

Abstract
Macrocyclic metal porphyrin complexes can act as shape‐selective catalysts mimicking the action of enzymes. To achieve enzyme‐like reactivity, a mechanistic understanding of the reaction at the molecular level is needed. We report a mechanistic study of alkene epoxidation by the oxidant iodosylbenzene, mediated by an achiral and a chiral manganese(V)oxo porphyrin cage complex. Both complexes convert a great variety of alkenes into epoxides in yields varying between 20–88 %. We monitored the process of the formation of the manganese(V)oxo complexes by oxygen transfer from iodosylbenzene to manganese(III) complexes and their reactivity by ion mobility mass spectrometry. The results show that in the case of the achiral cage complex the initial iodosylbenzene adduct is formed on the inside of the cage and in the case of the chiral one on the outside of the cage. Its decomposition leads to a manganese complex with the oxo ligand on either the inside or outside of the cage. These experimental results are confirmed by DFT calculations. The oxo ligand on the outside of the cage reacts faster with a substrate molecule than the oxo ligand on the inside. The results indicate how the catalytic activity of the macrocyclic porphyrin complex can be tuned and explain why the chiral porphyrin complex does not catalyze the enantioselective epoxidation of alkenes.
Keywords: Catalysis, Manganese(V)oxo complex, Mass spectrometry, Porphyrin cage, Supramolecular chemistry
Ion mobility mass spectrometry reveals that a manganese (III) porphyrin cage reacts with the oxidant iodosylbenzene to form an inside iodosylbenzene complex, which is converted into a manganese(V)oxo complex in which the oxo group is located inside the cage (left). It reacts within the cage with alkenes to form epoxides. When the cage is blocked by methyl groups the reaction with iodosylbenzene and the formation of the manganese(V)oxo complex occurs at the outside of the cage (right). Subsequent reaction with alkenes takes place at the outside.

Introduction
Cytochrome P450 is one of the most extensively studied oxygen transferring enzymes. [1] In its active site a heme structure is present, which is capable of catalyzing oxidation reactions. [2] The binding of a substrate initiates a series of chemical events, in which molecular oxygen is activated by the heme to generate a high valent iron‐oxo complex, which transfers its oxygen to the bound substrate. [3] The enzyme contains a binding pocket, which facilitates the formation of the oxo‐species and increases the catalytic efficiency of the reaction.[ 4 , 5 , 6 ] Such a cavity‐driven catalysis[ 7 , 8 ] is an important aspect that should be considered if one wants to design and construct synthetic systems mimicking the action of cytochrome P450 enzymes. [9]
A possible mimic of a binding pocket with a nearby catalytic reaction site is the porphyrin cage compound H2C (Figure 1).[ 10 , 11 , 12 ] It is based on the concave building block glycoluril, which is capped with a porphyrin ligand. The glycoluril cage compounds can bind low‐molecular weight substrates [10] and thread and bind onto polymers.[ 13 , 14 ] After insertion of a manganese center into the porphyrin, the resulting metallocage (MnC, Figure 1) can epoxidize alkene substrates including polymeric ones when sodium hypochlorite, [10] iodosylbenzene,[ 10 , 13 , 15 , 16 ] the combination of molecular oxygen and an aldehyde [17] or hydrogen peroxide (Table S1) are used as oxidants. The latter two oxidants are environmentally more friendly than the former two. An important question is whether the epoxidation reaction occurs inside the cavity of the porphyrin cage or whether it proceeds at the outside, where no effects of the cavity on substrate binding and on the selectivity of the reaction are expected.
Figure 1.

