

Abstract
Previous studies investigating innate leukocyte recruitment into the brain after cerebral ischemia have shown conflicting results. Using distinct cell surface and intracellular markers, the current study evaluated the contributions of innate immune cells to the poststroke brain following 1‐h middle cerebral artery occlusion (tMCAO) or permanent MCAO (pMCAO), and assessed whether these cells ascribed to an inflammatory state. Moreover, we examined whether there is evidence for leukocyte infiltration into the contralateral (CL) hemisphere despite the absence of stroke infarct. We observed the recruitment of peripheral neutrophils, monocytes and macrophages into the hemisphere ipsilateral (IL) to the ischemic brain infarct at 24 and 96 h following both tMCAO and pMCAO. In addition, we found evidence of increased leukocyte recruitment to the CL hemisphere but to a lesser extent than the IL hemisphere after stroke. Robust production of intracellular cytokines in the innate immune cell types examined was most evident at 24 h after pMCAO. Specifically, brain‐associated neutrophils, monocytes and macrophages demonstrated stroke‐induced production of tumor necrosis factor‐α (TNF‐α) and interleukin (IL)‐1β, while only monocytes and macrophages exhibit a significant expression of arginase 1 (Arg1) after stroke. At 96 h after stroke, brain‐resident microglia demonstrated production of TNF‐α and IL‐1β following both tMCAO and pMCAO. At this later timepoint, neutrophils displayed TNF‐α production and brain‐associated macrophages exhibited elevation of IL‐1β and Arg1 after tMCAO. Further, pMCAO induced significant expression of Arg1 and IL‐1β in monocytes and macrophages at 96 h, respectively. These results revealed that brain‐associated innate immune cells display various stroke‐induced inflammatory states that are dependent on the experimental stroke setting.
Keywords: heterogeneity, ischemia, neuroinflammation, neutrophils, stroke
Using distinct cell surface and intracellular markers, our study evaluated the contributions of innate immune cells to the poststroke brain, and assessed whether these cells ascribed to an inflammatory state. Our findings revealed that brain‐infiltrating innate immune cells display various stroke‐induced inflammatory states that are dependent on the experimental stroke setting and timing.

INTRODUCTION
Ischemic stroke is a devastating cerebrovascular event that remains one of the three major contributors to death and disability worldwide. 1 Novel preclinical therapies that aim to, and successfully, limit the neuroinflammatory damage of ischemic stroke has centered on the use of inflammatory inhibitors or immune cell depletions to reduce infarct volumes and restore neurological functions. Despite promising results from these novel therapeutic avenues, 2 the discoveries made in preclinical stroke research have fallen short of translating to beneficial clinical outcomes. 3 , 4 , 5 , 6 This conundrum has thus prompted a re‐evaluation of our understanding into the neuroinflammatory processes after stroke.
One possible phenomenon to explain this lack of bench‐to‐bedside translatability is the emerging experimental evidence indicating the existence of brain‐associated neutrophils and monocytes with dynamic functional capacity, enabling them to elicit both pro‐ and anti‐inflammatory effects depending on the disease and stage of inflammation. 6 , 7 , 8 , 9 Adoption of the classical “M1” versus nonclassical “M2” designations from macrophages is now more widely being applied to neutrophils. Coined as being “N2” or “N2‐like,” these neutrophils are positive for the expression of the M2 marker, arginase 1 (Arg1), which has been associated with immunosuppression following cerebral ischemia. 10 However, the specific functional role for the potential anti‐inflammatory capacity of these cells is not defined. Furthermore, the existence of neutrophils in the brain parenchyma following cerebral ischemia has often been difficult to fully elucidate. There are conflicting results indicating that neutrophils adhere only to the luminal side of the cerebrovasculature and do not enter the brain parenchyma, 11 whereas others have shown novel and intimate interactions indicated by the engulfment of neutrophils by microglia following ischemic stroke. 12 Possible explanations behind the conflicted contribution of peripheral innate immune cells in the establishment of poststroke neuroinflammation include the diverse animal models utilized between studies which differ in animal strains, presence of reperfusion, the localization and size of the resultant infarct to the cortex and expansion into the striatum.
In addition, previous stroke research has often struggled to delineate specific contributions of infiltrating immune cells to the neuroinflammatory component after stroke. This could be because of either the paucity of novel markers to distinguish these populations or the use of nonspecific markers such as GR‐1, which identifies cell types encompassing neutrophils, monocytes and other granulocytes. As such, the distinct contribution of these peripheral granulocytes infiltrating the brain and their potential inflammatory capacity after stroke is unclear. In this study, we developed a strategy to delineate the specific contributions of innate leukocytes to poststroke neuroinflammation using two widely accepted models of stroke induced by the intraluminal occlusion of middle cerebral artery (MCA). We investigated whether the severity of stroke alters the recruitment of various innate immune cells to the ischemic and contralateral (CL) brain hemispheres following reperfused or nonreperfused ischemic stroke. In addition, we assessed the intracellular cytokine production and expression of tumor necrosis factor‐α (TNF‐α), interleukin (IL)‐1β and Arg1 in these innate immune cells to reveal possible functional phenotypes after ischemic stroke.
RESULTS
Stroke‐induced infiltration of neutrophils is model specific
We previously published the degree of brain infarct developed at 24 h following reperfused 1‐h temporary middle cerebral artery occlusion (tMCAO; 15.36 μm ± 4.84) and nonreperfused permanent MCAO (pMCAO; 64.79 μm ± 11.30) models, which allow for the generation of moderate and severe strokes, respectively. 13 Here, we performed flow cytometry of the brain at 24 h after surgery to examine the number and phenotype of various immune populations. Neutrophils were identified as live CD45hiLy6G+ (Figure 1a) and comprised ~15% of the peripheral leukocyte population in the brains of sham‐operated mice irrespective of cerebral hemisphere (Figure 1b). At 24 h following tMCAO, the number of neutrophils had a significant increase in the injured ipsilateral (IL) hemisphere, but not in the uninjured CL compared with respective sham (tMCAO IL: 34 970 ± 20 355 cells versus sham: 3528 ± 685.1, P = 0.0185; Figure 1c). Interestingly, in mice that underwent the more severe pMCAO model, both the CL and IL hemispheres had significant increases in neutrophils at 24 h after stroke, especially in the injured IL hemisphere compared with their respective sham controls (pMCAO CL: 5609 ± 1302 cells versus sham CL: 2932 ± 347.3 cells, P = 0.0427; pMCAO IL: 45 886 ± 10 188 cells versus sham IL: 3528 ± 685.1, P = <0.0001; Figure 1c). Despite increased neutrophil infiltration following tMCAO, these neutrophils did not show any significant changes in their intracellular expression of IL‐1β, TNF‐α or Arg1 compared with sham (Figure 1d–f). However, following pMCAO stroke, neutrophils recruited to the ischemic hemisphere demonstrated significant expression of IL‐1β (pMCAO IL: 8.6 ± 2.4% versus sham IL: 0.7 ± 0.4%, P = 0.0159; Figure 1d) and TNF‐α (pMCAO IL: 21.3 ± 3.9% versus sham IL: 6.2 ± 1.9%, P = 0.0146; Figure 1e), but not Arg1 (Figure 1f), compared with their sham‐operated counterparts. Collectively, these data suggest that while both models of stroke resulted in significant neutrophil recruitment to the ischemic hemisphere, only after a more severe stroke do neutrophils exhibit a proinflammatory phenotype at 24 h. Moreover, we revealed that neutrophils are recruited to the CL hemisphere of the brain following pMCAO albeit to a lesser extent than the IL hemisphere at this timepoint.
Figure 1.
