no interaction effects between the four groups (p = .072), but a statistically significant effect of hypoxia (p = .007).
Figure 6.
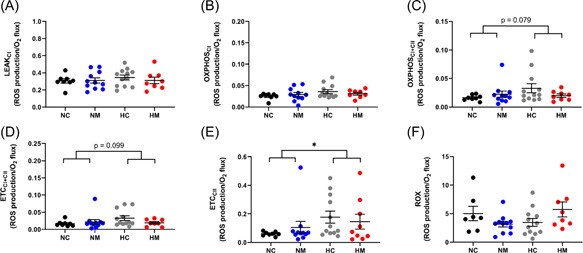
Developmental hypoxia significantly increases H2O2 production in rat heart mitochondria from male fetuses. Each panel represents a different respiratory state. n = 8 (NC from 6 litters), 11 (NM from 6 litters), 12 (HC from 7 litters), 8 (HM from 6 litters). *p< .05. Significance was assessed using a linear mixed model (nested). Error bars show mean ± SEM. CI, complex I; CII, complex II; ETC, electron transport capacity; HC, hypoxia control; HM, hypoxia melatonin; NC, normoxia control; NM, normoxia melatonin; OXPHOS, oxidative phosphorylation; ROX, residual oxygen consumption; SUIT, substrate–uncoupler–inhibitor–titration.
Figure 7.
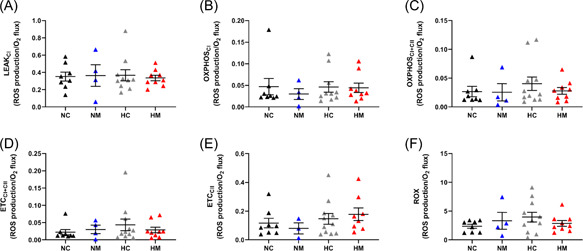
Developmental hypoxia does not change H2O2 production in rat heart mitochondria from female fetuses. Each panel represents a different respiratory state. n = 8 (NC from 7 litters), 4 (NM from 3 litters), 11 (HC from 8 litters), 9 (HM from 7 litters). Significance was assessed using a linear mixed model (nested). Error bars show mean ± SEM. CI, complex I; CII, complex II; ETC, electron transport capacity; HC, hypoxia control; HM, hypoxia melatonin; NC, normoxia control; NM, normoxia melatonin; OXPHOS, oxidative phosphorylation; ROX, residual oxygen consumption; SUIT, substrate–uncoupler–inhibitor–titration.
4. DISCUSSION
This is the first study to determine the effects of developmental hypoxia and maternal melatonin treatment on mitochondrial respiration and ROS production in the fetal heart. The principle findings of this study were that hypoxia reduced mitochondrial respiratory capacity in the hearts from male, but not female, fetuses. Male cardiac mitochondria showed increased rates of H2O2 production/O2 consumption, but this was not associated with increased lipid peroxidation. Therefore, the data show that developmental hypoxia has sex‐specific effects on cardiac mitochondria in the fetus, which may explain the differential risk to cardiac metabolic disease in later life in male and female offspring.
4.1. Effects of hypoxia and maternal melatonin treatment on fetal and maternal biometry
We found that hypoxia at 13% O2 from Days 6 to 20 had no effect on fetal body weight, but did cause an increase in placental weight and decrease in placental efficiency, which is consistent with previous reports of hypoxic pregnancy. 7 , 55 , 56 These findings reinforce the idea that in response to hypoxia in early pregnancy, placental growth is enhanced to maintain fetal growth, with the reduction in placental efficiency reflecting this. 6 , 56 Maternal melatonin treatment did not influence fetal or placental weight outcomes in this study.
4.2. Effects of hypoxia on heart mitochondrial respiratory capacity and H2O2 production in male fetuses
Male fetuses exposed to developmental hypoxia exhibited a mild but significant reduction in heart mitochondrial respiratory capacity and an increase in H2O2 production/O2 consumption. Importantly, we recently found a more pronounced version of the same phenotype in adult male mice from hypoxic pregnancies. 31 Therefore, combined, our studies suggest pathways involved in mitochondrial function and ROS production can be programmed by gestational hypoxia, and the phenotype may become more pronounced with aging. These changes in mitochondrial function in the heart may contribute to the range of cardiac abnormalities that are evident in adult male offspring from hypoxic pregnancies, including diastolic dysfunction, 9 sympathetic dominance, 6 and sensitivity to ischemia‐reperfusion injury. 16 , 17 In this regard, future work should investigate the effects of gestational hypoxia to stress‐tolerance in offspring cardiac mitochondria.
