

Abstract
Uveal melanoma (UM) is the most common primary malignancy of the adult eye but lacks any FDA‐approved therapy for the deadly metastatic disease. Thus, there is a great need to dissect the driving mechanisms for UM and develop strategies to evaluate potential therapeutics. Using an autochthonous zebrafish model, we previously identified MITF, the master melanocyte transcription factor, as a tumor suppressor in GNAQQ209L‐driven UM. Here, we show that zebrafish mitfa‐deficient GNAQQ209L‐driven tumors significantly up‐regulate neural crest markers, and that higher expression of a melanoma‐associated neural crest signature correlates with poor UM patient survival. We further determined how the mitfa‐null state, as well as expression of GNAQQ209L, YAPS127A;S381A, or BRAFV600E oncogenes, impacts melanocyte lineage cells before they acquire the transformed state. Specifically, examination 5 days post‐fertilization showed that mitfa‐deficiency is sufficient to up‐regulate pigment progenitor and neural crest markers, while GNAQQ209L expression promotes a proliferative phenotype that is further enhanced by YAPS127A;S381A co‐expression. Finally, we show that this oncogene‐induced proliferative phenotype can be used to screen chemical inhibitors for their efficacy against the UM pathway. Overall, this study establishes that a neural crest signature correlates with poor UM survival, and describes an in vivo assay for preclinical trials of potential UM therapeutics.
Keywords: cell differentiation, drug screening assay, melanocyte, MITF, uveal melanoma, zebrafish
Uveal melanoma (UM) arises from transformed melanocytes in the uvea of the eye, which consists of the choroid, iris, and ciliary body (Jager et al., 2020). The primary tumor is treatable, however, approximately 50% of patients develop fatal metastases, predominately to the liver, for which there are no FDA‐approved treatments (Jager et al., 2020). Unfortunately, therapies that have been successful for cutaneous melanoma (CM), such as targeted therapies or immunotherapies, have not improved UM survival (Jager et al., 2020). Thus, there is a great need to gain a deeper understanding of UM, as well as to develop effective in vivo chemical screening strategies to identify therapeutic candidates.
The vast majority (~90%) of UM patients present with an activating mutation in GNAQ/11, or another component of its pathway (Fallico et al., 2021). Two major signaling axes downstream of GNAQ/11 have been implicated in UM: PLCβ4‐RasGRP3‐MAPK and Trio‐Rho/Rac‐FAK‐YAP (Chen et al., 2017; Feng et al., 2014, 2019; Moore et al., 2018; Yu et al., 2014). Notably, using an autochthonous zebrafish model, we have previously established that active YAP, and not PLCβ4, is sufficient to rapidly drive UM tumorigenesis (Phelps et al., 2022). This is in stark contrast to CM, which is largely driven by activating mutations in NRAS or BRAF, and is highly dependent upon MAPK pathway activation (Lee et al., 2011).
In many tumor types, dedifferentiation is a frequent hallmark of more aggressive disease, as dedifferentiated cells are often more plastic and proliferative (Jögi et al., 2012; Tang, 2012). This concept is well established for CM, which is particularly deadly if dedifferentiated (Müller et al., 2014; Rabbie et al., 2019). In the case of UM, BAP1 loss has been strongly implicated in poor prognosis (Harbour et al., 2010), and modeling BAP1 loss in UM cells in vitro caused dedifferentiation (Matatall et al., 2013). Consequently, BAP1 is hypothesized to function as a UM tumor suppressor by blocking dedifferentiation.
We previously developed a zebrafish model of UM in which a GNAQQ209L transgene is expressed in the melanocytic lineage using the promoter of the zebrafish master melanocyte transcription factor mitfa (Perez et al., 2018; Phelps et al., 2022). When combined with a tp53 mutant allele (tp53 M214K/M214K , herein simplified to tp53 −/−) the resulting Tg(mitfa:GNAQ Q209L );tp53 −/− transgenics develop tumors with all the core characteristics of human UM tumors (Perez et al., 2018; Phelps et al., 2022). We recently found that mitfa behaves as a potent tumor suppressor in this model (Phelps et al., 2022). Specifically, when combined with mitfa w2/w2 mutant alleles (henceforth simplified to mitfa −/−), the Tg(mitfa:GNAQ Q209L );tp53 −/− transgenic zebrafish develop tumors more rapidly, and with increased invasive phenotypes, compared to mitfa +/+ controls (Phelps et al., 2022). Moreover, mitfa deficiency was sufficient to cooperate with GNAQQ209L in UM formation even in the presence of wildtype tp53 (Phelps et al., 2022). Notably, mitfa is essential for melanocyte differentiation and survival, but also fully dispensable for the establishment and maintenance of melanocyte stem cells (Johnson et al., 2011), as well as for activation of the mitfa promoter that drives GNAQQ209L expression in our transgenics (Figure S1; Perez et al., 2018; Phelps et al., 2022). Thus, we hypothesize that MITF deficiency causes melanocyte lineage cells to be locked in a more progenitor‐like state, which somehow enables UM tumorigenesis.
We began this current study by asking whether our zebrafish mitfa‐deficient GNAQQ209L tumors are more dedifferentiated than their mitfa‐wildtype counterparts. For this, we analyzed bulk RNA‐sequencing data from Tg(mitfa:GNAQ Q209L );tp53 −/− (abbreviated to Qpm+), Tg(mitfa:GNAQ Q209L );tp53 −/− ;mitfa −/− (Qpm−), and Tg(mitfa:GNAQ Q209L );mitfa −/− (Qm−) UM tumors, as well as zebrafish Tg(mitfa:BRAF V600E );tp53 −/− (Bpm+) CM tumors for comparison (GSE190802). We determined the relative expression of various melanocyte lineage markers (Table S1) by first calculating the z‐score for each gene in the considered gene list, and then calculating the average z‐score for all genes in each gene set for each tumor (Figure 1a). We first examined the relative expression of differentiated melanocyte markers and, as expected, found them significantly down‐regulated in mitfa‐deficient UM tumors (Qpm− and Qm−), compared to mitfa‐wildtype UM (Qpm+) and also CM (Bpm+) tumors (Figure 1a, left panel). We then extended our analyses to consider gene sets associated with progressively more dedifferentiated states: pigment progenitors and neural crest cells. We observed that Bpm+ tumors had significantly higher expression of both programs compared to Qpm+ (Figure 1a, middle and right panels, respectively). This is consistent with prior literature that BRAFV600E‐driven zebrafish CMs re‐express the neural crest marker crestin at melanoma initiation (Kaufman et al., 2016). Importantly, these melanocytic progenitors and neural crest programs were also upregulated significantly in Qpm− and Qm− tumors compared to Qpm+ (Figure 1a, middle and right panels), with the neural crest program showing the highest degree of significance. Thus, in agreement with our prior histopathological observations (Phelps et al., 2022), these data establish that mitfa‐deficient GNAQQ209L‐driven tumors are more dedifferentiated than their mitfa‐WT counterparts, and show enrichment of both pigment progenitor and neural crest markers. We hypothesize that these dedifferentiation phenotypes serve to enable the accelerated onset and enhanced progression of GNAQQ209L‐driven tumorigenesis caused by the mitfa‐deficient background.
