

Abstract
The effects of aging on the brain are widespread and can have dramatic implications on the overall health of an organism. Mitochondrial dysfunction is a hallmark of brain aging, but, the interplay between mitochondrial quality control, neuronal aging, and organismal health is not well understood. Here, we show that aging leads to a decline in mitochondrial autophagy (mitophagy) in the Drosophila brain with a concomitant increase in mitochondrial content. We find that induction of BCL2-interacting protein 3 (BNIP3), a mitochondrial outer membrane protein, in the adult nervous system induces mitophagy and prevents the accumulation of dysfunctional mitochondria in the aged brain. Importantly, neuronal induction of BNIP3-mediated mitophagy increases organismal longevity and healthspan. Furthermore, BNIP3-mediated mitophagy in the nervous system improves muscle and intestinal homeostasis in aged flies, indicating cell non-autonomous effects. Our findings identify BNIP3 as a therapeutic target to counteract brain aging and prolong overall organismal health with age.
Keywords: Autophagy, Mitophagy, Neuronal aging, Muscle aging, Intestinal barrier dysfunction, Intestinal stem cell, Mito-QC
Introduction
Brain function declines with age, manifesting as impairments in learning and memory, attention, decision-making speed, sensory perception, cognitive function and motor coordination 1–3. In addition, aging is the major risk factor for the development of neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease. As the burden of age-related neurodegenerative disorders increases at an exponential rate all over the world 4, there is a considerable need for a better understanding of the mechanisms of brain aging and relationship to organismal health and longevity. Examination of the molecular and cellular changes that occur during brain aging indicate significant overlap with the hallmarks of aging in other organ systems 5,6. Indeed, one of the most extensively studied hallmarks of brain aging is mitochondrial dysfunction, which has also been implicated in organismal aging and neurodegenerative diseases 5,7,8. Neurons are particularly vulnerable to mitochondrial dysfunction given that they are post-mitotic differentiated cells relying almost exclusively on the oxidative phosphorylation system to sustain their high energy needs. Hence, identifying interventions that could prevent the accumulation of dysfunctional mitochondria in the aging brain could provide potential approaches to counteract age-related health decline.
Autophagy, a lysosomal degradation pathway that plays essential roles in development, tissue homeostasis and disease pathogenesis 9, has emerged as an important modulator of tissue and organismal aging 10. In this process, cellular materials (referred to as autophagic cargo) are sequestered within double-membrane vesicles known as autophagosomes, and delivered to the lysosome for degradation 11. Mitochondrial autophagy (mitophagy) is a type of cargo-specific autophagy that mediates the removal of dysfunctional mitochondria 12,13. Studies in diverse species have reported an age-related decline in mitophagy or mitophagy-related gene expression 14–16. Moreover, studies in invertebrate models have shown that interventions that facilitate mitophagy can prolong lifespan and improve tissue homeostasis during aging 16–20. Hence, there is an emerging understanding that mitophagy represents a key pathway to preserve mitochondrial function, and, hence, cell and tissue health, during aging 8,10,21–23. Indeed, in recent years, it has been shown that mitophagy can restrain neuroinflammation 24 and represents an important therapeutic target to counteract Alzheimer’s disease pathogenesis 25.
The molecular mechanisms of mitophagy involve coordinating autophagy induction with mitochondrial priming for autophagic recognition 26,27. BNIP3 is a Bcl-2 family protein with an atypical BH3 domain that primarily localizes at the mitochondrial outer membrane. It has become apparent that BNIP3 can exert multiple cellular effects involving direct or indirect interactions with mitochondria 28. Initial studies reported that BNIP3 can act as a pro-apoptotic protein, inducing cell death and mitochondrial dysfunction 29–32. However, it has also been shown that BNIP3 can act as a potent inducer of autophagy/mitophagy without inducing cell death in multiple cell types 28,33–35. More specifically, BNIP3 has been shown to serve as an autophagy receptor for the binding of mitochondria to ATG8/LC3 on the autophagosome via its N-terminal LC3-interacting region 26,36,37. It has also been reported that BNIP3 interacts with PINK1 to suppress its cleavage, leading to enhanced mitochondrial clearance via mitophagy 35. BNIP3-mediated mitophagy has been reported to exert pro-survival effects in certain pathological conditions 35,38–41. However, the impact of BNIP3 induction during aging on mitochondrial homeostasis and organismal health are not known.
The role of BNIP3 in mitophagy induction prompted us to determine whether BNIP3 could modulate neuronal and/or organismal aging. First, we examined whether BNIP3 can improve mitochondrial homeostasis in the aging Drosophila brain. We show that in control flies there is a striking accumulation of dysfunctional mitochondria in aged brains. Up-regulation of BNIP3 in the adult nervous system is sufficient to induce mitophagy and prevent the accumulation of dysfunctional mitochondria in an autophagy-dependent manner. In assessing organismal aging, neuronal BNIP3-mediated mitophagy is sufficient to prolong lifespan and improve several markers of healthspan in aged flies. Interestingly, we find that neuronal BNIP3 induction improves markers of both mitochondrial homeostasis and proteostasis in aged muscle in an autophagy-dependent manner. In addition, neuronal BNIP3 induction delays intestinal stem cell aging and maintains intestinal barrier function, also, in an autophagy-dependent manner. Together, these findings indicate that functional up-regulation of BNIP3 in the aging brain may represent a therapeutic strategy to promote healthy aging in older adults.
Results
Neuronal BNIP3 induction improves mitochondrial function
With many parallels to human physiology, the fruit fly Drosophila is an excellent model to study the role of mitochondria homeostasis in aging and lifespan determination 42. Here, we set out to examine the effects of BNIP3 induction on mitophagy and, accordingly, mitochondrial homeostasis in the aging fly brain. To do so, we used Gene-Switch driver lines to express a UAS-hBNIP3-HA (hereafter BNIP3) transgene created by Zhang et al35. Crosses in an experiment share genetic background and developmental conditions; the inducing agent (RU486) or vehicle (ethanol) is provided in food during adulthood to drive gene expression in target tissues in a time- and dose-dependent manner 43. To investigate the effects of BNIP3 induction in adult neurons, we used the pan-neuronal Elav-Gene-Switch (elavGS) driver line 44. RU486-dependent transgene expression in elavGS>UAS-BNIP3 flies was validated by immunofluorescence (IF) microscopy (Extended Data Fig. 1a,b) and Western blot analysis (Extended Data Fig. 1c). IF microscopy revealed expression of BNIP3 in proximity to mitochondria in the brain with frequent points of colocalization (Extended Data Fig. 1d). Previous studies in Drosophila have reported age-related alterations in mitochondrial morphology and function in muscle tissue and in mitochondria isolated from whole animals 16,18,19,45–47. However, little is known about possible changes to mitochondrial homeostasis in the aging fly brain. Using IF microscopy, we began by investigating mitochondrial morphology and content in aging Drosophila brains. Compared to brains from young adult flies, aged brains showed a striking accumulation of mitochondrial content (Fig. 1a,b). Neuronal-specific induction of BNIP3 significantly reduced brain mitochondrial content to levels similar to those detected in young animals (Fig. 1a,b), supporting a role for BNIP3 in mitochondrial homeostasis. The human brain shrinks with age in older adults5; we noted an analogous reduction in the number of nuclei observed in aged Drosophila optic lobes (Extended Data Fig. 2a,b) and following isotropic fractionation of whole brains (Extended Data Fig. 2c,d). Remarkably, neuron-specific up-regulation of BNIP3 counteracted loss of brain nuclei detected in age-matched controls (Extended Data Fig. 2a,b,c,d). Since BNIP3 has been reported to promote apoptosis in certain cell types and conditions 29–32, we stained brains for cleaved (activated) caspase-3 to detect cells undergoing apoptosis. Although very few cleaved caspase-3 positive cells were detected per brain when imaging entire optic lobes, there was a significant increase in aged samples compared to young controls (Extended Data Fig. 2e,f). Importantly, neuronal BNIP3 induction was associated with fewer apoptotic cells in the aged brain as detected by cleaved caspase-3 staining (Extended Data Fig. 2e,f). Cumulatively, these results suggest that neuronal BNIP3 induction can improve mitochondrial homeostasis while being neuroprotective.
Figure 1. Neuronal BNIP3 induction prevents accumulation of dysfunctional mitochondria in the aged brain.
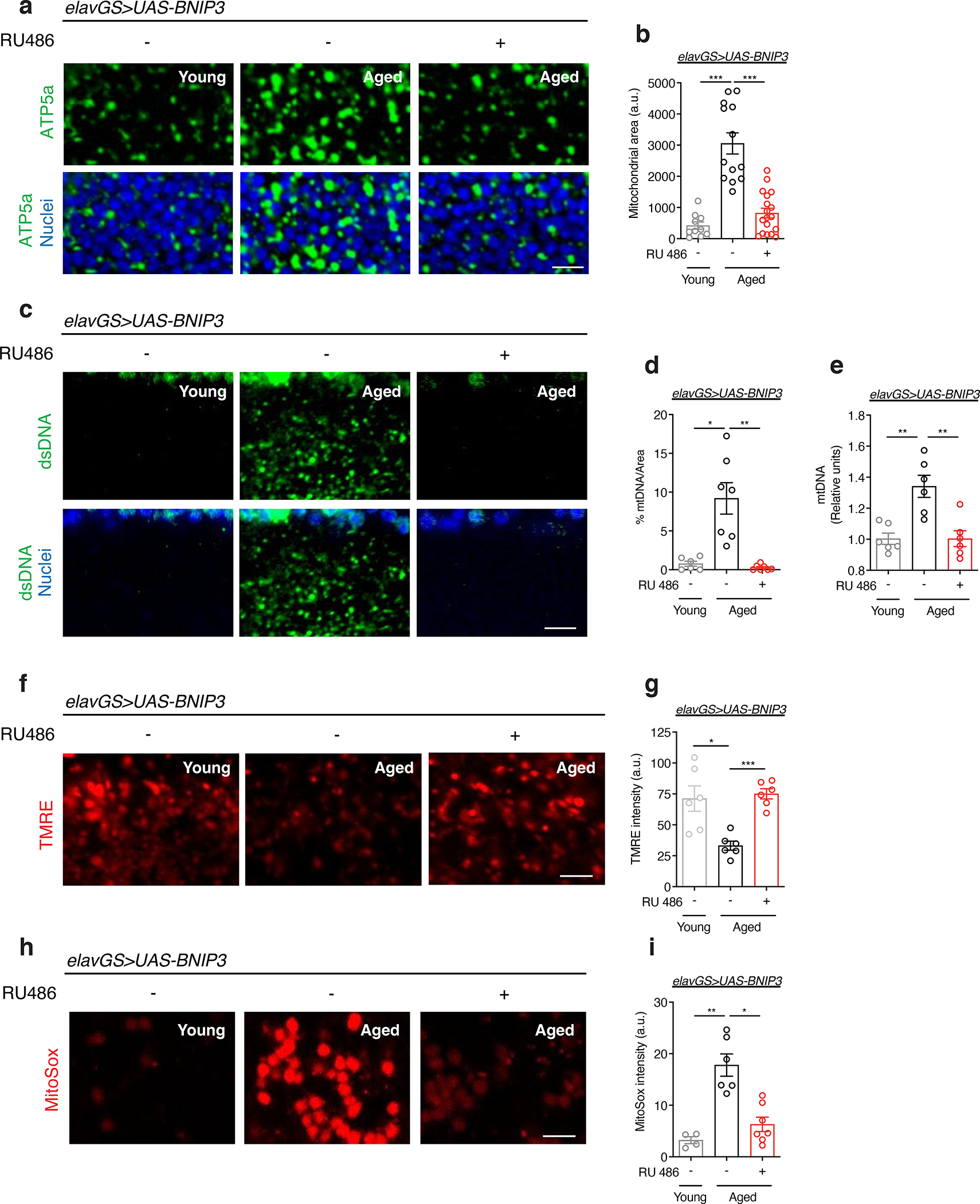
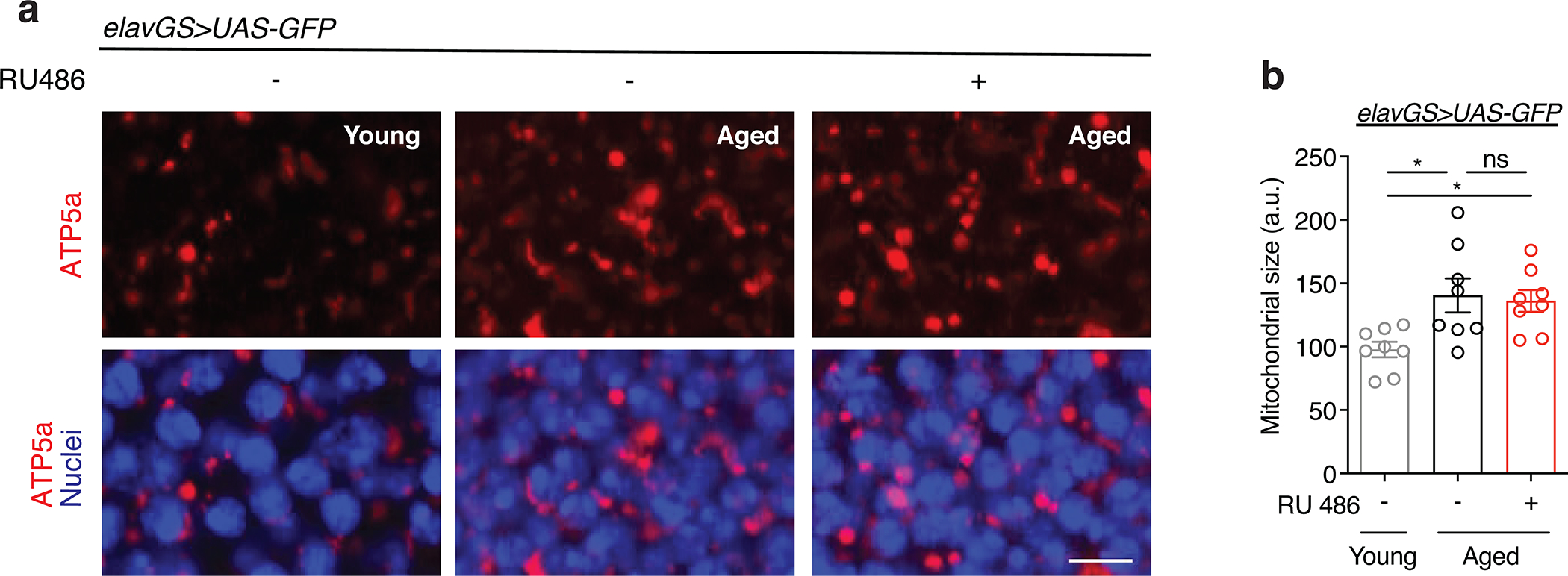
(a) Immunostaining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial morphology (green channel, anti-ATP5a) and nuclear DNA (blue channel, stained with To-Pro-3). Scale bar is 5 μm.
(b) Quantification of mitochondrial area in brain as shown in (a). n = 10 young, 13 aged RU−, and 18 aged RU+ biologically independent animals per condition, as indicated. ***p<0.0001 (young vs. aged RU−), ***p=0.0001 (aged RU− vs. aged RU+); Kruskal-Wallis test/Dunn’s multiple comparisons test.
(c) Immunostaining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial DNA (green channel, anti-dsDNA) and nuclear DNA (blue channel, stained with To-Pro-3). Scale bar is 5 μm.
(d) Quantification of mitochondrial DNA in brain as shown in (c). n = 6 young, 7 aged RU−, and 7 aged RU+ biologically independent animals per condition, as indicated. *p=0.0182, **p=0.0017; Kruskal-Wallis test/Dunn’s multiple comparisons test.
(e) Mitochondrial DNA (mtDNA) amount in brains of young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies as determined by quantitative PCR. **p=0.0015; one-way ANOVA/Tukey’s multiple comparisons test. Units are relative to the amounts of a nuclear DNA (nDNA) amplicon. n = 6 biologically independent replicates generated from pooling 10 samples per condition each.
(f) Staining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing TMRE fluorescence. Scale bar is 5 μm.
(g) Quantification of mitochondrial membrane potential measured by TMRE staining as shown in (f). n = 6 biologically independent animals per condition. *p=0.0345, ***p<0.0001; Brown-Forsythe and Welch ANOVA/Dunett’s T3 multiple comparisons test.
(h) Staining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing MitoSox fluorescence. Scale bar is 5 μm.
(i) Quantification of mitochondrial reactive oxygen species (ROS) measured by MitoSox staining as shown in (h). n = 4 young, 6 aged RU−, and 7 aged RU+ biologically independent animals per condition, as indicated. *p=0.0265, **p=0.0038; Kruskal-Wallis test/Dunn’s multiple comparisons test. RU486 or vehicle was provided in the media at a concentration of 5 μg/ml in the indicated treatment groups. Data are presented as scatter plots overlaying mean values +/− SEM.
