

Abstract
Objective
We performed a population‐based study on inclusion body myositis with the primary aims to define the prevalence, survival rate, and incidence, and to investigate the symptom profiles associated with disease duration and sex over a 33‐year period.
Methods
Patients diagnosed between 1985 and 2017 in Region Västra Götaland, Sweden, were identified according to the European Neuromuscular Centre diagnostic criteria from 2011.
Results
We identified 128 patients, 89 men and 39 women, with the strict clinicopathological definition of inclusion body myositis. The prevalence was 32 per million inhabitants, 19 per million women and 45 per million men, by December 31, 2017. Mean incidence was 2.5 per million inhabitants and year. Mean age at symptom onset was 64.4 years with quadriceps weakness being the most common presenting symptom followed by finger flexor weakness. Dysphagia was a common presenting symptom being more frequent in women (23%) than men (10%) and was during the disease course reported in 74% of men and 84% of women. Seventy‐three patients were deceased, with a mean survival of 14 years from symptom onset. Survival rates from both diagnosis date and symptom onset were decreased compared to the matched population.
Twenty‐one percent of the patients had an additional autoimmune disease. A cross‐sectional analysis of autoantibodies in 50 patients and 28 matched controls showed autoantibodies to cytosolic 5′‐nucleotidase 1A in 40% of the patients and 3.6% of controls.
Interpretation
Inclusion body myositis is an autoimmune disease with decreased survival rate and with marked sex differences in both prevalence and clinical manifestations. ANN NEUROL 2022;92:201–212
Inclusion body myositis (IBM), a slowly progressive inflammatory myopathy, has been the subject of many studies since the introduction of the term in 1971. 1 The clinical pattern of late‐onset progressive weakness and atrophy of the long finger flexors and quadriceps muscles, often combined with swallowing difficulties, and muscle biopsy showing inflammation, rimmed vacuoles, protein aggregates, and mitochondrial alterations are characteristic for the diagnosis (Fig 1). Due to the protractive course and lack of response to immunosuppressive treatment, 2 IBM has a major impact on the activity of daily living in a substantial group of elderly individuals.
FIGURE 1.
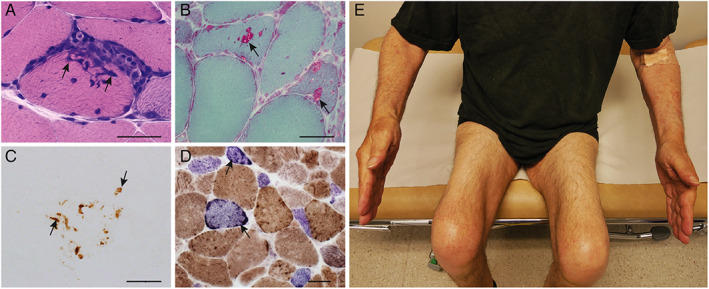
Inclusion body myositis. Typical muscle biopsy findings (A–D) and typical distribution of muscle atrophy (E). (A) Inflammatory cells in the endomysium surround and invade a muscle fiber (arrows; hematoxylin and eosin). (B) Muscle fibers with rimmed vacuoles (arrows; Gomori trichrome). (C) p62‐positive protein aggregates (arrows) in a muscle fiber. (D) Cytochrome c oxidase (COX)‐deficient fibers (arrows) that appear blue in combined staining for COX and succinate dehydrogenase. (E) Ten years after onset of symptoms, this patient with IBM shows prominent quadriceps muscle atrophy and mild atrophy of the long finger flexor muscles mainly in the left arm. Scale bars correspond to 30 μm. IBM = inclusion body myositis.
Early diagnostic criteria emphasized the histopathological changes in IBM muscle, 3 but more recent criteria include typical clinical signs and symptoms. 4 , 5 , 6 , 7 Although many studies on IBM patient cohorts include epidemiological aspects, there are only approximately a dozen population‐based investigations on IBM prevalence, and large differences ranging from 0.68 patients with IBM per million inhabitants in Turkey to 50.5 per million inhabitants in South Australia, both studies using local criteria, 8 , 9 and 182 per million inhabitants over 50 years old in Minnesota, in the United States, using the 2011 European Neuromuscular Centre (ENMC) criteria, have been described. 4 , 10 The effect of IBM on survival is unclear and although earlier studies have not shown any impact of IBM on life expectancy, 11 , 12 , 13 three more recent studies report a reduced survival rate. 10 , 14 , 15
An autoimmune pathogenesis of IBM is indicated by the auto‐invasion of inflammatory cytotoxic CD8+ T‐cells in IBM muscle, 16 the strong association with the human leukocyte antigen (HLA)‐DR3 haplotype 17 and a high prevalence of the autoantibody anti‐cytosolic 5′‐nucleotidase 1A (cN1A). 18 , 19 , 20 , 21 Other myositis‐associated antibodies have not been shown to be increased. 22 An alternative hypothesis implying that IBM is a primary degenerative disorder is mainly based on the unresponsiveness to immunomodulating treatment and presence of protein aggregates in muscle fibers. 23 Signs of mitochondrial dysfunction with cytochrome c oxidase (COX) deficient fibers have been attributed to both hypotheses. 24
We aimed to describe the epidemiological data of IBM, including survival, prevalence, and incidence in a retrospective study covering 33 years, in a well‐defined region where most of the patients with IBM probably have been identified. We also aimed to describe clinical parameters and perform a cross‐sectional analysis of immunological factors.
Materials and Methods
Workflow and Population Data
This study was performed in Region Västra Götaland (VGR), which is located in western Sweden and had 1,709,814 inhabitants by December 31, 2017 (Fig 2A). 25 The regional referral centers for neuromuscular disorders and muscle biopsy diagnostics are located at the Department of Neurology and Department of Pathology, both at Sahlgrenska University Hospital, Gothenburg. The Swedish public health system, which provides cost‐reduced health care for all inhabitants, the centralized care of patients with IBM, and the availability to detailed population statistics, makes VGR a suitable region for population‐based studies on neuromuscular disorders.
FIGURE 2.