A reduction in mitochondrial respiration is a common response in adult mammals 27 , 57 and ectothermic vertebrates 58 , 59 , 60 subjected to chronic hypoxia. In particular, lowlander humans acclimatizing to hypoxia during an ascent to Mount Everest have reduced mitochondrial respiratory capacity in skeletal muscle, and similar to our study, this is only evident with CII substrates. 61 The physiological significance of reducing respiratory capacity in hypoxia is debatable. At first sight, the response may seem counterintuitive because ATP production will be reduced when ATP demand already outstrips supply. However, a lower respiratory rate also serves to reduce electron leak from the electron transport chain, which should limit ROS production. 27 While H2O2 production was higher in the hypoxic cohort when H2O2 was normalized to O2 consumption, absolute levels of H2O2 in males were similar between groups when normalized to protein content. These results support the notion that by reducing respiratory capacity in hypoxia, absolute ROS production is maintained, representing an adaptive response to limit overall ROS production. Reducing flux through CII is particularly relevant, as this enzyme is heavily implicated in the overproduction of ROS during ischemia and reperfusion injury. 62 Nevertheless, lower respiratory capacity has negative consequences for ATP production, and while previous work suggests humans at high altitudes overcome this problem by reducing proton leak pathways to increase mitochondrial efficiency, 61 the present study found no effects of hypoxia on leak respiration rate or coupling efficiency (RCR) in male fetuses. In addition, our study on adult male mice from hypoxic pregnancies showed the reduction in respiratory capacity persists into adulthood and worsens, which is associated with an even greater basal H2O2 production. 31 Therefore, while mitochondrial remodeling during fetal hypoxia may be protective in the short term, it may become maladaptive in the long term.
Our experimental design also provides insight into the mechanism reducing respiratory capacity in male fetuses from hypoxic pregnancies. The lower respiratory rate was not associated with any changes in CS activity, which suggests decreased mitochondrial content is unlikely to be the underlying mechanism. 63 However, since the reduced rate was still evident in the presence of a CI inhibitor (rotenone), and absent when electrons were donated directly to CIV, our results suggest that the mechanism is likely to involve CII and/or CIII. It is possible that protein expression of these complexes is reduced by hypoxia exposure. Indeed, previous work on a guinea pig model of chronic hypoxia in late gestation (10.5% oxygen from GD 50 for 14 days) has shown male offspring to have reduced cardiac protein expression of complexes I, III, and V. 34 , 35 Furthermore, our recent study on adult male mice from hypoxic pregnancies (13% oxygen from GD 6–18; term in mice is ~GD 19) showed that the reduction in respiratory capacity was associated with reduced protein expression of complexes I, II and IV. 31 Therefore, the expression of mitochondrial proteins could be permanently modified by exposure to hypoxia, presumably via epigenetic mechanisms. In support of this concept, recent work has shown gestational hypoxia induces global changes in DNA methylation in the fetal heart, and many of the gene pathways were related to metabolism and mitochondrial function. 64 , 65
Our experimental design also allows us to speculate on the mechanism that underlies the increased H2O2 production in males from hypoxic pregnancies. Given that the effect was apparent when CI was inhibited with rotenone, and it disappeared when CIII was inhibited with antimycin A, our data indicate that the site of H2O2 production is the ubiquinone redox site, Qi, of CIII—the site bound by antimycin A. 66 CIII is a major producer of ROS within the mitochondrial respiratory chain, and production of ROS at this complex is linked with a wide variety of cardiac disease models associated with hypoxia. 67 , 68 Nevertheless, the decrease in respiratory capacity in male fetuses likely compensates for the increased H2O2 production, which is consistent with the lack of any lipid peroxidation. It is also possible that oxidative stress was limited by increasing antioxidant capacity, as previous metabolomic research has shown the potent antioxidant bilirubin is increased in the hypoxic fetal heart. 69 The lack of hypoxia‐induced oxidative stress contrasts with previous work where prenatal hypoxia increased markers of oxidative stress in the rat, sheep, and guinea pig heart. 35 , 69 , 70 However, these studies used a more severe level of hypoxia (10%–11% O2), which is likely to cause greater ROS production and may also affect maternal nutrition, which could independently promote oxidative stress. 71 , 72 Clearly, more research is needed to unravel the effects of hypoxia on fetal ROS signaling in the heart.