To provide an additional marker of mitochondrial content, we examined mitochondrial DNA (mtDNA) levels in the brains of young and aged flies. Consistent with an age-related accumulation of mitochondria, we observed an increase in mtDNA in aged brains, by IF, that was prevented with neuronal-specific induction of BNIP3 (Fig. 1c,d). We confirmed these changes to mtDNA levels in fly heads using quantitative polymerase chain reaction (qPCR) to detect cytochrome c oxidase subunit I (MT-CO1/COI) encoded in mitochondrial DNA (Fig. 1e). To assess changes to mitochondrial function in aging brains, we examined mitochondrial membrane potential using the potentiometric dye tetramethylrhodamine, ethyl ester (TMRE). While brain mitochondria showed reduced TMRE intensity with age, neuronal induction of BNIP3 resulted in significantly greater mitochondrial membrane potential compared to age-matched controls (Fig. 1f,g). Furthermore, MitoSOX staining showed an increase in mitochondrial reactive oxygen species (ROS) in aged brains that was significantly reduced by neuronal BNIP3 induction (Fig. 1h,i). Importantly, RU486 administration and expression of a control transgene in elavGS>UAS-GFP flies had no effect on mitochondria accumulation in aging brains (Extended Data Fig. 3a,b). Collectively, these data demonstrate that there is an increase in dysfunctional mitochondria in the aged fly brain that can be counteracted by upregulation of BNIP3 in adult neurons.
Neuronal BNIP3 induction promotes mitophagy
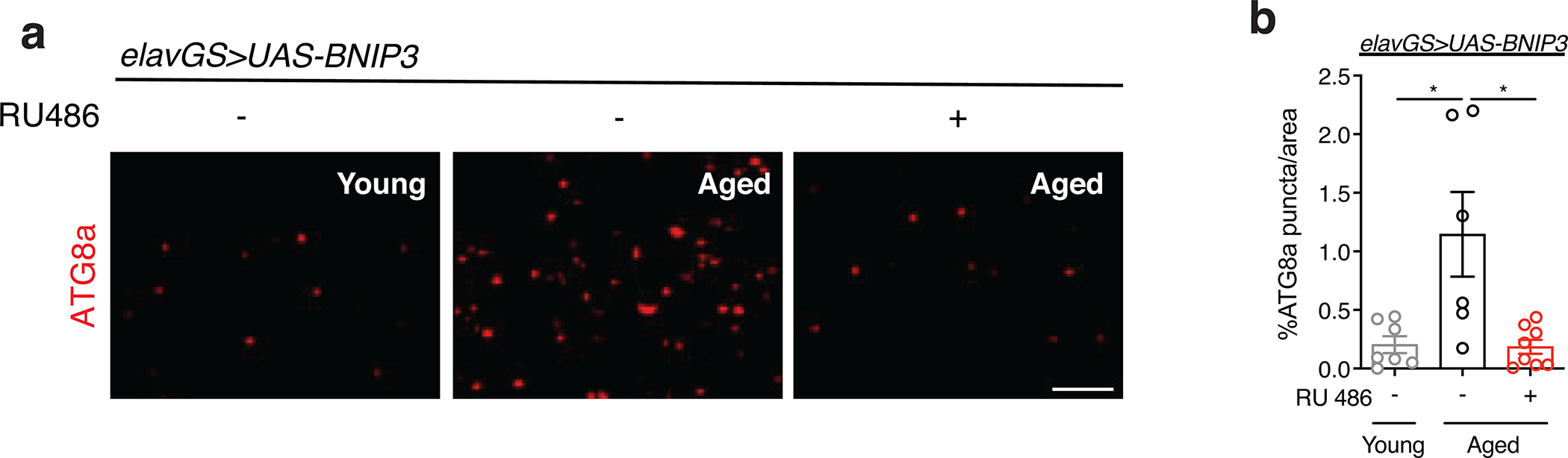
BNIP3 has been proposed to function as a mitophagy receptor 26. However, the relationships between BNIP3, mitophagy and mitochondrial function during neuronal aging are unknown. Endogenous LC3/ATG8 can be used as a marker of steady-state autophagy and allow the visualization of autophagosomes 48. Studies in both worms 49 and flies 18 have reported an age-related increase in autophagosomes, reflective of a decline in autophagic flux. To explore the interplay between BNIP3 induction and autophagy, we examined the levels of endogenous ATG8a in aging brains using IF microscopy. Aged fly brains revealed an accumulation of ATG8a that was abrogated with neuronal BNIP3 induction (Extended Data Fig. 4a,b). To further evaluate changes to aging brain autophagy with BNIP3 induction, we used a reporter line expressing GFP-mCherry-ATG8a ubiquitously under the control of the endogenous ATG8a promoter 50. Due to the pH-sensitive properties of GFP, ATG8a in acidic environments—autolysosomes—will display mCherry-only foci. When overexpressing BNIP3 in neurons, we detected significantly more red-only puncta compared to aged controls, indicating an increase in autolysosomes (Fig. 2a,b). Next, to evaluate changes more specifically to mitophagy, we used the recently characterized mitophagy reporter line, mito-QC, that encodes a tandem GFP-mCherry fusion protein that is targeted to the outer mitochondrial membrane 51. With this tool, mitochondria degraded in the acidic microenvironment of lysosomes (mitolysosomes) will appear as mCherry-only puncta 52. In agreement with its reported role as a mitophagy receptor, up-regulation of BNIP3 in neurons resulted in significantly more mitolysosomes compared to controls (Fig. 2c,d). Notably, neuronal induction of BNIP3 at midlife, for both one and two weeks (days 30–37 or 30–44), was sufficient to increase the number of detected mitolysosomes in aged brains (Extended Data Fig. 5a,b,c,d).
Figure 2. Neuronal BNIP3 induction induces mitophagy to improve mitochondrial homeostasis.
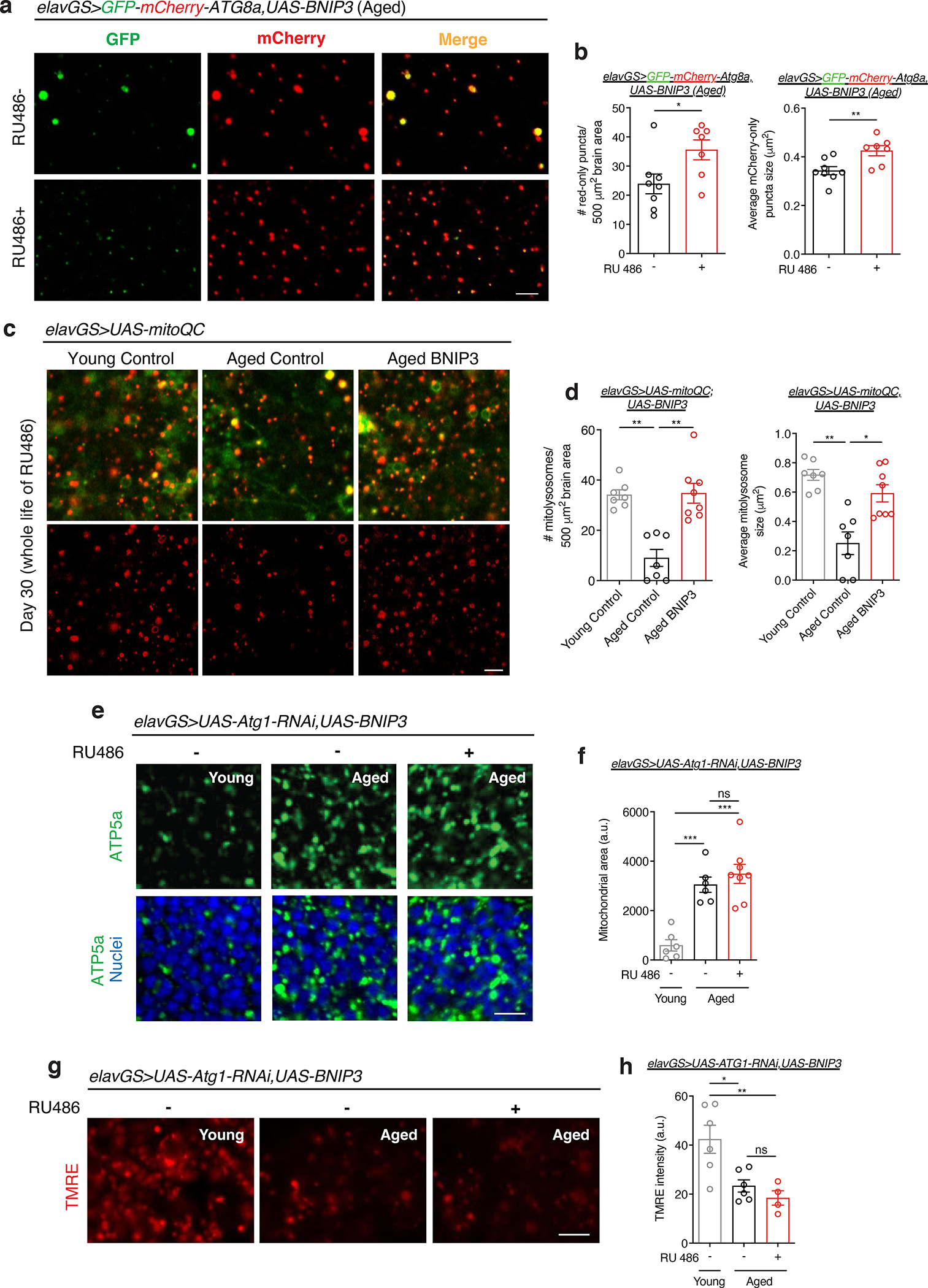
(a) GFP-mCherry-ATG8a of brains from 51-day-old elavGS>GFP-mCherry-ATG8a,UAS-BNIP3 flies. Images shown of GFP, mCherry, and merged GFP-mCherry channels. Scale bar is 5 μm.
(b) Quantification of autolysosomes (red-only puncta) area per μm2 and average size (μm2) as shown in (a). n = 8 RU− and 7 RU + biologically independent animals per condition, as indicated. Area *p=0.0186; size **p=0.0084; unpaired t tests.
(c) mito-QC of brains from 30-day-old flies. Genotypes analyzed were elavGS>UAS-mito-QC,UAS-lacZ, as a control, and elavGS>UAS-mito-QC,UAS-BNIP3. RU486-mediated transgenes were induced from day 5 onwards. Images shown of merged GFP and mCherry along with punctate mCherry-only foci (from merged images where GFP has been quenched; mitolysosomes). Scale bar is 5 μm.
(d) Quantification of mitolysosome area per μm2 and average size (μm2) as shown in (a). n = 7 young, 7 aged control, and 8 aged BNIP3+ biologically independent animals per condition, as indicated. Area **p=0.0022 (young vs. aged control), **p=0.0089 (aged control vs. aged BNIP3+); size *p=0.0448, **p=0.0010; Kruskal-Wallis tests/Dunn’s multiple comparisons tests.
(e) Immunostaining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial morphology (green channel, anti-ATP5a) and nuclear DNA (blue channel, stained with To-Pro-3). Scale bar is 5 μm.
(f) Quantification of mitochondrial area in brain as shown in (e). n = 6 young, 6 aged RU−, and 8 aged RU+ biologically independent animals per condition, as indicated. ***p=0.0004 (young vs. aged RU−), ***p<0.0001 (young vs. aged RU+), non-significant (n.s.); one-way ANOVA/Tukey’s multiple comparisons test.
(g) Staining of brains from young (10-day-old) and aged (51-day-old) elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing TMRE fluorescence. Scale bar is 5 μm.
(h) Quantification of mitochondrial membrane potential measured by TMRE staining as shown in (g). n = 6 young, 6 aged RU−, 4 aged RU+ biologically independent animals per condition, as indicated. *p=0.0140, **p=0.0064, non-significant (n.s.); one-way ANOVA/Tukey’s multiple comparisons test. RU486 or vehicle was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
A key prediction of the mitophagy model is that BNIP3-mediated changes to mitochondrial homeostasis will depend on autophagy. Atg1 (Autophagy-related 1, the Drosophila homolog of mammalian ULK1) is a Ser/Thr protein kinase essential in the initiation of autophagosome formation 53. To test if the observed improvements to mitochondrial homeostasis associated with neuronal BNIP3 induction (Fig. 1) are dependent upon autophagy, we generated elavGS>UAS-Atg1-RNAi,UAS-BNIP3 flies. Gene induction was confirmed in brains of elavGS>UAS-Atg1-RNAi,UAS-BNIP3 flies upon RU486 treatment (Extended Data Fig. 6a,b). Brains from flies with concomitant neuronal induction of Atg1-RNAi and BNIP3 showed age-related accumulation of mitochondria similar to age-matched controls (Fig. 2e,f). Furthermore, flies expressing both Atg1-RNAi and BNIP3 in neurons showed impaired mitochondrial membrane potential in aged brains (Fig. 2g,h) in contrast to observations made in flies with neuronal BNIP3 upregulation without Atg1-RNAi induction (Fig. 1f,g). These findings indicate an autophagy-dependent mechanism to BNIP3-induced changes to mitochondrial homeostasis in aging neurons.
Neuronal BNIP3 induction improves healthspan and longevity
Having observed that induction of BNIP3 in neurons improves mitochondrial homeostasis in aged brains, we decided to investigate the potential impact on organismal aging. Remarkably, adult neuronal BNIP3 induction resulted in flies with significantly longer lifespans compared to controls (Fig. 3a, Main Table 1). Interestingly, midlife neuronal BNIP3 induction was also sufficient to extend maximum lifespan of flies (Extended Data Fig. 5e). Conversely, expression of BNIP3-RNAi ubiquitously or specifically in neurons in adult flies resulted in shortened lifespans compared to controls (Extended Data Fig. 8d,e,f). To confirm that lifespan extension was a result of BNIP3 induction and not an artifact of transgene expression and/or RU486 treatment, elavGS>UAS-GFP flies were tested. However, adult induction of GFP in neurons failed to prolong lifespan (Extended Data Fig. 7a). Since changes to food intake can influence aging, we tested if neuronal BNIP3 upregulation affected feeding behavior. Using the Con-Ex feeding assay 54, we observed no alterations in food consumption and excretion upon neuronal BNIP3 induction (Extended Data Fig. 7d). Likewise, neuronal expression of GFP had no observable effect on feeding behavior (Extended Data Fig. 7b). To understand if the lifespan benefits of neuronal BNIP3 expression could be repeated when targeting BNIP3 induction in other cell types, daughterless (da)GS, Act88FGS, and 5966GS drivers were used to express BNIP3 ubiquitously, in muscles, and in intestinal enterocytes, respectively. However, none of these other interventions increased fly lifespan (Extended Data Fig. 8a,b,c). Hence, we conclude that upregulation of BNIP3 specifically in neurons increases Drosophila longevity.
Figure 3. Neuronal BNIP3 induction extends lifespan and improves healthspan.