Map of Region Västra Götaland and flow chart of the study. (A) Region Västra Götaland (VGR; grey) and Gothenburg (dot). Adapted from Region Västra Götaland, VGR Mediebank.25 (B) Flow chart describing the study (men:women). IBM = inclusion body myositis.
The aim was to identify all patients with IBM in VGR diagnosed from January 1, 1985, to December 31, 2017, with the date for diagnosis defined as the date for the first biopsy needed to fulfill all inclusion criteria. Patients were identified from the muscle pathology registry at the Department of Pathology and the outpatient clinic registry at the Department of Neurology, both at Sahlgrenska University Hospital. Muscle biopsy records and muscle biopsy specimens from all patients with suspected IBM were re‐examined. Complimentary examinations on the oldest archival biopsy specimens were performed when needed to establish the diagnosis and the date for clinicopathological diagnosis. This was repeated for all subsequent muscle biopsies from the same individual until the ENMC criteria for clinicopathological IBM were met or all biopsies were re‐evaluated.
Medical records from VGR up to June 30, 2018, were reviewed for all deceased patients and for all living patients consenting to participate, aiming to include at least six months of progress after diagnosis for all patients. Laboratory data were retrieved for the first visit after symptom onset if not stated otherwise. An additional blood sample was collected for analysis of myositis‐associated autoantibodies and complementary analysis of antinuclear antibody (ANA) screen and anti‐neutrophil cytoplasmic antibody (ANCA) if needed. Assessment of muscle weakness distribution for evaluation of diagnostic criteria was based on medical records of clinical examination and, when available, myometer values, both performed by a few experienced investigators.
Comorbidities were evaluated by manual review of medical records, including diagnosis codes. Malignancies were studied using the definition in NORDCAN, including basal‐cell carcinoma. 26 , 27 For autoimmune diseases, the definition of 3 possible or probable autoimmune diseases from Eaton et al 28 were used, including among other diseases Sjögren's syndrome, hypothyroidism, coeliac disease, polymyalgia rheumatica, type 1 diabetes, and seropositive rheumatoid arthritis, but excluding myositis. Positive autoimmune serology alone was not used for the evaluation of autoimmune diseases.
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the regional ethical review board of Gothenburg, approval number 633‐15 and T215‐16. Written informed consent was obtained from all living participants.
Inclusion and Exclusion Criteria
Inclusion criteria were (1) fulfillment of the ENMC IBM Research Diagnostic Criteria 2011 for clinicopathologicall IBM 4 ; (2) diagnostic muscle biopsy performed between January 1, 1985, and December 31, 2017; and (3) living in VGR any time during the studied time period.
Exclusion criteria were (1) high probability of another or additional muscle disease despite fulfilling the ENMC criteria (for example, cases of genetically verified Welander distal myopathy); and (2) initial analysis not fulfilling inclusion criteria and lack of sufficient muscle biopsy material for re‐analysis.
A flow chart describing the identification of the study group is presented in Figure 2B.
Muscle Biopsies and Morphology
Muscle biopsies were performed as an open biopsy from the vastus lateralis, deltoid, biceps, or tibialis anterior muscles. The muscle specimens were fresh frozen and stored at −80 degrees Celcius for analysis of morphology, enzyme histochemistry, and immunohistochemistry in accordance with standard protocols. 29 A separate specimen was processed for electron microscopy. New sections were prepared from frozen archival muscle tissue when needed. Rimmed vacuoles were assessed by Gomori trichrome and hematoxylin and eosin staining. Congo red, electron microscopy, and p62 immunohistochemistry were used to assess typical filamentous protein accumulation including amyloid.
Serum Samples
Blood samples were incubated at room temperature for 1 hour and centrifuged at 2,000 times gravity (xg) for 10 minutes within 4 hours. Serum was aliquoted and frozen at −20 degrees Celsius for 24 hours followed by transfer to −80 degrees Celsius for further storage.
Detection of Myositis‐Specific and Myositis‐Associated Antibodies
Serum samples from a group of 50 patients with IBM and a control group of blood donors from VGR matched for age and sex were investigated.
The commercial myositis line blot assay EUROLINE Autoimmune Inflammatory Myopathies 16 Ag (IgG) Profile (Euroimmun AG, Lübeck, Germany) was used, consisting of a membrane strip coated with autoantigens nucleosome remodeling deacetylase α (Mi‐2α), nucleosome remodeling deacetylase β (Mi‐2β), transcriptional intermediary factor 1 gamma (TIF1γ), melanoma differentiation‐associated gene 5 (MDA5), nuclear matrix protein‐2 (NXP2), small ubiquitin‐like modifier activating enzyme subunit 1 (SAE1), DNA‐dependent protein kinase regulatory subunit (Ku), exosome protein complex 100 (PM‐Scl‐100), exosome protein complex 75 (PM‐Scl‐75), histidyl‐tRNA synthetase (Jo‐1), signal recognition particle (SRP), threonyl‐tRNA synthetase (PL‐7), alanyl‐tRNA synthetase (PL‐12), glycyl‐tRNA synthetase (EJ), isoleucyl‐tRNA synthetase (OJ), and Sjögren's syndrome‐related antigen A (SSA/Ro‐52). The incubated strips were automatically photographed, and the band intensity was evaluated by the EUROLine‐Scan program. Results that were borderline according to the manufacturer's system were considered negative.
Antibodies against cytosolic 5′‐nucleotidase 1A (cN1A) were measured using a commercially available enzyme‐linked immunosorbent assay (ELISA) kit (Euroimmun AG, Lübeck, Germany). Antibodies of IgG subclass against 3‐Hydroxy‐3‐Methylglutaryl Coenzyme A (HMGCR) were measured using QUANTA Lite HMGCR ELISA assays (Inova Diagnostics, Inc., San Diego, CA, USA) according to the manufacturer's instructions. The ratio ≥1 was considered positive for anti‐cN1A and the quantitative values ≥20 units were considered positive for anti‐HMGCR.
