

Abstract
Heat shock protein 104 (Hsp104) protein disaggregases are powerful molecular machines that harness the energy derived from ATP binding and hydrolysis to disaggregate a wide range of protein aggregates and amyloids, as well as to assist in yeast prion propagation. Little is known, however, about how Hsp104 chaperones recognize such a diversity of substrates, or indeed the contribution of the substrate‐binding N‐terminal domain (NTD) to Hsp104 function. Herein, we present a NMR spectroscopy study, which structurally characterizes the Hsp104 NTD‐substrate interaction. We show that the NTD includes a substrate‐binding groove that specifically recognizes exposed hydrophobic stretches in unfolded, misfolded, amyloid and prion substrates of Hsp104. In addition, we find that the NTD itself has chaperoning activities which help to protect the exposed hydrophobic regions of its substrates from further misfolding and aggregation, thereby priming them for threading through the Hsp104 central channel. We further demonstrate that mutations to this substrate‐binding groove abolish Hsp104 activation by client proteins and keep the chaperone in a partially inhibited state. The Hsp104 variant with these mutations also exhibited significantly reduced disaggregation activity and cell survival at extreme temperatures. Together, our findings provide both a detailed characterization of the NTD‐substrate complex and insight into the functional regulatory role of the NTD in protein disaggregation and yeast thermotolerance.
Keywords: Hsp104, molecular chaperones, NMR spectroscopy, protein disaggregation
Heat shock protein 104 (Hsp104) chaperones form powerful molecular machines, capable of disaggregating a wide range of protein aggregates and amyloid fibres. Hsp104 N‐terminal domain contains a hydrophobic substrate‐binding groove that enables the chaperone to interact with unfolded, misfolded, amyloid and prion substrates. Mutations to this groove abolish Hsp104 activation by client proteins and keep the chaperone in a partially inhibited state, significantly reducing Hsp104 disaggregation activity and cell survival at extreme temperatures.
Abbreviations
- CSP
chemical shift perturbation
- FFL
firefly luciferase
- Hsp104
heat shock protein 104
- Hsp70
heat shock protein Hsp70
- HSQC
heteronuclear single quantum coherence spectroscopy
- MFI
mean fluorescence intensity
- mNG
mNeonGreen
- NBD
nucleotide binding domain
- NTD
N‐terminal domain
- PhoA
alkaline phosphatase
- ThT
Thioflavin‐T
- YPD
yeast extract peptone dextrose
Introduction
The process of folding is a seminal event in the life of a protein, as it is essential for its proper function. Incorrect folding, or misfolding, will not just lead to a near certain loss of function, but often also to the accumulation of protein aggregates that can harm cellular homeostasis [1, 2, 3, 4, 5, 6]. Indeed, aggregates are a hallmark of stressed, aged and disease‐state cells, posing danger not only upon their accumulation, but also mainly during the process of their formation [7, 8]. To protect cells from this type of damage, a specialized group of proteins termed molecular chaperones helps to ensure the proper folding of other proteins, acting as the first line of defence against protein misfolding and aggregation [9, 10, 11, 12]. The majority of stress‐inducible molecular chaperones, though, are unable to recognize or remodel protein aggregates once they have formed. Cells have therefore evolved powerful ATP‐driven protein disaggregases with the remarkable ability to rescue stress‐damaged proteins even from an aggregated state [13, 14, 15, 16, 17].
The heat shock protein 104 (Hsp104) is the main protein disaggregase in yeast, plants and the mitochondria of all eukaryotic cells, and thus is essential for cell survival during severe stress [14, 18, 19, 20]. It belongs to the AAA+ (ATPases Associated with various cellular Activities) family of molecular machines, which utilize ATP hydrolysis to facilitate diverse cellular functions [21, 22, 23]. In the case of Hsp104 disaggregases, the energy derived from ATP binding and hydrolysis drives protein unfolding and disaggregation, enabling the chaperones to solubilize both amorphous and ordered protein aggregates.
Hsp104, however, cannot generally solubilize aggregates on its own and requires the synergistic interaction with the heat shock protein Hsp70 (Hsp70) chaperone system [18, 24, 25]. There, Hsp70 chaperones, aided by J‐domain protein co‐chaperones, first perform initial remodelling of the aggregated substrates before presenting them to Hsp104. The Hsp70‐bound, partially‐unfolded polypeptides are then recognized by the Hsp104 hexamers, initiating the translocation process into the Hsp104 channel [25, 26]. In parallel, the binding of Hsp70 enhances Hsp104 ATP hydrolysis rates, thereby activating the disaggregase [27, 28, 29]. The activated Hsp104 then threads the polypeptide chains, one at a time, through the central pore of its hexameric ring [25, 30], thus unravelling the target aggregates.
In addition to its function as a disaggregase, the yeast Hsp104 is also essential for the propagation of prions [PSI+] [31], [URE3] [32] and [RNQ1+] [33], which are self‐replicating, amyloid‐like aggregates of the normal functional proteins. Hsp104 helps yeast prions to replicate mainly by fragmenting the amyloid‐like proteins into smaller pieces, thereby breeding new propagons, or seeds [34, 35]. Interestingly, the Hsp104 prion propagation function might not require collaboration with the Hsp70 chaperone system [36, 37].