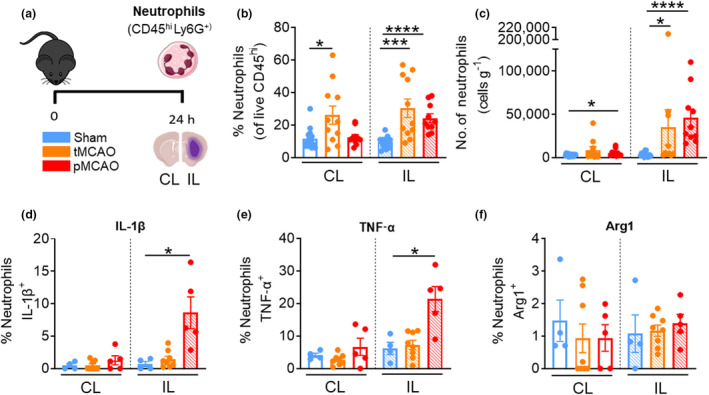
Stroke‐induced infiltration of neutrophils is model specific. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 24 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Neutrophils were defined as CD45hiLy6G+. (b) Proportion of neutrophils from live CD45+ cells. (c) Numbers of neutrophils normalized to the corresponding cerebral hemispheric weight. Percentage of neutrophils positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b, c) n = 10 or 15 per group, (d–f) n = 4 or 7 per group of two or three independent experiments. Data in b, c and f were analyzed by the Mann–Whitney U‐test. Data in d and e were analyzed by the unpaired t‐test per normality. *P < 0.05, ***P < 0.001, ****P < 0.0001. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Brain‐associated monocytes exhibit diverse phenotypes
Monocytes were defined as live CD45hiCD11b+Ly6C+ (Figure 2a) and comprised ~10% of the peripheral leukocyte population in the brains of sham‐operated mice. In a stepwise manner, we observed a significant increase in the proportion of monocytes in both the CL and IL hemispheres at 24 h following tMCAO and pMCAO (tMCAO IL: 22.8 ± 1.7% versus sham IL: 7.5 ± 0.7%, P < 0.0001; tMCAO CL: 15.5 ± 1.9% versus sham CL: 8.1 ± 0.6%, P = 0.0011; pMCAO IL: 32.9 ± 2.0% versus sham, P < 0.0001; pMCAO CL: 22.4 ± 3.6% versus sham, P = 0.0026; Figure 2b). In light of this, monocyte numbers were significantly increased in the IL damaged side of the brain following either stroke models compared with sham (tMCAO IL: 18 269 ± 8066 cells versus sham IL: 2791 ± 539 cells, P = 0.0004; pMCAO IL: 49 970 ± 7116 cells versus sham, P < 0.0001, Figure 2c). Of interest was the notable elevation of monocytes in the CL side of the brain following pMCAO, a seemingly or supposedly uninjured region (pMCAO CL: 11 762 ± 2572 cells versus sham CL: 2128 ± 208, P = 0.0021; Figure 2c). In fact, this elevation was to a similar level to that of the damaged hemisphere in mice following tMCAO. Nevertheless, the IL hemisphere displayed a significant ~17‐fold increase in monocyte infiltration after pMCAO compared with sham (Figure 2c). In concordance with elevated monocyte recruitment to the IL hemisphere of tMCAO mice, the proportion of monocytes that expressed TNF‐α was significantly increased in the IL hemisphere by ~5‐fold compared with the respective sham controls (tMCAO IL: 4.9% ± 1.0% versus sham IL: 0.9 ± 0.3%, P = 0.0240; Figure 2e). Interestingly, a significant proportion of monocytes also expressed Arg1 by 16‐fold compared with the respective sham control (tMCAO IL: 16.2 ± 6.4% versus sham IL: 1.1 ± 0.8%, P = 0.0263; Figure 2f), but not IL‐1β (Figure 2d), indicating a mixed phenotype at 24 h after transient ischemic injury. Conversely, mice that underwent pMCAO saw a significant ~5‐fold increase in the monocyte population expressing IL‐1β in the CL hemisphere compared with sham (pMCAO CL: 4.9 ± 0.4% versus sham CL: 1.1 ± 0.4%, P <0.0001; Figure 2d) and a significant ~27‐fold increase in the IL hemisphere compared with sham (pMCAO IL: 26.4 ± 2.5% versus sham IL: 1.4 ± 0.8%, P = 0.0006; Figure 2d). Similarly, a significant ~26‐fold increase of monocytes in the ischemic hemisphere of post‐pMCAO mice expressed TNF‐α compared with the sham control at 24 h following stroke onset (pMCAO IL: 20.55 ± 3.2% versus sham IL: 0.9 ± 0.3%, P = 0.0009; Figure 2e). Unlike neutrophils, a ~34‐fold increase in Arg1+ monocytes was observed in the IL hemisphere of pMCAO compared with the sham control (pMCAO IL: 31.2 ± 8.3% versus sham IL: 1.0 ± 0.8%, P = 0.0159; Figure 2f). Therefore, our data suggest that while monocytes are elevated in both the damaged and undamaged sides of the brain at 24 h after tMCAO and pMCAO, those infiltrating the injured hemisphere in both models demonstrated increased production of both pro‐ and anti‐inflammatory factors at this timepoint.
Figure 2.
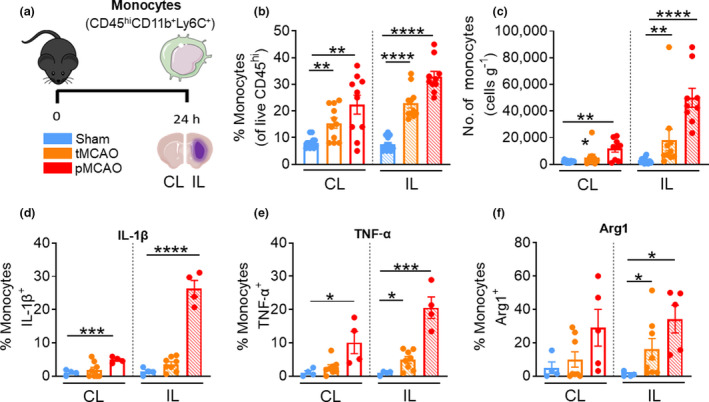
Brain‐associated monocytes exhibit diverse phenotypes. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 24 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Monocytes were defined as CD45hi Ly6G−CD11b+Ly6C+. (b) Proportion of monocytes from live CD45+ cells. (c) Numbers of monocytes normalized to the corresponding cerebral hemispheric weight. Percentage of monocytes positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b–f) n = 3 or 12 per group of two or three independent experiments. Data in b, c and f were analyzed by the Mann–Whitney U‐test, Data in d and e were analyzed by the unpaired t‐test per normality. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Macrophage heterogeneity at 24 h after stroke
Brain‐associated macrophages were defined as live CD45hiCD11b+Ly6C− (Figure 3a), 14 , 15 and comprised ~25% of the peripheral leukocyte population in the brains of sham‐operated mice (Figure 3b). In contrast to our findings in neutrophils and monocytes, we found that the proportions of macrophages in both the CL and IL hemispheres of tMCAO mice, and the IL hemisphere of pMCAO mice, were decreased compared with corresponding sham‐operated counterparts (tMCAO CL: 16.9% ± 2.2% versus sham CL: 27.1% ± 1.9%, P = 0.0016; tMCAO IL: 16.6% ± 2.4% versus sham IL: 27.8% ± 1.5%, P = 0.0005; pMCAO IL: 18.8% ± 2.1% versus sham IL, P = 0.0017; Figure 3b). Despite this, we did not observe changes in macrophage numbers in the hemispheres of tMCAO mice, while both the undamaged and damaged sides of the brain in mice following the more severe pMCAO exhibited significantly elevated macrophages at 24 h after stroke (pMCAO CL: 10 736% ± 1170 cells versus sham CL: 6845 ± 1030 cells, P = 0.0235; pMCAO IL: 27 549 ± 3606 cells versus sham IL 7814 ± 1180 cells, P = <0.0001; Figure 3c). Similar to brain‐associated monocytes, we found evidence for macrophages in the undamaged and damaged sides of the brain after pMCAO that displayed significantly increased expression of IL‐1β compared with sham (pMCAO CL: 9.8% ± 1.2% versus sham CL: 4.5% ± 1.3%, P = 0.0211; pMCAO IL: 24.12% ± 1.8% versus sham CL: 4.5% ± 1.3%, P = 0.0002; Figure 3d). Moreover, a significant proportion of infiltrating macrophages in the damaged IL side of the brain after pMCAO were positive for TNF‐α (pMCAO IL: 15.2% ± 1.1% versus sham IL: 3.9% ± 1.5%, P = 0.0159; Figure 3e) or Arg1 (pMCAO IL: 9.4 ± 0.5% versus sham IL: 0.7 ± 0.2%, P < 0.0001; Figure 3f). Taken together, these data suggest that macrophages infiltrate both cerebral hemispheres at 24 h following the more severe pMCAO model of stroke, and these cells display diverse phenotype similar to monocytes at this timepoint.