FIGURE 1.
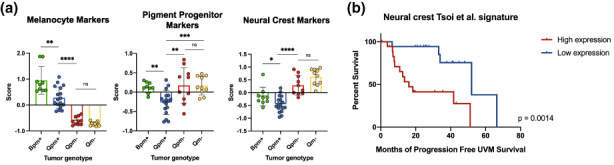
mitfa deficiency correlates with the upregulation of dedifferentiation markers in GNAQQ209L‐driven tumors. Genotypes (with abbreviations) for zebrafish tumor RNA‐sequencing data are as follows: Tg(mitfa:BRAF V600E );tp53 −/− (Bpm+), Tg(mitfa:GNAQ Q209L );tp53 −/− (Qpm+), Tg(mitfa:GNAQ Q209L );tp53 −/− ;mitfa −/− (Qpm−), and Tg(mitfa:GNAQ Q209L );tp53 −/− ;mitfa −/− (Qm−). (a) Expression of melanocyte, pigment progenitor, or neural crest marker genes in Bpm+, Qpm+, Qpm−, or Qm− zebrafish tumors. Zebrafish lineage marker gene lists were from Howard IV et al., 2021 (Table S1). Z‐scores were calculated for each gene within the gene set and then summed for each sample, and divided by the total number of genes in the list. Data are presented as mean ± SD. Statistical analyses were determined by Student's unpaired t‐test. Left panel: Melanocyte marker comparisons: Bpm+ vs. Qpm+ p = 0.0017**, Qpm+ vs. Qpm− or Qm− p < 0.0001****, Qpm− vs. Qm− p = 0.0532 n.s. Middle panel: Pigment progenitor comparisons: Bpm+ vs. Qpm+ p = 0.0017, Qpm+ vs. Qpm− p = 0.0065**, Qpm+ vs. Qm− p = 0.0008***, Qpm− vs. Qm− p = 0.9808 n.s. Right panel: Neural crest marker comparisons: Bpm+ vs. Qpm+ p = 0.0417*, Qpm+ vs. Qpm− or Qm− p < 0.0001****, Qpm− vs. Qm− p = 0.0511 n.s. (b) Application of a melanoma‐associated neural crest signature (Tsoi et al., 2018) to UVM patient survival obtained from TCGA (n = 80 patients). Kaplan–Meier curve of progression‐free UVM survival of patients with higher expression of this signature (upper quartile, n = 20 patients) compared to lower expression (bottom quartile, n = 20 patients). p = 0.0014 (determined by log‐rank test).
To determine if our findings are relevant to human UM, we interrogated RNA‐sequencing data from the single UM patient cohort (UVM cohort, n = 80) present in the Tumor Cancer Genome Atlas (TCGA). Previous work has identified unique transcriptional signatures of four different stages of melanocyte differentiation in human CM by comparing sequencing data from melanocytes with that from human embryonic stem cells induced to differentiate into neural crest cells, melanocyte progenitors, and melanocytes (Tsoi et al., 2018). This study found that the melanoma neural crest signature was enriched in invasive CM. We examined the melanoma neural crest signature in the UVM patient cohort (Figure 1b) and found that higher expression (upper quartile; n = 20) correlated with significantly decreased progression‐free UVM survival, compared to lower expression (lower quartile; n = 20). Overall, our data show that expression of neural crest markers correlates with accelerated UM development and shortened lifespan in our zebrafish UM model, as well as poor prognosis of human UM.
The development and progression of tumors require additional genetic and epigenetic changes beyond the initial tumor‐initiating mutation. The RNA‐sequencing analyses above were conducted in tumors, and thus reflect the holistic effect of accumulated genetic and epigenetic changes. Given this, we sought to determine how mitfa‐deficiency, with or without the expression of the UM‐driving oncogenes, GNAQQ209L and YAPS127A;S381A (henceforth simplified to YAPAA), or the CM‐driving BRAFV600E, impact melanocyte lineage cells before they acquire the transformed state. We had previously shown that GNAQQ209L alters the biological phenotypes of melanocytes in zebrafish larvae 5 days post‐fertilization (dpf; Perez et al., 2018), and thus selected this early time point for our analyses. For this study, we crossed no‐oncogene, Tg(mitfa:GNAQ Q209L ), or Tg(mitfa:BRAF V600E ) zebrafish to a Tg(mitfa:EGFP) reporter, to allow isolation of the melanocyte lineage cells, or utilized Tg(mitfa:YAP AA ;mitfa:EGFP) zebrafish, which already carry an EGFP marker. The YAPAA‐expressing zebrafish were on a tp53 +/− background, while the other genotypes were all tp53 +/+. Although tp53 +/− should be phenotypically equivalent to tp53 +/+, we included both tp53 +/− and tp53 +/+ no‐oncogene samples as controls. We conducted crosses to generate 500–1000 5 dpf zebrafish larvae for all of the genotypes (listed in Figure 2a along with their abbreviations), with n = 3–4 independent pools for each. For every sample, we removed the eyes, because they contain contaminating mitfa‐expressing RPE cells, and then dissociated the larvae and used fluorescence‐activated cell sorting (FACS) to isolate the mitfa:GFP+ cells. These samples were then subjected to bulk RNA‐sequencing (GSE197927) and computational analyses.
FIGURE 2.