Overexpression of Hsp104 in yeast, however, dissolves these seeds completely and, accordingly, cures the [PSI+] prion state [38], suggesting that Hsp104 could potentially serve as a therapeutic approach against amyloidogenic neurodegenerative diseases in humans, such as Alzheimer's, Parkinson's and Huntington's disease [39, 40, 41, 42]. Indeed Hsp104‐bearing potentiating mutations were shown to disassemble (in an Hsp70 independent manner) amyloid conformers of several proteins associated with neurodegenerative disease, including α‐synuclein, TDP‐43, FUS, polyglutamine, amyloid‐beta and tau [37, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52].
Structurally, Hsp104 and its bacterial homolog ClpB, form hexameric spiral assemblies [53, 54, 55, 56, 57, 58], with each protomer comprising an N‐terminal domain (NTD); and two nucleotide‐binding domains (NBD1 and NBD2), separated by a unique regulatory coil–coil domain [59] (Fig. 1), which serves as the Hsp70‐binding site and is thus essential for Hsp104 activation [28, 60, 61]. NBD1 and NBD2 both contain conserved Walker A and B motifs and an arginine finger residue, all of which are critical for ATP hydrolysis, as well as flexible tyrosine ‘pore loops’ that bind substrates and are required for their translocation into the Hsp104 hexameric chamber [62, 63, 64] (Fig. 1C).
Fig. 1.
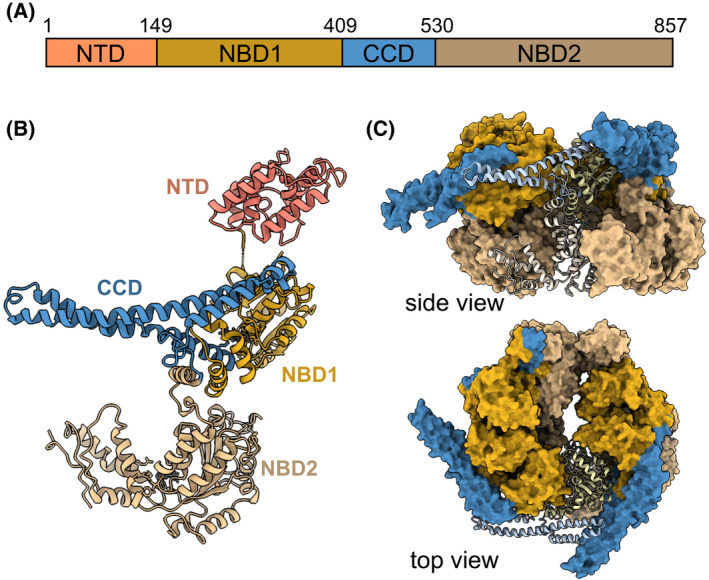
Structure and domain organization of Hsp104. (A, B) Domain organization (A) and protomeric structure (B) of Hsp104 (PDB 6N8T [65]). The Hsp104 protomer consists of an NTD (salmon), two NBDs (NBD1, NBD2 ‐ dark and light yellow, respectively), and a CCD insertion (blue). (C) The monomers assemble into asymmetric hexamers (PDB 5VY8 [54]) consisting of three layers ‐ the NTDs (top layer‐ absent from the presented structures), NBD1‐CCD (light yellow‐blue), and NBD2 (dark yellow), all enclosing the central pore. All structures were visualized using UCSF ChimeraX (https://www.cgl.ucsf.edu/chimerax).
In recent years, several detailed Hsp104 and ClpB cryo‐EM structures [53, 54, 55, 56, 57, 58, 65] have greatly advanced our understanding of not just the hexameric structure of Hsp104, but also of the mechanism of substrate translocation during the disaggregation reaction. Despite these recent advances, though, a key part of this mechanism has remained poorly understood –how Hsp104 initially recognizes and binds its diverse set of client proteins.
Several studies have pointed to the Hsp104/ClpB NTD as the initial client recognition site [66, 67, 68]. The NTD is a globular, 150‐residue α‐helical domain connected by a long unstructured linker to NBD1 [69] (Fig. 1). It was proposed to form a substrate entrance channel that both stabilizes the unfolded state of the polypeptide and helps to position it above NBD1 [58, 66], thus facilitating substrate translocation into the Hsp104/ClpB central pore. The flexibility of the NTD, however, has precluded it from high‐resolution structural characterization by cryo‐EM, leaving its orientation and location relative to the hexameric chamber largely unknown.
In addition, it is still not fully clear how the NTD recognizes the many Hsp104 clients, or whether this interaction is important for Hsp104 cellular functions. While many of the early reports show that this domain is entirely dispensable for disaggregation [67, 70, 71], others found that without it, the disaggregation activity is affected [61, 68, 72, 73]. Similarly, while the NTD is dispensable for prion propagation [71, 74, 75], some studies found it to be required for prion curing (dissolution) [38, 70], adding to the controversy regarding the function and importance of this domain.
