

Abstract
The molecular networks that regulate natural killer (NK) cell functions are not completely understood. Here, we present a workflow for efficient delivery of siRNA into human NK cells without compromising viability. This methodology represents a promising approach for rapidly interrogating gene functions in primary human NK cells.
Keywords: Gene regulation, Molecular biology, NK cells
Natural killer (NK) cells are innate lymphocytes that participate in immune responses against virus‐infected cells and tumors [1]. The functions of NK cells can be therapeutically exploited by adoptive transfer, which represents a promising therapy option against cancer [2, 3]. Our understanding of how NK cells sense their surroundings, recognize aberrant cells, and integrate receptor input has progressed considerably [4, 5, 6]. However, the molecular networks generating and maintaining their functional capacity remain incompletely understood and elucidating NK cell‐intrinsic regulatory networks holds promise for improving NK cell therapy.
Genetic manipulation of NK cells by electroporation, lipofection, or viral transduction has been limited by variable delivery efficiencies and impaired viability (reviewed in [7]). Genetic engineering using CRISPR/Cas9 has been described for NK cells with varying efficiency, ranging from 24% to 90% [8, 9, 10], and such approaches typically include vigorous activation in vitro, thereby precluding the study of genes that are expressed only prior to activation or dynamically regulated upon activation.
RNA interference‐mediated knock‐down of gene expression is a valuable tool for investigating molecular mechanisms that underlie cellular function and chemically modified small interfering RNA (siRNA) allow for passive uptake, providing an opportunity to study gene functions in cells that are otherwise challenging to manipulate.
As a starting point, we exposed purified NK cells to 6‐carboxyfluorescein (FAM)‐labeled siRNA with no homology to human genes (non‐targeting, control siRNA) for 96 h, which resulted in high fluorescence signal in NK cells while maintaining viability (Fig. 1A, Supporting Information Fig. S1A). We screened treatment conditions spanning four siRNA concentrations and four concentrations of IL‐15 in culture medium or commercially available siRNA delivery medium (Accell, Horizon Discovery) and evaluated uptake (measured as mean fluorescence intensity [MFI]) as well as viability across all 32 conditions (Fig. 1B). Next, we in‐depth profiled conditions in multiple donors to examine the protocol's robustness. We found that siRNA delivery medium consistently augmented siRNA uptake (Fig. 1C), that an siRNA concentration of 1.0 μM resulted in higher signal intensity than 0.25 μM (Fig. 1D), and that addition of 1 ng/mL IL‐15 improved cell viability and recovery in all donors (Fig. 1E). We thus selected 1 μM siRNA in siRNA delivery medium supplemented with 1 ng/mL IL‐15 as optimized condition for further evaluation.
Figure 1.
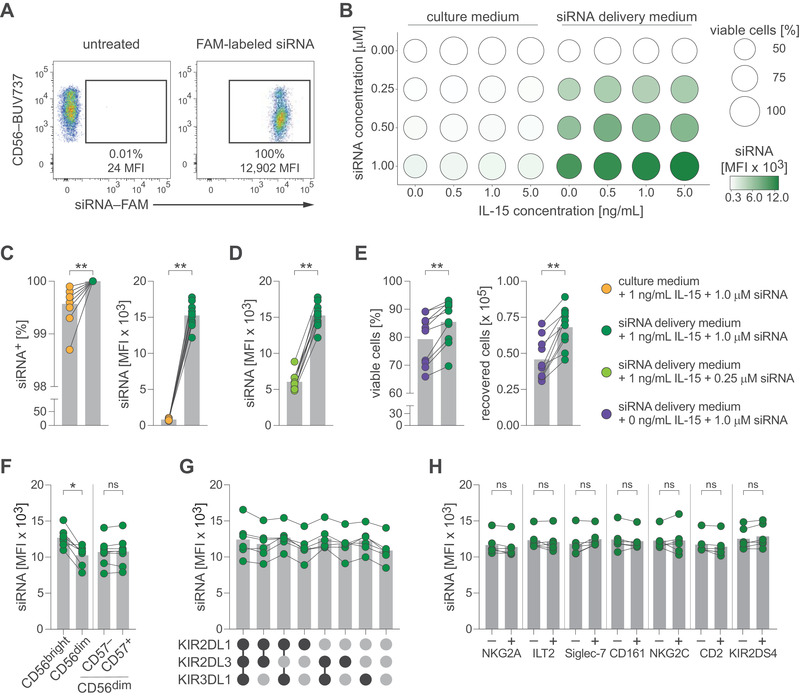