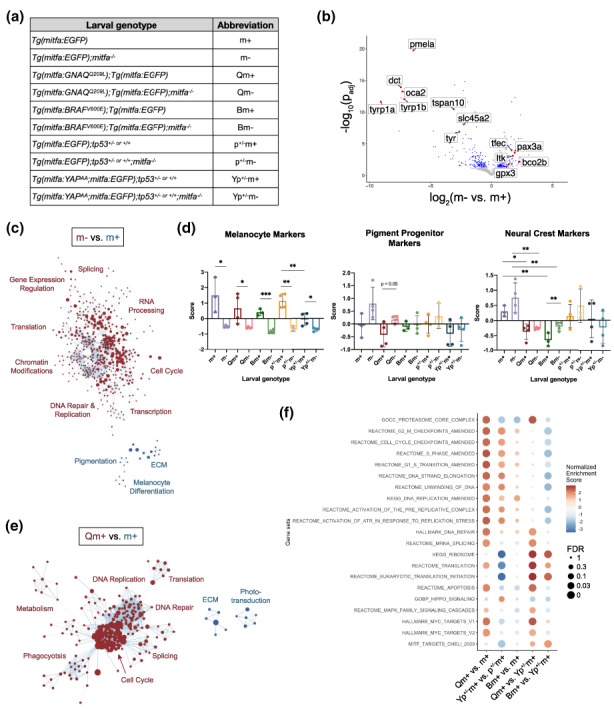
mitfa deficiency and GNAQQ209L expression drive proliferation programs in larval zebrafish melanocyte lineage cells. (a) Larval genotypes and abbreviations used in bulk RNA‐sequencing of 5 dpf mitfa:GFP+ cells. mitfa −/− = mitfa w2/w2 . Samples labeled tp53 +/− (= tp53 +/M214K ) were all derived from an intercross of tp53 +/− and tp53 +/+ fish and thus are a 50:50 mix of these two genotypes. (b) Volcano plot depicting differentially expressed genes between m+ and m− larval cells. Statistically significant (adjusted p‐value < 0.05) genes in blue or red, statistically insignificant (adjusted p‐value > 0.05) in grey. (c) Cytoscape enrichment map depiction of re‐occurring differential cellular processes in m+ vs. m− larval cells, determined by Gene Set Enrichment Analysis (GSEA), filtered to FDR q‐value < 0.05 (full results in Table S2). Red are programs upregulated in m+, blue are programs up in m−. The circle size denotes the number of genes in each gene set, length of connecting line depicts the amount of gene set overlap. (d) Expression of neural crest lineage markers in larval melanocyte lineage cells; genotype abbreviations listed in (a). Data are presented as mean ± SD and statistical analyses determined by Student's unpaired t‐test. Left panel: Melanocyte marker expression. m+ vs. m− p = 0.0139*, Qm+ vs. Qm− p = 0.0398*, Bm+ vs. Bm− p = 0.0002***, p+/−m+ vs. p+/−m− p = 0.0015**, Yp+/−m+ vs. Yp+/−m− p = 0.0212*, Yp+/−m+ vs p+/−m+ p = 0.0081**. Middle panel: Pigment progenitor marker expression. Statistical analyses were determined by Student's unpaired t‐test. m+ vs. m− p = 0.1075, Qm+ vs. Qm− p = 0.0506 n.s. Right panel: Neural crest marker expression. m+ vs. m− p = 0.1953, Qm+ vs. m+ p = 0.0113*, Qm− vs. m− p = 0.006**, Bm+ vs. Bm− p = 0.0215*, Bm+ vs. m+ p = 0.0045**, Bm− vs. m− p = 0.0095**, Yp+/−m− vs p+/−m− p = 0.1394 n.s. (e) Cytoscape enrichment map depiction of re‐occurring differential cellular processes in Qm+ vs. m+ larvae, determined by Gene Set Enrichment Analysis (GSEA), filtered to FDR q‐value < 0.05 (full results in Table S2). Red are programs upregulated in Qm+, blue are programs upregulated in m+. The circle size denotes the number of genes in each gene set, length of connecting line depicts the amount of gene set overlap. (f) Dot plot summary of GSEA results of different pair‐wise comparisons from the Tg(mitfa:EGFP) larval RNA‐sequencing dataset. Color represents NES and dot size represents FDR (full results and amended gene lists in Table S2).
We first performed principal component analysis on all samples and observed good co‐clustering of the genotype samples in most cases (Figure S2). Notably, the mitfa +/+ and mitfa −/− no‐oncogene samples each co‐clustered regardless of whether they were tp53 +/− or tp53 +/+, exactly as expected (Figure S2). Most importantly, PC1 largely divided samples based on mitfa status, and this separation was further enhanced by the added expression of the GNAQQ209L, YAPAA, or BRAFV600E oncogenes. Thus, mitfa‐deficiency has a profound impact on gene expression in untransformed melanocyte lineage cells. Given this finding, we looked closely at the specific genes differentially expressed between mitfa deficient (m−) versus wildtype (m+) larvae, in the absence of an oncogene (Figure 2b). As expected, melanocyte differentiation and pigmentation genes (pmela, dct, tyrp1a, tyrp1b, oca2, tspan10, tyr, and slc45a2; Howard IV et al., 2021) were significantly down‐regulated in m− versus m+ larvae. Moreover, markers of neural crest and/or pigment progenitors (pax3a, gpx3, tfec, ltk, and bco2b; Howard IV et al., 2021; Nikaido et al., 2021; Petratou et al., 2021) were significantly up‐regulated in m− versus m+, indicating that mitfa‐expressing m− cells are less differentiated. As anticipated, these differences were also largely observed in the no oncogene m− versus m+ samples that included tp53 +/− cells [Tg(mitfa:EGFP);tp53 +/− or +/+;mitfa −/− (abbreviated p+/−m−) versus Tg(mitfa:EGFP);tp53 +/− or +/+ (abbreviated p+/−m+)] (Figure S3). We note that some of these neural crests and/or pigment progenitors marker genes are also expressed in other zebrafish pigmented cell types, including iridophores (tfec, ltk; Petratou et al., 2021) and xanthophores (bco2b; Saunders et al., 2019). Iridophores do not express mitfa (Curran et al., 2010), and thus would not have been isolated in our study since they would be negative for EGFP. In contrast, prior single‐cell sequencing data revealed that a subset of xanthophores does express mitfa (Saunders et al., 2019). Thus, it is possible that xanthophores contribute some of the bco2b signals in our study, but it seems unlikely that these cells account for the differential expression levels between the mitfa wildtype and deficient states, as other known xanthophore markers (xdh, pax7a, gch2, aox5, and pax7b; Howard IV et al., 2021) were not significantly different between the mitfa‐wildtype and deficient larval cells. Consequently, the observed up‐regulation of pigment progenitor and/or neural crest marker genes strongly suggests that mitfa‐deficiency increases the representation of larval cells in a melanocyte precursor or neural crest state. Importantly, this mirrors the expression patterns of m− UM tumors, consistent with the notion that mitfa‐deficiency accelerates UM development and progression, at least in part, by enabling these less differentiated states.