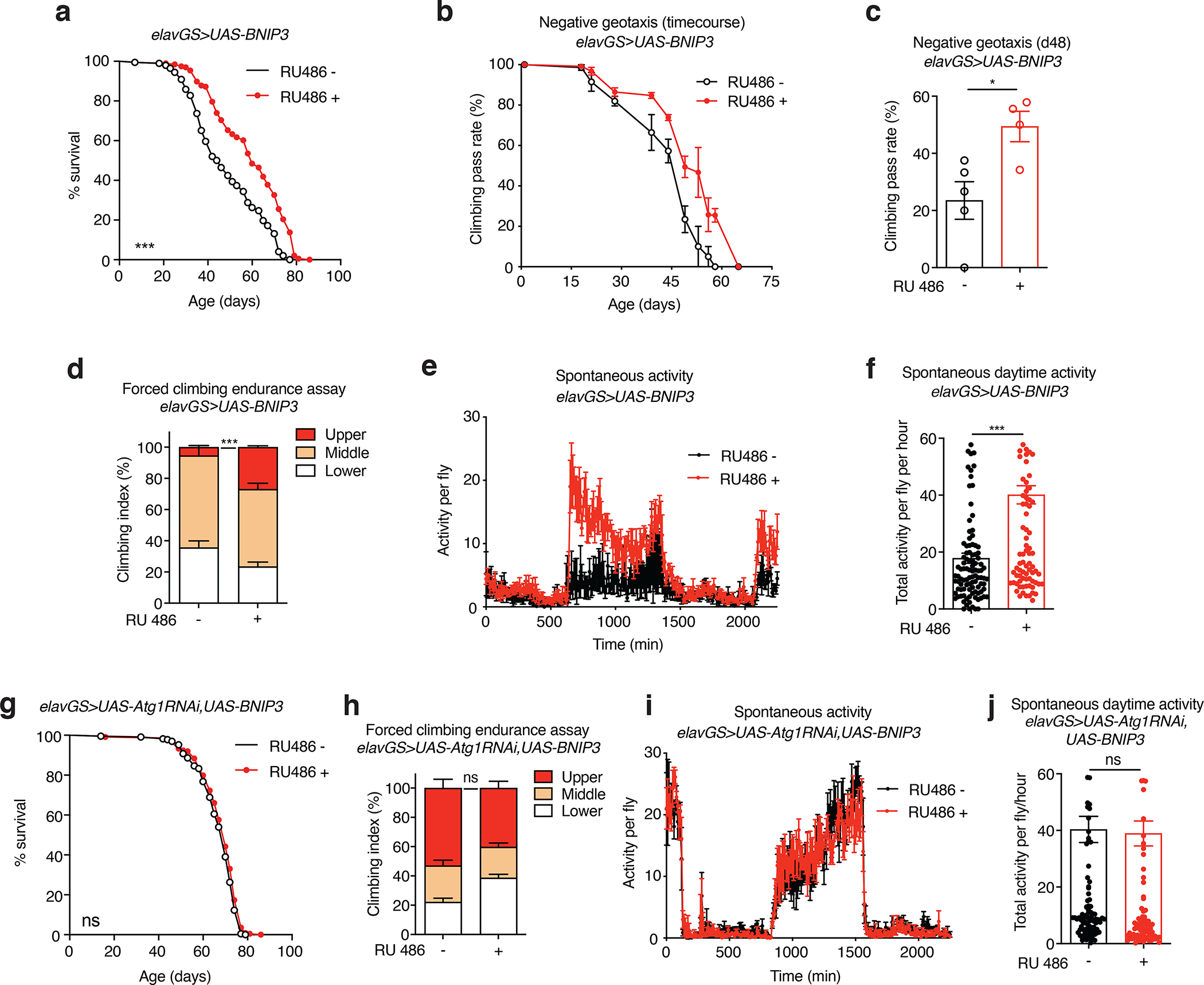
(a) Survival curves of elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 198 RU− and 196 RU+ biologically independent animals. ***p<0.0001; log-rank test.
(b) Climbing pass rate of elavGS>UAS-BNIP3 flies during aging with or without RU486-mediated transgene induction from day 5 onward; n = 150 RU− and 120 RU− biologically independent animals per condition at start of assay maintained in groups of 30.
(c) Climbing pass rate of 48-day-old elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 150 RU− and 120 RU+ biologically independent animals per condition measured in groups of 30. *p=0.0217; unpaired t test.
(d) Climbing index as a measure of endurance of 37-day-old elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 5 replicates of RU− and 4 replicates of RU+ with 100 biologically independent animals per replicate. ***p < 0.0001; unpaired t test.
(e) Spontaneous physical activity of 37-day-old elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 3 vials of 10 flies per condition.
(f) Quantification of total activity per fly per hour from spontaneous activity graphs (e). n = 3 vials of 10 biologically independent animals per condition. ***p<0.0001; Mann-Whitney test.
(g) Survival curves of elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 227 RU− and 224 RU+ biologically independent animals. Non-significant (n.s.); log-rank test.
(h) Climbing index as a measure of endurance of 51-day-old elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 8 biological replicates with 100 biologically independent animals per replicate. non-significant (n.s.); unpaired t test.
(i) Spontaneous physical activity of 51-day-old elavGS>UAS-Atg1RNAi;UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 3 vials of 10 biologically independent animals per condition
(j) Quantification of total activity per fly per hour from spontaneous activity graphs (i). n = 3 vials of 10 biologically independent animals per condition. Non-significant (n.s.); unpaired t test. RU486 or vehicle was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
Main Table 1.
Related to Figure 3. Neuronal-specific BNIP3 induction extends lifespan.
| Median Survival | Maximum Survival | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| RU− | RU+ | % median survival increase | RU− | RU+ | % median survival | p value | n RU− | n RU+ | |
| Exp 1 | 45 | 49 | 8.9 | 70 | 79 | 12.9 | <0.0001 | 146 | 147 |
| Exp 2 | 40 | 45 | 12.5 | 53 | 59 | 11.3 | <0.0001 | 149 | 145 |
| Exp 3 | 44 | 52 | 18.2 | 55 | 68 | 23.6 | <0.0001 | 153 | 125 |
| Exp 4 | 32 | 35 | 9.4 | 53 | 58 | 9.4 | <0.0001 | 291 | 270 |
| Exp 5 | 35 | 37 | 5.7 | 58 | 63 | 8.6 | 0.001 | 229 | 223 |
| Exp 6 | 35 | 39 | 11.4 | 77 | 77 | - | 0.0315 | 257 | 255 |
| Exp 7 | 35 | 44 | 25.7 | 65 | 81 | 24.6 | <0.0001 | 253 | 253 |
To assess how aging health could be affected by neuronal BNIP3 induction, we tested several behavioral readouts. Throughout the course of their lifespan, RU486-treated elavGS>UAS-BNIP3 flies showed a delayed reduction in locomotor activity associated with aging (Fig. 3b,c). Furthermore, these flies showed significant improvements to climbing endurance assays (Fig. 3d). No change was detected in climbing ability during aging in control flies expressing GFP in neurons (Extended Data Fig. 7c). Neuronal BNIP3 induction also conferred an increase in spontaneous daytime activity with no detectable nighttime restlessness in aged flies (Fig. 3e,f). Some lifespan extension strategies, including dietary restriction, are associated with reduced reproductive fitness 55. However, we did not detect a change in fertility in flies with neuronal BNIP3 induction (Extended Data Fig. 7e). Overall, these data reveal that upregulating BNIP3 in neurons prolongs not only lifespan but also several indicators of healthspan.
With changes to mitochondrial homeostasis in the brains of flies up-regulating BNIP3 in neurons being dependent on Atg1 (Fig. 2e–h), we decided to test if lifespan and healthspan improvements were also contingent on autophagy. Notably, concomitant induction of BNIP3 and Atg1-RNAi in neurons prevented the lifespan extension associated with BNIP3 induction alone (Fig. 3g). Likewise, RU486-treated elavGS>UAS-Atg1-RNAi,UAS-BNIP3 flies showed no detectable improvement in climbing endurance assays (Fig. 3h) or in spontaneous activity (Fig. 3i,j) compared to control flies. Hence, the ability of neuronal BNIP3 induction to prolong healthspan and extend lifespan is also dependent on autophagy.
Neuronal induction of BNIP3 slows muscle aging
Aging is a systemic process associated with the physiological decline of multiple organ systems. There is an emerging understanding that modulating components of neuronal aging can impact systemic aging 56,57. Hence, we set out to explore whether facilitating mitophagy in aged neurons could impact hallmarks of muscle aging. Previous work has revealed a midlife shift in Drosophila flight muscle toward an elongated mitochondrial morphology which is linked to impaired mitophagy and the accumulation of dysfunctional mitochondria 16,18. Remarkably, IF microscopy revealed that neuronal induction of BNIP3 resulted in smaller mitochondria in aged flight muscles (Fig. 4a,b). Inducing GFP in neurons, as a control, had no effect on mitochondrial size in aged muscle (Extended Data Fig. 9a,b). Consistent with a decrease in mitochondrial content, neuronal induction of BNIP3 reduced the amount of mtDNA detected in aged muscle (Fig. 4c,d). There is an emerging understanding the mitochondrial fission is a prerequisite for mitophagy 16–18,58,59. Interestingly, we observed that neuronal BNIP3 induction is linked to increased expression of Drp1, a Dynamin-related protein that promotes mitochondrial fission, in the thorax of middle-aged flies (Extended Data Fig. 10a). Next, we used TMRE to understand how changes in mitochondrial morphology and content in aged muscle related to mitochondrial function. Importantly, neuronal BNIP3 upregulation significantly improved mitochondrial membrane potential in aged muscle (Fig. 4e,f).
Figure 4. Neuronal BNIP3 induction improves mitochondrial homeostasis and proteostasis in aged muscle.
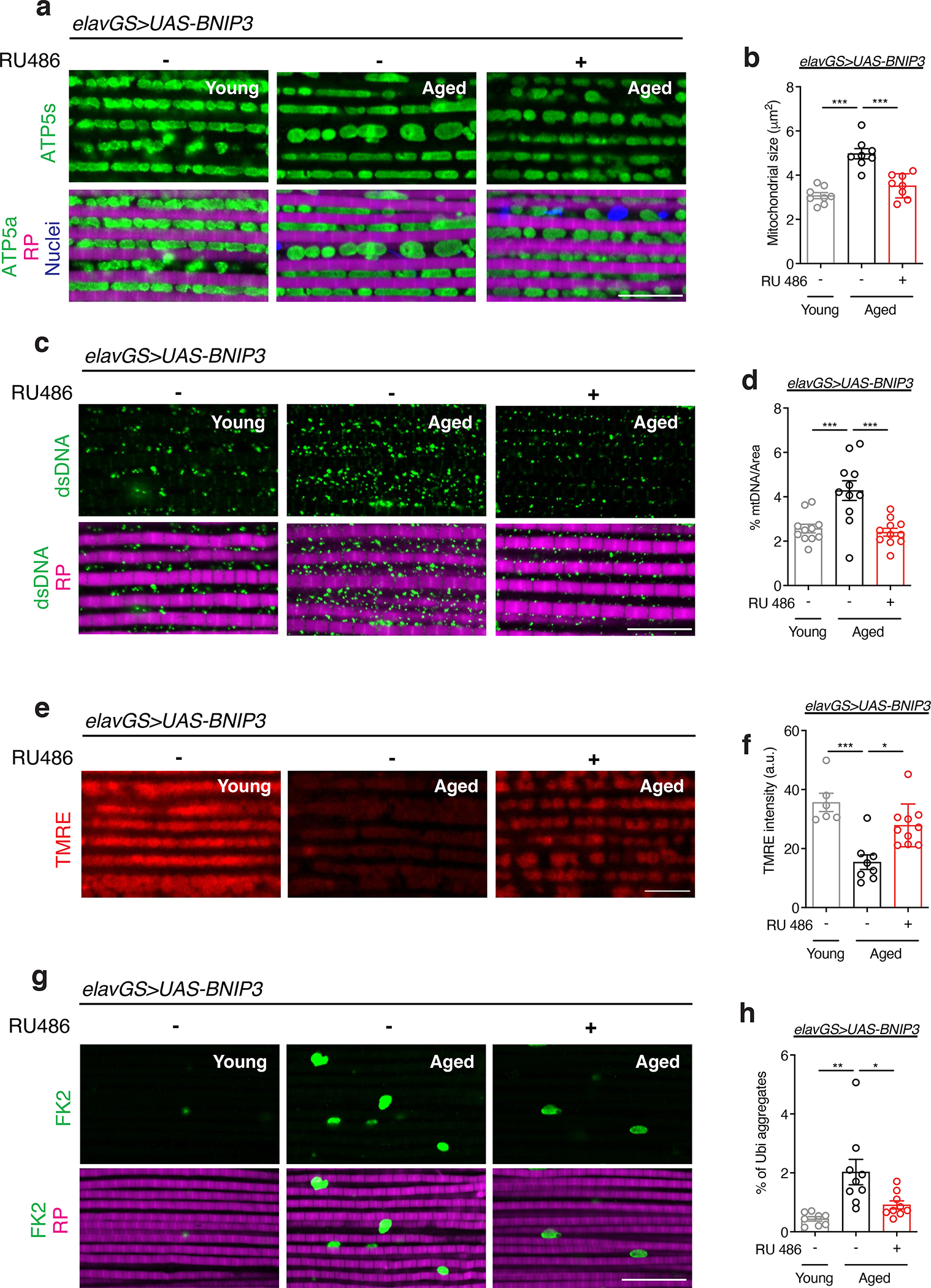
(a) Immunostaining of indirect flight muscles from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial morphology (green channel, anti-ATP5a), rhodamine phalloidin (RP, magenta channel), and nuclear DNA (blue channel, stained with DAPI). Scale bar is 10 μm.
(b) Quantification of mitochondrial size in muscle as shown in (a). n = 8 biologically independent animals per condition. ***p<0.0001; one-way ANOVA/Tukey’s multiple comparisons test.
(c) Immunostaining of indirect flight muscles from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial DNA (green channel, anti-dsDNA) and rhodamine phalloidin (magenta channel). Scale bar is 10 μm.
(d) Quantification of mitochondrial DNA in muscle as shown in (c). n =11 biologically independent animals per condition. ***p=0.0009 (young vs. aged RU−), ***p=0.0003 (aged RU− vs. aged RU+); one-way ANOVA/Tukey’s multiple comparisons test.
(e) Staining of indirect flight muscles from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing TMRE fluorescence. Scale bar is 10 μm. (f) Quantification of mitochondrial membrane potential measured by TMRE staining as shown in (e). n = 6 young, 8 aged RU−, and 10 aged RU+ biologically independent animals, as indicated. *p=0.0400, ***p=0.0006; Kruskal-Wallis test/Dunn’s multiple comparisons test.
(g) Immunostaining of indirect flight muscles from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing polyubiquitinated aggregates (green channel, anti-FK2) and rhodamine phalloidin (magenta channel). Scale bar is 10 μm.
(h) Quantification of polyubiquitin aggregates in muscle as shown in (g). n = 8 young, 9 aged RU−, and 9 aged RU+ biologically independent animals, as indicated. *p=0.0204, **p=0.0014; one-way ANOVA/Tukey’s multiple comparisons test. RU486 or vehicle was provided in the media at a concentration of 5 μg/ml in the indicated treatment groups. Data are presented as scatter plots overlaying mean values +/− SEM.
Another major cellular hallmark of aging is the loss of protein homeostasis (proteostasis) 6. Drosophila flight muscle accumulates aggregates of ubiquinated proteins during aging, consistent with a decline in proteostasis 16,18,19,60–62. Here, we observed that neuronal BNIP3 induction significantly reduced age-associated protein aggregates in flight muscle (Fig. 4g,h). Together, these data reveal that BNIP3 induction in neurons results in non-cell autonomous changes to aging muscles in flies.
Neuronal BNIP3 induction improves intestinal homeostasis
Intestinal homeostasis is essential in maintaining organismal health and longevity 63,64. In Drosophila, intestinal aging is associated with altered intestinal stem cell (ISC) behavior, microbial dysbiosis, and loss of barrier function 64–67. During aging, the number of mitotic cells in the Drosophila midgut increases due to ISC hyperproliferation and misdifferentiation 67,68. This age-induced hyperplasia can be assayed by scoring phosphorylated histone H3 (pH3) in the intestine. To examine the impact of neuronal BNIP3 upregulation on intestinal homeostasis, we examined the number of pH3+ cells in young and aged intestines. Remarkably, we found that inducing BNIP3 in neurons significantly reduced pH3 counts in the posterior midgut compared to control flies (Fig. 5a,b).
Figure 5. Neuronal BNIP3 induction improves intestinal homeostasis during aging.