4.3. Effects of hypoxia on mitochondrial respiratory capacity and H2O2 production in female fetuses
Additional data in the present study show that the effects of gestational hypoxia on fetal cardiac mitochondrial respiration were sex‐dependent, with females being more protected than males. Mitochondrial respiration was very similar between normoxic and hypoxic female fetuses across respiratory states, and H2O2 production was also unaffected when normalized to either protein or oxygen consumption, which is consistent with no change in lipid peroxidation. Interestingly, adult female mice from hypoxic pregnancies have increased respiratory capacity and lower H2O2 production, which was associated with the increased enzymatic activity of CIV. 31 Therefore, fetal cardiac mitochondria in females exposed to hypoxia appear to be on a different developmental trajectory to their male counterparts. This plasticity in mitochondrial function may be one of the reasons that female adult offspring from hypoxia are less susceptible to cardiac dysfunction than males, 32 and appear to recover better from ischemia‐reperfusion injury. 16
Thus far, there have been limited studies investigating sex‐dependent effects of hypoxia at the fetal life stage, particularly with regard to mitochondrial function. However, one study has shown late‐onset gestational hypoxia (10.5% oxygen from GD 50 for 14 days) reduces female fetal guinea pig mitochondrial protein expression of complexes I, III, and V in the heart, but in contrast to males, this was not associated with any change in enzymatic activity of complexes IV. 34 Interestingly, the same study showed that the timing of hypoxia was important in the guinea pig response, with early onset hypoxia (10.5% oxygen from GD 25 for 39 days) having no effects on female mitochondrial protein expression or activity. While the level of hypoxia was more severe in the guinea pig study (10.5% vs. 13% oxygen), the collective data suggest that female fetuses have a milder response to gestational hypoxia and maintain respiratory rates. These results align well with previous studies on various intrauterine stressors that show sex‐dependent differences in mitochondrial responses. 73 , 74 , 75 , 76 , 77 One possible explanation is differential gene expression by sex‐dependent epigenetic regulation. Indeed, previous work has found differences in the expression levels of DNA methyltransferase enzymes between males and females during gestation, 78 , 79 and Huang et al. found sex‐dependent methylation patterns in response to prenatal hypoxia. 65 Clearly, more work is necessary to identify the mechanisms behind sex‐dependent mitochondrial programming in the hypoxic fetus.
4.4. Effects of melatonin treatment
In the present study, we used melatonin as our antioxidant, because this hormone is not only safely taken by humans, but it can be transported into the mitochondria, and it is also synthesized within the mitochondrial matrix. 43 In this way, melatonin may be considered a mitochondria‐targeted antioxidant, 44 , 45 , 46 which scavenges ROS at the main site of free radical synthesis. 48 In the present study, we show that maternal treatment with melatonin in the drinking water led to a significant increase in the maternal and fetal plasma melatonin concentration. These data confirm that melatonin crosses the placenta in rodents when administered to the maternal drinking water. Interestingly, there was a trend toward an increased maternal melatonin concentration in the hypoxic dams compared to normoxic. Similar results have been reported in the same model elsewhere, 11 and while the underlying reason is unclear, we speculate that hypoxia could be altering metabolism, which may affect circulating melatonin concentrations. Nevertheless, the present study shows that prenatal hypoxia did not affect the fetal plasma concentrations of melatonin.
The final concentration of melatonin in maternal and fetal plasma following melatonin treatment was similar to that measured in other studies using pregnant rats, 10 , 80 , 81 and is within the range or lower than plasma concentrations in men and women following melatonin intake to avoid jet lag. 82 However, maternal melatonin treatment had no effects on respiratory capacity, H2O2 production, or lipid peroxidation in fetal hearts in normoxic or hypoxic pregnancies. It is possible that melatonin could have a protective effect on mitochondrial pathways and properties that were not measured in this study, such as the permeability transition pore, DNA mutations, membrane stability, or mitochondrial dynamics. 43 Furthermore, our study only investigated the chronic effects of fetal hypoxia on mitochondrial respiration and ROS production. Acute reoxygenation of heart tissue poses a significant challenge to hypoxic fetuses, and therefore, melatonin may have shown greater protective effects against cellular injury if measured at the reperfusion phase. Nevertheless, our study suggests that maternal melatonin treatment in normoxic pregnancies does not have any negative effects on mitochondrial respiration or ROS production in the fetal heart. This is important information for developing this antioxidant as a safe intervention for pregnancy complications, particularly as melatonin is currently being trialed in human pregnancies complicated by fetal growth restriction and pre‐eclampsia. 46 , 49
4.5. Study limitations
There are some limitations related to our study design and protocol. First, the relatively small sample size may have limited the power of our study, particularly in terms of revealing the melatonin interaction effects. Indeed, there were often trends within our data that melatonin was preventing mitochondrial remodeling, and larger sample size may have resolved these effects. In addition, although we chose a dose of melatonin that was clinically relevant 42 and previously shown to be protective, 11 the concentration may not have been high enough to achieve metabolic protection. With respect to respiration measurements, we provided OXPHOS substrates at a saturating concentration, and therefore our results do not account for potential differences in substrate availability and/or preference in vivo. Furthermore, any posttranslational modifications, such as nitrosylation of the electron transport chain, would have been lost as a result of the homogenization process, and therefore any potential effects would not have been detected. Finally, our model of hypoxia caused a reduction in maternal weight gain, which may have had independent stress on the developing fetus even in the absence of overt growth restriction.