We then further analyzed our sequencing data to identify cellular processes that differ between m− and m+ cells without oncogenic mutations. For this, we performed Gene Set Expression Analysis (GSEA) and display the significant results in a Cytoscape Enrichment Map (Figure 2c, full results in Table S2). This shows that m+ cells are enriched for pigmentation, melanocyte differentiation, and extracellular matrix programs (shown in blue), whereas m− cells show a striking enrichment for cell cycle, DNA repair and replication, chromatin modification, transcription, splicing, and translation programs (shown in red; Figure 2c). Importantly, these are the same programs that we previously found to be enriched in tumors that were Qpm− compared to Qpm+ (Phelps et al., 2022). Thus, we can now conclude that the pro‐proliferation and pro‐biosynthesis signatures observed in Qpm− tumors exist, at least to a certain extent, in the m− untransformed state, and are also independent of tp53‐status. Overall, our data show that the melanocytic lineage of m‐ larvae significantly up‐regulate neural crest and pigment progenitor marker genes, as well as pro‐proliferative processes, and these programs are hallmarks of the resulting mitfa‐deficient tumors.
Next, we evaluated the relative melanocyte differentiation states across all of our larval genotypes, including those carrying oncogenes (Figure 2a) using the method described in Figure 1a. Unsurprisingly, melanocyte markers were significantly down‐regulated in mitfa −/− larval samples, relative to their mitfa +/+ counterpart, regardless of the oncogenic driver (Figure 2d, left panel). Interestingly, larval samples that were mitfa +/+ and carried constitutively active YAP [Tg(mitfa:YAP AA ;mitfa:EGFP);tp53 +/− or +/+ (abbreviated Ym+)] expressed significantly lower levels of melanocyte markers than any of the other m+ genotypes, suggesting that YAPAA leads to a less differentiated state in the mitfa WT background (Figure 2d, left panel). Consistent with the differentiation gene analyses above (Figure 2b), the no‐oncogene m− and p+/−m− samples both displayed pigment progenitor and neural crest marker gene scores that trended higher than no‐oncogene m+ and p+/−m+ (Figure 2d, middle and right panels). Somewhat unexpectedly, the added presence of the GNAQQ209L, YAPAA, or BRAFV600E oncogenes tended to lower the expression of these dedifferentiation markers, for both the m− and m+ states (Figure 2d, middle and right panels). This was particularly striking for neural crest markers, which were significantly lowered by the presence of either GNAQQ209L or BRAFV600E in both the m− and m+ background, compared to their no‐oncogene controls (Figure 2d, right panel). Since the oncogenes did not increase the levels of melanocyte markers, compared to their no‐oncogene controls, it seems unlikely that the oncogenes serve to promote differentiation. An alternative possibility is that they perturb melanocyte differentiation, such that the cells diverge from the typical melanocytic, pigment progenitor, and neural crest programs and/or display a hybrid state. Clearly, additional experiments will be required to assess this possibility.
We also considered the impact of oncogenic GNAQ on cellular processes in melanocyte lineage cells in the larvae in a mitfa WT background. We performed GSEA and show key significant results in a Cytoscape enrichment map (Figure 2e, complete results in Table S2). Notably, expression of GNAQQ209L alone had a similar effect on cellular processes in larval cells as mitfa deficiency alone; Tg(mitfa:GNAQ Q209L );Tg(mitfa:EGFP; abbreviated Qm+) showed significant upregulation of cell cycle, DNA replication, transcription, splicing, and translation programs relative to no‐oncogene m+ controls, which is strikingly similar to the impact of mitfa‐deficiency on larval cells in the no‐oncogene context (Figure 2c). Qm+ were also particularly enriched for proteasome‐related genes, and we noted that these are abundant within GSEA proliferation gene sets. Thus, we removed proteasome‐related genes from those proliferation gene sets (marked “_Amended”, Table S2, Figure 2f) and re‐ran GSEA to specifically consider changes in the expression of proliferation regulators. These amended gene sets were significantly enriched in Qm+, showing that the proliferation signature enrichment truly reflected induction of cell cycle programs, and was not simply driven by proteasome genes (Figure 2f). This offers a mechanistic explanation for our prior observations that GNAQQ209L expression leads to melanocyte defects, including an increased number of overall melanocytes, in zebrafish larvae at 5 dpf (Perez et al., 2018). In concert with the up‐regulation of proliferation and translation gene sets, our analyses showed that the GNAQQ209L‐expressing cells, as well as the no‐oncogene mitfa‐deficient cells, are significantly enriched for MYC target genes relative to no‐oncogene m+ controls (Figure 2f, complete results in Table S2). This is highly reminiscent of the upregulation of MYC targets which is a hallmark of Qpm− tumors relative to their Qpm+ counterparts (Phelps et al., 2022). This observation, as well as the finding that GNAQQ209L expression or mitfa‐deficiency alone in un‐transformed larval cells leads to upregulation of similar programs as Qpm− tumors relative to Qpm+ counterparts, provides a possible mechanistic understanding of how the combination of GNAQQ209L expression and mitfa‐deficiency accelerates tumorigenesis.
We also considered how YAPAA and BRAFV600E modulate m+ melanocyte lineage cells, in comparison to GNAQQ209L (Figure 2f, complete results in Table S2). YAPAA also induced significant up‐regulation of cell cycle and DNA replication programs, including the amended (Δ proteosome) gene sets, albeit to a lesser extent than GNAQQ209L, whereas BRAFV600E had no significant effect on these gene sets (Figure 2f). YAPAA diverged from GNAQQ209L in other regards. Neither it, nor BRAFV600E, up‐regulated the splicing, translation, or MYC target gene data sets that were induced by GNAQQ209L (Figure 2f). Indeed, translation processes were significantly down‐regulated in YAPAA relative to its no‐transgene control (Figure 2f). In contrast to either GNAQQ209L or BRAFV600E, YAPAA up‐regulated Hippo signaling, although this was not significant. Additionally, as described above (see Figure 2d, left panel), YAPAA significantly down‐regulated MITF target genes (Figure 2f). Finally, and somewhat unexpectedly, the BRAFV600E‐expressing cells were not enriched for MAPK signaling, compared to the no‐transgene control (Figure 2f). Since MAPK signaling is the predominant effector pathway in BRAFV600E‐driven CM, including in zebrafish tumors, we conclude that this must be up‐regulated to enable these cells to transition into the tumorigenic state.