Figure 3.
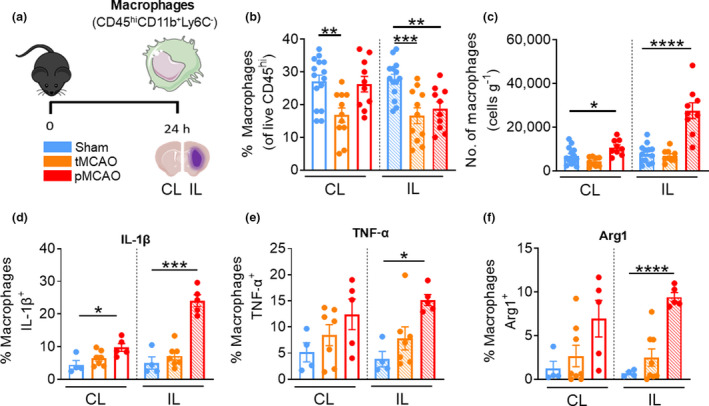
Macrophages are heterogeneous 24 h after stroke. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 24 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Macrophages were defined as CD45hiLy6G−CD11b+Ly6C−. (b) Proportion of macrophages from live CD45+ cells. (c) Numbers of macrophages normalized to the corresponding cerebral hemispheric weight. Percentage of macrophages positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b) n = 10 or 13, (c) n = 9 or 15, (d–f) n = 4 or 8 per group of two or three independent experiments. Data in b–f were analyzed by the unpaired t‐test and e and f by the Mann–Whitney U‐test per normality. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Resident microglia number largely unchanged after stroke at 24 h
To examine the contributions of brain‐resident microglia to poststroke neuroinflammation, we quantified the number of live CD45midCD11b+ cells after experimental stroke (Figure 4a). We observed a significant decrease in the proportions of microglia in the IL hemispheres at 24 h following both tMCAO and pMCAO (tMCAO IL: 92.4 ± 1.2%, versus sham IL: 95.3 ± 0.9%, P = 0.0387, pMCAO IL: 80.39 ± 4.0% versus sham IL, P < 0.0001; Figure 4b), likely owing to the contributions of the other brain‐associated innate leukocytes described earlier. We did not observe a significant change in the number of microglia in either hemisphere following tMCAO or pMCAO compared with sham mice (Figure 4c). Moreover, microglia in the ischemic hemisphere of poststroke mice displayed unchanged expression of IL‐1β, TNF‐α and Arg1 compared with sham controls despite reduced expression of Arg1 in the uninjured region following tMCAO (Figure 4d–f). These data suggest that the effect of stroke on the number and inflammatory state of microglia is much less prominent compared with the infiltrating innate immune cells at this acute timepoint.
Figure 4.
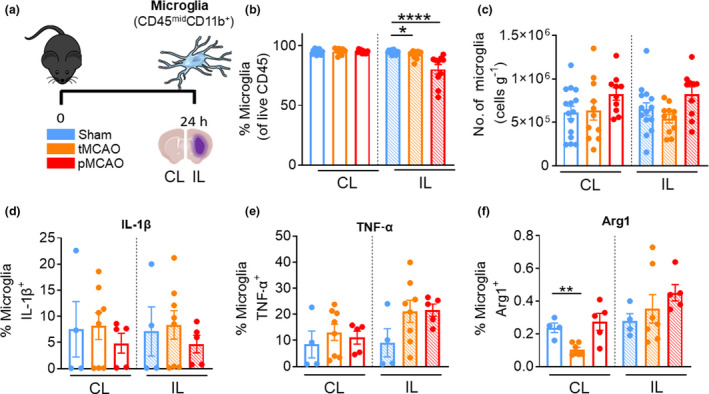
Resident microglia number remained largely unchanged 24 h after stroke. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 24 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Microglia were defined as CD45midCD11b+. (b) Proportion of microglia from live CD45+ cells. (c) Numbers of microglia normalized to the corresponding cerebral hemispheric weight. Percentage of microglia positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b, c) n = 9 or 15, (d–f) n = 4 or 8 per group of two or three independent experiments. Data in b and f were analyzed by the Mann–Whitney U‐test per normality. *P < 0.05, **P < 0.01, ****P < 0.0001. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Neutrophil infiltration and phenotype at 96 h are model dependent
We previously published the degree of brain infarct developed at 96 h following reperfused 1‐h tMCAO (3.57 μm ± 0.98) and nonreperfused pMCAO (65.07 μm ± 18.95) models. 13 To characterize the prolonged recovery effects of transient and permanent cerebral ischemia on brain innate leukocytes, we performed identical experiments assessing the population and their cytokine production and expression of TNF‐α, IL‐1β and Arg1 at 96 h after stroke onset. By 96 h after surgery, neutrophils consist of ~7% of the cerebral immune component in sham mice (Figure 5b). Despite infiltration at 24 h following tMCAO and pMCAO in the IL hemisphere, only the number of neutrophils were significantly higher in the IL hemisphere of post‐pMCAO animals compared with sham control at 96 h (pMCAO IL: 15 955 ± 7420 cells versus sham IL: 1475 ± 117, P = 0.0029; Figure 5c). In addition, the acute neutrophil expression of IL‐1β and TNF‐α in pMCAO mice at 24 h was no longer evident at this later timepoint (Figure 5d, e), but we found neutrophils of post‐tMCAO mice displayed elevated TNF‐α expression in both hemispheres compared with their sham‐operated counterparts at 96 h (tMCAO IL: 11.35 ± 1.2% versus sham IL: 2.3 ± 0.03%, P = 0.0012; tMCAO CL: 7.1 ± 0.9% versus sham CL: 1.7 ± 0.9%, P = 0.0077; Figure 5e). Again, we did not observe significant expression of Arg1 in neutrophils of poststroke mice at this later timepoint (Figure 5f). Collectively, these data suggest that neutrophils are still actively recruited to the ischemic hemisphere at 96 h after severe pMCAO stroke, while neutrophils of mice after tMCAO produce TNF‐α in both hemispheres.
Figure 5.