Statistical Analysis
Life expectancy in VGR, the population size in VGR, and the expected survival at 65 years of age in Sweden were gathered from Statistics Sweden. 30
For survival calculations, mortality data for the VGR population from Statistics Sweden were used. 30 For each year of survival, each patient with IBM was matched to the corresponding population in VGR based on calendar year, age, and sex. Observed, expected, and relative survival was calculated, with censoring for patients relocating from VGR during the follow‐up time. Log–log transformation was used for calculation of 95% confidence intervals for observed and relative survival. These calculations were made in SAS for Windows version 9.14 (SAS Institute, Cary, NC, USA).
Cancer prevalence data for the population in the Western Region of Sweden was gathered from Nordcan Registry in February 2020. 26 , 27
Descriptive statistics for clinical data, laboratory analyses, and autoantibodies were calculated using Microsoft Excel for Mac 2011 (Microsoft Corporation, Redmond, WA, USA).
Results
The registry search identified 184 patients with possible IBM living in VGR during the studied period. A total of 128 patients fulfilled the clinicopathological inclusion criteria, and an additional 14 patients fulfilled the criteria of clinically defined or probable IBM. Nine patients with symptom onset before 45 years of age, but otherwise fulfilling the clinicopathological criteria, were excluded (Fig 2B). 4
Of the 128 patients with clinicopathological IBM, three patients declined to participate in examination of clinical records and additional blood sampling. The mean follow‐up time for patients alive on June 30, 2018, was 7.7 years (median = 6.3, range = 0.6–24 years) from the date of diagnosis to June 30, 2018, and for all patients 8.0 years (median = 8.2, range = 0.2–24 years).
Epidemiology
On December 31, 2017, the prevalence of clinicopathological IBM in VGR was 31.9 patients per million inhabitants and the total prevalence including clinically defined and probable IBM was 33.7 patients per million inhabitants (Table 1). The mean incidence of clinicopathological IBM during the studied time period was 2.5 patients per million inhabitants and year.
TABLE 1.
Prevalence of Clinicopathological IBM in VGR (Per Million Inhabitants)
| All inhabitants | Women | Men | ||||
|---|---|---|---|---|---|---|
| Year | All ages | ≥50 | All ages | ≥50 | All ages | ≥50 |
| 1997 | 16.2 | 8.0 | 24.5 | |||
| 2007 | 33.6 | 92.4 | 18.0 | 47.2 | 49.3 | 142.5 |
| 2017 | 31.9 | 85.2 | 19.0 | 48.9 | 44.7 | 124.1 |
IBM = inclusion body myositis; VGR = Region Västra Götaland.
Mean survival from presenting symptom among the 73 deceased patients with clinicopathological IBM was 13.8 years. Mean expected remaining survival in VGR for persons 65 years old was 17 years for men and 20 for women for years 1998 to 2017 (data not available before 1998), 30 resulting in an expected age of death at 82 years for men and 85 for women in the general population.
For survival analysis, the number of matched controls for each patient varied from 85 to 11,866 controls for men and 348 to 11,473 controls for women depending on age and year. Observed survival of patients with IBM from both diagnosis date and symptom onset showed a decrease compared to the expected survival. Consequently, the relative survival decreased. A persisting decreased cumulative relative survival was seen from year 3 after diagnosis for women and year 13 after diagnosis for men (Fig 3A, B). From symptom onset, a persisting decreased cumulative relative survival was seen from year 12 for women and year 20 for men.
FIGURE 3.

Cumulative survival, age distribution, and presenting symptoms. (A, B) Sex specific life table estimates of cumulative observed and expected patient survival after IBM diagnosis using period analysis. Effective number at risk shown above the x‐axis. Vertical lines represent 95% confidence intervals. (C) Age at symptom onset and diagnosis. (D) Presenting symptom.IBM = inclusion body myositis.
Presenting Symptom
The patient characteristics are summarized in Table 2.
TABLE 2.
Patient Characteristics
| All patients (n = 128) | Men (n = 89) | Women (n = 39) | ||
|---|---|---|---|---|
| General data (mean; median (range)) | ||||
| Years of age at symptom onset a | 64.4; 64 (46–86) | 64.8; 65 (46–84) | 63.3; 63 (46–86) | |
| Years of age at diagnosis | 69.5; 70 (49–91) | 69.5; 72 (49–90) | 69.4; 69 (58–91) | |
| Diagnostic delay (years from symptom onset to diagnosis) a | 5.3; 4 (0–29) | 4.8; 4 (0–16) | 6.3; 5 (0–29) | |
| Deceased patients December 31, 2017 (n; %) | 73; 57 | 50; 56 | 23; 59 | |
| Years of age at death | 79.7; 80 (58–95) | 79.6; 80 (58–95) | 79.9; 81 (63–91) | |
| Years survival from symptom onset b | 13.8; 14 (2.3–40) | 13.5; 14 (2.8–29) | 14.3; 13 (2.3–40) | |
| Presenting symptom (n; %) (see Fig 3) | Quadriceps weakness | 74; 58 | 57; 64 | 17; 44 |
| Finger flexor weakness | 20; 16 | 13; 15 | 7; 18 | |
| Dysphagia | 18; 14 | 9; 10 | 9; 23 | |
| Other | 12; 9 | 9; 10 | 3; 8 | |
| n/a | 4; 3 | 1; 1 | 3; 8 | |
| All (n = 125) c | Men (n = 86) c | Women (n = 39) | ||
| Dysphagia (n; %) | ||||
| Dysphagia any time | 96; 77 | 64; 74 | 32; 84 | |
| Invasive treatment of dysphagia d | 31; 25 | 15; 17 | 16; 42 | |
| >1 invasive treatment of dysphagia d | 15; 12 | 8; 9 | 7; 18 | |
| Gastrostomy | 17; 14 | 8; 9 | 9; 24 | |
| Years from symptom onset to first invasive treatment of dysphagia n = 28 a , d (mean; median (range)) | 9.5; 8 (0–34) | |||
| Respiratory aids and assistive devices | ||||
| Ventilation assistance (n; %) | 10; 8 | |||
| Years from symptom onset to first reported use of ventilation assistance (mean; median (range)) | 11; 13 (3–19) | |||
| Wheelchair use (n; %) | 76; 61 | 54; 63 | 22; 56 | |
| Years from symptom onset to first reported use of wheelchair (mean; median (range)) a | 11; 9 (2–36) | 10; 9 (2–24) | 12; 9 (4–36) | |
| Comorbidities (n; %) | ||||
| Malignancy | 39; 31 | 27; 31 | 12; 31 | |
| Autoimmune diseases 28 | 27; 21 | 13; 15 | 14; 36 | |
| Immunomodulating treatment | ||||
| Tried any length of time | 82% | 81% | 82% | |
| Corticosteroid treatment any time after symptom onset | 50% | |||
One man and one woman missing.