We previously reported that GNAQQ209L expression leads to an increased number of mature melanocytes in zebrafish larvae by 5 dpf, suggesting that GNAQQ209L promotes melanocyte proliferation (Perez et al., 2018). To further explore this, we utilized FACS to determine the representation of mitfa:GFP+ (herein referred to as “GFP+”) melanocyte lineage cells in dissociated 5 dpf larvae expressing GNAQQ209L, YAPAA, or no‐oncogene (Figure 3a). Qm+ larvae contain a significantly higher proportion of GFP+ cells than no‐oncogene m+ controls, consistent with the previously observed increase in mature melanocytes (Perez et al., 2018), and also the up‐regulation of proliferation programs in Qm+ larvae (Figure 3a, left panel). In contrast, the YAPAA expressing (Ym+) larvae did not display a higher representation of GFP+ cells (Figure 3a, right panel). Since the mitfa:EGFP reporter differs between our YAPAA and GNAQQ209L models (i.e., has distinct integration sites), we cannot rule out that the Ym+ cells have weaker GFP expression, and thus are not as well detected by FACS. However, the lower representation of GFP+ cells in YAPAA versus GNAQQ209L is entirely consistent with the more modest upregulation of proliferation gene sets in Yp+/−m+ versus Qm+ GFP+ cells (Figure 3a).
FIGURE 3.
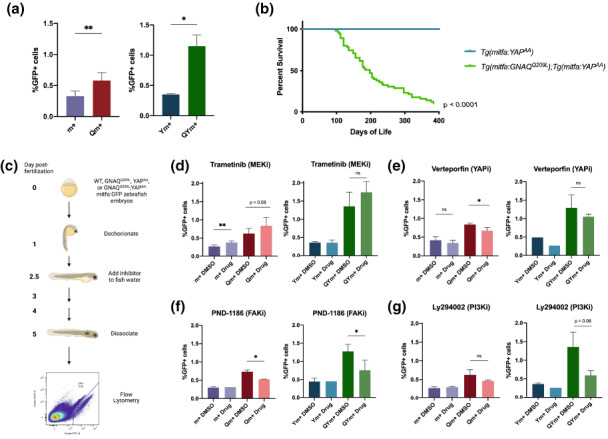
GNAQQ209L and YAPAA cooperate to drive tp53‐WT tumorigenesis and YAP pathway inhibitors decrease the proportion of mitfa‐expressing cells in GNAQQ209L larvae. Genotype abbreviations for larvae are in Figure 2a. (a) The proportion of mitfa:GFP+ in m+, Qm+, Ym+, or QYm+ larvae as determined by flow cytometry. Data are presented as mean ± SD. Number of replicates: m+ n = 4, Qm+ n = 10, Ym+ n = 2, QYm+ n = 2. Statistical analyses determined by Student's unpaired t‐test: Qm+ vs. m+ p = 0.0036**, QYm+ vs. Ym+ p = 0.0225. (b) Kaplan–Meier of overall survival. Cohort sizes: QYm+ n = 55 zebrafish, Ym+ n = 60 zebrafish. Survival statistics determined by log‐rank test: QYm+ vs. Ym+ p < 0.0001. (c) Experimental protocol of the zebrafish larvae chemical inhibitor assay (schematic created with BioRender.com). (d–g) m+, Qm+, Ym+, and QYm+ were treated with DMSO or a chemical inhibitor following the protocol depicted in (c). The number of replicates for each panel is as follows. Trametinib: m+ DMSO n = 6, Drug n = 5; Qm+ DMSO n = 6, Drug n = 5; Ym+ DMSO n = 2, Drug n = 2; QYm+ DMSO n = 4, Drug n = 3. Verteporfin: m+ DMSO n = 4, Drug n = 6; Qm+ DMSO n = 3, Drug n = 6; Ym+ DMSO n = 1, Drug n = 1; QYm+ DMSO n = 2, Drug QY n = 3. PND‐1186: m+ DMSO n = 3, Drug n = 1; Qm+ DMSO n = 2, Drug Qm+ n = 2; Ym+ DMSO n = 2, Drug n = 1; QYm+ DMSO n = 5, Drug n = 2. LY294002: m+ DMSO n = 6, Drug m+ n = 2; Qm+ DMSO n = 6, Drug Qm+ n = 2; Ym+ DMSO n = 2, Drug n = 1; QYm+ DMSO n = 4, Drug QYm+ n = 2. Data are presented as mean ± SD. Statistical analyses (determined by Student's unpaired t‐test) are as follows. m+ Drug vs. m+ DMSO: Trametinib p = 0.0014**, Verteporfin p = 0.19 n.s., PND‐1186 p = 0.6914 n.s., Ly294002 p = 0.4507 n.s. Qm+ Drug vs. Qm+ DMSO: Trametinib p = 0.0909 n.s, Verteporfin p = 0.0146*, PND‐1186 p = 0.0272*, Ly294002 p = 0.1795 n.s. Ym+ Drug vs. Ym+ DMSO: Trametinib p = 0.9397 n.s., Verteporfin n = 1, PND‐1186 p = 0.9717 n.s., Ly294002 p = 0.2123 n.s. QYm+ Drug vs. QYm+ DMSO: Trametinib p = 0.2125 n.s., Verteporfin p = 0.3039 n.s., PND‐1186 p = 0.0328, Ly294002 p = 0.0643 n.s.
Since GNAQQ209L and YAPAA both upregulate proliferation programs, and active GNAQ and nuclear YAP occur concurrently in patient UM, we wondered whether GNAQQ209L and YAPAA could cooperate to drive melanocytic lineage proliferation. We generated Tg(mitfa:YAP AA ;mitfa:EGFP); Tg(mitfa:GNAQ Q209L );tp53‐WT;mitfa‐WT (QYm+) larvae and found that the representation of GFP+ cells was increased approximately threefold compared to Ym+, and twofold compared to Qm+, indicating that co‐expression of these oncogenes amplifies the proliferation phenotype (Figure 3a, right panel). Given this finding, we wondered whether these combined oncogenes would also promote tumorgenicity. Previously, we found that Qm+ zebrafish develop tumors rarely, and with long latency, but in combination with tp53‐loss develop UM with complete penetrance (Perez et al., 2018). Similarly, Ym+ only develops tumors in combination with tp53‐loss (Figure S4). In stark contrast, GNAQQ209L and YAPAA were sufficient to drive rapid tumorigenesis in the presence of wildtype tp53 (Figure 3b; p < 0.0001 for QYm+ vs. Ym+). Thus, we conclude that GNAQQ209L and YAPAA cooperate to increase the proportion of melanocyte lineage cells and rapidly drive tumorigenesis. This suggests that the increased proportion of melanocytes at 5 dpf may be an indicator of tumorigenicity.