Structures of the porphyrin cage compounds and model compounds used in this study. In the Mn complexes X the counter ion is Cl− or PF6 −, see text.
In this paper we report a mechanistic study, particularly using mass spectrometry, of alkene epoxidation by MnC and a new (chiral) manganese porphyrin cage MnC*, which has sterically encumbered linkers connecting the porphyrin to the glycoluril framework (Figure 1). [18] For our studies, we chose iodosylbenzene (PhIO) as the oxidant, as it is the most frequently used oxygen donor for the epoxidation of alkenes by manganese porphyrin catalysts. [19] In order to give the mechanistic studies a firm basis we studied in detail the epoxidation activities of MnC and MnC* and compared these with the activities of the model compounds manganese meso‐tetraphenylporphyrin (MnTPP) and manganese meso‐(tetrakis‐2‐methoxyphenyl) porphyrin (MnTMPP), see Figure 1.
Results and Discussion
Catalytic epoxidation. The manganese porphyrin catalysts shown in Figure 1 were compared in epoxidation reactions of a variety of alkenes with the aim to identify the effects of the cage structure on the efficiency and selectivity of the reaction (Table 1). The compounds TPP, TMPP, [20] the porphyrin cages H2C [21] and H2C*, [18] and the corresponding manganese complexes were prepared according to our previously reported procedures. We selected PhIO as the oxidant for the catalytic epoxidation reactions, as it is usually regarded to be a mechanistically clean oxygen donor compared to other alternatives.[ 16 , 22 ] Initial experiments were performed with styrene (1) as the substrate and MnC (X=Cl) as the catalyst in the presence of an excess (300 equiv.) of the bulky axial ligand 4‐tert‐butylpyridine (tBuPy) in dichloromethane (DCM) as the solvent. The tBuPy ligand preferentially binds to the outer face of the porphyrin cage and should favor the reaction to take place inside the cavity. [20] For comparison, we performed the same experiments also in the absence of tBuPy and with porphyrin catalysts that have a non‐coordinating counter ion X−=PF6 − (Table 2). The optimal conditions for styrene epoxidation yielding 1 a in 68 % are listed in Table 1, (see also Table S1). Control experiments proved that PhIO could not oxidize olefin substrates without a porphyrin catalyst (Table S1, entry 22) and that it did not oxidize tBuPy to the corresponding N‐oxide (Figure S1).
Table 1.
Epoxidation of alkenes by manganese porphyrin catalysts.[a]
|
Entry |
Substrate |
Product |
Yield [%] |
|||
|---|---|---|---|---|---|---|
|
MnC |
MnC* |
MnTPP |
MnTMPP |
|||
|
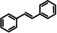
1 |
|
|
68 |
51 |
75 |
76 |
|
|
1 |
1 a |
|
|
|
|
|
2 |
|
|
78 |
|
74 |
|
|
|
2 |
2 a |
|
|
|
|
|
3 |
|
|
70 |
|
71 |
|
|
|
3 |
3 a |
|
|
|
|
|
4 |
|
|
50 |
52 |
70 |
77 |
|
|
4 |
4 a |
|
|
|
|
|
5 |
|
|
87 |
|
65 |
|
|
|
5 |
5 a |
|
|
|
|
|
6 |
|
|
20 |
62 |
86 |
89 |
|
|
6 |
6 a |
|
|
|
|
|
7 |
|
|
70 |
51[b] |
71 |
|
|
|
7 |
7 a |
|
|
|
|
|
8 |
|
|
88 |
|
78 |
|
|
|
8 |
8 a |
|
|
|
|
|
9 |
|
|
68 |
|
79 |
|
|
|
9 |
9 a |
|
|
|
|
|
10 |
|
|
47 |
53 |
92 |
88 |
|
|
10 |
10 a |
|
|
|
|
|
11 |
|
|
70 |
64 |
72 |
80 |
|
|
11 |
11 a |
|
|
|
|
|
12 |
|
|
46 |
41 |
77 |
36 |
|
|
12 |
12 a |
|
|
|
|
|
13 |
|
|
68 |
|
86 |
|
|
|
13 |
13 a |
|
|
|
|
|
14 |
|
|
52 |
|
81 |
|
|
|
14 |
14 a |
|
|
|
|
|
15 |
|
|
76 |
|
74 |
|
|
|
15 |
15 a |
|
|
|
|
|
16 |
|
|
8 (cis) 2 (trans) |
14 (cis) 19 (trans) |
46 (cis) 35 (trans) |
62 (cis) 9 (trans) |
|
|
16 |
16 a |
|
|
|
|
|
17 |
|
|
62 |
50[b] |
50 |
|
|
|
17 |
17 a |
|
|
|
|
|
18 |
|
|
trace |
|
trace |
|
|
|
18 |
18 a |
|
|
|
|
|
19 |
|
|
trace |
|
trace |
|
|
|
19 |
19 a |
||||





