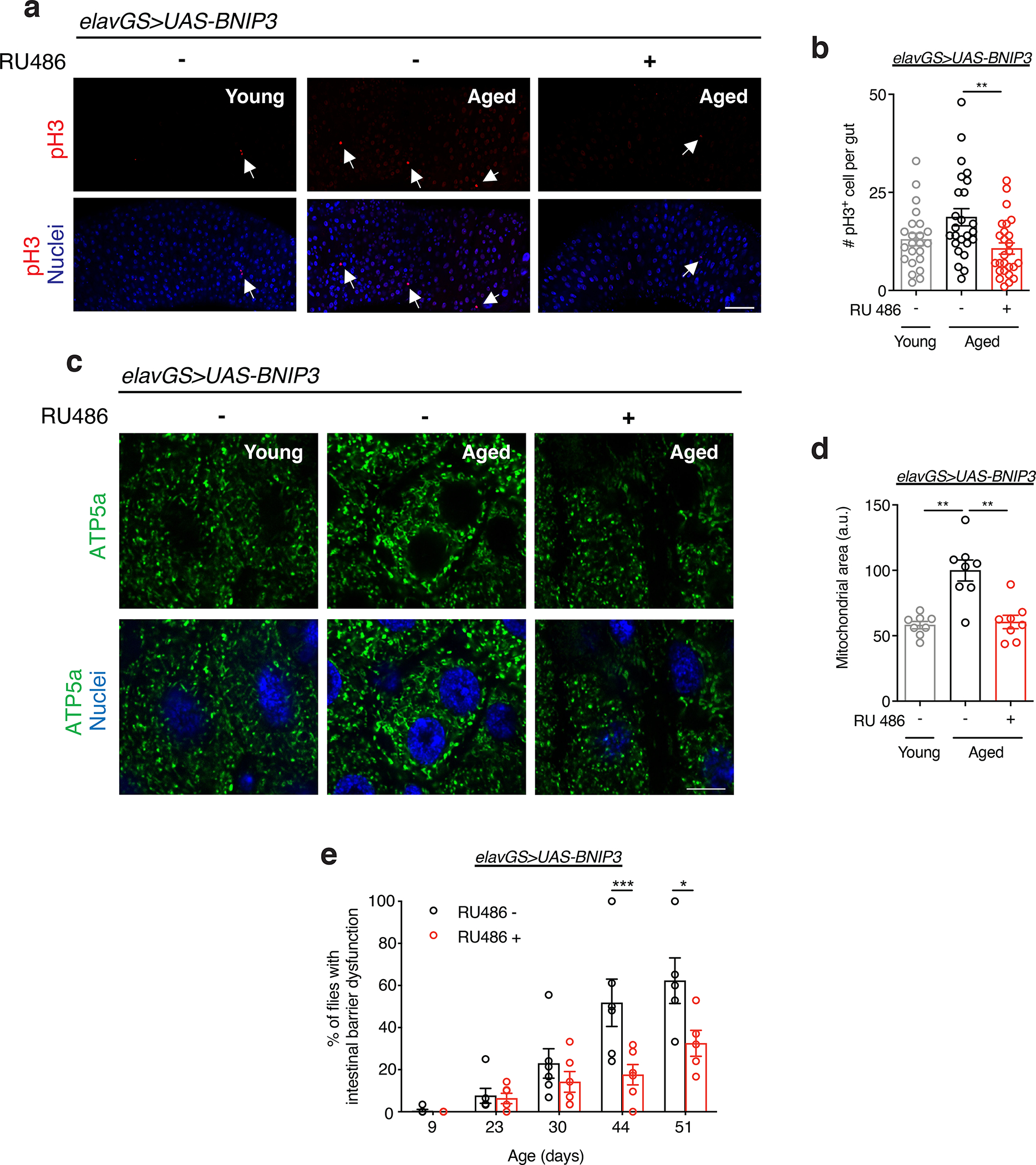
(a) Immunostaining of guts from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing intestinal stem cell proliferation (red channel, anti-PH3, white arrows) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 50 μm.
(b) Quantification of total number of PH3+ cells in gut as shown in (a). n = 23 young, 25 aged RU−, and 26 aged RU+ biologically independent animals, as indicated. **p=0.0051; one-way ANOVA/Tukey’s multiple comparisons test.
(c) Immunostaining of guts from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial morphology (green channel, anti-ATP5a) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 5 μm.
(d) Quantification of mitochondrial area in gut as shown in (c). n = 8 biologically independent animals per condition. ***p=0.0001 (young vs. aged RU−), ***p=0.0002 (aged RU− vs. aged RU+); one-way ANOVA/Tukey’s multiple comparisons test.
(e) Intestinal integrity during aging of elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 5 vials with 30 biologically independent animals per vial on day 9. *p=0.0120, ***p=0.0010; two-way ANOVA/Šídák’s multiple comparisons test. RU486 or vehicle was provided in the media at a concentration of 5 μg/ml in the indicated treatment groups. Data are presented as scatter plots overlaying mean values +/− SEM.
Little is known about changes to enterocyte mitochondrial dynamics as organisms age. Using IF microscopy, we found that mitochondrial content significantly increased in the enterocytes of the posterior midgut of aged flies compared to young controls (Fig. 5c,d). Interestingly, neuronal induction of BNIP3 resulted in reduced mitochondrial content in aged intestinal enterocytes compared to controls (Fig. 5c,d). Flies expressing control GFP in neurons showed no change to enterocyte mitochondrial content compared to those in age-matched guts (Extended Data Fig. 9c,d). Consistent with findings in the thorax, we observed that neuronal BNIP3 induction also led to increased Drp1 expression in the middle-aged intestine (Extended Data Fig. 10b). Intestinal barrier dysfunction has emerged as a conserved characteristic of aging that has been linked to systemic inflammation, organismal health decline and mortality 65,66,69–73. To determine whether neuronal BNIP3 induction can impact the intestinal barrier, we examined intestinal integrity during aging via the ‘Smurf assay’ 66,72. Remarkably, we observed a delay in the onset of intestinal barrier dysfunction in flies upregulating BNIP3 in neurons (Fig. 5e). To determine whether improved intestinal barrier function was linked to changes in gut bacteria, we performed qPCR with universal primers to bacterial 16S rRNA to characterize alterations in microbiota dynamics in response to neuronal BNIP3 expression. However, we did not detect a significant change in gut bacterial load in aged flies with neuronal BNIP3 induction compared to controls (Extended Data Fig. 10c). Together, these findings indicate that upregulation of neuronal BNIP3 can significantly delay markers of both muscle aging and intestinal aging.
Neuronal BNIP3 induction slows systemic aging via autophagy
Neuronal upregulation of BNIP3 improved mitochondrial homeostasis in the brain in an autophagy-dependent manner. Here, we set out to determine whether the impact of neuronal BNIP3 induction on muscle and intestinal aging also depends on neuronal autophagy. Remarkably, Atg1-RNAi expression in neurons prevented BNIP3-associated reduction in mitochondria size in the indirect flight muscle (Fig. 6a,b). Furthermore, mitochondria in muscles of flies with adult induction of Atg1-RNAi and BNIP3 in neurons showed significantly reduced membrane potential compared to young flies (Fig. 6c,d). Therefore, neuronal BNIP3 up-regulation requires neuronal autophagy to delay the accumulation of dysfunctional mitochondria in aged muscle.
Figure 6. Neuronal BNIP3 induction requires autophagy to delay systemic aging.
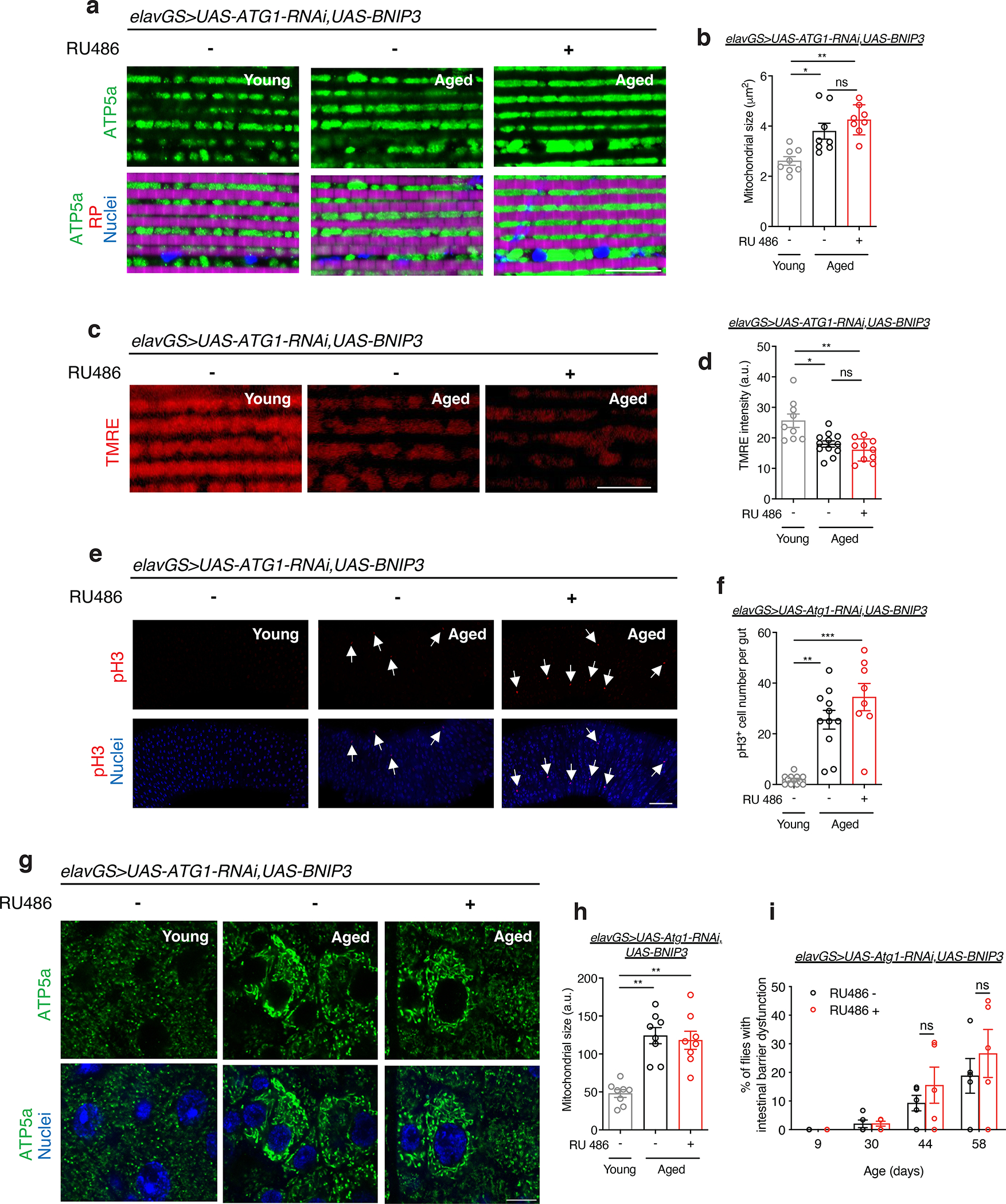
(a) Immunostaining of indirect flight muscles from young (10-day-old) and aged (30-day-old) elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial morphology (green channel, anti-ATP5a), rhodamine phalloidin (RP, magenta channel), and nuclear DNA (blue channel, stained with DAPI). Scale bar is 10 μm.
(b) Quantification of mitochondrial size in muscle as shown in (a). n = 8 biologically independent animals per condition. *p=0.0400, **p=0.0011, non-significant (n.s.); Kruskal-Wallis test/Dunn’s multiple comparisons test.
(c) Staining of indirect flight muscles from young (10-day-old) and aged (51-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing TMRE fluorescence. Scale bar is 10 μm.
(d) Quantification of mitochondrial membrane potential measured by TMRE staining as shown in (c). n = 9 young, 12 aged RU−, and 10 aged RU+ biologically independent animals. *p=0.0281, **p=0.0018, non-significant (n.s.); Kruskal-Wallis test/Dunn’s multiple comparisons test.
(e) Immunostaining of guts from young (10-day-old) and aged (51-day-old) elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing intestinal stem cell proliferation (red channel, anti-PH3, white arrows) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 50 μm.
(f) Quantification of total number of PH3+ cells in gut as shown in (e). n = 12 young, 11 aged RU−, and 8 aged RU+ biologically independent animals, as indicated **p=0.0011, ***p<0.0001, non-significant (n.s.); Kruskal-Wallis test/Dunn’s multiple comparisons test.
(g) Immunostaining of guts from young (10-day-old) and aged (51-day-old) elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial morphology (green channel, anti-ATP5a) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 5 μm.
(h) Quantification of mitochondrial area in gut as shown in (g). n = 8 biologically independent animals per condition. **p=0.0014 (young vs. aged RU−), **p=0.0044 (young vs. aged RU+); Kruskal-Wallis test/Dunn’s multiple comparisons test.
(i) Intestinal integrity during aging of elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 5 vials with 30 biologically independent animals per vial on day 10. n.s. = non-significant; two-way ANOVA/Šídák’s multiple comparisons test. RU486 or vehicle was provided in the media at a concentration of 5 μg/ml in the indicated treatment groups. Data are presented as scatter plots overlaying mean values +/− SEM.
In a similar manner, we examined whether the impact of neuronal BNIP3 induction on intestinal homeostasis during aging requires autophagy. pH3 counts in the posterior midgut were significantly higher in both aged control flies and in aged flies expressing Atg1-RNAi and BNIP3 in neurons compared to young controls (Fig. 6e,f). In addition, we found that co-expression of Atg1-RNAi with BNIP3 in adult neurons prevented neuronal BNIP3-associated reduction in the accumulation of mitochondria in aged intestinal cells (Fig. 6g,h). Finally, we also found that the neuronal BNIP3-associated impact on intestinal barrier function during aging are autophagy-dependent. Adult flies with induction of Atg1-RNAi and BNIP3 in neurons showed no difference in the number of ‘Smurf’ flies compared to age-matched controls (Fig. 6i). Together, these data reveal that observed changes to cellular and physiological hallmarks of muscle and intestinal aging associated with BNIP3 induction in neurons is dependent on neuronal autophagy.
Discussion
Mitochondria produce ATP and are, therefore, critical for neuronal health. Mitochondrial dysfunction is one of the most well-studied cellular hallmarks of brain aging and age-onset neurodegenerative diseases 5,7,8. Decades of research in model organisms and clinical studies have revealed a decline in mitochondrial function in aged brain tissue. However, the underlying mechanisms that lead to a loss of mitochondrial activity with age are not yet understood. One potential explanation for an age-related decline in mitochondrial function would be a loss of mitochondrial content with age. However, at present, a clear understanding of how mitochondrial content changes during brain aging is missing. Using the fruit fly Drosophila as a model organism, we find that there is a striking increase in mitochondrial content in the aged brain. Consistent with previous studies, which have reported an age-related decline in mitochondrial respiratory function in whole flies 46 and dissected muscle tissue 47, we find that mitochondria that accumulate in the aged brain show reduced mitochondrial membrane potential. Our findings were, in part, focused on a specific region of the fly brain (the optic lobe). It is beyond the scope of this study to document age-related changes in mitochondrial content and function in all brain regions. However, in future work, it would be interesting to examine whether there is regional or cell-type specificity in this regard. This could provide insight into the question of how neuronal mitophagy modulates systemic aging. Defects in mitophagy, a mitochondrial quality control mechanism enabling the degradation of damaged and superfluous mitochondria, have been implicated in a number of pathological contexts, including age-onset neurodegeneration 74,75. Here, we have observed that brain aging is linked to a decline in mitophagy in Drosophila. These findings lead us to conclude that a decline in mitochondrial quality control, rather than a loss of mitochondrial content, is a major factor underlying age-onset mitochondrial dysfunction in the fly brain. A logical extension of this idea is that targeting mitophagy to improve mitochondrial homeostasis in the aging brain may prove to be a viable strategy to forestall brain aging phenotypes. One potentially interesting avenue to explore would be the impact of BNIP3-mediated mitophagy on cognitive function in aged animals.
The major finding of this study is the identification of BNIP3 as a therapeutic target to counteract mitochondrial dysfunction in the aging brain and prolong healthy lifespan. BNIP3 has been reported to play roles in various cellular processes, including mitochondrial dysfunction, mitochondrial fragmentation, mitophagy and apoptosis 29,76. Hence, BNIP3 cannot be indiscriminately categorized as pro- or anti-apoptotic in function. Furthermore, BNIP3 is expressed in various tissues and is regulated by several different molecules under a variety of conditions 77. In the present study, we find that up-regulation of BNIP3 in adult neurons counteracts the accumulation of dysfunctional mitochondria in the aged brain, with no detectable induction of apoptosis in the aged brain. Interestingly, we observe that neuronal BNIP3 expression also counteracted age-related neuronal loss and brain caspase-3 activation. Hence, it would appear unlikely that a pro-apoptotic mechanism underlies the observed effects on mitochondrial content. Most importantly, we find that the ability of BNIP3 to prevent the accumulation of mitochondria is dependent upon the autophagy pathway. Moreover, BNIP3- mediated improvements in organismal healthspan also require autophagy. The simplest interpretation of these findings is that BNIP3 induces mitophagy to improve mitochondrial homeostasis in the aged brain, delaying organismal health decline and mortality. Interestingly, we find that neuronal up-regulation of BNIP3 in middle-aged flies is sufficient to facilitate mitophagy and promote longevity. This could prove important when considering the development of interventions based upon our findings. At the same time, however, we note that the prolongevity benefits of BNIP3 up-regulation may be specific to neuronal manipulations. Indeed, we failed to detect lifespan extension upon BNIP3 up-regulation using ubiquitous, muscle-specific, or intestine-specific manipulations. It is interesting to speculate that BNIP3 activation in non-neuronal cells may compromise tissue and organismal health via pro-apoptotic mechanisms. This may be important to consider in the context of studies implicating BNIP3 in cardiomyocyte cell death 78 and that mitophagy may regulates tumor survival in certain contexts 79.
It is intriguing that neuronal BNIP3 up-regulation produced alterations in cellular and physiological markers of aging in aged muscle and intestine. More specifically, neuronal BNIP3 was able to improve both mitochondrial homeostasis and proteostasis in aging muscle. In addition, neuronal BNIP3 was able to delay markers of intestinal stem cell aging and mitochondrial accumulation in the aged intestine. Finally, neuronal BNIP3 up-regulation delayed the onset of intestinal barrier dysfunction in aged flies. We interpret these findings to support a model in which healthy mitochondrial function in the aging brain is essential to maintain muscle and intestinal health. One potential future research direction would be to uncover the potential role of inter-organ signaling in mediating these effects.