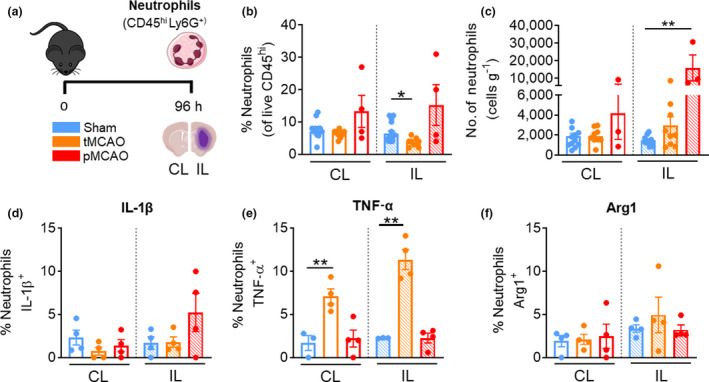
Neutrophils remain elevated in pMCAO, but not in tMCAO after 96 h. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 96 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Neutrophils were defined as CD45hiLy6G+. (b) Proportion of neutrophils from live CD45+ cells. (c) Numbers of neutrophils normalized to the corresponding cerebral hemispheric weight. Percentage of neutrophils positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b, c) n = 3 or 14, (d–f) n = 3 or 4 per group of one to three independent experiments. Data in b and c were analyzed by the Mann–Whitney U‐test and in d and e by the unpaired t‐test per normality. *P < 0.05, **P < 0.01. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Monocytes infiltration remains elevated at 96 h after stroke
Monocytes consisted approximately 10% of the CD45hi leukocyte population at 96 h after surgery in sham and poststroke mice (Figure 6b). Despite this, monocyte infiltration into the IL hemisphere was still evident at this latter timepoint in both models of stroke (tMCAO IL: 10 129 ± 3924 cells versus sham IL: 2780 ± 359 cells, P = 0.0235; pMCAO IL: 33 041 ± 12 461 cells versus sham IL, P = 0.0005; Figure 6c). Moreover, a significant proportion of monocytes that were recruited into the ischemic hemisphere of pMCAO were Arg1 positive (pMCAO IL: 4.8 ± 1.5% versus sham IL: 0.2 ± 0.2%, P = 0.0222; Figure 6f). These data suggests that brain‐associated monocytes after pMCAO at 96 h no longer exhibit elevated TNF‐α and IL‐1β but have switched to an anti‐inflammatory phenotype as indicated by the increased Arg1 expression.
Figure 6.
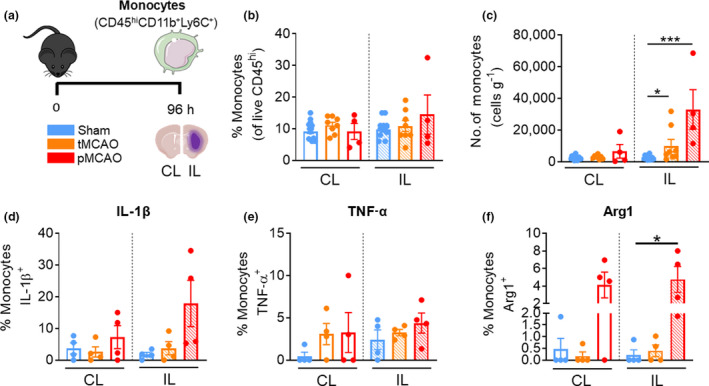
Monocytes remain significantly increased in both tMCAO and pMCAO after 96 h. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 96 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Monocytes were defined as CD45hiLy6G−CD11b+Ly6C+. (b) Proportion of monocytes from live CD45+ cells. (c) Numbers of monocytes normalized to the corresponding cerebral hemispheric weight. Percentage of monocytes positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b, c) n = 4 or 15, (d–f) 3 or 4 per group of one to three independent experiments. Data in c were analyzed by the Mann–Whitney U‐test, in d–f by the unpaired t‐test per normality. *P < 0.05, ***P < 0.001. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Macrophages remain heterogeneous at 96 h following tMCAO
We observed no significant changes in the proportion of macrophages in either the CL or IL hemispheres at 96 h following tMCAO or pMCAO (Figure 7b). Despite this, macrophage numbers remained significantly increased in the IL hemisphere at 96 h after tMCAO and pMCAO mice compared with sham (tMCAO IL: 15 784 ± 1922 cells versus sham IL: 7960 ± 1071 cells, P = 0.0029; pMCAO IL: 47 502 ± 193 cells versus sham IL, P = 0.0025; Figure 7c). In addition, the ischemic hemisphere of the brain in both tMCAO and pMCAO mice saw significant increases in the proportions of macrophages positive for IL‐1β (tMCAO IL: 9.8 ± 1.6% versus sham IL: 3.8 ± 0.5%, P = 0.0286; pMCAO IL: 14.6 ± 4.5% versus sham IL, P = 0.0286; Figure 7d). There was no significant change in macrophage expression of TNF‐α in the brains of post‐tMCAO or pMCAO mice compared with their sham‐operated counterparts (Figure 7e). However, there was a significant increase in the proportion of Arg1‐positive macrophages in both hemispheres of tMCAO, but not in pMCAO mice at 96 h after stroke compared with sham (tMCAO CL: 3.7 ± 0.6% versus sham CL: 0.8 ± 0.5%, P = 0.0080; tMCAO IL: 5.4 ± 0.9% versus sham IL: 0.5 ± 0.2%, P = 0.0020; Figure 7f). These data suggest that brain‐associated macrophages have adopted an Arg1+ phenotype in the CL hemisphere following tMCAO, but a mixed pro‐ and anti‐inflammatory state in the IL hemisphere at this later stage of reperfused experimental stroke.
Figure 7.
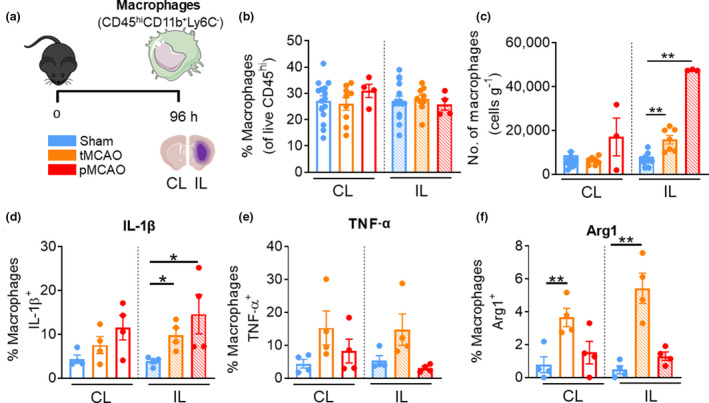
Macrophages remain significantly elevated 96 h after reperfusion injury stroke. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 96 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Macrophages were defined as CD45hiLy6G−CD11b+Ly6C−. (b) Proportion of macrophages from live CD45+ cells. (c) Numbers of macrophages normalized to the corresponding cerebral hemispheric weight. Percentage of macrophages positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b, c) n = 3 or 15, (d–f) n = 4 per group of one to three independent experiments. Data in c and f were analyzed by the unpaired t‐test, in d by the Mann–Whitney U‐test per normality. *P < 0.05, **P < 0.01. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Prolonged stroke induces microglial production of proinflammatory cytokines
The proportion of microglia relative to all immune cells was significantly decreased in the IL hemisphere of both tMCAO and pMCAO mice compared with sham counterparts at 96 h (tMCAO IL: 91.2 ± 1.2% versus sham IL: 95.45 ± 0.5%, P = 0.0011; pMCAO IL: 76.9 ± 5.6%, P = <0.0001; Figure 8b). However, similar to 24 h following stroke, microglial numbers remained unchanged at 96 h (Figure 8c). Instead, we observed a significant increase in the proportion of microglia positive for IL‐1β and TNF‐α in the IL hemispheres of tMCAO and pMCAO mice (IL‐1β—tMCAO IL: 22.7 ± 3.2% versus sham IL: 14.7 ± 0.4%, P < 0.0001; pMCAO IL: 19.6 ± 1.2% versus sham IL, P = 0.0084; Figure 8d and TNF‐α—tMCAO IL: 21.8 ± 2.8% versus sham IL: 11.5 ± 1.8%, P = 0.0221; pMCAO IL: 30.9 ± 3.7% versus sham IL, P = 0.0032; Figure 8e). Of note was the absence of significant Arg1+ microglia (Figure 8f). These data suggest that as the innate leukocytes infiltrate the ischemic hemisphere, microglia take time to develop into a primarily proinflammatory phenotype after stroke. A graphical summary in Figure 9 illustrates the significant phenotypic changes of each cell examined in the IL hemisphere following both models of experimental stroke.