Here, we have used NMR to structurally characterize the interaction of the Hsp104 NTD with multiple types of client proteins, and designed specific mutations abolishing these interactions. Moreover, by studying the activity of these mutants, we have been able to elucidate the functional role of the NTD in protein disaggregation and prion propagation. Our results demonstrate that the NTD contains a substrate‐binding groove that specifically recognizes hydrophobic residues exposed in unfolded or aggregated clients, as well as in amyloid‐ and prion‐forming proteins. Mutations to this NTD substrate‐binding groove have a dramatic effect on the activation of Hsp104 ATP hydrolysis, resulting from its interaction with client proteins, as well as on the overall disaggregation activity of the chaperone. Furthermore, we show that interaction with clients is essential for activating Hsp104 disaggregation and thermotolerance.
Together, our findings provide molecular insight into the NTD‐substrate complex as well as into the functional role of the NTD in both protein disaggregation, prion propagation and thermotolerance.
Results
Substrate interactions with the Hsp104 N‐terminal domain
Previous studies have shown that the NTD of Hsp104 bacterial homolog, ClpB, contains a hydrophobic groove that interacts with the exposed hydrophobic residues in unfolded proteins and polypeptides [66, 67, 68]. Hsp104, however, does not interact solely with unfolded proteins, but also with thermally and chemically induced protein aggregates, as well as prion proteins [35, 36, 76, 77]. It therefore remained unclear whether the Hsp104 NTD contains a distinct binding site for each of these diverse client types, and whether the hydrophobic groove also plays a role in these interactions.
To address this, we performed a series of NMR‐binding experiments studying the interaction of Hsp104 NTD with its diverse clients. First, intrinsically disordered α‐casein and fragments of reduced alkaline phosphatase (PhoA) were used as unfolded polypeptide clients, and their binding to uniformly 15N‐labelled NTD was tested by recording 1H–15N heteronuclear single quantum coherence spectroscopy (HSQC) spectra (Fig. 2). All three clients bound to Hsp104 NTD and caused chemical shift perturbations (CSPs), on a fast exchange time scale, to a distinct, yet similar subset of NTD residues.
Fig. 2.
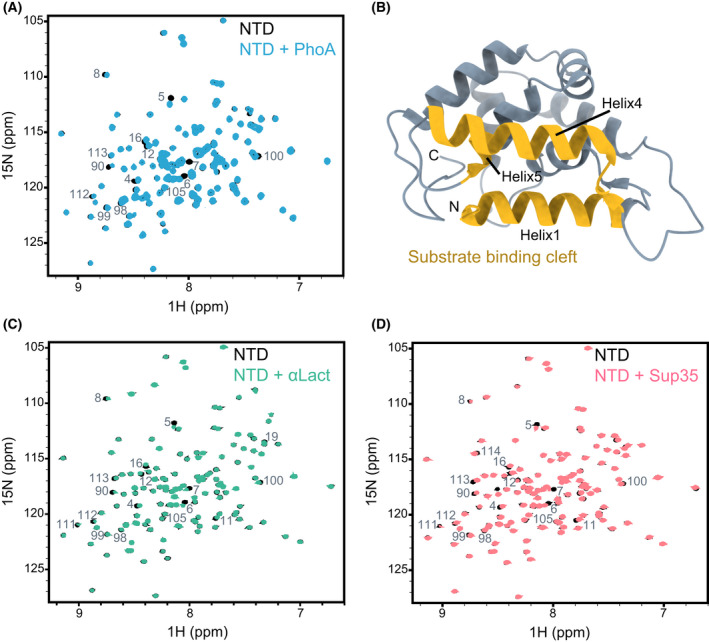
Interaction of Hsp104 NTD with client proteins. (A) 1H‐15N HSQC spectra of 200 µm NTD alone (black), and in complex with 400 µm unfolded PhoA protein fragment, resides 349–471 (blue). (B) Cartoon representation of Hsp104 NTD X‐ray structure (PDB 5U2U [69]), with the residues found in our NMR experiments to bind to client proteins highlighted in yellow. The largest CSPs were observed for residues D3–S23 (Helix1), T87–N105 (Helix4) and I111–L115 (Helix5). The figure was generated with UCSF ChimeraX. (C, D) 1H‐15N HSQC spectra of 200 µm of NTD alone (black), and in complex with 400 µm of heat‐denatured α‐lactalbumin (C, green), and 450 µm of Sup35 NM prion protein (D, light red). The NTD residues that are broadened significantly upon interaction with each client are indicated. All three substrates bind to the same regions of the NTD.
In order to map this site on the NTD, though, we first had to assign the domain's spectrum. To this end, we recorded a set of HNCACB, CBCA(CO)NH, HNCA, HN(CA)CO and HNCO 3D NMR experiments. Combined, these allowed us to unambiguously assign 70% of the non‐proline residues, with the relatively low degree of assignment coverage being due, in part, to severe peak overlap in our spectra. To overcome this, we used a ‘reverse isotope’ labelling approach [78], where 14N, 12C, 1H‐labelled lysine, arginine, isoleucine or leucine amino acids were incorporated into 15N‐labelled proteins (Fig. 3). The incorporation of 14N amino acids reduced spectral overlap, as well as allowed for unambiguous assignment of amino acid types. Using this strategy, we obtained 95% coverage of the non‐proline residues in the Hsp104 NTD.