Methods
Fly stocks
The fly strain UAS-hBNIP3-HA was kindly provided by Z. Zhang (Central South University, Changsha, Hunan, China). Elav–GeneSwitch (ElavGS) was provided by H. Keshishian (Yale University, New Haven, CT, USA), daughterless-GeneSwitch (daGS) was provided by H. Tricoire (Université Paris Diderot–Paris7, Paris, France), 5966-GeneSwitch (5966GS) was provided by H. Jasper (Genentech, San Francisco, CA, USA) and Actin88F-GeneSwitch (Act88FGS) was provided by F. Demontis (St. Jude Children’s Research Hospital, Memphis, TN, USA). GFP-mCherry-Atg8a was provided by Eric Baehrecke (University of Massachusetts Medical School, Worcester, MA, USA). UAS-mito-QC was provided by Alexander J. Whitworth (University of Cambridge, UK). UAS-Atg1-RNAi (16133) line was received by Vienna Drosophila RNAi Center (VDRC).
Fly Husbandry and Lifespan Analysis
Flies were maintained in vials containing cornmeal medium (1% agar, 3% yeast, 1.9% sucrose, 3.8% dextrose, 9.1% cornmeal, 1.1% acid mix, and 1.5% methylparaben, all concentrations given in wt/vol). Flies were collected under light nitrogen-induced anesthesia and housed at a density of 30 female flies per vial. All flies were kept in a humidified, temperature-controlled incubator with a 12h:12h dark:light cycle at 25 °C. RU486 was dissolved in ethanol and administered in the media as indicated while preparing food. Flies were flipped to fresh vials every 2–3 days and scored for death.
Immunostaining and Image analysis
For brain and muscle immunostaining, flies were fixed in 3.7% formaldehyde in phosphate buffered saline (PBS) for 20 min. After fixation, hemi-thoraces and brains were dissected and fixed again for 5 min. For gut immunostaining, intact adult guts were dissected and fixed for 30 min in 4% formaldehyde in PBS for 30 min, dehydrated for 5 min in each 50%, 75%, 87.5%, and 100% methanol, and rehydrated for 5 min in each 50%, 25%, and 12.5% methanol in 0.2% Triton X-100 in PBS (PBST) as previously described. Samples were then rinsed 3 times for 10 min with PBST and blocked in 3% BSA in PBST (PBST-BSA) for 1 hour. Primary antibodies were diluted in PBST-BSA and incubated overnight at 4°C. Primary antibodies used were: mouse-anti-ATP5a 1:250 (15H4C4, abcam); rabbit-anti-HA 1:250 (3724, CellSignaling); rabbit-anti-hBNIP3 1:400 (D7U1T Cell Signaling)); mouse-anti-FK2 1:250 (BML-PW8810–0500, ENZO); rabbit-anti-atg8a 1:250 (Rana et al., 2017). mouse-anti-dsDNA 1:250 (ab27156, abcam); rabbit-anti-cleaved-caspase-3 1:400 (D175 Cell Signaling); rabbit-anti-PH3 1:1000 (06–570, Millipore). Samples were then rinsed 3 times in PBST for 10 min. and incubated with the secondary antibodies and/or stained at room temperature for 3 hours. Secondary antibodies and stains used were: anti-rabbit or anti-mouse AlexaFluor-488 1:500 (A-11001 or A-11008, Thermo Fisher Scientific); anti-rabbit or anti-mouse AlexaFluor-568 1:500 (A-11031 or A-11036, Thermo Fisher Scientific); To-Pro-3 DNA 1:500 (T3605, Thermo Fisher Scientific); 4,6-diamidino-2- phenylindole (DAPI) 1:2000; phalloidin AlexaFlour-568 1:250 (A12380, Thermo Fisher Scientific). Finally, samples were rinsed 3 times with PBST for 10 min and mounted in Vectashield Mounting Medium (Vector Lab). Images were taken using Zeiss LSM780 or LSM880 confocal microscope and analyzed using ImageJ software to measure intensity, mitochondrial area, and aggregate sizes. The image of mitochondria in guts were taken from the posterior midgut R5 region by Buchon et al. The number of PH3+ cells were counted in whole midguts, defined from R1 to R5.
TMRE staining
Flies were anesthetized and dissected in cold Drosophila Schneider’s Medium (DSM). Brain and hemi-thoraces were incubated in TMRE staining solution (100nm TMRE (T669, Thermo Fisher Scientific) in DSM) for 12 min at room temperature. After staining, samples were rinsed once in wash solution (25nm TMRE in DSM) for 30 s before being mounted in wash solution. Images were acquired using a Zeiss LSM880 confocal microscope and TMRE intensity was quantified using ImageJ software.
GFP-mCherry-Atg8a tandem and Mito-QC staining
Flies were anesthetized and dissected in cold Drosophila Schneider’s Medium (DSM). Brains were mounted in DSM solution. Images were acquired on a Zeiss LSM780 or LSM880 confocal microscope and autolysosomes or mitolysosomes (mCherry-only foci) were quantified using ImageJ software.
Intestinal barrier dysfunction (Smurf) assay
Intestinal integrity assays were performed as previously described 66. Flies were aged to the indicated time points with standard RU− or RU+ food as indicated. To conduct the “Smurf” assay, flies were then transferred to new vials containing standard medium with 2.5% wt/vol F&D blue dye # 1 (SPS Alfachem) for 16 hours. The number of flies per vial with dye coloration outside the gut (Smurf flies) were then tallied and quantified.
Mitochondrial DNA measurement
Total cellular DNA from 10 heads were prepared by homogenization in 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.1% Triton X-100 and 10 ug/ml proteinase K. Following a 60 min incubation at 37 °C, Proteinase K was heat inactivated at 95 °C for 5 min. Mitochondrial DNA was quantified relative to nuclear DNA by the ratio of amplicons of cytochrome oxidase subunit I (COI) to amplicons of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in quantitative real-time PCRs. Primer sequences: COI, GAATTAGGACATCCTGGAGC and GCACTAATCAATTTCCAAATCC; GAPDH, GACGAAATCAAGGCTAAGGTCG and AATGGGTGTCGCTGAAGAAGTC.
Climbing activity assay
In negative geotaxis assays, flies were gently tapped to the bottom of 10 cm vials. After 10 seconds, the number of flies that climbed above 5 cm were recorded. For forced climbing assays, 100 adult female flies from each treatment group were placed in 200 ml glass cylinders. The cylinders were tapped quickly and the flies were allowed to settle for 2 min. This step was repeated nine times. 1 min after the final tap, the number of flies in the upper, middle, and lower 1/3rd part of the cylinder was recorded.
Spontaneous physical activity assay
Vials containing 10 adult female flies were placed inside a Drosophila activity monitor (TriKinetics). Movements were recorded continuously under normal culturing conditions for 36 h on a 12h:12h dark:light cycle. Graphs represent mean activity per fly per hour and the scatterplot shows spontaneous activity per fly during a 12h:12h dark:light cycle. Triplicate samples were used for each activity measurement
Consumption-Excretion (Con-Ex) assay
Con-EX assays were performed as previously described 54. Adult flies were transferred to new empty vials (10 flies per vial with a total of 6 vials) and fed from feeder caps containing standard medium with 2.5% wt/vol F&D blue dye # 1 for 20h at 25 °C. Feeder caps were discarded at the conclusion of feeding. For checking internal (consumed) dye, flies were homogenized in 500 ul of ddH2O and pellet debris were removed by centrifugation. The dye excreted by the flies on the walls of the vials was collected by adding 1ml of ddH2O to each vial and vortexing. Samples were quantified using an Epoch BioTek microplate reader and compared to a serially diluted standard.
Fecundity assay
Flies were kept on standard food media in a humidified, temperature- controlled incubator with 12h:12h dark:light cycle at 25 °C. Eggs laid per fly in 24 h were counted.
Quantitative real-time PCR.
Total RNA was extracted from samples using TRIzol reagent (Invitrogen) following manufacturer protocols. Samples were treated with DNAse before cDNA synthesis was performed using the First Strand cDNA Synthesis Kit from Fermentas. qPCR was performed using Power SYBR Green master mix (Applied Biosystems) on a BioRad Real-Time PCR system. Cycling conditions were as follows: 95°C for 10 min; 95°C for 15 s then 60°C for 60 s, cycled 40 times. Equalized amplicons of GAPDH were used as a reference to normalize. BioRad CFX Manager ver. 3.1 was used to collect and analyze qRT-PCR data. Primer sequences used are as follows:
GAPDH, GCGGTAGAATGGGGTGAGAC and TGAAGAGCGAAAACAGTAGC
DRP1, ATTGTTGTTCTAGGCAGTCAG and GAACTCTTGCCGGAGCT
Quantitative PCR for 16 S ribosomal RNA
Genomic DNA was extracted using the PowerSoil DNA isolation kit (MoBio). All flies were surface sterilized as previously described prior to sample preparation. To ensure consistent homogenization, 10 flies were pre-homogenized in 150 ul of solution from the PowerSoil bead tube using a motor pestle. Then the homogenate was returned to the bead tube and the manufacturers protocol was followed. The PCR was performed with PowerUP SYBR Green Master Mix (Ref#A25777, Applied Biosystems) on a CFX96Real Time PCR system. Cycling conditions were as follows: 95°C for 15 s then 60°C for 60 s, cycled 40 times. The 16S gene expression values were normalized to the value of the loading control gene Actin5C. Primer sequences: Act5C, TTGTCTGGGCAAGAGGATCAG and ACCACTCGCACTTGCACTTTC.
Universal primers for the 16 S ribosomal RNA gene were against variable regions 1 (V1F) and 2 (V2R), as previously published 65.
Isotropic Fractionation.
Protocol was adapted from 80,81. Flies were fixed in 3.7% formaldehyde in phosphate buffered saline (PBS) for 20 min at room temperature. After fixation, brains were dissected and fixed again for 1.5 h at 4°C. Samples were then rinsed 3 times for 10 min with PBST and blocked in 3% BSA in PBST (PBST-BSA) for 1 hour. Primary anti-ELAV (7E8A10 DSHB) was diluted in PBST-BSA and incubated for 48 h at 4°C on a nutator. Samples were washed 3 times for 10 min with PBST at RT. Brains were then incubated with secondary goat-anti-rat-488 1:500 (Invitrogen) for 48h at 4°C on a nutator. Following incubation with secondary antibodies, To-Pro-3 DNA 1:500 (T3605, Thermo Fisher Scientific) was added to samples for 30 min. Tissues were then washed three times for 10 min with PBST at RT. Brains from the indicated treatments were pooled into groups of 3 and transferred into a solution containing 40 mM sodium citrate and 1% triton X-100. Samples were heated for 10 min at 60°C and centrifuged at 14000g for 3 min before removing the dissociation solution. Brains were gently homogenized by micropestle and rinsed with 20 ul PBS. Homogenates were mixed thoroughly by pipette for 30 sec to further promote dissociation of nuclei before imaging at 20x using a Zeiss LSM 700 Imager M2 using Zen 2009 (Carl Zeiss).
Western blot assay.
Samples were collected as indicated and lysates were separated by SDS page using standard protocols. Membranes were probed with antibodies or antisera against anti-actin (1:15000 dilution, PA-16914, Thermo Fisher Scientific), anti-hBNIP3 (1:1000 dilution, D7U1T Cell Signaling). Mouse antibodies were detected using horseradish peroxidase-conjugated anti-mouse IgG antibodies (1:2000 dilution, Sigma). Amersham ECL Prime Western Blotting Detection Reagent (GE Life Sciences) was used to visualize the presence of horseradish peroxidase, with the chemiluminescent signal recorded using Syngene Pxi Western Blot Imager. Image analysis was done via ImageJ software.
Statistics
GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA) was used to perform statistical analysis and graphical display of data. Significance is expressed as p values as determined by two-tailed, unpaired, parametric, or non-parametric tests as indicated in figure legends. When comparing two groups, unpaired t-tests were used when data met criteria for parametric analysis and Mann-Whitney tests were used for non-parametric analysis. To compare more than two groups when parametric tests were appropriate, one-way ANOVAs with Tukey’s multiple comparisons tests were performed. To compare more than two groups sampled from a Gaussian distribution without assuming equal variances, Welch and Brown-Forsythe ANOVAs were used. To analyze more than two groups when data did not meet requirements for parametric tests, Kruskal-Wallis tests with Dunn’s multiple comparisons post hoc tests were used. When performing grouped analyses with multiple comparisons, two-way ANOVAs with Šídák’s multiple comparisons test were performed. Bar graphs depict mean ± standard error of the mean (SEM). The number (n) of biological samples used in each experiment can be found in figure legends. Log-rank (Mantel-Cox) tests were used to compare survival curves. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications 16,18. Blinding was performed when possible, specifically when conducting microscopy for TMRE, MitoSox, ATG8a-tandem, and mitoQC. Blinding was not always possible during experimental setup due to the need to carefully document the genotypes of flies when generating crosses or to track groups assigned RU486 vs. vehicle throughout lifespans. All experiments were conducted under the same conditions, and control and experimental samples were treated equally and in parallel to exclude bias. Additionally, all images were taken in the same location and depth in each tissue type. Parents of experimental flies were randomly grouped into mating vials with 10 virgin females to 7 mature males. Upon eclosion, experimental flies were randomly assigned to mating bottles (10 vials per bottle) for 3 days. These bottles were then sorted into vials containing 30 mated females each before evenly distributing these vials assigned randomly into treatment and control groups. No animals or data points were excluded from the analyses. The difference between two groups was defined as statistically significant for the following p values: *p<0.05, **p<0.01, ***p< 0.001 (and non-significant when p>0.05).
Data availability.
All data generated or analyzed during this study are included in the figures and text with representative images accompanying quantified results where applicable unless otherwise noted. Further information is available from the corresponding author upon reasonable request.
Extended Data
Extended Data Figure 1. Related to Figure 1. RU486 induces BNIP3 expression in the brain of elavGS>UAS-BNIP3 flies.
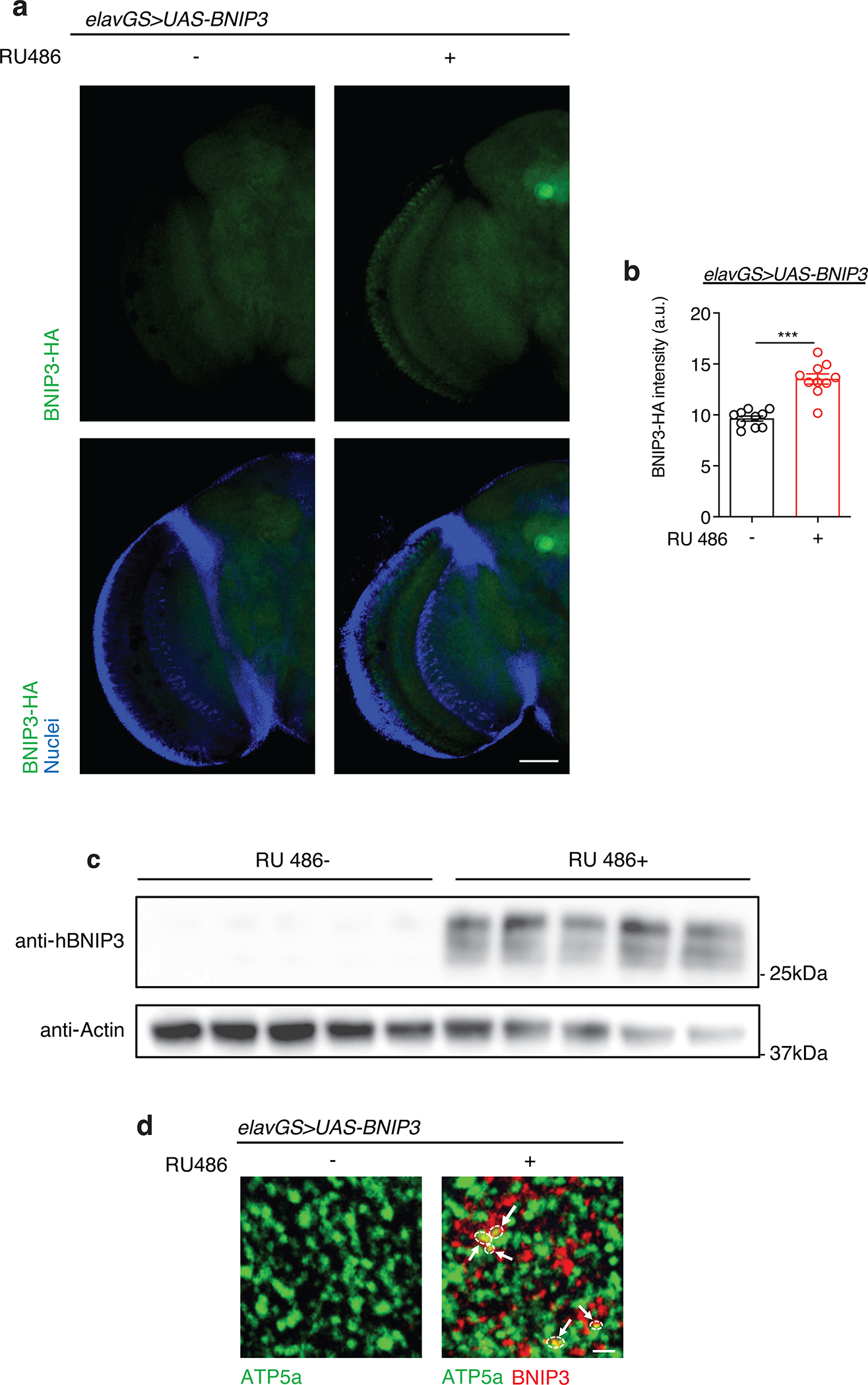
(a) Immunostaining of brains from 10-day-old elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing BNIP3 expression level (green channel, anti-HA) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 50 μm.