Given the desperate need for UM therapies, we asked whether the larval proliferative phenotype could be used as a readout for in vivo drug screens. Specifically, we wanted to identify inhibitors that would suppress the elevated representation of GFP+ cells in Qm+ and QYm+, but not the m+ control, larvae. Additionally, as YAP seems to be the dominant effector pathway in UM (Phelps et al., 2022), we also included Ym+ larvae. For this assay, we crossed zebrafish to yield 60–80 embryos for each genotype and sample, which were placed in Petri dishes and then treated at 30 hours post‐fertilization (hpf) with chemical inhibitors or vehicle by addition to the fish water. The representation of GFP+ cells was quantified at 5 dpf using our dissociation/FACS protocol depicted in Figure 3c. We focused on compounds that had been previously tested in various UM contexts (cells, mouse models, and/or patients) to determine the degree to which our zebrafish assay would recapitulate previously observed results.
We first studied the impact of MEK inhibition on the representation of m+, Qm+, Y+, or QYm+ melanocyte lineage cells. Although PKC/MAPK inhibitors decreased the viability of human UM cell lines in some studies, they were found to be ineffective in both a UM mouse model and UM clinical trials (Gonçalves et al., 2020; Jager et al., 2020; Moore et al., 2018). Consistent with the mouse and human results, we recently reported that PLCβ‐ERK signaling is down‐regulated in GNAQQ209L‐driven tumors in the mitfa‐deficient background, suggesting that it is largely dispensable and that decreased expression of MAPK transcriptional targets correlates with poor UM patient prognosis (Phelps et al., 2022). Aligning with the mouse, zebrafish and human tumor results, our larval drug screen showed that Trametinib (MEKi) failed to decrease, and even trended towards increasing, the representation of GFP+ cells in Qm+ or QYm+ larvae, and did significantly increase their representation in m+ larvae (Figure 3d). These results raise the possibility that MEK/ERK signaling is anti‐proliferative in the larval melanocytic lineages. Moreover, they support the use of our larvae assay as a valid preclinical screen for UM inhibitors, as they mirror the known effects of MEKi in both mouse UM studies and clinical trials.
We next extended our proof‐of‐principle studies to determine the impact of inhibition of the major UM downstream effector, YAP. To do this, we first treated m+, Qm+, Y+, and QYm+ larvae with Verteporfin, which disrupts the YAP/TAZ‐TEAD interaction (Giraud et al., 2020; Liu‐Chittenden et al., 2012), but has also been found to be non‐specific and toxic (Zhang et al., 2015). Relative to their DMSO controls, Verteporfin reduced the representation of GFP+ cells in Qm+ larvae significantly (p = 0.0146), as well in both QYm+ and Ym+, although neither reached significance (Figure 3e). This finding is consistent with the notion that YAP/TAZ‐TEAD inhibitors can impede the increase in the proportion of melanocyte lineage cells driven by UM oncogenes but needs to be interpreted with caution given Verteporfin's known toxicity. Given this, we extended our analyses to another GNAQ‐YAP pathway inhibitor. Specifically, FAK was recently identified as an upstream activator of YAP in UM, and a FAK inhibitor, PND‐1186, has been shown to impede UM cell line and transplantation survival (Feng et al., 2019). We tested PND‐1186 in our assay and found that this significantly decreased the proportion of GFP+ cells in both Qm+ and QYm+ larvae, but not the m+ controls (Figure 3f). Notably, PND‐1186 also had no effect on Ym+, exactly as expected since YAPAA is constitutively active and thus FAK‐independent. This indicates that the GNAQ‐FAK‐YAP pathway is important for the GNAQ‐driven mitfa‐expressing proliferation phenotype, and argues strongly that the heightened proliferation phenotype in QYm+ relies, at least in part, on GNAQ activation of YAP/TAZ. Most importantly, our findings underscore previous conclusions that FAK inhibition is a promising UM therapeutic.
Finally, we sought to inhibit another downstream effector of GNAQ/11‐PLCβ4‐RasGRP3, PI3K‐Akt, using the PI3K inhibitor, Ly294002. This has not been previously tested in in vivo UM models but was shown to be a promising inhibitor against UM cell lines in vitro (Babchia et al., 2010; Farhan et al., 2021; Ye et al., 2008). In a preliminary experiment, Ly294002 decreased the proportion of GFP+ cells in both Qm+ and QYm+ larvae (Figure 3g). Further experiments are required, due to the small sample size, to determine if this particular PI3K inhibitor, or newer PI3K inhibitors not yet tested in UM cell lines, could be a promising in vivo therapy against UM.
In summary, this study determined that mitfa‐deficiency and/or GNAQ oncogenic signaling drive the expression of melanocyte lineage precursor genes and proliferation programs. It also revealed that high expression of a neural crest transcriptional signature correlates with decreased UM progression‐free patient survival, emphasizing the negative impact of dedifferentiation on UM prognosis. These findings are not entirely unexpected, given that dedifferentiation is a hallmark of aggressiveness in many cancers (Jögi et al., 2012). It also fits with prior observations in UM. First, loss of BAP1, which is located on chromosome 3, is thought to enable UM metastasis by promoting dedifferentiation (Matatall et al., 2013), although this has yet to be demonstrated in in vivo UM models. Second, we recently reported that the absence of MITF, which also resides on chromosome 3, promotes the development and aggressiveness of zebrafish UM tumors, which are associated with a more dedifferentiated state (Phelps et al., 2022). Our current study shows that this dedifferentiated phenotype is a very early consequence of mitfa‐deficiency and/or GNAQ oncogenic signaling, well before these cells achieved the transformed state. Finally, we have identified a proliferation phenotype in both GNAQQ209L and GNAQQ209L;YAPAA larvae, which can be harnessed to screen for candidate UM therapeutics in vivo. This assay corroborated prior findings that FAK, and not MEK, inhibition is an effective therapeutic against UM in in vivo mouse models (Feng et al., 2019; Moore et al., 2018) and suggests that PI3K inhibitors also merit further evaluation. We believe that this method will be a useful resource for the field to conduct preclinical trials of candidate UM therapeutics.
FUNDING INFORMATION
Funding support was from the Ludwig Center at MIT (J.A.L.); the National Cancer Institute for the Koch Institute Support (core) Grant P30‐CA14051; the NIH Pre‐Doctoral Training Grant T32GM007287 (G.B.P. and H.R.H.); an MIT School of Science Fellowship in Cancer Research (H.R.H.); and a David H. Koch Graduate Fellowship (G.B.P.). J.A.L. is the D.K. Ludwig Professor for Cancer Research at the Koch Institute for Integrative Cancer Research.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Appendix S1
Table S1
Table S2
ACKNOWLEDGEMENTS
We thank Sofia Hu and Clare L. Phelps for assistance with computational analyses, and the Koch Institute Swanson Biotechnology Center, particularly the Zebrafish, Flow Cytometry, and Integrated Genomics and Bioinformatics cores for technical support.