One woman missing.
For three men only data for presenting symptom, date and age for diagnosis, time from symptom onset to diagnosis, years survived, and age at death were included.
One woman excluded due to cancer surgery in head and neck region.
IBM = inclusion body myositis; n/a = not available; VGR = Region Västra Götaland.
Mean age at symptoms onset was 66.4 years (Fig 3C). The most common presenting symptom was quadriceps weakness, and swallowing difficulties was a more common presenting symptom among women than men (Fig 3D). The mean age at symptom onset for patients grouped by presenting symptom was 64.8 years for quadriceps weakness, 62.2 years for finger flexor weakness, and 67.7 years for swallowing difficulties, respectively.
For the deceased patients, data regarding symptom onset were available for 72 individuals. Quadriceps weakness was the presenting symptom in 43 of these patients, with a mean survival of 13.0 years from symptom onset. The 9 patients reporting finger flexor weakness as the presenting symptom had the longest mean survival of 15.1 years. Dysphagia as the presenting symptom was associated with a mean survival of 14.7 years in 12 patients, but when the outlier value 40.1 was omitted, the mean survival from symptom onset was 12.4 years. Wheelchair use was reported in 77% of the 73 deceased patients.
Two patients had received an amyotrophic lateral sclerosis diagnosis and eight patients had a polymyositis diagnosis before diagnosed with IBM. Twenty patients reported myalgia and patients reported arthralgia.
Dysphagia and Respiratory Aids
Over time, a majority of the patients developed swallowing difficulties and 25% of all patients needed invasive treatment of their swallowing difficulties, such as myotomy, botulinum toxin injection, cricopharyngeal dilatation, percutaneous endoscopic gastrostomy, and extirpation of food from the esophagus (see Table 2). Seven patients underwent more than five invasive treatments.
Ventilation assistance, including continuous positive airway pressure (CPAP) and BiLevel was reported in 10 patients (8%; see Table 2). Furthermore, one patient had a cough machine. No patient had more than one ventilation assistance device. Of the six patients that were prescribed BiLevel, three had never or very rarely used the device. Within one year after the initiation of BiLevel treatment, four of the six patients were deceased. CPAP was used regularly by three patients and one patient had a home ventilator. Four patients had sleep apnea as the cause for using ventilation assistance. A total of five of the 10 patients with reported ventilation assistance were deceased, corresponding to 7% of all deceased patients.
Comorbidities and Immunomodulating Treatment
For patients followed until 75 years of age or until death before 75 years of age (n = 97), a total of 22% had one or more malignancies, compared to the risk of malignancy before 75 years of age in the Western Region, Sweden, during the years 2012 to 2016 of 30.6% for men and 28.3% for women. 26 , 27
Among the patients with IBM, the prevalence of autoimmune diseases was 21% using the definition from Eaton et al 28 including 30 probable or possible autoimmune conditions after excluding myositis (see Table 2). Four patients with IBM had more than one autoimmune disease. The most common autoimmune disease was hypothyroidism (10 patients) followed by celiac disease (six patients) and psoriasis (four patients). Systemic lupus erythematosus was reported in one patient, and Sjögren's syndrome was reported in three patients. The other reported diseases were type 1 diabetes, autoimmune haemolytic anemia, ulcerative cholitis, polymyalgia rheumatica, and Graves’ disease. Nine women and six men with autoimmune disease were deceased, and mean age at death was 78.3 and 83.1 years, respectively. Twelve patients had symptoms or signs that were not included in this definition of autoimmune disease but are commonly regarded as autoimmune, including three patients with hypogammaglobulinemia, three patients who reported sicca symptoms, and six patients who reported Raynaud syndrome.
The most frequently utilized immunomodulating treatment (steroids not included) was methotrexate (30%) followed by mycophenolate mofetil (27%), and 56% had tried an immunomodulating treatment for at least 12 months. Thirteen patients had tried at least two different treatments for more than 12 months each. No clear difference was seen between treatment frequencies in men and women.
Autoantibodies in Patients and Controls
Fifty living patients participated in a cross‐sectional analysis of immunological factors with blood samples gathered November 2, 2016 to November 16, 2018 (Table 3). A panel of myositis‐specific and myositis‐associated autoantibodies was analyzed in the patients with IBM with a mean symptom duration of 12.1 years (range = 2.8–27 years), and in the 28 blood donor controls from VGR that matched for age and sex.
TABLE 3.
Autoantibodies in Patients and Controls
| Patients (n = 50) | Controls a (n = 28) | |
|---|---|---|
| General data | ||
| Men (n; %) | 34; 68 | 18; 64 |
| Women (n; %) | 16; 32 | 10; 36 |
| Years of age (mean (range)) | 74 (61–89) | 70 (60–82) |
| Immunomodulating treatment at time for test b (n; %) | 14; 28 | |
| Autoantibodies positive in patients or controls (n; %) | ||
| anti‐cN1A | 20; 40 | 1; 36 |
| anti‐HMGCR | 2; 4 | 0 |
| anti‐SRP | 1; 2 | 0 |
| anti‐TIF1γ | 1; 2 | 0 |
| anti‐PL‐12 | 0 | 1; 4 |
| anti‐Ku | 2; 4 | 0 |
| anti‐PM‐SCL‐75 | 1; 2 | 3; 11 |
| anti‐PM‐SCL‐100 | 0 | 1; 4 |
| anti‐SSA/Ro‐52 | 8; 16 | 0 |
Autoantibodies anti‐OJ, anti‐EJ, anti‐PL‐7, anti‐Jo‐1, anti‐SAE1, anti‐NXP2, anti‐MDA5, anti‐Mi‐2α, anti‐Mi‐2β were not positive in any patient or control.
Controls for autoantibodies recruited among blood donors.