[a] Conditions for all reactions: 0.633 mmol substrate, 0.082 mmol PhIO, 0.7 μmol porphyrin catalyst, 210 mmol tBuPy (300 equiv. with respect to catalyst), 0.9 ml dichloromethane at room temperature (20 °C) for 1 hour with 450 rpm stirring. The epoxide yield was calculated by 1H NMR spectroscopy and is based on the amount of PhIO consumed. The counterion of all manganese porphyrin catalysts was X=Cl. In the case of MnC* the (S,S,S,S)‐enantiomer was used for the catalysis experiments. [b] Isolated yield.
Table 2.
Epoxidation of styrene by manganese porphyrins as a function of counter ion and ligand.[a]
|
Entry |
Catalyst |
Ligand |
Counter ion |
Epoxide Yield [%] |
|---|---|---|---|---|
|
1 |
MnC |
tBuPy |
Cl |
68 |
|
2 |
MnC |
None |
Cl |
67 |
|
3 |
MnTPP |
tBuPy |
Cl |
75 |
|
4 |
MnTPP |
None |
Cl |
87 |
|
5 |
MnC* |
tBuPy |
Cl |
51 |
|
6 |
MnC* |
None |
Cl |
61 |
|
7 |
MnTMPP |
tBuPy |
Cl |
76 |
|
8 |
MnTMPP |
None |
Cl |
85 |
|
9 |
MnC |
tBuPy |
PF6 − |
57 |
|
10 |
MnC |
None |
PF6 − |
51 |
|
11 |
MnTPP |
tBuPy |
PF6 − |
63 |
|
12 |
MnTPP |
None |
PF6 − |
57 |
|
13 |
MnC* |
tBuPy |
PF6 − |
48 |
|
14 |
MnC* |
None |
PF6 − |
50 |
|
15 |
MnTMPP |
tBuPy |
PF6 − |
65 |
|
16 |
MnTMPP |
None |
PF6 − |
58 |
[a] Reaction conditions: 0.633 mmol styrene, 0.082 mmol PhIO, 0.7 μmol porphyrin catalyst, 300 equiv. tBuPy, 0.9 ml dichloromethane, room temperature (20 °C), stirring at 450 rpm for 1 hour. The epoxide yield was calculated by 1H NMR and is based on the amount of PhIO consumed. In the case of MnC* the (S,S,S,S)‐enantiomer was used for the catalysis experiments.
With these optimized reaction conditions in hand, we expanded the list of alkene substrates. Table 1 shows that MnC epoxidizes different types of substrates with yields varying from 20 % (compound 6) to 88 % (compound 8). Interestingly, with MnTPP (X=Cl) as the catalyst the yields were more uniform, i. e. roughly between 70 and 90 % (Table 1). For a selected series of substrates, we also performed reactions with the catalysts MnTMPP (X=Cl) and MnC* (X=Cl) (see Table 1, epoxides 1, 4, 6, 10, 11, 12 and 16). The former catalyst provided similar yields as MnTPP, except for the low yield of 12 a, which may be a result of the steric involvement of the methyl substituent of 1‐methylcyclohexene with the methoxy substituents of MnTMPP. The sterically congested catalyst MnC* generated similar yields as MnC except in the case of (Z)‐1‐phenylpropene 6, for which it displayed a significantly increased yield of epoxide 6 a (MnC* 62 %, MnC 20 %). The results in Table 1 demonstrate that the cavities of MnC and MnC* affect the reaction in a productive or unproductive way. Depending on the alkene reactant a cavity effect is operative, although other effects, e. g. electronic ones, may have an influence as well. When trans‐stilbene was used as a substrate only trace amounts of epoxide 19 a were obtained with MnTPP and MnC as catalysts, which is in line with the literature. [23] cis‐Stilbene, on the other hand, was converted by MnTPP into 81 % epoxide 16 a (46 % cis‐ and 35 % trans‐epoxide) and by MnC in only 10 % epoxide (8 % cis‐ and 2 % trans‐epoxide). In a similar competition experiment, using a 1 : 1 mixture of styrene and cis‐stilbene, MnC produced 60 % styrene oxide 1 a and 2 % epoxide 16 a (1.5 % cis‐ and 0.5 % trans‐epoxide). The low yield for the cis‐stilbene epoxidation by MnC is a result of the fact that this substrate is too bulky to be converted inside the cavity of this cage catalyst, but other effects may play a role as well. This phenomenon was not observed when MnTPP was used as the catalyst: in the same competition experiment, styrene oxide 1 a was formed in 32 % yield and epoxide 16 a in 52 % yield (26 % cis‐ and 26 % trans‐epoxide). Since MnTPP does not have a cage structure, the alkene double bond of cis‐stilbene can easily reach this porphyrin catalyst and be converted at the manganese center.
Next, we tested the effect of the counter ions and tBuPy on the reactivity of the complexes in the epoxidation of styrene (Table 2). Comparison of the reactivities in the presence of either coordinating (Cl−) or non‐coordinating (PF6 −) counter ions showed that the yields were slightly higher in the presence of Cl−. The effect of the tBuPy addition depended on the counter ion. For complexes with Cl−, the addition of tBuPy slightly impaired the reaction yields (except for MnC, where the effect was negligible). On the contrary, reactions of the porphyrin complexes with PF6 − counter ions proceeded generally with a slightly increased reaction yield in the presence of tBuPy (except for chiral MnC*, where the reaction yield slightly decreased).
Finally, we investigated whether the chirality of the MnC* cage could induce stereoselectivity in the epoxidation reaction. MnC* has methyl groups that can rotate inwards and outwards of the cage. We synthesized both enantiomers of MnC*, i. e. (R,R,R,R)− MnC* and (S,S,S,S)‐ MnC* from the corresponding resolved metal‐free compounds (R,R,R,R)‐ H2C* and (S,S,S,S)‐ H2C* (Figure 1). [18] They displayed similar but opposite Cotton effects in the circular dichroism (CD) spectra (Figure 2a), just like the chiral metal‐free cages. [18] We tested two prochiral alkene substrates, the terminal conjugated alkene 7 and the alkyl bridged terminal alkene 17, under standard conditions, i. e. with excess tBuPy present. After reaction, the products were analyzed by chiral HPLC, which revealed that only racemic mixtures of products had been formed in isolated yields of 51 and 50 %, respectively. In order to get information about the chiral environment around the manganese center, we solved the X‐ray structure of (R,R,R,R)‐ MnC*. Interestingly, the crystal structure contained a dimethylformamide (DMF) solvent molecule, which acted as a guest molecule in the cavity of (R,R,R,R)‐ MnC*. In a previous paper we showed that the methyl groups on the chiral carbon centers of the spacers in (R,R,R,R)‐ H2C* and (S,S,S,S)‐ H2C* are located inside the cavity. [18] As can be seen in the crystal structure of (R,R,R,R)‐ MnC* (Figure 2b) these methyl groups are now pushed out of the cavity, because the oxygen atom of DMF coordinates to the manganese center. Furthermore, the chiral centers do not induce any chiral twist in the relative orientation of the xylylene sidewalls of the glycoluril framework. These results suggest that an alkene substrate inside the cavity of MnC* will not experience an environment around the manganese center that is sufficiently chiral to allow it to be converted into a homochiral epoxide.
Figure 2.

Characterization of chiral porphyrin cage MnC* (X=Cl). a, CD spectra of the enantiomers of MnC*. b, Crystal structure of (R,R,R,R)‐ MnC* with a DMF molecule inside its cavity (white for hydrogen, grey for carbon, red for oxygen, blue for nitrogen, purple for manganese, and green for chlorine).
Mechanistic studies. Epoxidation of alkenes by manganese porphyrins and PhIO as oxidant is assumed to take place via a high‐valent manganese‐oxo species. We studied the possible formation of such a species by UV‐vis spectroscopy. The manganese (III) porphyrin cage MnC (X=Cl) in dichloromethane in the presence of 300 equiv. of tBuPy displayed a Soret band at 479 nm and two Q bands at 582 and 616 nm. On the addition of PhIO, these bands gradually decreased in intensity and new bands at 418 and 530 nm appeared, with isosbestic points being visible at 461, ∼495 and ∼555 nm (Figure 3a). This result indicates that the oxygen atom of PhIO is transferred to the manganese center generating a high valent oxo‐manganese species. Similar results were observed for MnC* (X=Cl) with increasing bands at 416 and 532 nm, decreasing bands at 479, 580 and 615 nm, and isosbestic points at 459, ∼500, and ∼555 nm (Figure 3b). The UV−vis spectral changes suggest the formation of an oxo‐manganese(V) species,[ 24 , 25 , 26 ] which we further studied and confirmed by mass spectrometry.
Figure 3.