Fig. 3.
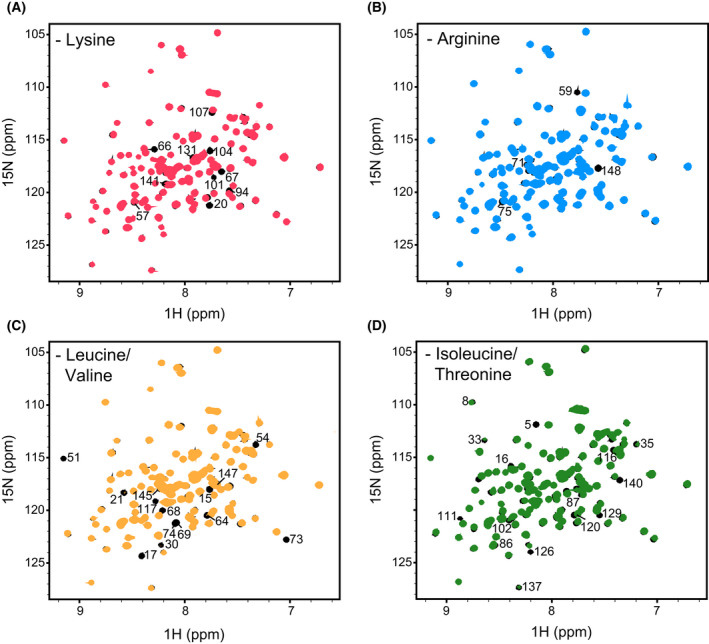
NMR ‘reverse’ isotopic labelling. (A–D) 1H‐15N HSQC spectra of Hsp104 NTD ‘unlabelled’ with 0.2 g·L−1 of 14N lysine (A, red), 1 g·L−1 of 14N arginine (B, blue), 0.4 g·L−1 of 14N leucine (C, yellow) or 0.2 g·L−1 of 14N isoleucine (D, green) overlaid on a uniformly 15N‐labelled NTD spectrum (black). The unlabelled residues are indicated. In the case of leucine reverse labelling, the intensity of peaks arising from valine were also greatly reduced. Similarly, isoleucine reverse labelling also affected threonine residues.
With the assignments in hand, we could now map the binding site for unfolded proteins on the NTD. The largest CSPs observed were for NTD residues D3–S23 (Helix1), T87–N105 (Helix4) and I111–L115 (Helix5). Mapping these CSPs onto the crystal structure of the Hsp104 NTD [69] revealed that unfolded proteins bind to the hydrophobic groove (Figs 2A–B and 4A–C), similarly to the interaction of the same substrates with ClpB [66]. Thus, the interaction of hydrophobic residues in the unfolded client proteins with the hydrophobic groove of the NTD is conserved between eukaryotic and prokaryotic disaggregases.
Fig. 4.
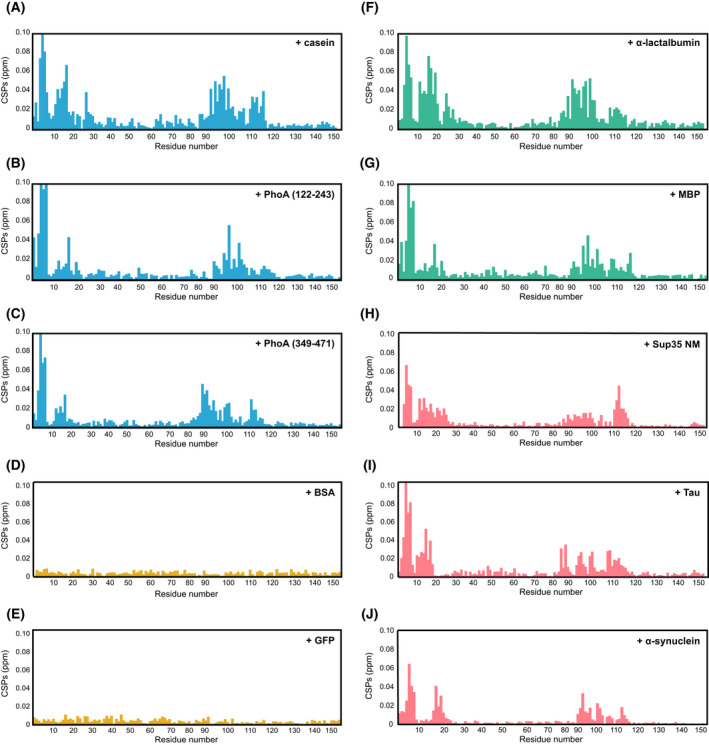
A diverse set of Hsp104 clients bind to the same hydrophobic groove in the NTD. (A–J) CSPs induced by casein (A), PhoA122–243 (B), PhoA349–471 (C), BSA (D), GFP (E), heat‐denatured aggregated α‐lactalbumin (F), heat‐denatured aggregated MBP (G), Sup35 prion protein NM domains (H), tau (I) or α‐synuclein (J) binding to 15N‐labelled Hsp104 NTD. Unfolded/unstructured client proteins are coloured blue, natively folded proteins yellow, aggregated protein green and prion/amyloid‐forming proteins light red. The unfolded, misfolded and aggregated clients, as well as prion proteins all bind to the same region of the NTD, which corresponds to the hydrophobic groove (Fig. 2D). Natively folded proteins, however, are not recognized by this domain.