5. CONCLUSION AND PERSPECTIVES
Our study shows that developmental hypoxia has significant sex‐dependent effects on fetal cardiac mitochondrial respiration and ROS production, which may have long‐term implications for cardiac metabolic health and disease. These findings strongly align with the emerging evidence that mitochondria represent a key cellular target that underpins developmental programming (reviewed in Gyllenhammer et al. 83 ). Indeed, multiple studies have demonstrated mitochondrial changes induced by intrauterine stress can persist into adulthood, 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 and even across generations. 93 , 94 , 95 Given the crucial relationship between mitochondrial function and cardiac health, intrauterine mitochondrial programming could be an important mechanism that drives cardiovascular disease susceptibility in adulthood. Our study also shows that maternal melatonin treatment has no effects on fetal cardiac mitochondrial respiration and ROS production, despite its ability to prevent cardiac programming in adult offspring of hypoxic pregnancies. 11 The fact that melatonin had no negative effects on mitochondrial parameters in normoxic pregnancies is encouraging and provides support for this antioxidant being used therapeutically in complicated pregnancies.
AUTHOR CONTRIBUTIONS
Kerri L. M. Smith, Agnieszka Swiderska, Gina L. J. Galli, Mitchell C. Lock, Lucia Graham, Wulan Iswari, Tashi Choudhary, Donna Thomas, Elizabeth C. Cottrell, and Hager M. Kowash all contributed to data acquisition and analysis. Michelle Desforges, Elizabeth C. Cottrell, Andrew W. Trafford, Dino A. Giussani, and Gina L. J. Galli contributed to the concept and experimental design. Kerri L. M. Smith, Agnieszka Swiderska, Mitchell C. Lock, Michelle Desforges, Elizabeth C. Cottrell, Andrew W. Trafford, Dino A. Giussani, and Gina L. J. Galli contributed to drafting and approval of the manuscript.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
This study was funded by a British Heart Foundation (BHF) PhD studentship awarded to Kerri Smith and a BHF project grant awarded to Dr Gina Galli, Prof. Andrew Trafford, and Prof. Dino Giussani (PG/18/5/33527). We would like to thank the staff of the Biological Services Facility, University of Manchester.
Smith KLM, Swiderska A, Lock MC, et al. Chronic developmental hypoxia alters mitochondrial oxidative capacity and reactive oxygen species production in the fetal rat heart in a sex‐dependent manner. J Pineal Res. 2022;73:e12821. 10.1111/jpi.12821
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Hutter D, Kingdom J, Jaeggi E. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int J Pediatr. 2010;2010:401323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen E, Baerts W, van Bel F. Brain‐sparing in intrauterine growth restriction: considerations for the neonatologist. Neonatology. 2015;108(4):269‐276. [DOI] [PubMed] [Google Scholar]
- 3. Giussani DA. The fetal brain sparing response to hypoxia: physiological mechanisms. J Physiol. 2016;594(5):1215‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Louey S, Jonker SS, Giraud GD, Thornburg KL. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol. 2007;580(Pt. 2):639‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ganguly E, Spaans F, Morton JS, et al. Placenta‐targeted treatment in hypoxic dams improves maturation and growth of fetal cardiomyocytes in vitro via the release of placental factors. Exp Physiol. 2020;105(9):1507‐1514. [DOI] [PubMed] [Google Scholar]
- 6. Romanowicz J, Dhari Z, Guerrelli D, et al. Chronic perinatal hypoxia delays cardiac maturation in a mouse model for cyanotic congenital heart disease. Am J Physiol Heart Circ Physiol. 2020;320(5):H1873‐H1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giussani DA, Camm EJ, Niu Y, et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS One. 2012;7(2):e31017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lawrence KM, Hennessy‐Strahs S, McGovern PE, et al. Fetal hypoxemia causes abnormal myocardial development in a preterm ex utero fetal ovine model. JCI Insight. 2018;3(24):e124338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niu Y, Kane AD, Lusby CM, et al. Maternal allopurinol prevents cardiac dysfunction in adult male offspring programmed by chronic hypoxia during pregnancy. Hypertension. 2018;72(4):971‐978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Herrera EA, Camm EJ, Cross CM, Mullender JL, Wooding FB, Giussani DA. Morphological and functional alterations in the aorta of the chronically hypoxic fetal rat. J Vasc Res. 2012;49(1):50‐58. [DOI] [PubMed] [Google Scholar]