UV‐vis spectra of manganese porphyrin cages. a, UV−vis spectral changes for MnC (X=Cl, 1.1×10−5 M) in the presence of 300 equiv. of tBuPy upon the addition of 120 equiv. of PhIO in CH2Cl2, showing the formation of a high‐valent manganese oxo‐species (inset isosbestic point at 460). b, UV‐vis spectral changes for (R,R,R,R)‐ MnC* (X=Cl, 1.1×10−5 M) during the same experiment (inset isosbestic point at 459 nm).
Experiments using electrospray ionization mass spectrometry [27] provided further information about the structure of the manganese porphyrin cages, of their adducts with PhIO, and the generation of the active species. [28] In the mass spectrometer only charged species can be studied. In addition, the presence of the tBuPy ligand complicates the interpretation of the mass spectra. However, as discussed above, the effect of counter ions and tBuPy are only minor. Therefore, we performed the experiments in the absence of tBuPy and studied the positively charged porphyrin complexes without the coordinated counterion. Electrospray ionization of a dichloromethane solution of MnC (X=Cl) and 20 equiv. PhIO led to the detection of ions corresponding to [MnIIIC]+, [MnIIIC(PhIO)]+, and [MnVC(O)]+. Similarly, when using MnC* (X=Cl) we could detect the species [MnIIIC*]+, [MnIIIC*(PhIO)]+ and [MnVC*(O)]+, albeit that the adduct with PhIO had a much smaller intensity in the complex with MnC* than with MnC (Figure S7). The porphyrin cages MnIIIC and MnIIIC* can form two types of adducts with PhIO, i. e. one in which the oxidant is bound on the inside of the cage and another one in which it is bound on the outside (see Figure 4a). We further investigated these possibilities by ion mobility separation experiments.[ 29 , 30 , 31 ] The results clearly showed that for both types of porphyrin cages only one single type of adduct with PhIO was generated (Figure 4b). We could unambiguously assign the [MnIIIC]+ adduct to the isomer in which PhIO is located on the inside of the cage, i. e. ([Mn III C(PhIO)in]+), because its mobility (1/K0) agreed well with those of other adducts of this cage having guest molecules bound inside the cavity (Figure S8). For a further control, we performed an addition reaction of the manganese porphyrin cages with 1‐(tert‐butylsulfonyl)‐2‐iodosylbenzene (tBuO2SArIO), which is a bulkier oxidant than PhIO, forcing it to coordinate on the outside of the cage. In agreement, the detected complexes of MnIIIC and MnIIIC* with tBuO2SArIO have much higher 1/K0 values than those with PhIO (see the grey mobilograms in Figure 4b). The results thus clearly show that we detect for both cages solely complexes with PhIO inside the cage and with tBuO2SArIO outside the cage. The absence of isomers with PhIO coordinated to the outside of the manganese porphyrin cages was puzzling. Therefore, we tried to detect the PhIO complexes with MnTPP (X=Cl) as well, but we could only observe the masses of [MnIIITPP]+ (and [Mn V TPP(O)]+) and not those of [MnIIITPP(PhIO)]+ (Figure S9). This result indicates that the PhIO complexes with manganese porphyrins easily dissociate towards formation of the manganese‐oxo complexes and thus do not survive the transfer to the gas phase, unless they are stabilized by interactions with the cavity walls of the cage ligands. The complexes with tBuO2SArIO are probably stabilized by an additional interaction of the sulfonyl group with the porphyrin ligand and therefore we were able to detect the complexes with these ligands at the outside of the cages. In analogy, the [MnIIITPP(tBuO2SArIO)]+ type ions were detected previously by electrospray or cryospray ionization mass spectrometry.[ 16 , 32 , 33 ]
Figure 4.