Next, we repeated the binding experiment using misfolded/aggregated clients. Addition of two‐molar equivalent of heat‐denatured α‐lactalbumin (Figs 2C and 4F) and MBP (Fig. 4G) to 15N‐labelled NTD also caused CSPs to the residues in the hydrophobic pocket (Fig. 2B), showing that this site in the NTD can recognize both misfolded and unfolded substrates.
In addition to recognizing misfolded or aggregated client proteins, Hsp104 is also an important factor in prion curing (dissolution), a process known to be dependent on a functional NTD [70, 73, 79]. We therefore tested whether the NTD can recognize and interact with the prion‐forming protein, Sup35. This translation‐termination factor is a prion‐forming protein that can change its conformation from a fully active soluble state, [psi−], to an inactive amyloidogenic state, [PSI+]. Sup35 protein consists of three domains: the aggregation‐prone NTD (N), rich in asparagines and glutamines (also termed as the ‘prion domain’); the highly charged middle domain (M), that modulates the prion state and the C‐terminal GTPase domain (C). Using NMR, we probed for an interaction between the prion‐forming NM domains (M domain presence was required to increase the solubility of the protein) and the Hsp104 NTD. Addition of two‐fold excess of Sup35 NM to 15N‐labelled Hsp104 caused significant chemical shift changes to residues in helices 1, 4 and 5, that correspond to the substrate‐binding hydrophobic groove of the NTD (Fig. 2B,D and Fig. 4H). Similar CSP patterns were also observed upon addition of the amyloid‐forming proteins tau and α‐synuclein (Fig. 4I,J). Thus, prion proteins do not have a unique binding site on the Hsp104 NTD, but rather are recognized by a similar site as unfolded and misfolded client proteins.
As a control, we decided to verify that folded globular proteins do not interact with Hsp104 NTD. As expected, addition of 4‐fold excess of BSA or GFP did not result in changes to the 1H‐15N HSQC spectra of the NTD, and only minimal CSPs were observed, thereby demonstrating that the Hsp104 NTD indeed does not recognize properly folded proteins (Fig. 4D,E).
Combined, our NMR experiments show that the NTD contains a conserved hydrophobic binding groove capable of recognizing and binding to unfolded, misfolded and prion proteins, but not to properly folded, native ones.
Mutations to the NTD
To investigate the functional importance of the interaction between the NTD hydrophobic groove and various client proteins, we generated Hsp104 NTD mutants that abolish its ability to interact with substrates.
Several studies have reported that the replacement of hydrophobic residues in the NTD‐binding pocket reduces the disaggregation activity of Hsp104 [66, 67, 68]. However, such mutations also greatly decreased the stability of the NTD, which may have contributed to this impairment of Hsp104 activity. Under our experimental conditions, mutations to helix 1 residues (F7A and L15A) significantly destabilized the NTD, with its melting temperature (T m) decreasing from 84 °C (WT NTD) to 71 °C in the NTDF7A, L15A mutant. Mutations to helix 4 (L96A) likewise decreased the T m value by 10 degrees, and mutations to helix 5 (L111A and L116A) by 12 degrees. Combined mutants, such as NTDF7A,L15A,L96A,I111A, NTDF7A,L15A,L96A,I116A, or NTDL12A,L15A,L96A,I111A,I116A, were highly unstable (T m ~ 45 °C) and displayed NMR spectra characteristic of partially unfolded and/or aggregated proteins.
To disrupt the hydrophobic NTD‐binding pocket without significantly affecting the stability of the protein, we instead designed two point‐mutations replacing the hydrophobic residues in NTD helices I and IV with charged residues (Fig. 5A). The resulting Hsp104 NTD L15D, L96R mutant (NTDDR) had similar stability to that of the wild type protein, with a melting temperature of 80 °C (Fig. 5B). Moreover, the 1H‐15N HSQC spectrum of NTDDR was well dispersed and of high quality, indicating that the protein was properly folded and that its overall conformation remained unchanged (Fig. 5C).
Fig. 5.