Figure 8.
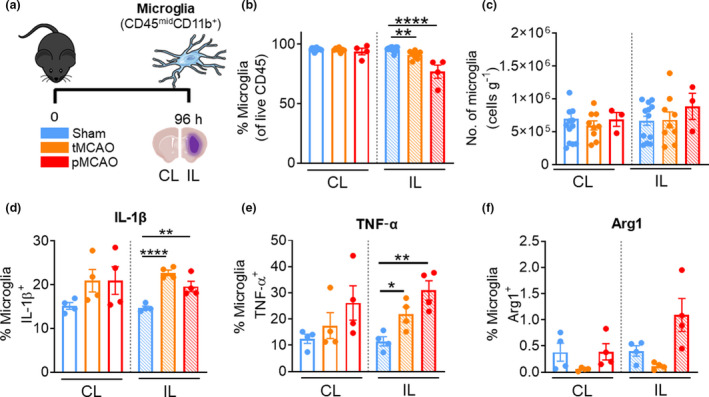
Microglia become proinflammatory 96 h after reperfusion injury stroke. (a) C57Bl/6J mice underwent sham surgery, 1‐h tMCAO or pMCAO and were culled after 96 h. The brain was separated into left (contralateral) and right (ipsilateral) hemispheres and underwent flow cytometric analysis to quantify live, unstimulated CD45+7‐AAD− leukocytes. Microglia were defined as CD45midCD11b+. (b) Proportion of microglia from live CD45+ cells. (c) Numbers of microglia normalized to the corresponding cerebral hemispheric weight. Percentage of microglia positive for (native) intracellular (d) pro‐IL‐1β, (e) TNF‐α and (f) Arg1 were assessed. Data are shown as mean ± s.e.m.: (b, c) n = 3 or 15, (d–f) n = 4 per group of one to three independent experiments. Data in b, d and e were analyzed by the unpaired t‐test per normality. *P < 0.05, **P < 0.01, ****P < 0.0001. 7‐AAD, 7‐aminoactinomycin D; Arg1, arginase 1; CL, contralateral; IL, ipsilateral; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 9.
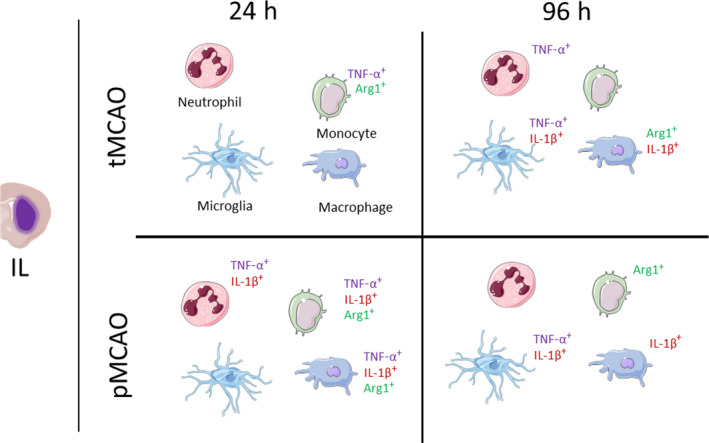
The global immune changes seen between tMCAO and pMCAO across time. Significant changes in the production of IL‐1β, TNF‐α and arginase 1 in the cells in the ipsilateral stroke hemisphere (left to right) of neutrophils, monocytes, microglia and macrophages. Arg1, arginase 1; IL‐1β, interleukin‐1β; pMCAO, permanent middle cerebral artery occlusion; tMCAO, temporary middle cerebral artery occlusion; TNF, tumor necrosis factor. [Colour figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
In this study, we demonstrated that peripheral neutrophils, monocytes and macrophages infiltrate the brain acutely and chronically after transient and permanent cerebral ischemia. In addition, we showed that brain‐associated innate leukocytes are increased in the cerebral hemisphere CL to the ischemic region at 24 h following permanent ischemia. Moreover, we provide evidence that immune cells in the IL hemisphere of the brain infarct show stroke model‐ or severity‐dependent expression of TNF‐α, IL‐1β and Arg1 over time.
Neutrophils
There is accumulating experimental and clinical evidence that suggests neutrophils exhibit functional heterogeneity, but our understanding of this notion in the context of stroke is only at its infancy. 16 We observed a significant increase in the number of neutrophils to the IL hemisphere at 24 h following 1‐h tMCAO. In a model of 45‐min tMCAO, Liu et al. 17 noted no significant increases in polymorphonuclear leukocytes (PMNs) at 48 h after stroke, while Herz et al. 18 also observed no significant changes in neutrophil numbers at 24 h after 20‐min tMCAO. As the severity of stroke is found to correlate with the length of occlusion experimentally and in patients with ischemic stroke, 6 this indicates that occlusion for less than 1 h may result in insufficient ischemic injury that would enable the infiltration of neutrophils to the cerebral hemisphere. To support this notion, we observed a further increase in neutrophil infiltration in both the CL and IL brain hemispheres of mice following the more severe model of experimental stroke in pMCAO.
A previous seminal work by Cuartero et al. 8 observed the presence of N2‐like neutrophils in the ischemic infarct core at 48 h after pMCAO based on their positive expression for classic M2 cellular markers Ym1 and CD206, but these cells were only scarcely positive for Arg1. In support of this, we did not observe changes in Arg1 expression in brain‐associated neutrophils at 24 or 96 h after stroke. However, we observed an increased proportion of neutrophils in the IL hemisphere of pMCAO mice that were primarily proinflammatory at 24 h, as indicated by marked elevations in their intracellular expression for IL‐1β and TNF‐α. A recent study demonstrated that neutrophil functionality can possibly be skewed in vitro toward an N1‐like phenotype triggering neuronal death, while N2‐skewed neutrophils did not affect neuronal viability to a significant extent during oxygen–glucose deprivation. 19 Although this experiment suggested that neutrophil phenotype can possibly be manipulated in vitro and have consequential effects on co‐cultured neurons, the classification of N1 versus N2 neutrophils based on mRNA expression for TNF‐α, and Arg1 and CD206, respectively, may not be reflective of in vivo settings. In this study, we did not detect a significant difference in Arg1 protein expression in the brain‐associated neutrophils following either experimental stroke models at 24 or 96 h. Instead, brain‐associated neutrophils acquired a predominantly proinflammatory phenotype, likely contributing to poststroke neuroinflammation.
Monocytes and macrophages
It was previously reported that monocytes and macrophages are recruited to the ischemic tissue 3–7 days after stroke. 20 In addition, work by Wattananit et al. using 30‐min tMCAO in mice observed the peak of monocyte infiltrate at day 3, with a dominant anti‐inflammatory phenotype by day 7. The authors also noted that the specific inhibition of monocyte recruitment to the brain using anti‐CCR2 prevented behavioral recovery and expression of anti‐inflammatory genes. 21 In contrast to this, we observed a proportion of monocytes and macrophages of diverse phenotype, as indicated by varying degrees of positivity for IL‐1β, TNF‐α and Arg1 at 24 and 96 h after stroke. Previous evidence suggests that monocytes exist in either classical (proinflammatory, IL‐1β and TNF‐α) or nonclassical (Arg1) subsets depending on their expression and retention of CCR2. It was suggested that decreases in the number of nonclassical monocytes are associated with post‐stroke infection, whereas increases in classical/intermediate monocytes correlate more closely with infarct severity and outcome. 22 Although we did not examine this modality, given the absence of a predominant pro‐ or anti‐inflammatory monocyte/macrophage phenotype in the less severe model of stroke (tMCAO), our data suggest that stroke severity may determine the observable polarization of monocytes and macrophages after stroke.