Steroids not included.
The most frequent autoantibody was anti‐cN1A (see Table 3). Among the 14 patients on immunomodulating treatment, anti‐cN1A was positive in 43%. Of the patients with positive anti‐cN1A, five were women and 15 men, corresponding to 31% and 44%, respectively, of the tested individuals. Mean symptom duration for the patients with positive anti‐cN1A was 12.3 years, and for patients with negative anti‐cN1A it was 12.0 years. Mean age at symptom onset was 63.0 and 61.9, respectively. The most common presenting symptom in patients both with and without anti‐cN1A antibodies was quadriceps weakness. Dysphagia occurred during the disease course in 80% for both groups. Other probable or possible autoimmune diseases occurred in 20% of patients with positive anti‐cN1A and 27% of patients with negative anti‐cN1A. 28 Of patients with anti‐cN1A antibodies, 35% were wheelchair users, compared to 40% of patients lacking antibodies.
Two patients were positive for anti‐HMGCR, both treated with statins after IBM diagnosis (22% of all patients had been using statins before and/or after diagnosis). One of the studied patients, that was earlier successfully treated for two malignancies, was positive for the cancer‐associated autoantibody TIF1γ. 31 Renewed clinical examination showed no indication of relapse or new malignancy (see Table 3).
Laboratory Features
The mean first documented creatine kinase (CK) was 2.1 times the upper limit of normal (ULN; range = 0.12–7.4) and the mean maximal CK 2.8 times the ULN (range = 0.36–7.8). The mean first troponin T (TnT) was 4.3 times the ULN (range = 0.17–31, values from admissions for probable myocardial infarction not included). The mean first documented creatinine level was 72 μmol/L (range = 28–136, reference interval 60–110 μmol/L), and the mean lowest creatinine level was 50 μmol/L (range = 10–110). Erythrocyte sedimentation rate (ESR) and C‐reactive protein (CRP) were normal or slightly elevated in the majority of patients.
ANA and ANCA were analyzed in 125 patients, 86 men and 39 women. ANA was positive in 18% of these patients, whereas ANCA was not positive in any of them.
Discussion
In this study, we analyzed epidemiological and clinical aspects of IBM in Region Västra Götaland, Sweden using a population‐based approach. We identified 128 patients with clinicopathological IBM and 14 patients with clinically defined or probable IBM diagnosed between 1985 and 2017. 4 Earlier population‐based prevalence studies published in 2000 or later including 20 or more patients are described in Table 4. 9 , 10 , 11 , 32 , 33 , 34 , 35
TABLE 4.
Prevalence of IBM Per Million Inhabitants in Population‐Based Studies Published 2000 or Later Including ≥20 Patients
| Prevalence | Years of age (mean) | |||||||
|---|---|---|---|---|---|---|---|---|
| First author | n (M:W) | Country and area | Diagnostic criteria | All ages | ≥50 yr | Year | Symptom onset | Diagnosis |
| Badrising 11 | 76 (50:26) | Netherlands | ENMC 19975 | 4.9 | 16 | 1999 |
M: 59 W: 60 |
67‐68 a |
| Felice 32 | 35 (23:12) | Connecticut | Griggs 19953 | 10.7 | 28.9 b | 1992–2002 | 64.3 | 70 |
| Needham 33 | 31 (19:12) | Western Australia | Needham and Mastaglia 6 | 14.9 | 51.3 | 2007 |
M: 60 W: 60.6 |
‐ |
| Tan 9 | 126 (60:66) | South Australia | Local criteria c | 50.5 | 139 | 2009 | ‐ | 67.5 |
| Dobloug 34 | 100 (60:40) | Southeast Norway | ENMC 20114 and 19975 | 35 | ‐ | 2012 |
M: 61 W: 61 |
M: 67 W: 67 |
| Lefter 35 | 149 | Republic of Ireland | Hilton‐Jones 7 | 32.5 a | 117 | 2013 | ‐ | ‐ |
| Shelly 10 | 21 (11:10)e | Olmsted County, Minnesota | ENMC 2011 4 | ‐ | 182 | 2010 | 67 | 68 |
| Present study |
128 (89:39) d 142 (94:48) e |
VGR, Sweden | ENMC 20114 |
31.9 d 33.7 e |
85.2 d 94.7 e |
2017 |
M: 65 W: 63 |
M: 70 W: 69 |
Years of age at symptom onset and diagnosis is described to facilitate comparison between studies.
Calculated from values in the manuscript.
≥45 years.
Local criteria: Rimmed vacuoles, inflammatory infiltrate of CD45+ lymphocytes and CD68+ macrophages in interstitial space, MHC1 sarcolemmal positivity (strong), EM findings: tubulofilamentous inclusions, TDP 43, Tau, amyloid immunoabnormalities (not seen in all).
Clinicopathological IBM.
Clinicopathological, clinically defined and probable IBM.
ENMC = European Neuromuscular Centre; M = men; W = women.
We describe a high prevalence of IBM in VGR, especially in men with a factor 2.5 times compared to women among inhabitants 50 years of age and older. The prevalence in VGR is similar to the described prevalence in the geographically and ethnically close region of southeastern Norway, 34 but lower than in South Australia, the Republic of Ireland, and Olmsted County, Minnesota, United States, among inhabitants ≥50 years. 9 , 10 , 35 It is difficult to assess if the differences in prevalence reflect a true difference between regions. The diagnostic criteria used in different studies are not identical. In the recent study from Olmsted County, Minnesota, United States (approximately 160,000 inhabitants), only 10 of the 20 included patients had clinicopathological IBM compared with 128 patients in our study, which may partly explain differences. Our results indicate an 100% increase in IBM prevalence from 1997 to 2007. The prevalence has then remained stable. We believe the increased prevalence is due to increased awareness and better diagnostic methods, rather than a true increase in prevalence. Even though the knowledge of IBM is high in VGR and the prevalence stable, our results might be an underestimation due to, for example, comorbidities masking symptoms of IBM, inattention of both patients and doctors of late‐onset weakness, and a reluctancy to perform extensive examinations, including muscle biopsy, in elderly patients.