Mass spectrometry and molecular modeling studies on PhIO adducts of manganese porphyrin cages. a, DFT calculated structures of [Mn III C(PhIO)]+ and [MnC III *(PhIO)]+ with PhIO coordinated inside (in) or outside (out) the cages. b, Ion mobility separation of mass‐selected [MnIIIC(PhIO)]+ (m/z 1617, left) and [MnC*III (PhIO)]+ (m/z 1673, right). In grey the corresponding traces for the adducts with tBuSO2PhIO. c, Collision‐induced dissociation of mass‐selected [MnIIIC(PhIO)]+ (m/z 1617, left) and [MnIIIC*(PhIO)]+ (m/z 1673, right).
To further support the mass spectrometry measurements, we carried out DFT calculations (see Supplementary Information), which suggested that PhIO should coordinate exclusively inside MnC (ΔΔG 298K DCM ([MnIIIC(PhIO)in]+)=0 kcal mol−1 vs. ΔΔG 298K DCM ([MnIIIC(PhIO)out]+=11.6 kcal mol−1, yielding “in” versus “out” abundances of 100:0). For MnC* the calculations revealed that outside PhIO coordination was preferred, but the inside coordination could still happen in circa 1.4 % of the complexes (ΔΔG 298K DCM ([MnIIIC*(PhIO)in]+)=2.5 kcal mol−1 vs. ΔΔG 298K DCM ([MnIIIC*(PhIO)out]+=0 kcal mol−1). The [MnIIIC*(PhIO)out] species was not detected in the mass spectrometer because of the fragmentation of this ion, as explained above. The coordination of PhIO to the inside of the cavity of MnC* forces the methyl groups of the spacers to rotate outwards in a similar way as shown in the crystal structure of the complex of MnC* with DMF (Figure 2b).
This outward rotation is associated with an increase of the volume of the complex, which is consistent with the somewhat larger 1/K0 value detected in the ion mobility experiment compared to that of MnC (1.89 vs. 1.85 V. s/cm2).
We further probed the structures of the detected [MnIIIC(PhIO)in]+ and [MnIIIC*(PhIO)in]+ complexes by collision‐induced dissociation experiments (Figure 4c). Both complexes lost predominantly PhI to form the oxo‐manganese(V) complexes followed by elimination of H2O. The H2O elimination from the primarily formed oxo‐manganese(V) species suggests that the oxo ligand can react internally with neighboring C−H bonds of the spacers of the cages upon collisional heating in the gas phase.
In a next series of mass spectrometry experiments, we compared the structures of the [MnVC(O)]+ and [MnVC*(O)]+ complexes (Figure 5). For both complexes, we clearly detected the two possible isomers, i. e. the one with the oxo ligand inside the cage and the one with the oxo ligand outside the cage. In both cases, the inside orientation of the oxo ligand occurred with a relatively smaller abundance and the disparity was more pronounced for the chiral [MnVC*(O)in]+ and [MnVC*(O)out]+ species. We also calculated the structures of the [MnVC(O)]+ and [MnVC*(O)]+ species by DFT (see Supplementary Information). The calculated relative abundances of the “in” vs. “out” isomers for [Mn V C(O)]+ were 52 % and 48 %, respectively, corresponding to ΔΔG 298K DCM([Mn V C(O)in]+)=0 kcal mol−1 vs. ΔΔG 298K DCM([Mn V C(O)out]+)=0.44 kcal mol−1. For [MnVC*(O)]+ the calculated relative abundances were 89 % for the ”in”‐isomer and 11 % for the “out“‐isomer, which corresponds to ΔΔG 298K DCM([MnVC*(O)in]+)=0 kcal mol−1 vs. (ΔΔG 298K DCM([MnVC*(O)out]+)=1.28 kcal mol−1. The relative energies of the inside vs. outside isomers for [Mn V C*(O)]+ do not relate with the experimental abundances. However, based on the relative energies of the “in” vs. “out” isomers of the precursor species [MnIIIC*(PhIO)]+, it appears that MnC* get preferentially oxidized on the outside in solution, yielding a higher abundance of [MnVC*(O)out]+. On the other hand, MnC gets preferentially oxidized on the inside, as mentioned above, and the “out” [Mn V C(O)out]+ ions likely originate from the dissociation of “out” precursor [MnIIIC(PhIO)out]+ during transfer from solution to the gas phase.
Figure 5.