Phelps, G. B. , Amsterdam, A. , Hagen, H. R. , García, N. Z. , & Lees, J. A. (2022). MITF deficiency and oncogenic GNAQ each promote proliferation programs in zebrafish melanocyte lineage cells. Pigment Cell & Melanoma Research, 35, 539–547. 10.1111/pcmr.13057
DATA AVAILABILITY STATEMENT
All data needed to evaluate the conclusions of the paper are present in the paper or Supporting Information. RNA‐seq data are available from the Gene Expression Omnibus under the accession number GSE197927.
REFERENCES
- Babchia, N. , Calipel, A. , Mouriaux, F. , Faussat, A.‐M. , & Mascarelli, F. (2010). The PI3K/Akt and mTOR/P70S6K signaling pathways in human uveal melanoma cells: Interaction with B‐Raf/ERK. Investigative Ophthalmology & Visual Science, 51(1), 421–429. 10.1167/iovs.09-3974 [DOI] [PubMed] [Google Scholar]
- Chen, X. , Wu, Q. , Depeille, P. , Chen, P. , Thornton, S. , Kalirai, H. , Coupland, S. E. , Roose, J. P. , & Bastian, B. C. (2017). RasGRP3 mediates MAPK pathway activation in GNAQ mutant uveal melanoma. Cancer Cell, 31(5), 685–696.e6. 10.1016/j.ccell.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, K. , Lister, J. A. , Kunkel, G. R. , Prendergast, A. , Parichy, D. M. , & Raible, D. W. (2010). Interplay between Foxd3 and Mitf regulates cell fate plasticity in the zebrafish neural crest. Developmental Biology, 344(1), 107–118. 10.1016/j.ydbio.2010.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallico, M. , Raciti, G. , Longo, A. , Reibaldi, M. , Bonfiglio, V. , Russo, A. , Caltabiano, R. , Gattuso, G. , Falzone, L. , & Avitabile, T. (2021). Current molecular and clinical insights into uveal melanoma (review). International Journal of Oncology, 58(4), 10. 10.3892/ijo.2021.5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan, M. , Silva, M. , Xingan, X. , Zhou, Z. , & Zheng, W. (2021). Artemisinin inhibits the migration and invasion in uveal melanoma via inhibition of the PI3K/AKT/mTOR signaling pathway. Oxidative Medicine and Cellular Longevity, 2021, 9911537. 10.1155/2021/9911537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, X. , Degese, M. S. , Iglesias‐Bartolome, R. , Vaque, J. P. , Molinolo, A. A. , Rodrigues, M. , Zaidi, M. R. , Ksander, B. R. , Merlino, G. , Sodhi, A. , Chen, Q. , & Gutkind, J. S. (2014). Hippo‐independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio‐regulated rho GTPase signaling circuitry. Cancer Cell, 25(6), 831–845. 10.1016/j.ccr.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, X. , Arang, N. , Rigiracciolo, D. C. , Lee, J. S. , Yeerna, H. , Wang, Z. , Lubrano, S. , Kishore, A. , Pachter, J. A. , König, G. M. , Maggiolini, M. , Kostenis, E. , Schlaepfer, D. D. , Tamayo, P. , Chen, Q. , Ruppin, E. , & Gutkind, J. S. (2019). A platform of synthetic lethal gene interaction networks reveals that the GNAQ uveal melanoma oncogene controls the hippo pathway through FAK. Cancer Cell, 35(3), 457–472.e5. 10.1016/j.ccell.2019.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud, J. , Molina‐Castro, S. , Seeneevassen, L. , Sifré, E. , Izotte, J. , Tiffon, C. , Staedel, C. , Boeuf, H. , Fernandez, S. , Barthelemy, P. , Megraud, F. , Lehours, P. , Dubus, P. , & Varon, C. (2020). Verteporfin targeting YAP1/TAZ‐TEAD transcriptional activity inhibits the tumorigenic properties of gastric cancer stem cells. International Journal of Cancer, 146(8), 2255–2267. 10.1002/ijc.32667 [DOI] [PubMed] [Google Scholar]
- Gonçalves, J. , Emmons, M. F. , Faião‐Flores, F. , Aplin, A. E. , Harbour, J. W. , Licht, J. D. , Wink, M. R. , & Smalley, K. S. M. (2020). Decitabine limits escape from MEK inhibition in uveal melanoma. Pigment Cell & Melanoma Research, 33(3), 507–514. 10.1111/pcmr.12849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour, J. W. , Onken, M. D. , Roberson, E. D. O. , Duan, S. , Cao, L. , Worley, L. A. , Council, M. L. , Matatall, K. A. , Helms, C. , & Bowcock, A. M. (2010). Frequent mutation of BAP1 in metastasizing uveal melanomas. Science (New York, N.Y.), 330(6009), 1410–1413. 10.1126/science.1194472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard, A. G., IV , Baker, P. A. , Ibarra‐García‐Padilla, R. , Moore, J. A. , Rivas, L. J. , Tallman, J. J. , Singleton, E. W. , Westheimer, J. L. , Corteguera, J. A. , & Uribe, R. A. (2021). An atlas of neural crest lineages along the posterior developing zebrafish at single‐cell resolution. eLife, 10, e60005. 10.7554/eLife.60005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager, M. J. , Shields, C. L. , Cebulla, C. M. , Abdel‐Rahman, M. H. , Grossniklaus, H. E. , Stern, M.‐H. , Carvajal, R. D. , Belfort, R. N. , Jia, R. , Shields, J. A. , & Damato, B. E. (2020). Uveal melanoma. Nature Reviews. Disease Primers, 6(1), 24. 10.1038/s41572-020-0158-0 [DOI] [PubMed] [Google Scholar]
- Jögi, A. , Vaapil, M. , Johansson, M. , & Påhlman, S. (2012). Cancer cell differentiation heterogeneity and aggressive behavior in solid tumors. Upsala Journal of Medical Sciences, 117(2), 217–224. 10.3109/03009734.2012.659294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, S. L. , Nguyen, A. N. , & Lister, J. A. (2011). Mitfa is required at multiple stages of melanocyte differentiation but not to establish the melanocyte stem cell. Developmental Biology, 350(2), 405–413. 10.1016/j.ydbio.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, C. K. , Mosimann, C. , Fan, Z. P. , Yang, S. , Thomas, A. , Ablain, J. , Tan, J. L. , Fogley, R. D. , van Rooijen, E. , Hagedorn, E. , Ciarlo, C. , White, R. , Matos, D. , Puller, A.‐C. , Santoriello, C. , Liao, E. , Young, R. A. , & Zon, L. I. (2016). A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science (New York, N.Y.), 351(6272), aad2197. 10.1126/science.aad2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.‐H. , Choi, J.‐W. , & Kim, Y.‐S. (2011). Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta‐analysis. British Journal of Dermatology, 164(4), 776–784. 10.1111/j.1365-2133.2010.10185.x [DOI] [PubMed] [Google Scholar]