Delivery of siRNA into human NK cells. (A) Purified NK cells were left untreated or treated with fluorescently labeled siRNA and analyzed by flow cytometry. Uptake of siRNA as frequency and MFI of single viable CD3– CD56+ NK cells. (B‐E) NK cells were exposed to different concentrations of fluorescently labeled siRNA and IL‐15 in culture medium or Accell siRNA delivery medium. (B) Screening matrix with bubble area indicating frequency of viable cells and bubble color indicating siRNA uptake as MFI. (C) Uptake of siRNA by NK cells treated with 1.0 μM siRNA in culture medium or Accell siRNA delivery medium (both containing 1 ng/mL IL‐15). Left: frequency, right: MFI. (D) Uptake of siRNA by NK cells treated with 0.25 μM siRNA or 1.0 μM siRNA (both in Accell siRNA delivery medium containing 1 ng/mL IL‐15). (E) Survival of NK cells treated with 1.0 μM siRNA in Accell siRNA delivery medium alone or containing 1 ng/mL IL‐15. Left: frequency of viable NK cells, right: absolute count of recovered viable NK cells (input before treatment: 1 × 105 NK cells). (F‐H) NK cells were treated with fluorescently labeled siRNA. Uptake of siRNA by NK cell sub‐populations based on (F) differentiation stages (G) KIR co‐expression pattern or (H) stratified for expression of activating or inhibitory NK cell receptors. Data are displayed as mean (B) or mean and individual datapoints (C‐H) and representative of n = 12 donors in four independent experiments (A) or pooled of n = 2 donors in one experiment (B), n = 10 donors in three independent experiments (C–E), n = 8 donors in three independent experiments (F, H), and n = 6 donors in three independent experiments (G). Symbols represent individual donors (C–H). Statistical significance was tested using Wilcoxon test (C–F, H). *p < 0.05, **p < 0.01.
Human NK cells can be divided into sub‐populations based on their differentiation stage or expression of inhibitory or activating receptors [11, 12]. When comparing siRNA uptake in sub‐populations, we found that CD56bright CD16– NK cells displayed slightly increased signal intensities, while no differences were found between CD57– and CD57+ cells of the CD56dim CD16+ population (Fig. 1F; Supporting Information Fig. S1B). Furthermore, uptake did not differ between CD56dim CD16+ subsets stratified for inhibitory kill‐cell immunoglobulin‐like receptor (KIR) co‐expression patterns (Fig. 1G) or when comparing expression of other NK cell receptors including NKG2A, ILT2, Siglec‐7, CD161, NKG2C, CD2, or KIR2DS4 (Fig. 1H; Supporting Information Fig. S1B).
We tested whether siRNA treatment alters NK cell functionality by treating NK cells with culture medium or siRNA delivery medium containing 1 μM control siRNA (both supplemented with 1 ng/mL IL‐15), followed by co‐culture with K562 leukemia cells or stimulation with IL‐12 and IL‐18. We did not detect consistent changes in degranulation (as measured by CD107a mobilization), TNF, or IFN‐γ expression (Supporting Information Fig. S1C and D).
To assess functional interference, we employed non‐FAM‐labeled siRNA molecules targeting transcripts of the beta‐2‐microglobulin (B2M) gene. Treatment resulted in clear down‐regulation of β2m protein and B2M transcripts (Fig. 2A–C). Furthermore, knock‐down of B2M resulted in near complete loss of HLA class I protein (Fig. 2D), demonstrating disruption of protein–protein interactions.
Figure 2.