- 11. Hansell JA, Richter HG, Camm EJ, et al. Maternal melatonin: effective intervention against developmental programming of cardiovascular dysfunction in adult offspring of complicated pregnancy. J Pineal Res. 2022;72(1):e12766. [DOI] [PubMed] [Google Scholar]
- 12. Hula N, Spaans F, Vu J, et al. Placental treatment improves cardiac tolerance to ischemia/reperfusion insult in adult male and female offspring exposed to prenatal hypoxia. Pharmacol Res. 2021;165:105461. [DOI] [PubMed] [Google Scholar]
- 13. Shah A, Matsumura N, Quon A, Morton JS, Dyck J, Davidge ST. Cardiovascular susceptibility to in vivo ischemic myocardial injury in male and female rat offspring exposed to prenatal hypoxia. Clin Sci (Lond). 2017;131(17):2303‐2317. [DOI] [PubMed] [Google Scholar]
- 14. Rueda‐Clausen CF, Morton JS, Lopaschuk GD, Davidge ST. Long‐term effects of intrauterine growth restriction on cardiac metabolism and susceptibility to ischaemia/reperfusion. Cardiovasc Res. 2011;90(2):285‐294. [DOI] [PubMed] [Google Scholar]
- 15. Xu Y, Williams SJ, O'brien D, Davidge ST. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J. 2006;20(8):1251‐1253. [DOI] [PubMed] [Google Scholar]
- 16. Xue Q, Zhang L. Prenatal hypoxia causes a sex‐dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: role of protein kinase cϵ. J Pharmacol ExpTher. 2009;330(2):624‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Investig. 2003;10(5):265‐274. [DOI] [PubMed] [Google Scholar]
- 18. Wojczakowski W, Kimber‐Trojnar Ż, Dziwisz F, Słodzińska M, Słodziński H, Leszczyńska‐Gorzelak B. Preeclampsia and cardiovascular risk for offspring. J Clin Med. 2021;10(14):3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang AM, Karumanchi SA. Offspring cardiovascular disease in preeclampsia. Hypertension. 2017;69(4):589‐590. [DOI] [PubMed] [Google Scholar]
- 20. Mamun AA, O'callaghan MJ, Williams GM, Najman JM. Maternal smoking during pregnancy predicts adult offspring cardiovascular risk factors—evidence from a community‐based large birth cohort study. PLoS One. 2012;7(7):e41106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thornburg KL, O'Tierney PF, Louey S. Review: the placenta is a programming agent for cardiovascular disease. Placenta. 2010;31(Suppl):S54‐S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gonzalez‐Candia A, Herrera EA. High altitude pregnancies and vascular dysfunction: observations from Latin American studies. Front Physiol. 2021;12:786038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moore LG. Hypoxia and reproductive health: reproductive challenges at high altitude: fertility, pregnancy and neonatal well‐being. Reproduction. 2021;161(1):F81‐F90. [DOI] [PubMed] [Google Scholar]
- 24. Papa S, Martino PL, Capitanio G, et al. The oxidative phosphorylation system in mammalian mitochondria. Adv Exp Med Biol. 2012;942:3‐37. [DOI] [PubMed] [Google Scholar]
- 25. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Rad Bio Med. 2016;100:14‐31. [DOI] [PubMed] [Google Scholar]
- 26. Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta. 2010;1797(6‐7):1171‐1177. [DOI] [PubMed] [Google Scholar]
- 27. Murray AJ, Horscroft JA. Mitochondrial function at extreme high altitude. J Physiol. 2016;594(5):1137‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murray AJ. Metabolic adaptation of skeletal muscle to high altitude hypoxia: how new technologies could resolve the controversies. Genome Med. 2009;1(12):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. West‐Eberhard M. Developmental Plasticity and Evolution. Oxford University Press; 2003. [Google Scholar]
- 30. Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56(2):130‐140. [DOI] [PubMed] [Google Scholar]
- 31. Hellgren KT, Premanandhan H, Quinn CJ, Trafford AW, Galli G. Sex‐dependent effects of developmental hypoxia on cardiac mitochondria from adult murine offspring. Free Radic Biol Med. 2021;162:490‐499. [DOI] [PubMed] [Google Scholar]