We detected an elevation of Arg1 expression in monocytes and macrophages after stroke, though at times this was at relatively lower frequency than TNF‐α+ and IL‐1β+ cells, but we believe Arg1+ cells would induce a significant biological effect. This is supported by numerous cell depletion or inhibition studies that have shown the importance of brain‐associated monocyte/macrophage in stroke despite their relatively low cell frequencies. 23 , 24 , 25 Furthermore, the conflicting results of whether brain‐associated monocytes and macrophages play pro‐ or anti‐inflammatory roles after stroke are possibly because of depletion strategies targeting specific subsets, thus resulting in their possible worsening or beneficial roles, respectively. 16 Nevertheless, of important distinction is whether the changes in macrophages observed here are derived from microglia or monocytes‐derived macrophages. Given that the cytokine signatures and changes in cell numbers between monocytes and macrophages were more similar than that of microglia at 24 h after stroke, we propose that monocytes might be the source of mixed phenotype in macrophages at this timepoint.
Microglia
Microglia have been shown to activate within minutes of stroke onset, 25 and may adopt classical M1 and nonclassical M2 phenotypes following stroke. Our data indicate that microglial numbers are unchanged in both the undamaged and ischemia‐affected hemispheres following either tMCAO or pMCAO. These data are inconsistent with other findings in pMCAO models noting the distinct increases in global gliosis 24 h after cerebral ischemia, 25 but this may not be reflected in cell numbers alone. In mice that underwent 90‐min tMCAO, Ritzel et al. 26 observed no significant changes in CD45midCD11b+ microglial numbers in the IL hemisphere at 24 h after stroke. This is consistent with our findings in the cohort of mice that underwent 1‐h tMCAO and assessed at the same timepoint. The authors also noted that the degree of microglial phagocytosis positively correlated with the production of TNF‐α, 26 which supports our findings of increased TNF‐α in the IL hemisphere following pMCAO at 96 h, but not at 24 h. Interestingly, microglia in the IL region of post‐pMCAO mice were IL‐1β positive at 96 h, but were not Arg1 positive. In exploring microglial phenotypes following tMCAO in rats, Rajan et al. 27 observed the significant presence of Arg1 in Iba1+ microglia and CD11b+ macrophages as assessed via immunofluorescence in the infarct core. In a mouse model of 1‐h tMCAO, Cai et al. 28 observed that deficiency of STAT6, an upstream modulator of Arg1, resulted in impairments to microglial efferocytosis and an increase in brain infarction, indicating the strong role for Arg1 in determining experimental poststroke outcome. Despite this, we did not observe significantly increased changes in Arg1 in our microglial population, once again highlighting the difficulty in fully characterizing specific cellular phenotypes following stroke and problems with simply designating multifaceted cells to distinct M1 or M2 phenotypes. Moreover, future in‐depth histological assessment is required to further understand the spatial location of different inflammatory leukocyte subsets in relation to infarct regions, brain microvasculature and connections to other brain parenchymal cells.
Contralateral hemisphere
To our knowledge, immune recruitment to the noninfarcted CL hemisphere is not routinely assessed after cerebral ischemia. Foundational stroke research has traditionally used the CL hemisphere of stroke animals as an experimental control on the basis that this tissue does not undergo cerebral ischemia, and is thus regarded as “healthy” tissue. However, we and others have demonstrated that stroke impacts distal sequelae including systemic inflammation and immunosuppression, mucosal barrier breakdown and lung infection. 13 , 29 Thus, we hypothesized that the CL hemisphere may also be prone to inflammatory response and immune cell recruitment. In this study, we frequently observed the elevations of neutrophils, monocytes, macrophages and microglia in the noninfarcted hemisphere, yet only following permanent but not transient cerebral artery occlusion. In some instances, the degree of infiltrate into the CL hemisphere of pMCAO mice was even directly comparable to those seen in the IL hemisphere of post‐tMCAO mice. Investigations into the contribution of CL hemisphere in poststroke neuroinflammation or recovery have been scarce. Clinically, the CL hemisphere undergoes increased excitatory neural activity and activation positively correlated with motor function. 30 Experimentally, Fury et al. 31 revealed overlapping of M1 and M2 macrophage‐related genes in both hemispheres after stroke, suggesting that the CL hemisphere may rely on some form of immune influence to remain functional or supportive to the ischemic regions of the brain. Interestingly, a recent study suggested that the inflammatory response elicited by the injured brain could lead to the secretion of growth factors and immune modulators able to affect neuronal progenitor proliferation and survival. 32 Whether neuronal synaptogenesis and functionality rely on peripheral leukocyte infiltrate is an interesting concept that remains to be fully examined.
Study design and limitations
In this study, we have utilized flow cytometry to examine the infiltration of various innate immune cells into the postischemic brain for its advantage in analyzing only live cells with the use of live/dead dye and avoiding nonspecific staining which can be abundant with immunohistochemistry of injured tissue. Furthermore, our single‐cell preparation of brain tissue was not obtained from perfused tissue, allowing us to examine the involvement of intravascular leukocytes. Numerous studies have demonstrated the importance of circulating leukocytes in mediating intravascular immunity and inflammation in various tissue beds. 33 In fact, there is an accumulating body of evidence to suggest that the interplay of intravascular neutrophils and platelets in the cerebrovasculature facilitates the production of inflammatory cytokines and neutrophil extracellular traps (NETs) after stroke. 34 , 35 Therefore, we believe that intravascular neutrophils or leukocytes are highly relevant in poststroke neuroinflammation and as such we did not perform transcardial perfusion prior to tissue dissociation. Another advantage of our flow cytometry protocol is the investigation of intracellular protein expression of TNF‐α, IL‐1β and Arg1 of infiltrating cells which would have been difficult or near impossible to detect with other methods such as immunohistochemistry. Nevertheless, a limitation of our study design is that we cannot comment on the spatial location of infiltrated cells in poststroke brain. Moreover, we cannot comment or conclude an increase of cell infiltration over time or between experimental models with our study design, as each experimental endpoint for each stroke model was compared with its sham‐operated control at that timepoint, not across time. Despite this, we have overcome batch‐to‐batch effects in cell counts and cytokine detection in our flow cytometry experiments with the use of counting microbeads to standardize between experimental samples and days. Furthermore, we used the same staining protocol, same antibodies (clone and batch) and techniques throughout our study to avoid staining or detection issues.
Conclusion
In summary, the findings of this study indicate that in both transient and permanent models of cerebral ischemia, neutrophils, monocytes and macrophages show increased recruitment to not only the ischemic hemisphere, but also to the CL hemisphere at 24 h after stroke. Following nonreperfused pMCAO, neutrophils demonstrated significant production of TNF‐α and IL‐1β, but not Arg1, while a significant proportion of monocytes and macrophages exhibited marked expression for all three markers examined after stroke. Indeed, designation of any possible N1‐like/M1 and N2‐like/M2 states is evident only in more severe nonreperfusion injury, and not following tMCAO. Our results shed light on the specific contributions of key innate leukocytes in the development of poststroke neuroinflammation.
METHODS
Mice
Adult C57Bl/6J male mice were obtained from the Monash Animal Research Platform and were housed at Monash Medical Centre Animal Facility (Clayton, VIC, Australia) in specific pathogen‐free conditions with access to food and water ad libitum. Mice were housed in temperature‐controlled rooms (22°C) under a standard 12‐h light–dark cycle. Prior to the start of experiments, mice were acclimatized for a minimum of 7 days before use. All procedures were approved by the Monash Medical Centre Animal Ethics Committee (MMCB/2018/002, Monash Medical Centre). This study was not aimed to examine sex‐dependent effects; therefore, male mice were used to avoid hormonal fluctuations that exist in female animals. In addition, male mice exhibit more consistent size and weight compared with female counterparts of the same age, and this is particularly important to ensure consistent size of monofilament used for arterial occlusion when inducing experimental stroke. A proportion of mice in the pMCAO 96‐h cohort required humane killing because of ethical endpoint of weight loss reaching >15%.