The survival rate in IBM has been debated and several earlier studies have not been able to demonstrate any effect of IBM on survival. 11 , 12 , 13 More recently, Dobloug et al 14 showed an increased mortality rate in 100 patients with IBM compared to 1,500 matched controls in Norway, and Shelly et al 10 showed a shorter life expectancy in 17 patients with IBM compared to 170 matched controls in Olmsted County, Minnesota, United States. Naddaf et al 15 showed a lower survival in 50 patients with IBM, five of whom were included in the study by Shelly et al, 10 compared to 300 controls in 27 counties in Minnesota and Wisconsin, United States. These studies analyzed survival from diagnosis date. In our study, the so far largest population‐based study of survival, including for each of the 128 patients 85 to 11,866 matched controls, we show a decreased cumulative survival in patients with IBM, both from diagnosis date and symptom onset. Analysis of survival from diagnosis date avoids overestimation of survival among patients but with a long follow‐up time and large groups of patients and controls, we were able to show a decreased cumulative survival also from onset of symptoms contrasting earlier studies. 11 , 12 , 13 The long follow‐up time and large group of patients also enabled separate analysis of survival for men and women and we could show that survival was decreased already from year three after the diagnosis date for women.
In line with earlier studies, the mean age at death was not apparently shorter for patients with autoimmune disease. 15 Due to the small number of patients in each subgroup, further subgroup analysis of comorbidities was not performed. A weakness of our study is the retrospective design, but the frequency of patients excluded due to lack of material or data was low. Cause of death was not analyzed due to insufficient validated data.
Swallowing difficulties were the presenting symptom in 14% of the patients in this study, but 77% of the patients reported dysphagia during the disease course and invasive treatment was common. Patients with IBM and swallowing difficulties as the presenting symptom had the shortest mean survival when an outlier value was excluded. Earlier studies report dysphagia in 40% to 80% of patients with IBM, 12 , 13 , 36 , 37 , 38 and swallowing difficulties were significantly more common as the presenting symptom in IBM than in other inflammatory myopathies in 62 patients with dysphagia associated with inflammatory myopathy. 39 A tendency of faster decrease in muscle strength in patients with IBM with onset of symptoms in bulbar muscles has also been described. 40
Our results suggest that women are affected by dysphagia earlier in the disease course and more frequently than men, whereas quadriceps muscle weakness is a more common early symptom among men (see Table 2, Figure 3D).
Decreased lung vital capacity has been reported in IBM, 32 and disorders in the respiratory system have been reported to be an over‐represented cause of death in patients with IBM. 13 The use of ventilatory support may have a large impact on quality of life for the individual patient but has not been studied in population‐based IBM cohorts. Four of the six patients in VGR that initiated BiLevel treatment were deceased within a year, suggesting that the need for ventilation assistance is a late and severe symptom. Patients with IBM visiting the regional referral center for neuromuscular disorders at the Department of Neurology at Sahlgrenska University Hospital are routinely screened for hypoventilation, but many patients in our cohort have their follow‐up at their local hospital or primary care unit and the frequency of hypoventilation might be underestimated.
Autoimmune diseases have been described in 13% of 99 patients with IBM in Maryland, in 16% of patients with IBM in Minnesota and Wisconsin, both in the United States, 15 , 41 and in 24% of 100 patients with IBM in Norway. 34 In the present study, 21% of patients with IBM in Region Västra Götaland, Sweden, had one or more autoimmune diseases, compared to a lifetime prevalence of 5.3% in the general population in Denmark described by Eaton et al 28 using the same definition with the exception of myositis. The Danish population can be regarded as similar to the population in VGR. Eaton et al used registered diagnostic codes for evaluation of prevalence, whereas this study used both diagnostic codes and manual review of medical records. A relative overestimation of autoimmune diseases in our study is possible, but a majority of the reported autoimmune diseases leads to medical treatment and are probably correctly registered. This suggests that autoimmune diseases are more common in patients with IBM than in the general population, supporting the concept that IBM is an autoimmune disease.
Another support for autoimmune pathogenesis is the high specificity of anti‐cN1A antibodies in IBM but the clinical correlation and the possible role in the pathogenesis is still unknown. 20 Anti‐cN1A was positive in 40% of our patients with IBM, which is in line with previous studies showing an anti‐cN1A frequency of 33% to 80% in IBM cohorts. 18 , 19 , 20 , 21 , 42 , 43 The frequency was higher in men than women. Although dysphagia was a more common presenting symptom in the group with anti‐cN1A, there were no clear differences in symptom duration or frequency of ongoing immunomodulating treatment in patients with and without expression of anti‐cN1A. Previous studies report more frequent dysphagia, reduced forced vital capacity, and more severe muscle weakness in patients with anti‐cN1A compared to patients without anti‐cN1A. 42 , 44 A European multicenter study, including 311 patients, indicated an increased mortality risk among patients with anti‐cN1A compared to patients without anti‐cN1A, 43 whereas an American study of 212 patients showed no such difference. 45 Therefore more data on anti‐cN1A and its clinical implications are warranted.
Immunomodulating treatment was tried in a total of 82% of the patients and 50% had tried corticosteroid treatment. A study on the true natural history in IBM can only include untreated patients, and this study can therefore not be regarded as such. However, whereas the individual patient might respond to treatment, no treatment has proven to be efficient at a group level in patients with IBM. The data presented in this study might, therefore, due to the large group of patients with IBM included, be considered as being close to the natural history.
Although some autoimmune diseases have low cancer prevalence, some of the inflammatory myopathies are known to have an association with malignancy. In this material, there was no apparent difference between the cancer risk for patients with IBM and the general population in Western Region, Sweden. This is similar to earlier studies in Minnesota, United States, and Norway. 10 , 14 On the contrary, an Australian study showed an increased risk for malignancy and a study from Minnesota and Wisconsin, United States, showed an increased risk for hematologic malignancies. 15 , 46 A more thorough study of cancer prevalence in a larger group of patients with IBM with matched population controls is needed to establish any possible increased risk of cancer of different types and influence on survival in patients with IBM.