- Liu‐Chittenden, Y. , Huang, B. , Shim, J. S. , Chen, Q. , Lee, S.‐J. , Anders, R. A. , Liu, J. O. , & Pan, D. (2012). Genetic and pharmacological disruption of the TEAD‐YAP complex suppresses the oncogenic activity of YAP. Genes & Development, 26(12), 1300–1305. 10.1101/gad.192856.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall, K. A. , Agapova, O. A. , Onken, M. D. , Worley, L. A. , Bowcock, A. M. , & Harbour, J. W. (2013). BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer, 13, 371. 10.1186/1471-2407-13-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, A. R. , Ran, L. , Guan, Y. , Sher, J. J. , Hitchman, T. D. , Zhang, J. Q. , Hwang, C. , Walzak, E. G. , Shoushtari, A. N. , Monette, S. , Murali, R. , Wiesner, T. , Griewank, K. G. , Chi, P. , & Chen, Y. (2018). GNA11 Q209L mouse model reveals RasGRP3 as an essential signaling node in uveal melanoma. Cell Reports, 22(9), 2455–2468. 10.1016/j.celrep.2018.01.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, J. , Krijgsman, O. , Tsoi, J. , Robert, L. , Hugo, W. , Song, C. , Kong, X. , Possik, P. A. , Cornelissen‐Steijger, P. D. M. , Geukes Foppen, M. H. , Kemper, K. , Goding, C. R. , McDermott, U. , Blank, C. , Haanen, J. , Graeber, T. G. , Ribas, A. , Lo, R. S. , & Peeper, D. S. (2014). Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nature Communications, 5, 5712. 10.1038/ncomms6712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido, M. , Subkhankulova, T. , Uroshlev, L. A. , Kasianov, A. J. , Sosa, K. C. , Bavister, G. , Yang, X. , Rodrigues, F. S. L. M. , Carney, T. J. , Schwetlick, H. , Dawes, J. H. P. , Rocco, A. , Makeev, V. , & Kelsh, R. N. (2021). Zebrafish pigment cells develop directly from persistent highly multipotent progenitors. bioRxiv. 10.1101/2021.06.17.448805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, D. E. , Henle, A. M. , Amsterdam, A. , Hagen, H. R. , & Lees, J. A. (2018). Uveal melanoma driver mutations in GNAQ/11 yield numerous changes in melanocyte biology. Pigment Cell & Melanoma Research, 31(5), 604–613. 10.1111/pcmr.12700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petratou, K. , Spencer, S. A. , Kelsh, R. N. , & Lister, J. A. (2021). The MITF paralog tfec is required in neural crest development for fate specification of the iridophore lineage from a multipotent pigment cell progenitor. PLoS ONE, 16(1), e0244794. 10.1371/journal.pone.0244794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps, G. B. , Hagen, H. R. , Amsterdam, A. , & Lees, J. A. (2022). MITF deficiency accelerates GNAQ‐driven uveal melanoma. Proceedings of the National Academy of Sciences of the United States of America, 119(19), e2107006119. 10.1073/pnas.2107006119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbie, R. , Ferguson, P. , Molina‐Aguilar, C. , Adams, D. J. , & Robles‐Espinoza, C. D. (2019). Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. The Journal of Pathology, 247(5), 539–551. 10.1002/path.5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, L. M. , Mishra, A. K. , Aman, A. J. , Lewis, V. M. , Toomey, M. B. , Packer, J. S. , Qiu, X. , McFaline‐Figueroa, J. L. , Corbo, J. C. , Trapnell, C. , & Parichy, D. M. (2019). Thyroid hormone regulates distinct paths to maturation in pigment cell lineages. eLife, 8, e45181. 10.7554/eLife.45181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, D. G. (2012). Understanding cancer stem cell heterogeneity and plasticity. Cell Research, 22(3), 457–472. 10.1038/cr.2012.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi, J. , Robert, L. , Paraiso, K. , Galvan, C. , Sheu, K. M. , Lay, J. , Wong, D. J. L. , Atefi, M. , Shirazi, R. , Wang, X. , Braas, D. , Grasso, C. S. , Palaskas, N. , Ribas, A. , & Graeber, T. G. (2018). Multi‐stage differentiation defines melanoma subtypes with differential vulnerability to drug‐induced iron‐dependent oxidative stress. Cancer Cell, 33(5), 890–904.e5. 10.1016/j.ccell.2018.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, M. , Hu, D. , Tu, L. , Zhou, X. , Lu, F. , Wen, B. , Wu, W. , Lin, Y. , Zhou, Z. , & Qu, J. (2008). Involvement of PI3K/Akt signaling pathway in hepatocyte growth factor–induced migration of uveal melanoma cells. Investigative Ophthalmology & Visual Science, 49(2), 497–504. 10.1167/iovs.07-0975 [DOI] [PubMed] [Google Scholar]
- Yu, F.‐X. , Luo, J. , Mo, J.‐S. , Liu, G. , Kim, Y. C. , Meng, Z. , Zhao, L. , Peyman, G. , Ouyang, H. , Jiang, W. , Zhao, J. , Chen, X. , Zhang, L. , Wang, C.‐Y. , Bastian, B. C. , Zhang, K. , & Guan, K.‐L. (2014). Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell, 25(6), 822–830. 10.1016/j.ccr.2014.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Ramakrishnan, S. K. , Triner, D. , Centofanti, B. , Maitra, D. , Győrffy, B. , Sebolt‐Leopold, J. S. , Dame, M. K. , Varani, J. , Brenner, D. E. , Fearon, E. R. , Omary, M. B. , & Shah, Y. M. (2015). Tumor‐selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Science Signaling, 8(397), ra98. 10.1126/scisignal.aac5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Table S1
Table S2
Data Availability Statement
All data needed to evaluate the conclusions of the paper are present in the paper or Supporting Information. RNA‐seq data are available from the Gene Expression Omnibus under the accession number GSE197927.