(b) Quantification of BNIP3 expression level in brain as shown in (a). n = 10 biologically independent animals per condition. ***p<0.0001; unpaired t test. Data are presented as scatter plots overlaying mean values +/− SEM.
(c) Western blot detection of BNIP3 transgene induction in the heads of day 14 elavGS>UAS-BNIP3 flies with or without 9 days of RU486 treatment. n = 5 biological replicates per condition with 10 flies pooled per replicate.
(d) Immunostaining of brains from young (10-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondrial morphology (green channel, anti-ATP5a) and BNIP3 (red channel). Arrows and outlines indicate sites of colocalization. Scale bar is 3 μm. RU486 was provided in the media at a concentration of 5 μg/ml. Images are representative of 5 samples treated with RU and 6 samples provided vehicle.
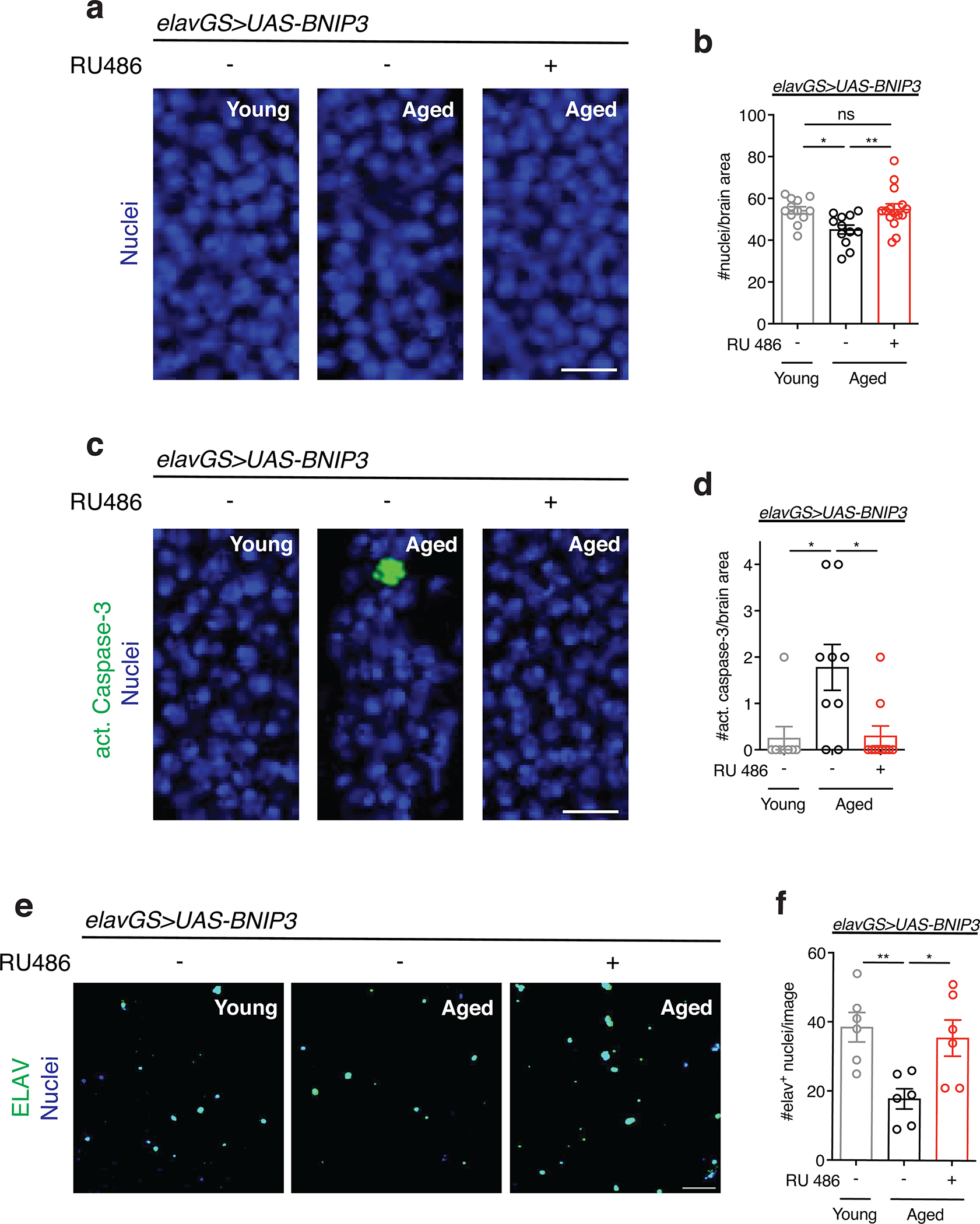
Extended Data Figure 2. Related to Figure 1. Neuronal BNIP3 induction prevents loss of neurons in aged brains.
(a) TOPRO staining of nuclei in brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. Scale bar is 5 μm.
(b) Quantification of nuclei per 350 μm2 of optic lobe as shown in (a). n = 12 young, 12 aged RU−, and 15 aged RU− biologically independent animals per condition, as indicated. *p=0.0219, **p=0.0097; one-way ANOVA/Tukey’s multiple comparisons test.
(c) Immunostaining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing cleaved (activated) caspase-3 (green channel) and nuclear DNA (blue channel, stained with To-Pro-3). Scale bar is 5 μm.
(d) Quantification of cleaved caspase-3 in one optic lobe per fly as shown in (a). n = 8 young, 9 aged RU−, and 10 aged RU+ biologically independent animals per condition, as indicated. *p=0.0140 (young vs. aged RU−), *p=0.0119 (aged RU− vs. aged RU+); one-way ANOVA/Tukey’s multiple comparisons test.
(e) Immunostaining of nuclei isolated via isotropic fractionation from brains of young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing ELAV (green channel) and DNA (blue channel, stained with To-Pro-3). Scale bar is 50 μm.
(f) Quantification of neuronal (elav+) nuclei isolated from brains via isotropic fractionation as shown in (e). n = 6 biological replicates per condition with 3 pooled brains per replicate. *p=0.0263, **p=0.0098; one-way ANOVA/Tukey’s multiple comparisons test. Data are presented as scatter plots overlaying mean values +/− SEM.
Extended Data Figure 3. Related to Figure 1. RU486 treatment in control flies has no effect on mitochondria homeostasis in aged brains.
(a) Immunostaining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-GFP flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondria morphology (red channel, anti-ATP5a) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 5 μm.
(b) Quantification of mitochondria area in brain as shown in (a). n = 8 biologically independent animals per condition. *p=0.0327 (both), non-significant (n.s.); Kruskal-Wallis test/Dunn’s multiple comparisons test. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
Extended Data Figure 4. Related to Figure 2. Neuronal specific BNIP3 induction reduces ATG8 levels in aged brains.
(a) Immunostaining of brains from young (10-day-old) and aged (30-day-old) elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing ATG8a levels (red channel, anti-ATG8a). Scale bar is 5 μm.
(b) Quantification of Atg8a levels in brain as shown in (a). n = 7 young, 6 aged RU−, and 8 aged RU+ biologically independent animals, as indicated. *p=0.0446 (young vs. aged RU−), *p=0.0169 (aged RU− vs. aged RU+); Kruskal-Wallis test/Dunn’s multiple comparisons test. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
Extended Data Figure 5. Related to Figures 2 and 3. Midlife neuronal induction of BNIP3 induces mitophagy and extends lifespan.
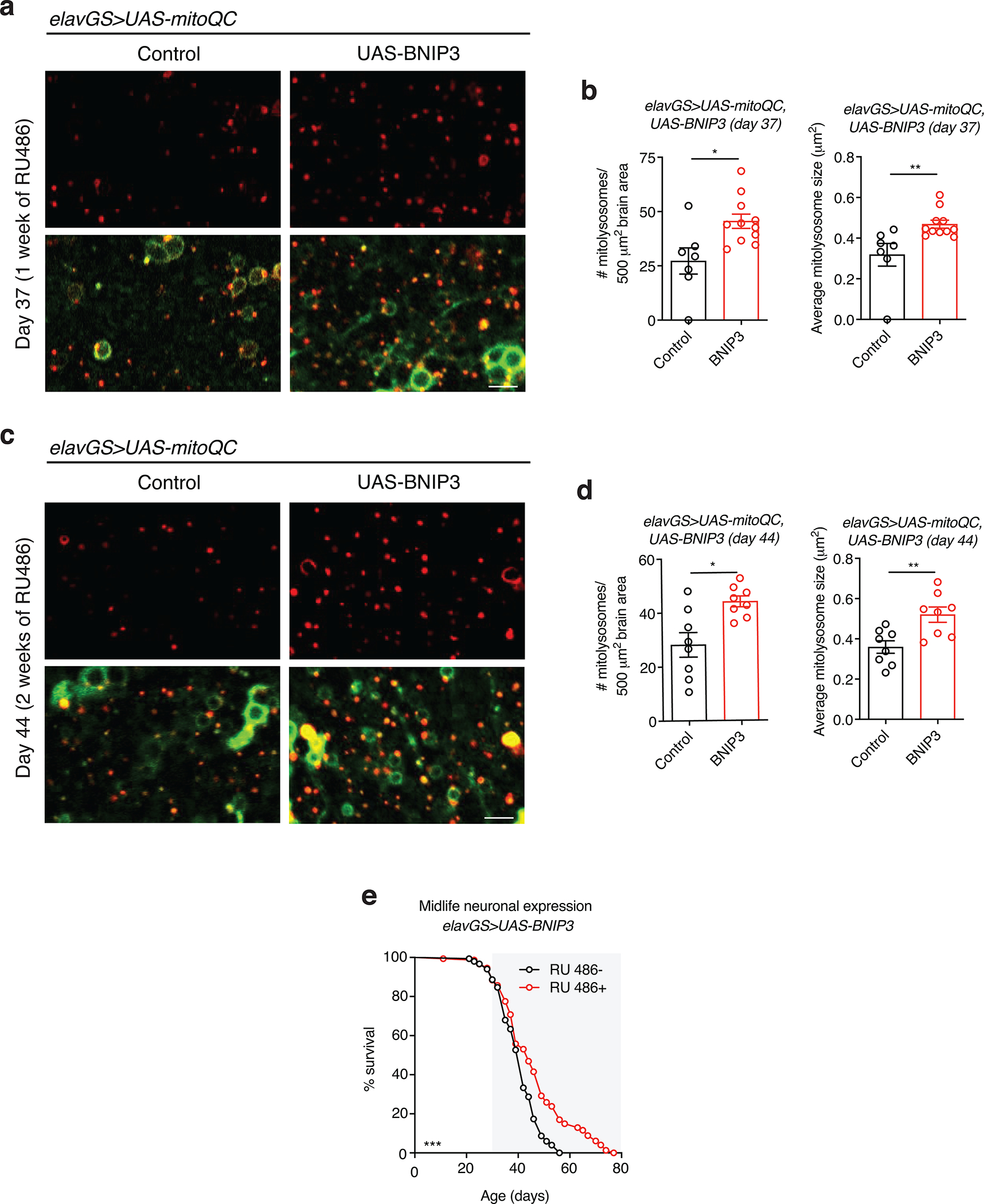
(a and c) mito-QC of brains from 37-day-old (a) and 44-day-old (c) flies. Genotypes analyzed were elavGS>UAS-mito-QC,UAS-lacZ, as a control, and elavGS>UAS-mito-QC,UAS-BNIP3. RU486-mediated transgenes were induced from day 30 to day 37 (a) or from day 30 to day 44 (c). Images shown of merged GFP and mCherry along with punctate mCherry-only foci (from merged images where GFP has been quenched; mitolysosomes). Scale bar is 5 μm.
(b and d) Quantification of the number of mitolysosomes per 500 μm2 brain area and average size (μm2) as shown in (a) and (c) at day 37 (b) and day 44 (d). (b) n = 7 control and 11 BNIP3+ biologically independent animals. *p=0.0102, **p=0.0089; unpaired t tests. (d) n = 8 biologically independent animals per condition. *p=0.0115, **p=0.0053; Mann-Whitney test (#), unpaired t tests (size).
(e) Survival curves of elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 30 onward. The shaded area indicates the duration of BNIP3 induction. ***p=0.0010; log-rank test; n = 150 RU− and 147 RU+ biologically independent animals. RU486 was provided in the media at a concentration of 25 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
Extended Data Figure 6. Related to Figure 2. RU486 induces BNIP3 expression in the brain of elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies.
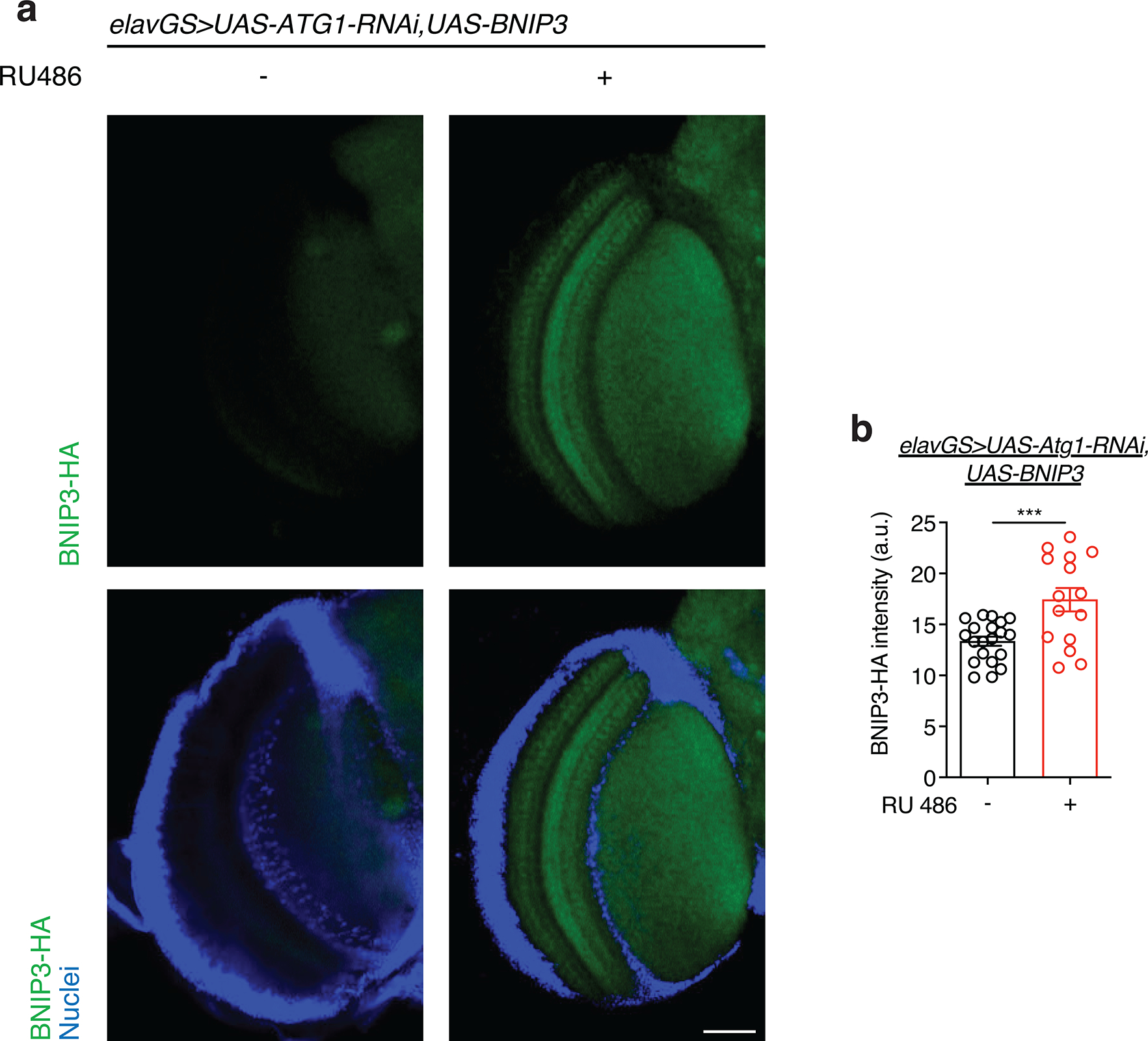
(a) Immunostaining of brains from 10-day-old elavGS>UAS-Atg1RNAi,UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward, showing BNIP3 expression level (green channel, anti-HA) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 50 μm.