Mass spectrometric and molecular modeling studies on oxo complexes of manganese porphyrin cages. a, DFT calculated structures of [Mn V C(O)]+ and [MnVC*(O)]+ with the oxo ligand coordinated inside (in) or outside (out) of the cages. b, Ion mobility separation of mass‐selected [Mn V C(O)]+ (m/z 1413, left) and [MnVC*(O)]+ (m/z 1469, right). c, Gas‐phase reactivity of ion‐mobility‐ and mass‐selected [Mn V C(O)]+ (m/z 1413 & 1/K 0 1.835 and m/z 1413 & 1/K0 1.855, left) and [MnVC*(O)]+ (m/z 1469 & 1/K0 1.855 and m/z 1469 & 1/K0 1.875, right). Note that the instrument does not permit unit mass‐selection at this mass range; therefore, the reactivity spectra contain also impurities denoted by the star symbol.
The ion mobility (1/K 0) of [MnVC(O)in]+ was found to be almost identical to the ion mobility of [MnIIIC(PhIO)in]+, attesting that the axial ligands (oxo and PhIO) are inside the cage and, hence, do not affect the overall size of the complexes significantly. In contrast, the ion mobilities of [MnVC*(O)in]+ and [MnVC*(O)out]+ were smaller than the ion mobility of [MnIIIC*(PhIO)in]+. This effect must be related to the orientation of the methyl groups attached to the spacers on the sidewalls of MnC*. For the oxo‐complexes, there is sufficient space for the methyl groups on the inside of the cage and these groups will, therefore, likely point inwards, making the overall size of the complex smaller than the size of the [MnIIIC*(PhIO)in]+ complex with its methyl groups pointing outwards.
In order to compare the gas‐phase reactivities of the oxo‐complexes of the manganese porphyrin cages, we studied their reactions with dimethylsulfide (Figure 5c). Dimethylsulfide is more reactive in oxygen transfer reactions than alkenes and therefore more suitable for gas phase studies. We tested the reactivity with styrene, but we observed only negligible yields for oxygen transfer on the time scale of the mass spectrometry experiment (Figure S10). With dimethylsulfide all the oxo complexes quickly transferred their oxygen atom to form dimethyl sulfoxide. The resulting complexes can eliminate the sulfoxide because the reaction is exothermic. However, we also detected the intact products, because they can be stabilized by subsequent collisions with the reactant gas (we worked under multi‐collision conditions). The stabilization is more effective if the sulfoxide forms inside the cage as evidenced by a larger relative intensity of the adduct signal (see Figure 5c). Finally, the empty cages, which are left after oxygen atom transfer, are observed to be capable of associating with another dimethylsulfide molecule. For both cages, the complexes with the oxo ligand at the outside of the cavity reacted faster than those with the oxo ligand at the inside, as evidenced by the nearly complete depletion of [MnVC(O)out]+ and [MnVC*(O)out]+ (Figure 5c). Note that ion mobility separation also revealed signals of self‐oxidized cage complexes at lower 1/K0 values, that did not show any reactivity with dimethylsulfide (see Figure S11 for more details).
In summary, the ion mobility mass spectrometry experiments show that MnC and MnC* differ in their ability to form complexes with PhIO. MnC gets preferentially oxidized on the inside of the cage, whereas MnC* gets preferentially oxidized on the outside. In addition, in the gas phase the oxo ligand at the inside of the MnC* cage is sterically protected by the inwards rotated methyl groups and the analogous situation can be expected in the non‐polar dichloromethane solvent. Hence, the epoxidation mediated by MnC* should almost exclusively take place at the outside of the cage, whereas the epoxidation mediated by MnC should proceed preferentially at the inside. This selectivity explains the lower epoxidation yields for the internal alkenes with the latter catalyst.
The gas phase experiments show that the complexes of MnC and MnC* with the oxo ligand at the outside react faster in oxygen transfer reactions than complexes with this oxo ligand on the inside. In solution the reactivity at the outside might be slowed down, if tBuPy is added, although the evidence for this is not strong. Accordingly, the MnC* catalyst gives somewhat smaller yields in oxidations of the terminal alkenes compared to MnC in the presence of tBuPy. In the absence of tBuPy, the yields obtained with the MnC* catalyst become comparable with the yields obtained with MnC and this is particularly clear in the experiments with non‐coordinating counter ions (compare Table 2, entries 2,6 and 10,14). This line of reasoning also explains the unexpected results for the epoxidation of internal alkenes (1‐phenylpropene and cis‐stilbene). Both alkenes are epoxidized in a larger yield with MnC* than with MnC. This is because the internal, sterically hindered alkenes are likely to be epoxidized at the outside of the cage, which is preferentially happening with MnC*. On the contrary, the original MnC cage has the oxo‐functionality preferentially at the inside of the cage, where the large alkene cannot enter.
Conclusion
In this paper we have reported a mechanistic study of the epoxidation of alkenes with iodosylbenzene, catalyzed by achiral and a chiral manganese (III)porphyrin cage complexes. We show that both catalysts give similar yields of epoxide (between 20 and 88 %). However, the achiral cage catalyst consistently shows a somewhat higher epoxidation yield for the terminal alkenes than the chiral one. In contrast, the sterically more congested chiral‐cage catalyst gives larger epoxidation yields for internal alkenes. At the same time, we did not detect any stereoselectivity. These findings have been rationalized by investigating the individual reactive complexes using ion mobility mass spectrometry. The experiments show that the first step in the catalytic epoxidation is the formation of the iodosylbenzene adduct between the manganese(III) porphyrin complex and the iodosylbenzene ligand and this adduct can be formed on either the inside or the outside of the cage. In the subsequent step, the elimination of iodobenzene provides the catalytically active manganese(V)oxo complex. The ion mobility experiments show that the achiral cage MnC favours the formation of the reactive manganese(V)oxo moiety at the inside of the cavity and hence the epoxidation of the substrate also occurs at the inside of the cavity. This is in line with our earlier studies on the epoxidation of polybutadienes by iodosyl benzene with MnC (X=Cl) as a catalyst.[ 13 , 15 ] This complex was shown to thread onto the polymer chain, and while held in a rotaxane fashion, it moved along the chain and epoxidized the polymer double bonds. The present study reveals that this inside reaction is an intrinsic feature of MnC and does not depend on the presence of tBuPy on the outside of the cage, as we initially thought. In the case of MnC* the steric influence of the asymmetric spacers attached to the walls of this cage catalyst results in the preferential formation of the manganese(V)oxo moiety at the outside of the cage. Hence, also the epoxidation reaction occurs at the outside of the cage catalyst and, therefore, is not enantioselective. In order to achieve a stereoselective epoxidation reaction other types of chiral porphyrin catalysts are probably needed. In a previous paper we showed that porphyrin cages displaying planar chirality bind the achiral guest N,N’‐dimethyl‐4,4’‐bipyridinium dihexafluorophosphate (methylviologen) in an enantioselective fashion, i. e. the guest rotates its pyridine groups along its long axis and binds in a distinct helical conformation, either (P)‐ or (M) depending on the chirality of the host.12 In future studies we will work this out further in the direction of an enantioselective catalytic epoxidation system. This study also shows the power of ion mobility mass spectrometry studies for unravelling molecular details of complex molecular reactions, such as the one reported here. The intermediates derived from porphyrin cage catalysts differing merely in the orientation of the oxygen atom can be nicely distinguished and selectively studied. This is not easily achieved with other techniques, such as NMR, UV‐Vis or IR.
Deposition Number 2115320 (for ( R,R,R,R)‐ MnC*) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Author Contributions
X.C. Data curation:Equal; Investigation:Equal
Q.D. Data curation:Equal
G.T. Data curation:Equal
P.G. Data curation:Equal; Investigation:Equal
D.P. Data curation:Supporting; Investigation:Supporting
P.T. Investigation:Supporting; Methodology:Supporting
J.E. Conceptualization:Equal; Data curation:Equal; Investigation:Equal; Methodology:Equal; Supervision:Equal; Writing – review & editing:Equal
J.R. Conceptualization:Equal; Data curation:Equal; Formal analysis:Equal; Funding acquisition:Equal; Investigation: Equal; Methodology:Equal; Writing – review & editing:Equal
R.N. Conceptualization:Equal; Data curation:Equal; Formal analysis:Equal; Funding acquisition:Equal; Investigation: Equal; Methodology:Equal; Writing – review & editing:Equal
Conflict of interest
The authors declare no conflict of interest.
1.
Biographical Information
Xiaofei Chen obtained his MSc in polymer chemistry from the Northeast Normal University in Changchun, China. Subsequently, he did his PhD studies at RWTH Aachen (Germany) in the group of Prof. Markus Albrecht on the topic of helicate‐based and cation‐triggered molecular springs. After earning his PhD with him in 2019, he moved to Radboud University in Nijmegen, The Netherlands, where he is currently working as a postdoctoral fellow with Prof. Roeland Nolte and Dr. Hans Elemans. His research interests include the supramolecular chemistry of helicates and metal porphyrin cage compounds, the latter for use in photo‐switchable enantioselective catalysis.