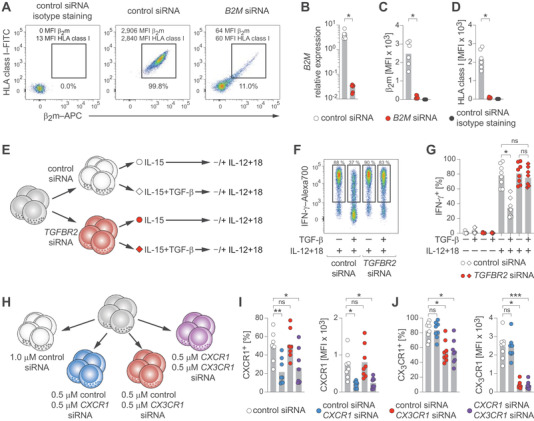
Functional gene analyses in human NK cells. (A–D) NK cells were treated with control siRNA or B2M siRNA. (A) Representative β2m and HLA class I protein expression determined by flow cytometry after the indicated treatments. (B) Summary of B2M mRNA expression relative to GAPDH as determined by RT‐qPCR. (C) Summary of β2m protein expression as MFI measured by flow cytometry. (D) Summary of HLA class I protein expression as MFI measured by flow cytometry. (E–G) NK cells were treated with control siRNA or TGFBR2 siRNA, exposed or not to TGF‐β, and stimulated with IL‐12 and IL‐18. (E) Experimental setup. (F) Representative IFN‐γ expression after the indicated treatments as determined by flow cytometry. (G) Summary of IFN‐γ expression. (H–J) NK cells were treated with control siRNA or with the indicated combinations of control siRNA, CXCR1 siRNA, and CX3CR1 siRNA and protein expression was analyzed by flow cytometry. (H) Treatment setup. (I) Summaries of CXCR1 expression. Left: frequency. Right: MFI. (J) Summaries of CX3CR1 expression. Left: frequency. Right: MFI. Data are displayed as mean and individual datapoints (B‐D, G, I, J) and representative of n = 6 donors in three independent experiments (A) and n = 8 donors in three independent experiments (F) or pooled from n = 6 donors in two independent experiments (B–D) and n = 8 donors in three independent experiments (G, I, J). Symbols represent individual donors (B–D, G, I, J). Statistical significance was tested using Wilcoxon test (B–D) or Friedman test with Dunn's test (G, I, J). *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we targeted TGFBR2, which is crucial for TGF‐β signaling [13], and functionally evaluated its effect by exposing NK cells to TGF‐β, followed by stimulation with IL‐12 and IL‐18 (Fig. 2E). We observed a fourfold reduction of TGFBR2 transcripts (Supporting Information Fig. S2A) and TGF‐β exposure reduced IFN‐γ expression in control‐treated NK cells, while NK cells treated with TGFBR2‐targeting siRNA were almost completely resistant to TGF‐β‐mediated suppression (Fig. 2F and G). Moreover, using varying siRNA concentrations, IFN‐γ responses were recovered in a dose‐dependent manner (Supporting Information Fig. S2B and C).
Finally, we investigated whether treatment with multiple siRNA molecules could enable simultaneous knock‐downs. To this end, we treated NK cells with combinations of siRNA targeting CXCR1 and CX3CR1 (Fig. 2H) and found that combined treatment markedly reduced expression of both chemokine receptors, similar to the effects observed in single targeting (Fig. 2I and J; Supporting Information Fig. S2D–F).
In summary, we present an experimental workflow that allows for rapid analyses of gene functions in human NK cells under near‐resting conditions (step‐by‐step protocol in the Supporting Information).
Rigorous validation of putative targets with complementary techniques remains required to exclude off‐target effects [14, 15] and variable knock‐down efficiencies across different targets, such as CXCR1 and CX3CR1 in the examples above, need to be considered during implementation.
Given the clinical potential of NK cell therapy, improved understanding of gene circuits regulating NK cell functions has important implications and identifying genes that control anti‐tumor responses may advance next‐generation immunotherapy.