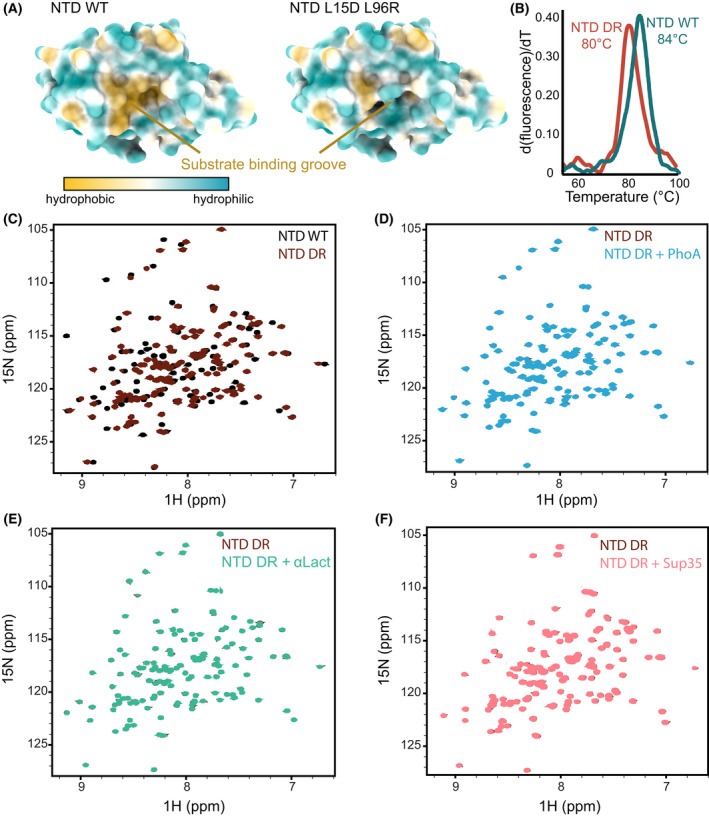
Mutations to the NTD hydrophobic groove impair substrate binding. (A) Structure of wild‐type NTD (left) and NTD L15D L96R (NTD DR) variant (right), coloured based on residue hydrophobicity using UCSF ChimeraX. Surface representation of wild‐type NTD shows the hydrophobic substrate‐binding groove that is eliminated by the introduction of L15D and L96R charge mutations. (B) Thermal unfolding curves of wild‐type NTD and NTD DR. The calculated melting temperatures (T m) are indicated. (C) Overlaid 1H‐15N HSQC spectra of wild‐type NTD (black) and NTD DR mutant (dark red). (D–F) Overlaid 1H‐15N HSQC spectra of 200 µm of NTD DR alone (dark red), and when incubated with either 4‐fold excess of PhoA fragment (D, blue), 2‐fold excess of heat‐denatured α‐lactalbumin (E, green), or 4‐fold excess of Sup35 NM prion protein (F, light red). No changes were observed in any of these binding spectra, indicating that NTD DR does not interact with these client proteins.
We next tested whether the NTDDR mutant was impaired in substrate binding. Addition of 6‐fold excess of either PhoA fragment, heat denatured α‐lactalbumin or Sup35 NM prion‐forming domain to 15N‐labelled NTDDR mutant did not result in any changes to the NMR spectra (Fig. 5D–F), indicating that this mutant indeed has a significantly reduced affinity towards client proteins. Having generated a stable NTDDR mutant, yet with highly reduced substrate‐binding capabilities, we could then test the effects of the NTD‐substrate interaction on Hsp104 functions.
Chaperoning functions of the NTD are impaired by the mutations
The NTD binds to its client proteins through exposed hydrophobic regions, stabilizing the substrate's unfolded state [66]. Thus, the NTD itself could potentially function as a ‘holdase’ chaperone, preventing the aggregation of certain client proteins. We therefore monitored the changes to the aggregation of misfolded α‐lactalbumin upon addition of this Hsp104 domain.
The formation of insoluble amorphous aggregates of α‐lactalbumin (300 µm) was induced by addition of DTT (2 mm), and monitored by light scattering. Under these conditions, α‐lactalbumin aggregation occurred following a lag period of ~ 30 min and reached a plateau, indicative of amorphous aggregate formation, after ~ 150 min (Fig. 6A, grey). Addition of Hsp104 NTD greatly increased the lag phase, inhibiting aggregation entirely over the course of 15 h (Fig. 6A, cyan). Addition of the same concentration of Hsp104 NTDDR (100 μm), however, had no effect on the aggregation (Fig. 6A, red), demonstrating that the NTD suppression of α‐lactalbumin aggregation is dependent on substrate interaction with its hydrophobic groove.
Fig. 6.