- 32. Thompson LP, Chen L, Polster BM, Pinkas G, Song H. Prenatal hypoxia impairs cardiac mitochondrial and ventricular function in guinea pig offspring in a sex‐related manner. Am J Physiol Regul Integr Comp Physiol. 2018;315(6):R1232‐R1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Al‐Hasan YM, Pinkas GA, Thompson LP. Prenatal hypoxia reduces mitochondrial protein levels and cytochrome c oxidase activity in offspring guinea pig hearts. Reprod Sci. 2014;21(7):883‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song H, Polster BM, Thompson LP. Chronic hypoxia alters cardiac mitochondrial complex protein expression and activity in fetal guinea pigs in a sex‐selective manner. Am J Physiol Regul Integr Comp Physiol. 2021;321:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Al‐Hasan YM, Evans LC, Pinkas GA, Dabkowski ER, Stanley WC, Thompson LP. Chronic hypoxia impairs cytochrome oxidase activity via oxidative stress in selected fetal guinea pig organs. Reprod Sci. 2013;20(3):299‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giussani D, Niu Y, Herrera E, et al. Heart disease link to fetal hypoxia and oxidative stress. In: Zhang L, Ducsay CA, eds. Advances in Fetal and Neonatal Physiology. Vol 814. Springer; 2014:77‐87. [DOI] [PubMed] [Google Scholar]
- 37. Kane AD, Herrera EA, Camm EJ, Giussani DA. Vitamin c prevents intrauterine programming of in vivo cardiovascular dysfunction in the rat. Circ J. 2013;77(10):2604‐2611. [DOI] [PubMed] [Google Scholar]
- 38. Giussani DA. Breath of life: heart disease link to developmental hypoxia. Circulation. 2021;144(17):1429‐1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Botting KJ, Skeffington KL, Niu Y, et al. Translatable mitochondria‐targeted protection against programmed cardiovascular dysfunction. Sci Adv. 2020;6(34):eabb1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Itani N, Skeffington KL, Beck C, et al. Protective effects of pravastatin on the embryonic cardiovascular system during hypoxic development. FASEB J. 2020;34(12):16504‐16515. [DOI] [PubMed] [Google Scholar]
- 41. Spiroski A‐M, Niu Y, Nicholas LM, et al. Mitochondria antioxidant protection against cardiovascular dysfunction programmed by early‐onset gestational hypoxia. FASEB J. 2021;35(5):e21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herxheimer A, Petrie KJ. Melatonin for the prevention and treatment of jet lag. Cochrane Database Syst Rev. 2002;2:Cd001520. [DOI] [PubMed] [Google Scholar]
- 43. Tan D‐X, Manchester LC, Qin L, Reiter RJ. Melatonin: A mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int J Mol Sci. 2016;17(12):2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hardeland R. Melatonin and the electron transport chain. Cell Mol Life Sci. 2017;74(21):3883‐3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre‐Jimenez M, Qin L. Melatonin as an antioxidant: under promises but over delivers. J Pineal Res. 2016;61(3):253‐278. [DOI] [PubMed] [Google Scholar]
- 46. Reiter RJ, Rosales‐Corral S, Tan DX, Jou MJ, Galano A, Xu B. Melatonin as a mitochondria‐targeted antioxidant: one of evolution's best ideas. Cell Mol Life Sci. 2017;74(21):3863‐3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. López A, García JA, Escames G, et al. Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J Pineal Res. 2009;46(2):188‐198. [DOI] [PubMed] [Google Scholar]
- 48. Lowes DA, Webster NR, Murphy MP, Galley HF. Antioxidants that protect mitochondria reduce interleukin‐6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth. 2013;110(3):472‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hobson SR, Gurusinghe S, Lim R, et al. Melatonin improves endothelial function in vitro and prolongs pregnancy in women with early‐onset preeclampsia. J Pineal Res. 2018;65(3):e12508. [DOI] [PubMed] [Google Scholar]
- 50. Alers NO, Jenkin G, Miller SL, Wallace EM. Antenatal melatonin as an antioxidant in human pregnancies complicated by fetal growth restriction—a phase I pilot clinical trial: study protocol. BMJ Open. 2013;3(12):e004141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fantasia I, Bussolaro S, Stampalija T, et al. The role of melatonin in pregnancies complicated by placental insufficiency: A systematic review. Authorea 2021. 10.22541/au.161790597.72492340/v1 [DOI] [PubMed]
- 52. Percie du Sert N, Hurst V, Ahluwalia A, et al. The arrive guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miyajima A, Sunouchi M, Mitsunaga K, Yamakoshi Y, Nakazawa K, Usami M. Sexing of postimplantation rat embryos in stored two‐dimensional electrophoresis (2‐DE) samples by polymerase chain reaction (PCR) of an Sry sequence. J Toxicol Sci. 2009;34(6):681‐685. [DOI] [PubMed] [Google Scholar]