Biographical Information
Quentin Duez obtained his PhD in chemistry in 2020 from the University of Mons (Belgium), where he worked under the supervision of Prof. Pascal Gerbaux and Dr. Jérôme Cornil. His PhD topic focused on conformational analyses of gaseous (co)polymer ions by ion mobility mass spectrometry and molecular dynamics simulations. He then joined the group of Prof. Jana Roithová at Radboud University in Nijmegen, The Netherlands as a postdoctoral researcher. He is currently investigating the potential of ion mobility and ion spectroscopy to unravel details of reaction mechanisms and chemical processes.

Biographical Information
Guilherme L. Tripodi obtained his MSc in chemistry in 2018 from the University of Campinas (Brazil), under the supervision of Prof. Marcos N. Eberlin. He is currently a PhD candidate at Radboud University in Nijmegen, The Netherlands where he works under the supervision of Prof. Jana Roithová. His research focuses on the development of methods for the monitoring of fast chemical reactions, with emphasis on the reactivity and spectroscopic properties of short‐lived reaction intermediates from C−H activation reactions.

Biographical Information
Pieter J. Gilissen studied chemistry at Radboud University in Nijmegen and obtained his MSc in chemistry in 2018. Currently, he is conducting his PhD research with Prof. Roeland Nolte and Dr. Hans Elemans. His research topics cover the synthesis of chiral porphyrin cages and molecular motors, and their use as functional molecular systems.

Biographical Information
Dimitrios Piperoudis did his master internship in the group of Prof. Roeland Nolte at Radboud University in Nijmegen, on the topic of chiral porphyrin cage compounds. As a junior researcher, he is currently working at the same university on the development of reversible photo‐switchable inhibitors for in situ studies by NMR and on reactive oxocarbenium ions.

Biographical Information
Paul Tinnemans was born in 1979 in The Netherlands and obtained his MSc in chemistry in 2005 at Radboud University in Nijmegen. In 2012, he completed his PhD studies on surface X‐ray diffraction at the European Synchrotron Radiation Facility (ESRF) in collaboration with the University of Twente and Radboud University. After a few years in industry, he returned to Radboud University as a crystallographer, focusing on the characterization of small (chiral) organic molecules using single‐crystal and powder X‐ray diffraction.

Biographical Information
Hans Elemans completed his PhD in supramolecular chemistry with Prof. Roeland Nolte at Radboud University in Nijmegen, The Netherlands, in 2001. He then developed a research line in which chemical reactivity is studied by scanning tunneling microscopy at the single molecule level. After postdoctoral work with Prof. Steven De Feyter at K. U. Leuven (Belgium) he returned to Radboud University as associate professor of molecular nanotechnology. His scientific interests involve functional supramolecular architectures, catalysis, and scanning probe microscopy.

Biographical Information
Jana Roithová graduated at Charles University in the Czech Republic (1998). Her PhD thesis focused on reaction dynamics (2003) and she studied mass spectrometry techniques with Prof. Schwarz (Berlin). In 2007–2018, she served as a lecturer and then professor at the Charles University. Since 2018, she holds a chair in spectroscopy and catalysis at Radboud University in the Netherlands. She develops mass spectrometry and ion spectroscopy techniques to study reaction mechanisms, with a particular focus on reactive intermediates in metal‐catalysed reactions. Her research interests span from reaction mechanisms of organometallic reactions and mechanisms of small molecules activation to new reactivity concepts and reaction design.