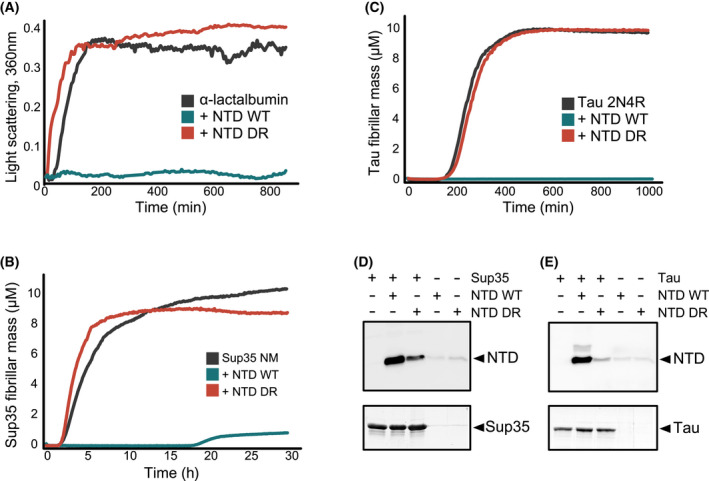
Hsp104 NTD can function as a bona fide chaperone. (A) Aggregation of α‐lactalbumin (300 μm) monitored by light scattering (OD 360 nm), either alone (grey) or when NTD WT (cyan) or NTD DR mutant (red) were added at the beginning of the aggregation reaction. NTD WT (100 μm) suppressed α‐lactalbumin aggregation for the entire course of the experiment, while the addition of NTD DR (100 μm) had no effect on the aggregation profiles. (B, C) ThT fluorescence assay of Sup35 NM (B) or tau 2N4R (C) aggregation reaction profiles, either alone (grey), or in the presence of NTD WT (cyan) or NTD DR (red). NTD WT can suppress the aggregation of Sup35 NM for ~ 20 h and of tau 2N4R for over 16 h, while NTD DR mutant is completely inactive in aggregation suppression. Data points represent the means of 3–5 independent measurements. (D, E) Co‐sedimentation of preformed Sup35 NM prions (15 μm, D) or tau amyloid fibrils (10 μm, E) in the presence of 20 μm Hsp104 NTD or Hsp104 NTDDR mutant. Pellet fractions after ultracentrifugation were analysed by SDS/PAGE and blotted with Hsp104 antibody.
After identifying that the NTD can prevent aggregation of amorphous aggregates, we next asked if it is also capable of preventing the formation of prion proteins and amyloid fibres. For this, Sup35 prion formation was initiated by addition of prion seeds, and then monitored by Thioflavin‐T (ThT) fluorescence [80], which is sensitive to the formation of cross‐β amyloid folds (Fig. 6B). Upon incubation at 37 °C, Sup35 aggregation occurred following a lag period of ~ 1.6 h. Addition of Hsp104 NTD greatly suppressed Sup35 aggregation for the course of ~ 19 h, while addition of NTDDR showed no such suppression, and even slightly accelerated Sup35 aggregation, potentially due to co‐aggregation with the prion fibres (Fig. 6B). In order to test if the NTD inhibits prion formation by interaction not only with Sup35 monomers but also with the NM fibrils, we performed co‐sedimentation assays. Preformed Sup35 NM fibres were incubated with NTD or NTDDR, and the insoluble fraction of these reactions, following ultracentrifugation, was blotted with Hsp104 antibody. Indeed, a large portion of NTD, but not of the mutant NTDDR, was detected in the insoluble fraction together with Sup35 (Fig. 6D), indicating a strong interaction between the NM prion fibrils and the wild‐type chaperone.
We next tested if the NTD can also suppress the aggregation of the amyloid‐forming protein, tau. Aggregation of tau 2N4R protein was initiated by addition of heparin polyanion (1:4 tau:heparin ratio), and monitored by ThT fluorescence [80]. As in the case of Sup35, addition of Hsp104 NTD completely inhibited the formation of tau fibrils for over 16 h (Fig. 6C), with no observable ThT signal being detected over this length of time. In contrast, aggregation in the presence of NTDDR was similar to that of tau alone (Fig. 6C). Also, as in the case of Sup35 fibres, co‐sedimentation experiments indicated a strong interaction between mature tau amyloid fibres and wild type, but not mutant NTD (Fig. 6E).
Thus, the ability of the NTD to prevent the formation of amorphous aggregates, prions and amyloid fibres is entirely dependent on the presence of a functional hydrophobic substrate‐binding groove.
Functional NTD is required for Hsp104‐substrate activation
After successfully generating an Hsp104 mutant in which we disrupted the binding and chaperoning functions of the NTD, we tested the impact of this mutation in the context of the full‐length Hsp104 hexameric disaggregase (Hsp104DR).
Hsp104 is an AAA+ ATPase whose activity is tightly regulated by association with client proteins and with Hsp70 chaperones. The basal ATPase activity of wild‐type Hsp104 can be stimulated 4‐fold following the addition of α‐casein as a substrate (Fig. 7A, cyan), and 3‐fold upon addition of Hsp70 activator (Fig. 7B, cyan), in agreement with previously published data [27, 66, 81].
Fig. 7.