(b) Quantification of BNIP3 expression level in brain as shown in (a). n = 20 RU− and 15 RU+ biologically independent animals. ***p=0.0008; unpaired t test. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
Extended Data Figure 7. Related to Figure 3. RU486 treatment in control flies has no effect on lifespan and healthspan, and neuronal-specific BNIP3 induction does not alter food consumption or fecundity.
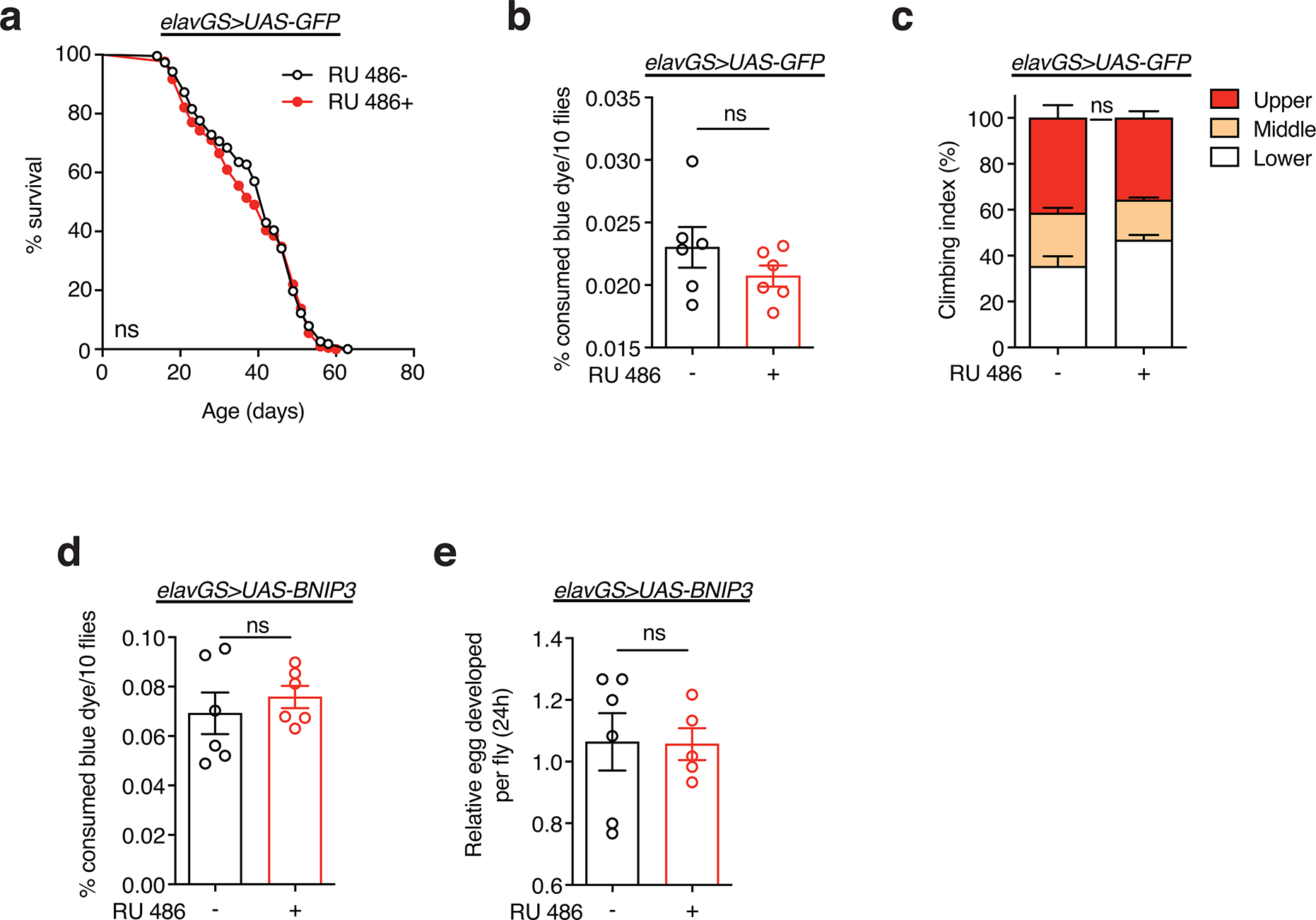
(a) Survival curve of elavGS>UAS-GFP flies with or without RU486-mediated transgene induction from day 5 onward. non-significant (n.s.); log-rank test. n = 228 RU− and 218 RU+ biologically independent animals.
(b) Con-Ex feeding assay of 10-day-old elavGS>UAS-GFP flies with or without RU486-mediated transgene induction from day 5 onward. n = 6 vials of 10 flies per condition. non-significant (n.s.); unpaired t test.
(c) Climbing index as a measure of endurance of 30-day-old elavGS>UAS-GFP flies with or without RU486-mediated transgene induction from day 5 onward. n = 8 biological replicates with 100 flies per replicate. non-significant (n.s.); unpaired t test. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as mean values +/− SEM.
(d) Con-Ex feeding assay of 10-day-old elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 6 vials of 10 flies per condition. non-significant (n.s.); unpaired t test.
(e) Fecundity of 37-day-old elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n =6 vials of 30 RU− biologically independent animals and 5 vials of 30 RU+ biologically independent animals with values normalized per fly, as indicated. non-significant (n.s.); unpaired t test. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM unless otherwise indicated.
Extended Data Figure 8. Related to Figure 3. Ubiquitous, gut-, or muscle-specific BNIP3 induction shortens lifespan.
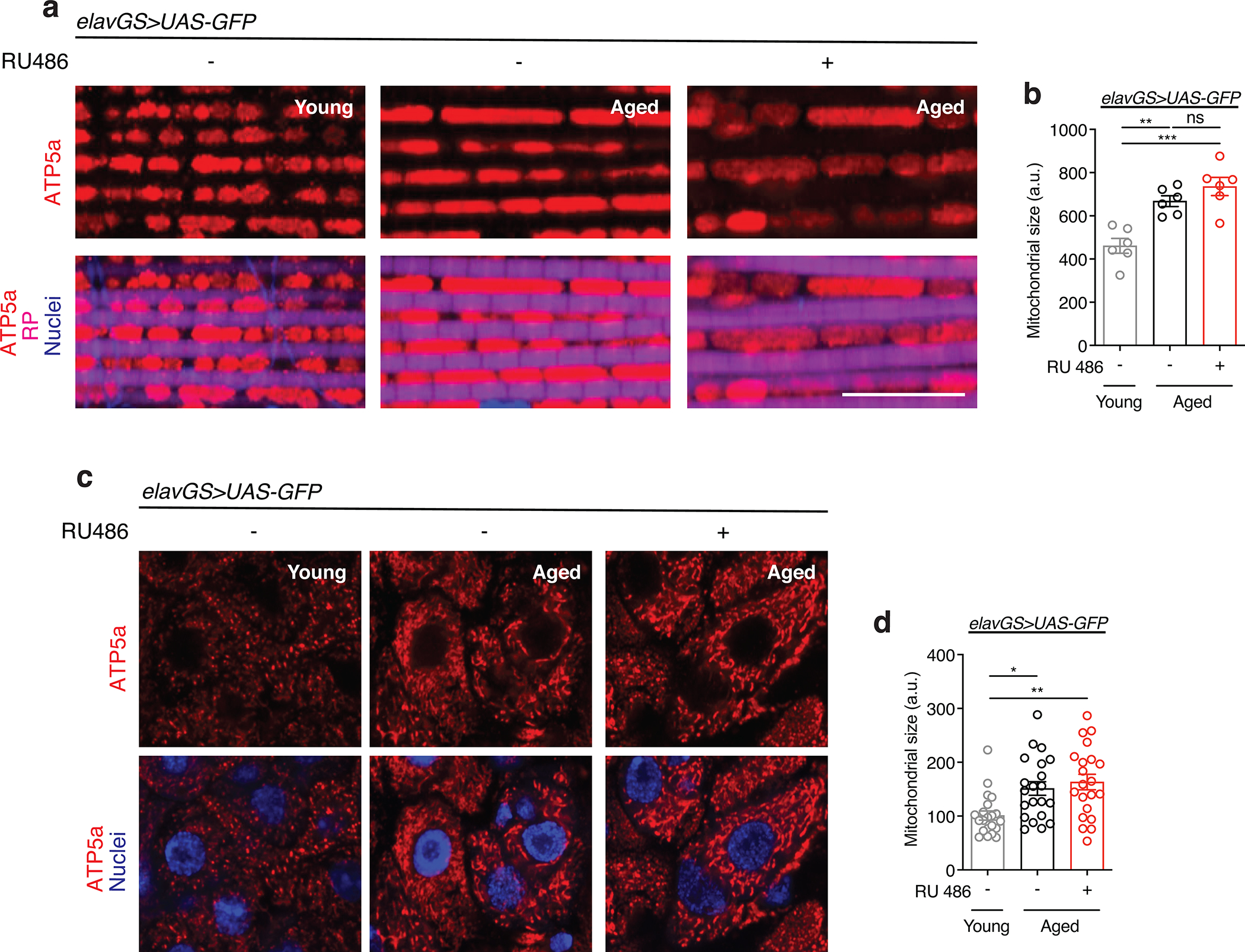
(a) Survival curves of daGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. ***p=0.0006 (RU0 vs. RU5) and ***p<0.0001 (RU0 vs. each other RU dose), (n.s.) non-significant; log-rank test; n = 208 RU−, 200 RU5, 204 RU10, 198 RU25, and 201 RU50 biologically independent animals.
(b) Survival curves of 5966GS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. ***p<0.0001 (RU0 vs. each RU dose); log-rank test; n = 201 RU−, 194 RU5, 197 RU10, 199 RU25, and 204 RU50 biologically independent animals.
(c) Survival curves of Act88FGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. ***p<0.0001 (RU0 vs. each RU dose); log-rank test; n = 239 RU−, 473 RU5, 233 RU10, 234 RU25, and 238 RU50 biologically independent animals.
(d) Survival curves of daGS>UAS-BNIP3-RNAi flies with or without RU486-mediated transgene induction from day 5 onward. ***p=0.0006; log-rank test; n = 209 RU− and 233 RU+ biologically independent animals.
(e) Survival curves of daGS>UAS-BNIP3-RNAi flies with or without RU486-mediated transgene induction from day 30 onward. ***p<0.001; log-rank test; n = 209 RU− and 202 RU+ biologically independent animals.
(f) Survival curves of elavGS>UAS-BNIP3-RNAi flies with or without RU486-mediated transgene induction from day 5 onward. *p=0.0203; log-rank test; n = 165 RU− and 180 RU+ biologically independent animals.
Extended Data Figure 9. Related to Figures 4 and 5. RU486 treatment in control flies has no effect on mitochondria homeostasis in aged muscle and gut.
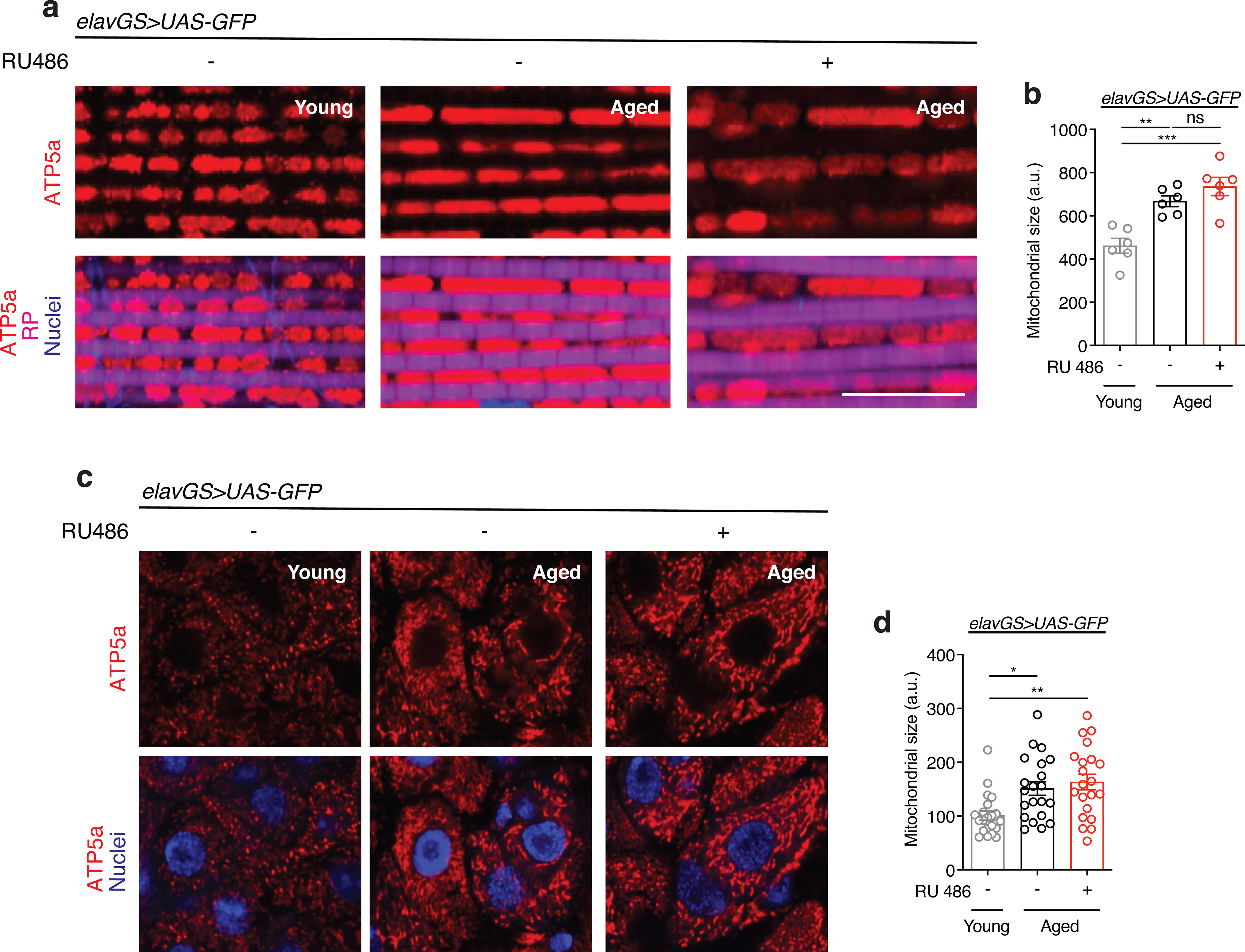
(a) Immunostaining of indirect flight muscles from young (10-day-old) and aged (30-day-old) elavGS>UAS-GFP flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondria morphology (red channel, anti-ATP5a), muscles (magenta channel, stained with phalloidin/F-Actin), and nuclear DNA (blue channel, stained with DAPI). Scale bar is 10 μm.
(b) Quantification of mitochondria area in muscle as shown in (a). n = 6 biologically independent animals per condition. **p=0.0020, ***p=0.0001, non-significant (n.s.); one-way ANOVA/Tukey’s multiple comparisons test.
(c) Immunostaining of guts from young (10-day-old) and aged (30-day-old) elavGS>UAS-GFP flies with or without RU486-mediated transgene induction from day 5 onward, showing mitochondria morphology (red channel, anti-ATP5a) and nuclear DNA (blue channel, stained with DAPI). Scale bar is 5 μm.
(d) Quantification of mitochondria area in gut as shown in (A). n = 21 biologically independent replicates per condition. *p=0.0131, **p=0.0017; one-way ANOVA/Tukey’s multiple comparisons test. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
Extended Data Figure 10. Related to Figure 4 and 5. Neuronal-specific BNIP3 upregulation induces midlife Drp1 expression in the thorax and gut but does not alter microbial dynamics in the gut.