An increased Troponin T level has been shown to remain over time by repeated sampling up to 17 months in patients with IBM. 47 Although cardiac muscle seems unaffected by IBM, TnT can leak from regenerating skeletal muscle or increase in patients with impaired renal function. 48 In this study, the mean first TnT was 4.3 time the ULN, and the mean lowest level of creatinine was 50 μmol/L (reference interval 60–110 μmol/L). However, as a surrogate measurement of muscle mass, creatinine is not fully reliable to assess renal function in this patient group. Further investigations would be of interest to explain the origin of the TnT increase in patients with IBM.
IBM is a disease with severe symptoms and reduced survival that is not uncommon in the elderly population. This emphasizes the importance of knowledge of the diagnosis and typical symptoms and signs to decrease the frequent very long delay from onset of symptoms to diagnosis. Increased awareness of treatable common complications, such as swallowing problems and respiratory dysfunction, as well as sex differences, is important and may improve the impaired survival rate. Our hope is that this, so far the largest population‐based study of epidemiology and clinical symptoms, will aid the health care providers in both the specialized and primary health care settings, and we believe that it also will be valuable when designing and evaluating future treatment studies.
Author Contributions
All authors contributed to the conception and design of the study, the acquisition and analysis of data, critically revised the manuscript for intellectual content and approved the final manuscript. U.L., R.P., and A.O. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Acknowledgments
This study was supported by the Swedish Research Council (no 2018‐02821), The Research Fund for Neuromuscular Disorders in West Sweden and the Swedish state under the agreement between the Swedish government and the county councils, the ALF‐agreement (ALFGBG‐716821, ALFGBG‐926621, ALFGBG‐872571, and ALFGBG‐965012).
The authors wish to thank Brith Leidvik, Jaana Lundgren, and Pernilla Olsson for methodological and technical assistance and Blanka Andersson for help in the recruitment of patients and technical assistance. The authors are grateful for statistical advice and calculations from Thomas Karlsson, Biostatistics, School of Public Health and Community Medicine, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
The authors wish to thank all the patients with IBM and their relatives in Region Västra Götaland, Sweden. Open access funding enabled and organized by Projekt DEAL.
Data availability
Data not provided in the article because of space limitations may be shared (anonymized) at the request of any qualified investigator for purposes of replicating procedures and results.
References
- 1. Yunis EJ, Samaha FJ. Inclusion body myositis. Lab Invest 1971;25:240–248. [PubMed] [Google Scholar]
- 2. Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol 2019;15:257–272. [DOI] [PubMed] [Google Scholar]
- 3. Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995;38:705–713. [DOI] [PubMed] [Google Scholar]
- 4. Rose MR, Amato AA, Engel A, et al. 188th ENMC international workshop: inclusion body myositis, 2‐4 December 2011, Naarden. The Netherlands Neuromuscul Disord 2013;23:1044–1055. [DOI] [PubMed] [Google Scholar]
- 5. Verschuuren J, Badrising UA, Wintzen AR, et al. Inclusion body myositis. In: diagnostic criteria for neuromuscular disorders. 2nd ed. London: Royal Society of Medicine Press, European Neuromuscular Centre, 1997. [Google Scholar]
- 6. Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol 2007;6:620–631. [DOI] [PubMed] [Google Scholar]
- 7. Hilton‐Jones D, Miller A, Parton M, et al. Inclusion body myositis: MRC Centre for neuromuscular diseases, IBM workshop, London, 13 June 2008. Neuromuscul Disord 2010;20:142–147. [DOI] [PubMed] [Google Scholar]
- 8. Oflazer PS, Deymeer F, Parman Y. Sporadic‐inclusion body myositis (s‐IBM) is not so prevalent in Istanbul/Turkey: a muscle biopsy based survey. Acta Myol 2011;30:34–36. [PMC free article] [PubMed] [Google Scholar]
- 9. Tan JA, Roberts‐Thomson PJ, Blumbergs P, et al. Incidence and prevalence of idiopathic inflammatory myopathies in South Australia: a 30‐year epidemiologic study of histology‐proven cases. Int J Rheum Dis 2013;16:331–338. [DOI] [PubMed] [Google Scholar]
- 10. Shelly S, Mielke MM, Mandrekar J, et al. Epidemiology and natural history of inclusion body myositis: a 40‐year population‐based study. Neurology 2021;96:e2653–e2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Badrising UA, Maat‐Schieman M, van Duinen SG, et al. Epidemiology of inclusion body myositis in The Netherlands: a nationwide study. Neurology 2000;55:1385–1387. [DOI] [PubMed] [Google Scholar]
- 12. Benveniste O, Guiguet M, Freebody J, et al. Long‐term observational study of sporadic inclusion body myositis. Brain 2011;134:3176–3184. [DOI] [PubMed] [Google Scholar]
- 13. Cox FM, Titulaer MJ, Sont JK, et al. A 12‐year follow‐up in sporadic inclusion body myositis: an end stage with major disabilities. Brain 2011;134:3167–3175. [DOI] [PubMed] [Google Scholar]
- 14. Dobloug GC, Garen T, Brunborg C, et al. Survival and cancer risk in an unselected and complete Norwegian idiopathic inflammatory myopathy cohort. Semin Arthritis Rheum 2015;45:301–308. [DOI] [PubMed] [Google Scholar]
- 15. Naddaf E, Shelly S, Mandrekar J, et al. Survival and associated comorbidities in inclusion body myositis. Rheumatology (Oxford) 2021;61:2016–2024. 10.1093/rheumatology/keab716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol 1984;16:193–208. [DOI] [PubMed] [Google Scholar]
- 17. Rojana‐udomsart A, James I, Castley A, et al. High‐resolution HLA‐DRB1 genotyping in an Australian inclusion body myositis (s‐IBM) cohort: an analysis of disease‐associated alleles and diplotypes. J Neuroimmunol 2012;250:77–82. [DOI] [PubMed] [Google Scholar]