Biographical Information
Roeland Nolte is emeritus professor of organic chemistry and emeritus Royal Netherlands Academy of Science professor at Radboud University in Nijmegen, The Netherlands. During his scientific career he worked on a wide range of topics at the interfaces of supramolecular chemistry, macromolecular chemistry, and biomimetic chemistry, focusing on the design of catalysts and (macro)molecular materials. After his retirement in 2010 he was awarded a special university chair in molecular nanotechnology. His current research interest is the encoding of digital information in single polymer chains by catalytic molecular machines.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was funded by the European Research Council (ERC Advanced Grant No. 74092 to R. J. M. N.), the Dutch Research Council (VI.C.192.044 and OCENW.KLEIN.348 to J.R), and by the Dutch Ministry of Education, Culture, and Science (Gravitation program 024.001.035). We thank Mr. Max T. G. M. Derks for using his automatized flow setup (labm8) to perform the mass spectrometry experiments.
X. Chen, Q. Duez, G. L. Tripodi, P. J. Gilissen, D. Piperoudis, P. Tinnemans, J. A. A. W. Elemans, J. Roithová, R. J. M. Nolte, Eur. J. Org. Chem. 2022, e202200280.
Contributor Information
Dr. Johannes A. A. W. Elemans, Email: J.Elemans@science.ru.nl, https://www.ru.nl/science/molecularnanotechnology/, https://www.ru.nl/science/spectroscopy‐and‐catalysis/.
Prof. Dr. Jana Roithová, Email: jana.roithova@ru.nl.
Prof. Dr. Roeland J. M. Nolte, Email: R.Nolte@science.ru.nl.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Que L., Tolman W. B., Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- 2. Denisov I. G., Makris T. M., Sligar S. G., Schlichting I., Chem. Rev. 2005, 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- 3. Huang X., Groves J. T., Chem. Rev. 2017, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Corey E., Noe M. C., J. Am. Chem. Soc. 1996, 118, 319–329. [Google Scholar]
- 5. Li A., Ye L., Yang X., Wang B., Yang C., Gu J., Yu H., ChemCatChem 2016, 8, 3229–3233. [Google Scholar]
- 6. Mann S. I., Nayak A., Gassner G. T., Therien M. J., DeGrado W. F., J. Am. Chem. Soc. 2021, 143, 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amouri H., Desmarets C., Moussa J., Chem. Rev. 2012, 112, 2015–2041. [DOI] [PubMed] [Google Scholar]
- 8. Chakrabarty R., Mukherjee P. S., Stang P. J., Chem. Rev. 2011, 111, 6810–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen K., Arnold F. H., Nat. Catal. 2020, 3, 203–213. [Google Scholar]
- 10. Elemans J. A. A. W., Nolte R. J. M., Chem. Commun. 2019, 55, 9590–9605. [DOI] [PubMed] [Google Scholar]
- 11. Elemans J. A. A. W., Claase M. B., Aarts P. P., Rowan A. E., Schenning A. P., Nolte R. J. M., J. Org. Chem. 1999, 64, 7009–7016. [Google Scholar]
- 12. Ouyang J., Swartjes A., Geerts M., Gilissen P. J., Wang D., Teeuwen P. C., Tinnemans P., Vanthuyne N., Chentouf S., Rutjes F. P. J. T., Naubron J.-V., Crassous J., Elemans J. A. A. W., Nolte R. J. M., Nat. Commun. 2020, 11, 4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thordarson P., Bijsterveld E. J., Rowan A. E., Nolte R. J. M., Nature 2003, 424, 915–918. [DOI] [PubMed] [Google Scholar]
- 14. Deutman A. B. C., Monnereau C., Elemans J. A. A. W., Ercolani G., Nolte R. J. M., Rowan A. E., Science 2008, 322, 1668–1671. [DOI] [PubMed] [Google Scholar]
- 15. Monnereau C., Ramos P. H., Deutman A. B. C., Elemans J. A. A. W., Nolte R. J. M., Rowan A. E., J. Am. Chem. Soc. 2010, 132, 1529–1531. [DOI] [PubMed] [Google Scholar]
- 16. Guo M., Dong H., Li J., Cheng B., q Huang Y., q Feng Y., Lei A., Nat. Commun. 2012, 3, 1190. [DOI] [PubMed] [Google Scholar]
- 17. Bernar I., Rutjes F. P. J. T., Elemans J. A. A. W., Nolte R. J. M., Catalysts 2019, 9, 195. [Google Scholar]
- 18. Gilissen P. J., Slootbeek A. D., Ouyang J., Vanthuyne N., Bakker R., Elemans J. A. A. W., Nolte R. J. M., Chem. Sci. 2021, 12, 1661–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collman J. P., Zeng L., Wang H. J. H., Lei A., Brauman J. I., Eur. J. Org. Chem. 2006, 2707–2714. [Google Scholar]
- 20. Elemans J. A. A. W., Bijsterveld E. J. A., Rowan A. E., Nolte R. J. M., Eur. J. Org. Chem. 2007, 751–757. [Google Scholar]
- 21. Gilissen P. J., Swartjes A., Spierenburg B., Bruekers J. P. J., Tinnemans P., White P. B., Rutjes F. P. J. T., Nolte R. J. M., Elemans J. A. A. W., Tetrahedron 2019, 75, 4640–4647. [Google Scholar]
- 22. Kitagishi H., Tamaki M., Ueda T., Hirota S., Ohta T., Naruta Y., Kano K., J. Am. Chem. Soc. 2010, 132, 16730–16732. [DOI] [PubMed] [Google Scholar]
- 23. Groves J. T., Nemo T. E., Myers R. S., J. Am. Chem. Soc. 1979, 101, 1032–1033. [Google Scholar]
- 24. Nam W., Kim I., Lim M. H., Choi H. J., Lee J. S., Jang H. G., Chem. Eur. J. 2002, 8, 2067–2071. [DOI] [PubMed] [Google Scholar]
- 25. Groves J. T., Lee J., Marla S. S., J. Am. Chem. Soc. 1997, 119, 6269–6273. [Google Scholar]
- 26. Zhang R., Horner J. H., Newcomb M., J. Am. Chem. Soc. 2005, 127, 6573–6582. [DOI] [PubMed] [Google Scholar]
- 27. Mehara J., Roithová J., Chem. Sci. 2020, 11, 11960–11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Polewski L., Springer A., Pagel K., Schalley C. A., Acc. Chem. Res. 2021, 54, 2445–2456. [DOI] [PubMed] [Google Scholar]
- 29. Lanucara F., Holman S. W., Gray C. J., Eyers C. E., Nat. Chem. 2014, 6, 281–294. [DOI] [PubMed] [Google Scholar]
- 30. Kruve A., Caprice K., Lavendomme R., Wollschläger J. M., Schoder S., Schröder H. V., Nitschke J. R., Cougnon F. B., Schalley C. A., Angew. Chem. Int. Ed. 2019, 58, 11324–11328; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11446–11450. [Google Scholar]
- 31. Ujma J., De Cecco M., Chepelin O., Levene H., Moffat C., Pike S. J., Lusby P. J., Barran P. E., Chem. Commun. 2012, 48, 4423–4425. [DOI] [PubMed] [Google Scholar]
- 32. Guo M., Lee Y.-M., Seo M. S., Kwon Y.-J., Li X.-X., Ohta T., Kim W.-S., Sarangi R., Fukuzumi S., Nam W., Inorg. Chem. 2018, 57, 10232–10240. [DOI] [PubMed] [Google Scholar]
- 33. Zhang L., Lee Y. M., Guo M., Fukuzumi S., Nam W., J. Am. Chem. Soc. 2020, 142, 19879–19884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.