Conflict of interest
K.‐J.M. is scientific advisor for as well as has funding from Fate Therapeutics and is a scientific advisor for Vycellix. The other authors declare no conflict of interest.
Author contributions
Conceptualization: K.‐J.M. and Q.H.; methodology: D.P., J.D., and Q.H.; formal analysis: D.P., E.H.A., and Q.H.; investigation: D.P., P.M., O.H., E.H.A., and Q.H.; writing–original draft: Q.H.; writing–review & editing: all authors; supervision: Q.H.; funding acquisition: K.‐J.M. and Q.H.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202149710.
Supporting information
Supporting informatioin
Acknowledgements
We thank Merete Thune Wiiger for technical assistance. This work was supported by EU H2020 (MSCA‐IF‐2018 838909 to Q.H. and ITN‐765104‐MATURE‐NK to K.‐J.M.), Knut and Alice Wallenberg Foundation (KAW 2018‐011 to K.‐J.M.), Swedish Research Council (2020‐02286 to K.‐J.M.), Swedish Children's Cancer Society (PR2020‐0159 to K.‐J.M.), Swedish Cancer Society (CAN 2017/676 to K.‐J.M.), Research Council of Norway (275469 to K.‐J.M.), South‐Eastern Norway Regional Health Authority (2021073 to K.‐J.M.), Norwegian Cancer Society (190386‐2017 to K.‐J.M.), Region Stockholm (2020‐0733 to P.M.), German Research Foundation (DU1964/1‐1 to J.D.), Petrus och Augustra Hedlunds Stiftelse (M2021‐1533 to Q.H.), Stiftelsen Clas Groschinskys Minnesfold (M21120 to Q.H.), and Stiftelsen Torspiran (to Q.H.), and Stiftelsen Lars Hiertas Minne (FO2021‐0263 to Q.H.).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon request.
References
- 1. Annunziato, F. et al., The 3 major types of innate and adaptive cell‐mediated effector immunity. J. Allergy Clin. Immunol. 2015. 135: 626–635. [DOI] [PubMed] [Google Scholar]
- 2. Demaria, O. et al., Harnessing innate immunity in cancer therapy. Nature 2019. 574: 45–56. [DOI] [PubMed] [Google Scholar]
- 3. Malmberg, K.‐.J. et al., Natural killer cell‐mediated immunosurveillance of human cancer. Semin. Immunol. 2017. 31: 20–29. [DOI] [PubMed] [Google Scholar]
- 4. Lanier, L. L. et al., Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 2008. 9: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hammer, Q. et al., Natural killer cell specificity for viral infections. Nat. Immunol. 2018. 19: 800–808. [DOI] [PubMed] [Google Scholar]
- 6. Quatrini, L. et al., Human NK cells, their receptors and function. Eur. J. Immunol. 2021. 51: 1566–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carlsten, M. et al. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front. Immunol. 2015. 6: 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lambert, M. et al., CRISPR/Cas9‐based gene engineering of human natural killer cells: protocols for knockout and readouts to evaluate their efficacy. Methods Mol. Biol. 2020. 2121: 213–239. [DOI] [PubMed] [Google Scholar]
- 9. Rautela, J. et al.,, Drug target validation in primary human natural killer cells using CRISPR RNP. J. Leukoc. Biol. 2020. 108: 1397–1408. [DOI] [PubMed] [Google Scholar]
- 10. Pomeroy, E. J. et al., A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol. Ther. 2020. 28: 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freud, A. G. et al., The broad spectrum of human natural killer cell diversity. Immunity 2017. 47: 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pfefferle, A. et al., Deciphering natural killer cell homeostasis. Front. Immunol. 2020. 11: 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horbelt, D. et al., A portrait of transforming growth factor Î2 superfamily signalling: background matters. Int. J. Biochem. Cell Biol. 2012. 44: 469–474. [DOI] [PubMed] [Google Scholar]
- 14. Lin, A. et al., CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on‐going clinical trials. Elife 2017. 6: e24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin, A. et al., Off‐target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019. 11: eaaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting informatioin
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.