- 18. Pluk H, van Hoeve BJ, van Dooren SH, et al. Autoantibodies to cytosolic 5′‐nucleotidase 1A in inclusion body myositis. Ann Neurol 2013;73:397–407. [DOI] [PubMed] [Google Scholar]
- 19. Larman HB, Salajegheh M, Nazareno R, et al. Cytosolic 5′‐nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 2013;73:408–418. [DOI] [PubMed] [Google Scholar]
- 20. Amlani A, Choi MY, Tarnopolsky M, et al. Anti‐NT5c1A autoantibodies as biomarkers in inclusion body myositis. Front Immunol 2019;10:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Muro Y, Nakanishi H, Katsuno M, et al. Prevalence of anti‐NT5C1A antibodies in Japanese patients with autoimmune rheumatic diseases in comparison with other patient cohorts. Clin Chim Acta 2017;472:1–4. [DOI] [PubMed] [Google Scholar]
- 22. Ronnelid J, Barbasso Helmers S, Storfors H, et al. Use of a commercial line blot assay as a screening test for autoantibodies in inflammatory myopathies. Autoimmun Rev 2009;9:58–61. [DOI] [PubMed] [Google Scholar]
- 23. Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion‐body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol 2012;71:680–693. [DOI] [PubMed] [Google Scholar]
- 24. Lindgren U, Roos S, Hedberg Oldfors C, et al. Mitochondrial pathology in inclusion body myositis. Neuromuscul Disord 2015;25:281–288. [DOI] [PubMed] [Google Scholar]
- 25. Region Västra Götaland , VGR Mediebank. Accessed 8 December 2020, https://mediebank.vgregion.se/fotoweb/archives/5099Kartor/Kartor/Geografisk%20grundkarta/Norden/Norden%20Baskarta/Ljusblå/
- 26. Danckert B, Ferlay J, Engholm G, et al. NORDCAN: cancer incidence, mortality, prevalence and survival in the Nordic countries. Association of the Nordic Cancer Registries. Danish Cancer Society, 2020. Accessed 24 February 2020, http://www.ancr.nu [Google Scholar]
- 27. Engholm G, Ferlay J, Christensen N, et al. NORDCAN–a Nordic tool for cancer information, planning, quality control and research. Acta Oncol 2010;49:725–736. [DOI] [PubMed] [Google Scholar]
- 28. Eaton WW, Rose NR, Kalaydjian A, et al. Epidemiology of autoimmune diseases in Denmark. J Autoimmun 2007;29:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dubowitz V, Sewry C, Oldfors A. Muscle biopsy ‐ a practical approach. 5th ed. Amsterdam: Elsevier, 2021. [Google Scholar]
- 30. Statistics Sweden . Accessed 24 February 2020, www.scb.se
- 31. Hida A, Yamashita T, Hosono Y, et al. Anti‐TIF1‐gamma antibody and cancer‐associated myositis: a clinicohistopathologic study. Neurology 2016;87:299–308. [DOI] [PubMed] [Google Scholar]
- 32. Felice KJ, North WA. Inclusion body myositis in Connecticut: observations in 35 patients during an 8‐year period. Medicine (Baltimore) 2001;80:320–327. [DOI] [PubMed] [Google Scholar]
- 33. Needham M, Corbett A, Day T, et al. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci 2008;15:1350–1353. [DOI] [PubMed] [Google Scholar]
- 34. Dobloug GC, Antal EA, Sveberg L, et al. High prevalence of inclusion body myositis in Norway; a population‐based clinical epidemiology study. Eur J Neurol 2015;22:672–e641. [DOI] [PubMed] [Google Scholar]
- 35. Lefter S, Hardiman O, Ryan AM. A population‐based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology 2017;88:304–313. [DOI] [PubMed] [Google Scholar]
- 36. Badrising UA, Maat‐Schieman ML, van Houwelingen JC, et al. Inclusion body myositis. Clinical features and clinical course of the disease in 64 patients. J Neurol 2005;252:1448–1454. [DOI] [PubMed] [Google Scholar]
- 37. Mohannak N, Pattison G, Hird K, Needham M. Dysphagia in patients with sporadic inclusion body myositis: management challenges. Int J Gen Med 2019;12:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oh TH, Brumfield KA, Hoskin TL, et al. Dysphagia in inclusion body myositis: clinical features, management, and clinical outcome. Am J Phys Med Rehabil 2008;87:883–889. [DOI] [PubMed] [Google Scholar]
- 39. Oh TH, Brumfield KA, Hoskin TL, et al. Dysphagia in inflammatory myopathy: clinical characteristics, treatment strategies, and outcome in 62 patients. Mayo Clin Proc 2007;82:441–447. [DOI] [PubMed] [Google Scholar]
- 40. Lindberg C, Oldfors A. Prognosis and prognostic factors in sporadic inclusion body myositis. Acta Neurol Scand 2012;125:353–358. [DOI] [PubMed] [Google Scholar]
- 41. Koffman BM, Rugiero M, Dalakas MC. Immune‐mediated conditions and antibodies associated with sporadic inclusion body myositis. Muscle Nerve 1998;21:115–117. [DOI] [PubMed] [Google Scholar]
- 42. Goyal NA, Cash TM, Alam U, et al. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J Neurol Neurosurg Psychiatry 2016;87:373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lilleker JB, Rietveld A, Pye SR, et al. Cytosolic 5′‐nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis 2017;76:862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lucchini M, Maggi L, Pegoraro E, et al. Anti‐cN1A antibodies are associated with more severe dysphagia in sporadic inclusion body myositis. Cell 2021;10:1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ikenaga C, Findlay AR, Goyal NA, et al. Clinical utility of anti‐cytosolic 5′‐nucleotidase 1A antibody in idiopathic inflammatory myopathies. Ann Clin Transl Neurol 2021;8:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy‐proven inflammatory myopathy. A population‐based cohort study. Ann Intern Med 2001;134:1087–1095. [DOI] [PubMed] [Google Scholar]
- 47. Lindberg C, Klintberg L, Oldfors A. Raised troponin T in inclusion body myositis is common and serum levels are persistent over time. Neuromuscul Disord 2006;16:495–497. [DOI] [PubMed] [Google Scholar]
- 48. Cox FM, Delgado V, Verschuuren JJ, et al. The heart in sporadic inclusion body myositis: a study in 51 patients. J Neurol 2010;257:447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data not provided in the article because of space limitations may be shared (anonymized) at the request of any qualified investigator for purposes of replicating procedures and results.