Middle cerebral artery occlusion surgery
The mouse model of ischemic stroke was performed as previously described, 13 whereby infarct is induced in the right cerebral hemisphere. In brief, mice were anesthetized by an intraperitoneal injection of ketamine (as hydrochloride, 150 mg kg‐1; Baxter, Deerfield, IL, USA) and xylazine (10 mg kg‐1; Troy Animal Healthcare, Glendenning, NSW, Australia). Once anesthetized, an incision at the top of the head was made to expose the skull, and a laser Doppler holder (PeriFlux System 5000, Stroke model kit 407; Perimed, Järfälla, Stockholm, Sweden) was glued to the right side of the skull to monitor blood flow above the right hemisphere of the brain. The fur at the site of the neck incision was removed and the area was sterilized with 80% ethanol. A 1–2‐cm incision was made along the midline of the throat to expose the trachea. The right common carotid artery, external carotid artery and internal carotid artery were dissected away from any connective tissue. A vessel clip was used on the right common carotid artery, and a slit was made in the external carotid artery. A silicone‐coated monofilament with a diameter of 0.21–0.23 mm (Doccol Corporation, Sharon, MA, USA) was immediately inserted into the external carotid artery and advanced into the internal carotid artery to occlude blood flow into the MCA. A successful occlusion was determined by a significant reduction in Doppler reading. The external carotid artery was tied off and the vessel clip removed. In this study, we utilized both reperfused and nonreperfused models of MCAO. In the reperfused model termed transient MCAO or tMCAO, the monofilament was retracted to allow for reperfusion of blood flow into the MCA following 1 h of occlusion. In the nonreperfused model termed permanent MCAO or pMCAO the monofilament was not retracted, and MCA was occluded for the duration of the experimental timepoint. After successful occlusion of MCA in either model, the neck incision was sutured, the Doppler probe removed and the head incision closed. As a control, potential surgical stress was mimicked in a sham surgery, whereby similar surgical procedures were performed without the insertion of the monofilament. Notably, mice that underwent stroke (tMCAO or pMCAO) was accompanied by at least one nonstroke control (sham‐operated) animal to ensure batch consistency between experiments. After surgery, all mice were allowed to recover overnight on a heat pad. Mice were then monitored daily until experimental endpoint.
Cerebral immune cell disassociation
At the experimental endpoint, mice were culled via anesthetic overdose and cervical dislocation, and the brains carefully dissected. Transcardial perfusion was not performed to include intravascular leukocytes in our analysis. With the cerebellum removed, the cerebrum was separated into left (CL) and right (IL) hemispheres using a scalpel and stored in ice‐cold phosphate‐buffered saline. Each hemisphere was cut into fine pieces and digested in 1 mL of collagenase digestion buffer consisting of collagenase XI (125 U mL‐1; Sigma‐Aldrich, St Louis, MO, USA), hyaluronidase (60 U mL‐1; Sigma‐Aldrich) and collagenase type I (450 U mL‐1; Sigma‐Aldrich) in Dulbecco’s phosphate‐buffered saline (Ca2+/Mg2+ supplemented; Thermo Fisher Scientific, Waltham, MA, USA) with agitation in an orbital shaker at 120 rpm at 37°C for 30 min. Samples were then passed through a 70‐μm nylon cell strainer along with Fluorescence‐Activated Cell Sorting (FACS) buffer [2% fetal bovine serum (Moregate Biotech, Bulimba, QLD, Australia) and 1 mm ethylenediaminetetraacetic acid] and kept on ice. Single‐cell suspensions were subjected to 30%/70% Percoll (GE Healthcare, Uppsala, Sweden) gradient and centrifuged at 600g for 20 min at room temperature (approximately 20–25°C). The fat layer was removed and the interphase containing the enriched mononuclear cells was collected and washed with FACS buffer.
Numeration of leukocyte infiltrate
Enriched mononuclear cells were treated with Fc receptor block (1:100, 2.4G2; BD Pharmingen, Franklin Lakes, NJ, USA) for 15 min on ice to prevent nonspecific binding of antibodies. Furthermore, we used unstained cerebral single cells and fluorescent beads as compensation controls to identify distinct populations and avoid the analysis of false‐positive signals. Samples were washed, then stained 1:200 with the following fluorescent antibodies in the dark on ice for 20 min: PE‐conjugated anti‐mouse CD45 (clone 30‐F11, eBioscience 12–0451‐82; Thermo Fisher Scientific), PE‐Cy7‐conjugated anti‐mouse CD11b (clone M1/70, eBioscience 25–0112‐82; Thermo Fisher Scientific), BV510‐conjugated anti‐mouse Ly6G (clone 1A8, 127633; BioLegend, San Diego, CA, USA), APC‐Cy7‐conjugated anti‐mouse Ly6C (clone AL‐21, 560596; BD Pharmingen), e450‐conjugated anti‐mouse B220 (clone RA‐6B2, eBioscience 48–0452‐80; Thermo Fisher Scientific) and fluorescein isothiocyanate‐conjugated anti‐mouse CD3e (clone 145‐2C11, eBioscience 11–0031‐85; Thermo Fisher Scientific), After staining, the cells were washed, resuspended with counting bead mixture (5000 beads/sample) and 7‐aminoactinomycin D live‐dead viability stain (1,50, 420404; BioLegend) in FACS buffer. Samples were analyzed using the BD LSRX Fortessa and analyzed using FlowJo (version 10.7.0). Cell numbers were normalized to the weight of the corresponding cerebral hemisphere. Immune cell populations of interest were identified according to the gating strategy illustrated in Supplementary figure 1.
Immune cell intracellular staining
To examine intracellular cytokine and protein expression, single‐cell suspensions were incubated in 2 mL Roswell Park Memorial Institute medium (Thermo Fisher Scientific; supplemented with 10% fetal bovine serum) containing Brefeldin A (1:1000; BD Biosciences, Franklin Lakes, NJ, USA) for 2 h at 37°C. Samples were pelleted and treated with Fc receptor block and first stained with extracellular surface markers (listed above, with the exception of CD3εe and B220), followed by fixation and permeabilization with 100 μL fixation solution (BD Cytofix/Cytoperm Plus, 555028; BD Biosciences) for 20 min in the dark on ice. Cells were washed with fix/wash solution and stained 1:200 with AlexaFluor‐488‐conjugated anti‐human/mouse Arg1 (clone A1exF5, eBioscience 53–3697‐80; Thermo Fisher Scientific,), 1:100 with APC‐conjugated anti‐mouse IL‐1β (Pro‐form, clone NJTEN3, eBioscience 17–7114‐80; Thermo Fisher Scientific), 1:100 with PE‐Dazzle (AF594)‐conjugated anti‐mouse TNF‐α (clone MP6‐XT22, 506 345; BioLegend) on ice for 30 min in the dark. Cells were washed and resuspended in 200 μL fix/wash solution for flow cytometric analysis. Samples were run using the BD LSR Fortessa and analyzed using FlowJo (version 9.3.0). Intracellular expression of TNF‐α, IL‐1β and Arg1 were expressed as percentage or proportion of each cell type.