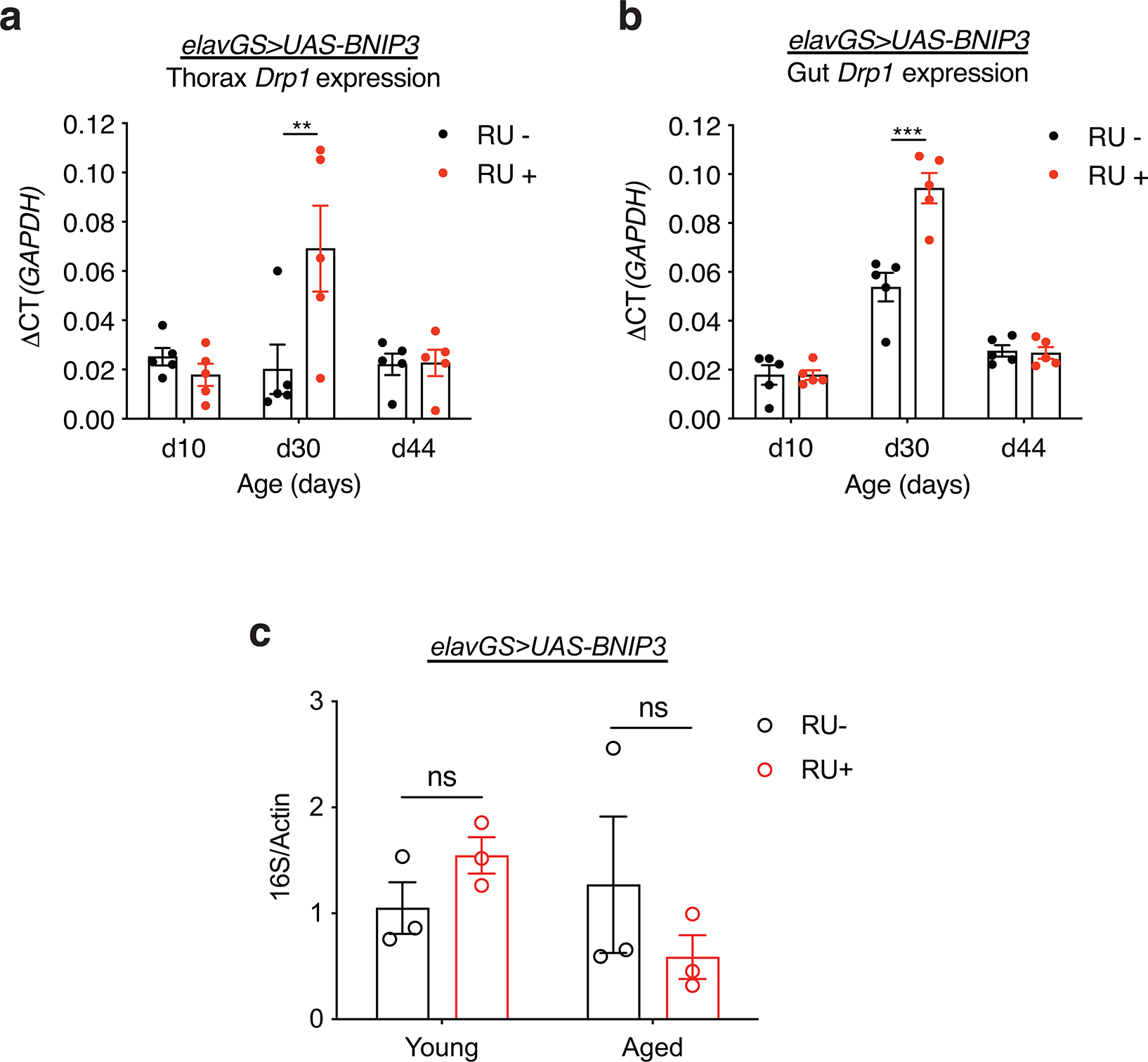
(a) qPCR analyses of Drp1 mRNA levels relative to GAPDH in the thorax on days 10, 30, and 44 in elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 5 biological replicates with 5 dissected thoraxes pooled per replicate. **p=0.0023; two-way ANOVA/Šídák’s multiple comparisons test.
(b) qPCR analyses of Drp1 mRNA levels relative to GAPDH in dissected guts on days 10, 30, and 44 in elavGS>UAS-BNIP3 flies with or without RU486-mediated transgene induction from day 5 onward. n = 5 biological replicates with 5 dissected guts pooled per replicate. ***p<0.0001; two-way ANOVA/Šídák’s multiple comparisons test. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
(c) Bacterial levels assayed by qPCR of 16S rRNA gene in surface sterilized, elavGS>UAS-BNIP3 flies with or without 5 μg/ml RU486 treatment from day 5 post-eclosion onwards. n.s.; not significant, two-way ANOVA/Šídák’s multiple comparisons test; n = 3 replicates of ten flies pooled per condition. RU486 was provided in the media at a concentration of 5 μg/ml. Data are presented as scatter plots overlaying mean values +/− SEM.
Supplementary Material
Acknowledgments
We thank Zhuohua Zhang (Xiangya Medical School), Eric Baehrecke (UMass Medical School), Vienna Drosophila RNAi Center, and the Bloomington Drosophila Stock Center (NIH P40OD018537) for fly stocks; José Armando Guerrero and Vishal Patel for help with fly work; Lesly Palacios Castillo and Ricardo Aparicio for technical assistance. We thank Nathanaël Prunet and the MCDB/BSCRC Microscopy Core for training and microscope facilities. This work was supported by NIH grants (R01AG037514, R01AG049157) to D.W.W. This research was conducted while D.W.W. was a Julie Martin Mid-Career Awardee in Aging Research supported by The Ellison Medical Foundation and AFAR.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Alexander GE et al. Characterizing cognitive aging in humans with links to animal models. Front Aging Neurosci 4, 21, doi: 10.3389/fnagi.2012.00021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dykiert D, Der G, Starr JM & Deary IJ Age differences in intra-individual variability in simple and choice reaction time: systematic review and meta-analysis. PLoS One 7, e45759, doi: 10.1371/journal.pone.0045759 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levin O, Fujiyama H, Boisgontier MP, Swinnen SP & Summers JJ Aging and motor inhibition: a converging perspective provided by brain stimulation and imaging approaches. Neurosci Biobehav Rev 43, 100–117, doi: 10.1016/j.neubiorev.2014.04.001 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Prince MJ et al. The burden of disease in older people and implications for health policy and practice. Lancet 385, 549–562, doi: 10.1016/S0140-6736(14)61347-7 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Mattson MP & Arumugam TV Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab 27, 1176–1199, doi: 10.1016/j.cmet.2018.05.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217, doi: 10.1016/j.cell.2013.05.039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimm A & Eckert A Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem 143, 418–431, doi: 10.1111/jnc.14037 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun N, Youle RJ & Finkel T The Mitochondrial Basis of Aging. Mol Cell 61, 654–666, doi: 10.1016/j.molcel.2016.01.028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine B & Kroemer G Autophagy in the pathogenesis of disease. Cell 132, 27–42, doi: 10.1016/j.cell.2007.12.018 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen M, Rubinsztein DC & Walker DW Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol 19, 579–593, doi: 10.1038/s41580-018-0033-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng Y, He D, Yao Z & Klionsky DJ The machinery of macroautophagy. Cell Res 24, 24–41, doi: 10.1038/cr.2013.168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickles S, Vigie P & Youle RJ Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol 28, R170–R185, doi: 10.1016/j.cub.2018.01.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youle RJ & Narendra DP Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12, 9–14, doi: 10.1038/nrm3028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drummond MJ et al. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison. J Gerontol A Biol Sci Med Sci 69, 1040–1048, doi: 10.1093/gerona/glu004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun N et al. Measuring In Vivo Mitophagy. Mol Cell 60, 685–696, doi: 10.1016/j.molcel.2015.10.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rana A et al. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat Commun 8, 448, doi: 10.1038/s41467-017-00525-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Amico D et al. The RNA-Binding Protein PUM2 Impairs Mitochondrial Dynamics and Mitophagy During Aging. Mol Cell 73, 775–787 e710, doi: 10.1016/j.molcel.2018.11.034 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aparicio R, Rana A & Walker DW Upregulation of the Autophagy Adaptor p62/SQSTM1 Prolongs Health and Lifespan in Middle-Aged Drosophila. Cell Rep 28, 1029–1040 e1025, doi: 10.1016/j.celrep.2019.06.070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rana A, Rera M & Walker DW Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A 110, 8638–8643, doi: 10.1073/pnas.1216197110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryu D et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22, 879–888, doi: 10.1038/nm.4132 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Vicente M Neuronal Mitophagy in Neurodegenerative Diseases. Front Mol Neurosci 10, 64, doi: 10.3389/fnmol.2017.00064 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palikaras K, Lionaki E & Tavernarakis N Coupling mitogenesis and mitophagy for longevity. Autophagy 11, 1428–1430, doi: 10.1080/15548627.2015.1061172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palikaras K, Lionaki E & Tavernarakis N Mitophagy: In sickness and in health. Mol Cell Oncol 3, e1056332, doi: 10.1080/23723556.2015.1056332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sliter DA et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262, doi: 10.1038/s41586-018-0448-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Fang EF et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22, 401–412, doi: 10.1038/s41593-018-0332-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Onishi M, Yamano K, Sato M, Matsuda N & Okamoto K Molecular mechanisms and physiological functions of mitophagy. EMBO J 40, e104705, doi: 10.15252/embj.2020104705 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pickrell AM & Youle RJ The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 85, 257–273, doi: 10.1016/j.neuron.2014.12.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorn GW 2nd. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3, 374–383, doi: 10.1007/s12265-010-9174-x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burton TR & Gibson SB The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ 16, 515–523, doi: 10.1038/cdd.2008.185 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kale J, Osterlund EJ & Andrews DW BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25, 65–80, doi: 10.1038/cdd.2017.186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubli DA, Ycaza JE & Gustafsson AB Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 405, 407–415, doi: 10.1042/BJ20070319 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regula KM, Ens K & Kirshenbaum LA Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91, 226–231, doi: 10.1161/01.res.0000029232.42227.16 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Rikka S et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 18, 721–731, doi: 10.1038/cdd.2010.146 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellot G et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29, 2570–2581, doi: 10.1128/MCB.00166-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang T et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J Biol Chem 291, 21616–21629, doi: 10.1074/jbc.M116.733410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanna RA et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287, 19094–19104, doi: 10.1074/jbc.M111.322933 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem 288, 1099–1113, doi: 10.1074/jbc.M112.399345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Sullivan TE, Johnson LR, Kang HH & Sun JC BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 43, 331–342, doi: 10.1016/j.immuni.2015.07.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glick D et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol 32, 2570–2584, doi: 10.1128/MCB.00167-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li R et al. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol 18, 229–243, doi: 10.1016/j.redox.2018.07.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang C et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis 10, 677, doi: 10.1038/s41419-019-1899-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho J, Hur JH & Walker DW The role of mitochondria in Drosophila aging. Exp Gerontol 46, 331–334, doi: 10.1016/j.exger.2010.08.010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osterwalder T, Yoon KS, White BH & Keshishian H A conditional tissue-specific transgene expression system using inducible GAL4. P Natl Acad Sci USA 98, 12596–12601, doi:DOI 10.1073/pnas.221303298 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poirier L, Shane A, Zheng J & Seroude L Characterization of the Drosophila gene-switch system in aging studies: a cautionary tale. Aging Cell 7, 758–770, doi: 10.1111/j.1474-9726.2008.00421.x (2008). [DOI] [PubMed] [Google Scholar]
- 45.Walker DW & Benzer S Mitochondrial “swirls” induced by oxygen stress and in the Drosophila mutant hyperswirl. Proc Natl Acad Sci U S A 101, 10290–10295, doi: 10.1073/pnas.0403767101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brandt T et al. Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophila. Elife 6, doi: 10.7554/eLife.24662 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferguson M, Mockett RJ, Shen Y, Orr WC & Sohal RS Age-associated decline in mitochondrial respiration and electron transport in Drosophila melanogaster. Biochem J 390, 501–511, doi: 10.1042/BJ20042130 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klionsky DJ et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17, 1–382, doi: 10.1080/15548627.2020.1797280 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang JT, Kumsta C, Hellman AB, Adams LM & Hansen M Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. Elife 6, doi: 10.7554/eLife.18459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee TV, Kamber Kaya HE, Simin R, Baehrecke EH & Bergmann A The initiator caspase Dronc is subject of enhanced autophagy upon proteasome impairment in Drosophila. Cell Death Differ 23, 1555–1564, doi: 10.1038/cdd.2016.40 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JJ et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol 217, 1613–1622, doi: 10.1083/jcb.201801044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McWilliams TG et al. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214, 333–345, doi: 10.1083/jcb.201603039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakatogawa H, Suzuki K, Kamada Y & Ohsumi Y Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 10, 458–467, doi: 10.1038/nrm2708 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Shell BC et al. Measurement of solid food intake in Drosophila via consumption-excretion of a dye tracer. Sci Rep 8, 11536, doi: 10.1038/s41598-018-29813-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Partridge L, Gems D & Withers DJ Sex and death: what is the connection? Cell 120, 461–472, doi: 10.1016/j.cell.2005.01.026 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Miller HA, Dean ES, Pletcher SD & Leiser SF Cell non-autonomous regulation of health and longevity. Elife 9, doi: 10.7554/eLife.62659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weir HJ & Mair WB SnapShot: Neuronal Regulation of Aging. Cell 166, 784–784 e781, doi: 10.1016/j.cell.2016.07.022 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Twig G & Shirihai OS The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 14, 1939–1951, doi: 10.1089/ars.2010.3779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho HM et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol Cell 73, 364–376 e368, doi: 10.1016/j.molcel.2018.11.009 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Demontis F & Perrimon N FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 143, 813–825, doi: 10.1016/j.cell.2010.10.007 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schinaman JM, Rana A, Ja WW, Clark RI & Walker DW Rapamycin modulates tissue aging and lifespan independently of the gut microbiota in Drosophila. Sci Rep 9, 7824, doi: 10.1038/s41598-019-44106-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ulgherait M, Rana A, Rera M, Graniel J & Walker DW AMPK Modulates Tissue and Organismal Aging in a Non-Cell-Autonomous Manner. Cell Rep 8, 1767–1780, doi: 10.1016/j.celrep.2014.08.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rera M, Azizi MJ & Walker DW Organ-specific mediation of lifespan extension: more than a gut feeling? Ageing Res Rev 12, 436–444, doi: 10.1016/j.arr.2012.05.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jasper H Exploring the physiology and pathology of aging in the intestine of Drosophila melanogaster. Invertebr Reprod Dev 59, 51–58, doi: 10.1080/07924259.2014.963713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark RI et al. Distinct Shifts in Microbiota Composition during Drosophila Aging Impair Intestinal Function and Drive Mortality. Cell Rep 12, 1656–1667, doi: 10.1016/j.celrep.2015.08.004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rera M, Clark RI & Walker DW Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc Natl Acad Sci U S A 109, 21528–21533, doi: 10.1073/pnas.1215849110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H & Jasper H Gastrointestinal stem cells in health and disease: from flies to humans. Dis Model Mech 9, 487–499, doi: 10.1242/dmm.024232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Biteau B, Hochmuth CE & Jasper H JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell 3, 442–455, doi: 10.1016/j.stem.2008.07.024 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu DJ & Jasper H Epithelia: Understanding the Cell Biology of Intestinal Barrier Dysfunction. Curr Biol 27, R185–R187, doi: 10.1016/j.cub.2017.01.035 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Dambroise E et al. Two phases of aging separated by the Smurf transition as a public path to death. Sci Rep 6, 23523, doi: 10.1038/srep23523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kavanagh K et al. Microbial translocation and skeletal muscle in young and old vervet monkeys. Age (Dordr) 38, 58, doi: 10.1007/s11357-016-9924-z (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rera M et al. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab 14, 623–634, doi: 10.1016/j.cmet.2011.09.013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thevaranjan N et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 21, 455–466 e454, doi: 10.1016/j.chom.2017.03.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palikaras K, Lionaki E & Tavernarakis N Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20, 1013–1022, doi: 10.1038/s41556-018-0176-2 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Lou G et al. Mitophagy and Neuroprotection. Trends Mol Med 26, 8–20, doi: 10.1016/j.molmed.2019.07.002 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Zhang J & Ney PA Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 16, 939–946, doi: 10.1038/cdd.2009.16 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao A, Jiang J, Xie F & Chen L Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin Chim Acta 506, 72–83, doi: 10.1016/j.cca.2020.02.024 (2020). [DOI] [PubMed] [Google Scholar]
- 78.Ney PA Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta 1853, 2775–2783, doi: 10.1016/j.bbamcr.2015.02.022 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Jung J et al. Mitochondrial NIX Promotes Tumor Survival in the Hypoxic Niche of Glioblastoma. Cancer Res 79, 5218–5232, doi: 10.1158/0008-5472.CAN-19-0198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raji JI & Potter CJ The number of neurons in Drosophila and mosquito brains. PLoS One 16, e0250381, doi: 10.1371/journal.pone.0250381 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Herculano-Houzel S & Lent R Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. J Neurosci 25, 2518–2521, doi: 10.1523/JNEUROSCI.4526-04.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in the figures and text with representative images accompanying quantified results where applicable unless otherwise noted. Further information is available from the corresponding author upon reasonable request.