- 54. Makrecka‐Kuka M, Krumschnabel G, Gnaiger E. High‐resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules. 2015;5(3):1319‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nuzzo AM, Camm EJ, Sferruzzi‐Perri AN, et al. Placental adaptation to early‐onset hypoxic pregnancy and mitochondria‐targeted antioxidant therapy in a rodent model. Am J Pathol. 2018;188(12):2704‐2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Richter HG, Camm EJ, Modi BN, et al. Ascorbate prevents placental oxidative stress and enhances birth weight in hypoxic pregnancy in rats. J Physiol. 2012;590(6):1377‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heather LC, Cole MA, Tan JJ, et al. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol. 2012;107(3):268. [DOI] [PubMed] [Google Scholar]
- 58. Galli GLJ, Ruhr IM, Crossley J, Crossley DA. The long‐term effects of developmental hypoxia on cardiac mitochondrial function in snapping turtles. Front Physiol. 2021;12:689684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. St‐Pierre J, Boutilier RG. Aerobic capacity of frog skeletal muscle during hibernation. Physiol Biochem Zool. 2001;74(3):390‐397. [DOI] [PubMed] [Google Scholar]
- 60. Hickey AJ, Renshaw GM, Speers‐Roesch B, et al. A radical approach to beating hypoxia: depressed free radical release from heart fibres of the hypoxia‐tolerant epaulette shark (Hemiscyllum ocellatum). J Comp Physiol B. 2012;182(1):91‐100. [DOI] [PubMed] [Google Scholar]
- 61. Horscroft JA, Kotwica AO, Laner V, et al. Metabolic basis to sherpa altitude adaptation. Proc Natl Acad Sci USA. 2017;114(24):6382‐6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia‐reperfusion injury. Physiol Rev. 2008;88(2):581‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Larsen S, Nielsen J, Hansen CN, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590(14):3349‐3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen X, Zhang L, Wang C. Prenatal hypoxia‐induced epigenomic and transcriptomic reprogramming in rat fetal and adult offspring hearts. Sci Data. 2019;6(1):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang L, Chen X, Dasgupta C, et al. Foetal hypoxia impacts methylome and transcriptome in developmental programming of heart disease. Cardiovasc Res. 2019;115(8):1306‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Huang L‐S, Cobessi D, Tung EY, Berry EA. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen‐bonding pattern. J Mol Biol. 2005;351(3):573‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen Y‐R, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114(3):524‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bleier L, Dröse S. Superoxide generation by complex iii: from mechanistic rationales to functional consequences. Biochim Biophys Acta. 2013;1827(11):1320‐1331. [DOI] [PubMed] [Google Scholar]
- 69. Gao Y, Dasgupta C, Huang L, Song R, Zhang Z, Zhang L. Multi‐omics integration reveals short and long‐term effects of gestational hypoxia on the heart development. Cells. 2019;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li H, Hu J, Liu Y, et al. Effects of prenatal hypoxia on fetal sheep heart development and proteomics analysis. Int J Clin Exp Pathol. 2018;11(4):1909‐1922. [PMC free article] [PubMed] [Google Scholar]
- 71. Williams SJ, Campbell ME, McMillen IC, Davidge ST. Differential effects of maternal hypoxia or nutrient restriction on carotid and femoral vascular function in neonatal rats. Am J Physiol Regul Integr Comp Physiol. 2005;288(2):R360‐R367. [DOI] [PubMed] [Google Scholar]
- 72. Turan S, Aberdeen GW, Thompson LP. Chronic hypoxia alters maternal uterine and fetal hemodynamics in the full‐term pregnant guinea pig. Am J Physiol Regul Integr Comp Physiol. 2017;313(4):R330‐R339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pereira SP, Tavares LC, Duarte AI, et al. Sex‐dependent vulnerability of fetal nonhuman primate cardiac mitochondria to moderate maternal nutrient reduction. Clin Sci (Lond). 2021;135(9):1103‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Woodman AG, Mah R, Keddie D, et al. Prenatal iron deficiency causes sex‐dependent mitochondrial dysfunction and oxidative stress in fetal rat kidneys and liver. FASEB J. 2018;32(6):3254‐3263. [DOI] [PubMed] [Google Scholar]
- 75. Jiang S, Teague AM, Tryggestad JB, Aston CE, Lyons T, Chernausek SD. Effects of maternal diabetes and fetal sex on human placenta mitochondrial biogenesis. Placenta. 2017;57:26‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Larsen TD, Sabey KH, Knutson AJ, et al. Diabetic pregnancy and maternal high‐fat diet impair mitochondrial dynamism in the developing fetal rat heart by sex‐specific mechanisms. Int J Mol Sci. 2019;20(12):3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nadal‐Casellas A, Bauzá‐Thorbrügge M, Proenza AM, Gianotti M, Lladó I. Sex‐dependent differences in rat brown adipose tissue mitochondrial biogenesis and insulin signaling parameters in response to an obesogenic diet. Mol Cell Biochem. 2013;373(1):125‐135. [DOI] [PubMed] [Google Scholar]