Data handling and statistical analysis
No specific system was used to place mice into respective experimental groups; however, littermates from the same cage were randomly assigned to different experimental groups. Experimenters were not blind to group or outcome assignment. In addition, data were not processed randomly nor blocked; however, the gating strategies used for flow cytometry were applied to all samples without bias. Sample sizes were determined from our previous and extensive experience of both stroke models. All statistical analyses were performed using GraphPad Prism 8 Software (La Jolla, CA, USA). The normality of data was first assessed using a Shapiro–Wilk normality test. It is of note that we did not perform all three groups (sham, tMCAO and pMCAO) in any single independent experiment, thus statistical comparisons were kept solely between sham and either stroke model. We analyzed using either an unpaired t‐test (parametric data) or Mann–Whitney U‐test (nonparametric data), as required. Outliers were identified using Grubb’s test α ≤ 0.05 on a per cell‐type basis to avoid repeated removal of data. Where outliers were removed for a single hemisphere, the corresponding value for the biologically respective CL or IL were also removed. All values are presented as mean ± s.e.m. A value of P ≤ 0.05 was considered statistically significant.
AUTHOR CONTRIBUTIONS
Brooke Wanrooy: Data curation; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Shu Wen Wen: Conceptualization; data curation; investigation; methodology; supervision; writing – original draft; writing – review and editing. Raymond Shim: Data curation; methodology. Jenny L Wilson: Data curation; investigation. Kathryn Prame Kumar: Data curation; investigation. Connie Wong: Conceptualization; project administration; supervision; writing – original draft; writing – review and editing.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ETHICS APPROVAL
All experimental animal procedures were approved by the Monash Medical Centre B Animal Ethics Committee (MMCB/2018/02).
Supporting information
Figure S1
ACKNOWLEDGMENTS
We thank the Monash Medical Centre Animal Facility staff for the care of animals, and Michael Thompson from the Monash FlowCore facility. This work is supported by the CSL Centenary Fellowship. The financial supports have no role in conducting the research and/or preparation of the article. Open access publishing facilitated by Monash University, as part of the Wiley ‐ Monash University agreement via the Council of Australian University Librarians.
DATA AVAILABILITY STATEMENT
All data presented in this study are included in the manuscript.
REFERENCES
- 1. Donkor ES. Stroke in the 21(st) century: a snapshot of the burden, epidemiology, and quality of life. Stroke Res Treat 2018; 2018: 3238165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kitagawa K, Matsumoto M, Mabuchi T, et al. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J Cereb Blood Flow Metab 1998; 18: 1336–1345. [DOI] [PubMed] [Google Scholar]
- 3. Enlimomab Acute Stroke Trial Investigators . Use of anti‐ICAM‐1 therapy in ischemic stroke: results of the Enlimomab acute stroke trial. Neurology 2001; 57: 1428–1434. [DOI] [PubMed] [Google Scholar]
- 4. Krams M, Lees KR, Hacke W, et al. Acute stroke therapy by inhibition of neutrophils (ASTIN): an adaptive dose‐response study of UK‐279,276 in acute ischemic stroke. Stroke 2003; 34: 2543–2548. [DOI] [PubMed] [Google Scholar]
- 5. Becker KJ. Anti‐leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr Med Res Opin 2002; 18(Suppl 2): s18–s22. [DOI] [PubMed] [Google Scholar]
- 6. Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 2015; 35: 888–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xie X, Shi Q, Wu P, et al. Single‐cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat Immunol 2020; 21: 1119–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cuartero MI, Ballesteros I, Moraga A, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARγ agonist rosiglitazone. Stroke 2013; 44: 3498–3508. [DOI] [PubMed] [Google Scholar]
- 9. Cai W, Wang J, Hu M, et al. All trans‐retinoic acid protects against acute ischemic stroke by modulating neutrophil functions through STAT1 signaling. J Neuroinflammation 2019; 16: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sippel TR, Shimizu T, Strnad F, Traystman RJ, Herson PS, Waziri A. Arginase I release from activated neutrophils induces peripheral immunosuppression in a murine model of stroke. J Cereb Blood Flow Metab 2015; 35: 1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Otxoa‐de‐Amezaga A, Gallizioli M, Pedragosa J, et al. Location of neutrophils in different compartments of the damaged mouse brain after severe ischemia/reperfusion. Stroke 2019; 50: 1548–1557. [DOI] [PubMed] [Google Scholar]
- 12. Neumann J, Sauerzweig S, Ronicke R, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci 2008; 28: 5965–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shim R, Wen SW, Wanrooy BJ, et al. Stroke severity, and not cerebral infarct location, increases the risk of infection. Transl Stroke Res 2020; 11: 387–401. [DOI] [PubMed] [Google Scholar]
- 14. Posel C, Moller K, Boltze J, Wagner DC, Weise G. Isolation and flow cytometric analysis of immune cells from the ischemic mouse brain. J Vis Exp 2016; 108: 53658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amarilla AA, Santos‐Junior NN, Figueiredo ML, et al. CCR2 plays a protective role in Rocio virus‐induced encephalitis by promoting macrophage infiltration into the brain. J Infect Dis 2019; 219: 2015–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wanrooy BJ, Wen SW, Wong CH. Dynamic roles of neutrophils in post‐stroke neuroinflammation. Immunol Cell Biol 2021; 99: 924–935. [DOI] [PubMed] [Google Scholar]
- 17. Liu Q, Johnson EM, Lam RK, et al. Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nat Immunol 2019; 20: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herz J, Sabellek P, Lane TE, Gunzer M, Hermann DM, Doeppner TR. Role of neutrophils in exacerbation of brain injury after focal cerebral ischemia in hyperlipidemic mice. Stroke 2015; 46: 2916–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai W, Liu S, Hu M, et al. Functional dynamics of neutrophils after ischemic stroke. Transl Stroke Res 2020; 11: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim E, Yang J, Beltran CD, Cho S. Role of spleen‐derived monocytes/macrophages in acute ischemic brain injury. J Cereb Blood Flow Metab 2014; 34: 1411–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wattananit S, Tornero D, Graubardt N, et al. Monocyte‐derived macrophages contribute to spontaneous long‐term functional recovery after stroke in mice. J Neurosci 2016; 36: 4182–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. ElAli A, Jean LBN. The role of monocytes in ischemic stroke pathobiology: new avenues to explore. Front Aging Neurosci 2016; 8: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma Y, Li Y, Jiang L, et al. Macrophage depletion reduced brain injury following middle cerebral artery occlusion in mice. J Neuroinflammation 2016; 13: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Planas AM. Role of immune cells migrating to the ischemic brain. Stroke 2018; 49: 2261–2267. [DOI] [PubMed] [Google Scholar]
- 25. Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol 2013; 2013: 746068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ritzel RM, Patel AR, Grenier JM, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation 2015; 12: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rajan WD, Wojtas B, Gielniewski B, Gieryng A, Zawadzka M, Kaminska B. Dissecting functional phenotypes of microglia and macrophages in the rat brain after transient cerebral ischemia. Glia 2019; 67: 232–245. [DOI] [PubMed] [Google Scholar]
- 28. Cai W, Dai X, Chen J, et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 2019; 4: e131355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stanley D, Mason LJ, Mackin KE, et al. Translocation and dissemination of commensal bacteria in post‐stroke infection. Nat Med 2016; 22: 1277–1284. [DOI] [PubMed] [Google Scholar]
- 30. Buetefisch CM. Role of the Contralesional hemisphere in post‐stroke recovery of upper extremity motor function. Front Neurol 2015; 6: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fury W, Park KW, Wu Z, et al. Sustained increases in immune transcripts and immune cell trafficking during the recovery of experimental brain ischemia. Stroke 2020; 51: 2514–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cuartero MI, de la Parra J, Perez‐Ruiz A, et al. Abolition of aberrant neurogenesis ameliorates cognitive impairment after stroke in mice. J Clin Invest 2019; 129: 1536–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McDonald B, Kubes P. Neutrophils and intravascular immunity in the liver during infection and sterile inflammation. Toxicol Pathol 2012; 40: 157–165. [DOI] [PubMed] [Google Scholar]
- 34. Denorme F, Portier I, Rustad JL, et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J Clin Invest 2022; 132: e154225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang L, Yu H, Yang X, et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun 2020; 11: 2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
All data presented in this study are included in the manuscript.