- 78. Bermejo‐Alvarez P, Rizos D, Rath D, Lonergan P, Gutierrez‐Adan A. Epigenetic differences between male and female bovine blastocysts produced in vitro. Physiol Genomics. 2008;32(2):264‐272. [DOI] [PubMed] [Google Scholar]
- 79. Galetzka D, Weis E, Tralau T, Seidmann L, Haaf T. Sex‐specific windows for high mrna expression of DNA methyltransferases 1 and 3a and methyl‐cpg‐binding domain proteins 2 and 4 in human fetal gonads. Mol Reprod Dev. 2007;74(2):233‐241. [DOI] [PubMed] [Google Scholar]
- 80. Okatani Y, Wakatsuki A, Kaneda C. Melatonin increases activities of glutathione peroxidase and superoxide dismutase in fetal rat brain. J Pineal Res. 2000;28(2):89‐96. [DOI] [PubMed] [Google Scholar]
- 81. Tamura H, Takayama H, Nakamura Y, Reiter RJ, Sugino N. Fetal/placental regulation of maternal melatonin in rats. J Pineal Res. 2008;44(3):335‐340. [DOI] [PubMed] [Google Scholar]
- 82. Deacon S, Arendt J. Melatonin‐induced temperature suppression and its acute phase‐shifting effects correlate in a dose‐dependent manner in humans. Brain Res. 1995;688(1‐2):77‐85. [DOI] [PubMed] [Google Scholar]
- 83. Gyllenhammer LE, Entringer S, Buss C, Wadhwa PD. Developmental programming of mitochondrial biology: A conceptual framework and review. Proc Biol Sci. 2020;287(1926):20192713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zander‐Fox DL, Fullston T, McPherson NO, et al. Reduction of mitochondrial function by FCCP during mouse cleavage stage embryo culture reduces birth weight and impairs the metabolic health of offspring. Biol Reprod. 2015;92:5. [DOI] [PubMed] [Google Scholar]
- 85. Fetterman JL, Pompilius M, Westbrook DG, et al. Developmental exposure to second‐hand smoke increases adult atherogenesis and alters mitochondrial DNA copy number and deletions in apoE−/− mice. PLoS One. 2013;8(6):e66835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bruin JE, Petre MA, Raha S, Morrison KM, Gerstein HC, Holloway AC. Fetal and neonatal nicotine exposure in wistar rats causes progressive pancreatic mitochondrial damage and beta cell dysfunction. PLoS One. 2008;3(10):e3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Peterside IE, Selak MA, Simmons RA. Impaired oxidative phosphorylation in hepatic mitochondria in growth‐retarded rats. Am J Physiol Endocrinol Metab. 2003;285(6):E1258‐E1266. [DOI] [PubMed] [Google Scholar]
- 88. Alfaradhi MZ, Fernandez‐Twinn DS, Martin‐Gronert MS, Musial B, Fowden A, Ozanne SE. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am J Physiol Regul Integr Comp Physiol. 2014;307(1):R26‐R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. de Velasco PC, Chicaybam G, Ramos‐Filho DM, et al. Maternal intake of trans‐unsaturated or interesterified fatty acids during pregnancy and lactation modifies mitochondrial bioenergetics in the liver of adult offspring in mice. Br J Nutr. 2017;118(1):41‐52. [DOI] [PubMed] [Google Scholar]
- 90. Tarry‐Adkins JL, Fernandez‐Twinn DS, Chen JH, et al. Poor maternal nutrition and accelerated postnatal growth induces an accelerated aging phenotype and oxidative stress in skeletal muscle of male rats. Dis Model Mech. 2016;9(10):1221‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mdaki KS, Larsen TD, Wachal AL, et al. Maternal high‐fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am J Physiol Heart Circ Physiol. 2016;310(6):H681‐H692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Burgueño AL, Cabrerizo R, Gonzales Mansilla N, Sookoian S, Pirola CJ. Maternal high‐fat intake during pregnancy programs metabolic‐syndrome‐related phenotypes through liver mitochondrial DNA copy number and transcriptional activity of liver ppargc1a. J Nutr Biochem. 2013;24(1):6‐13. [DOI] [PubMed] [Google Scholar]
- 93. Andreas E, Reid M, Zhang W, Moley KH. The effect of maternal high‐fat/high‐sugar diet on offspring oocytes and early embryo development. Mol Hum Reprod. 2019;25(11):717‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Saben JL, Boudoures AL, Asghar Z, et al. Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep. 2016;16(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hanafi MY, Abdelkhalek TM, Saad MI, Saleh MM, Haiba MM, Kamel MA. Diabetes‐induced perturbations are subject to intergenerational transmission through maternal line. J Physiol Biochem. 2016;72(2):315‐326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.