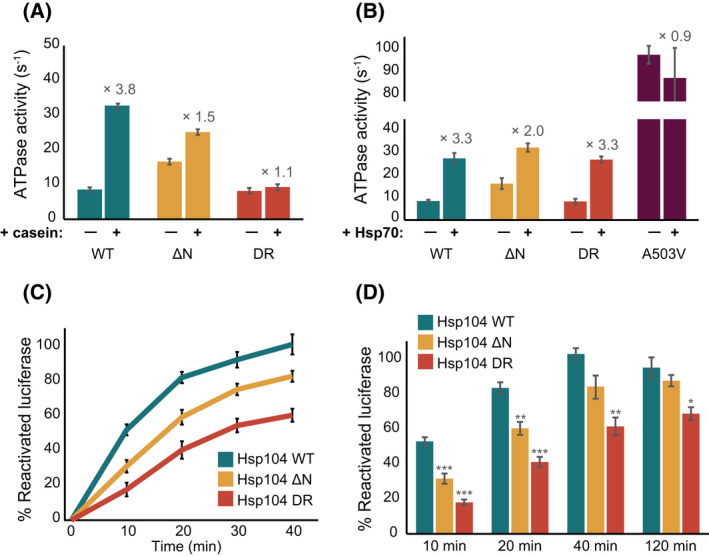
Mutations in NTD affect Hsp104 disaggregation activity. (A) Steady state ATPase activity of Hsp104WT (cyan), Hsp104ΔN (yellow) and Hsp104DR (red) variants measured alone, and in the presence of α‐casein substrate. Substrate activation of Hsp104 is shown to be highly dependent on the presence of a functional NTD. Data are means ± SEM (n = 3). (B) ATPase activity of all Hsp104 variants is enhanced through interaction with Hsp70. Rates of Hsp104WT (cyan), Hsp104ΔN (yellow) and Hsp104DR (red) ATP hydrolysis were measured in the absence and presence of Hsp70T204A. Hsp104A503V which does not bind Hsp70, was not stimulated in the presence of Hsp70 beyond its higher basal ATPase activity [37] (purple). Data are means ± SEM (n = 3). (C) Disaggregation kinetics of chemically‐induced aggregates of FFL by Hsp104 WT (cyan), Hsp104ΔN (yellow) or Hsp104DR (red) in the presence of Ssa1 and Ydj1. Data are means ± SEM (n = 5). (D) Yields of FFL disaggregation at various time points across the FFL disaggregation reactions for Hsp104 WT (cyan), Hsp104ΔN (yellow) or Hsp104DR (red) variants. One‐way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001 (n = 5).
Interestingly, an Hsp104 variant in which the entire NTD was removed (residues 1–156; Hsp104ΔNTD) exhibited two‐fold higher basal ATP hydrolysis rates (Fig. 7A,B, yellow) than the WT, in agreement with recent findings that in the Hsp104 bacterial homologue, ClpB, the NTD suppresses activity by restricting coiled‐coil domain (CCD) motions [82]. Furthermore, the higher basal activity of Hsp104ΔNTD was only moderately increased upon the addition of casein (1.5‐fold; Fig. 7A, yellow), demonstrating that it is indeed the direct interaction of the NTD with substrates that releases its inhibition of Hsp104 ATPase activity. In contrast, in the presence of Hsp70 chaperones, similar hydrolysis rates were reached by Hsp104 and Hsp104ΔNTD (27.2 ± 2.3 s−1 and 31.9 ± 1.9 s−1, respectively, Fig. 7B). Combined, these results suggest that the activity of the NTD‐truncated Hsp104 variant is regulated by Hsp70 chaperones, but less so by its interaction with client proteins.
We next measured the ATPase activity of the mutant Hsp104 impaired in NTD‐substrate binding (Hsp104DR). Hsp104DR hexamers displayed basal ATPase activity very similar to that of the wild‐type protein, and a similar degree of activation by Hsp70 (Fig. 7B, red). These variants, however, showed no increase in ATPase hydrolysis rates upon addition of casein (Fig. 7A, red). This, then, indicates that the ATPase activity enhancement observed for the wild‐type Hsp104 in the presence of casein indeed relies on the direct interaction of the substrate with the Hsp104 NTD.
Thus, the NTD serves both as an initial substrate‐binding site and as a steric repressor of Hsp104 activity. Interaction of NTD with substrates restricts the mobility of the NTD [66], with this potentially driving the release of the inhibition and the subsequent activation of the Hsp104 disaggregase. In the absence of such an interaction, as in the Hsp104DR mutant that does not bind to client proteins through their NTD, the chaperones are retained in a partially inhibited state.
The substrate‐binding groove in the NTD plays a role in Hsp104 disaggregation activity
The role of the NTD in Hsp104‐dependent protein disaggregation has been the subject of many conflicting reports. While some claim it is entirely dispensable for disaggregation [67, 70, 71], others found that without it, the disaggregation activity is affected [68, 73]. Identifying that the NTD, in fact, performs two roles – one involving directly interacting with substrates, and the other, suppressing Hsp104 ATPase activity, we wanted to understand if these contradictory findings perhaps stem from the inability to separate the two NTD functions. Having designed an Hsp104 mutant that is only deficient in substrate binding, but is still capable of repressing Hsp104 activity, we had a tool to directly test the role of NTD‐substrate binding in protein disaggregation.
To this end, we monitored the disaggregation activity of wild‐type Hsp104, Hsp104ΔNTD and Hsp104DR variants, aided by the Hsp70 chaperone system (Ssa1/Ydj1) and using chemically denatured firefly luciferase (FFL) as a substrate. Neither Ssa1/Ydj1 nor Hsp104 alone, as expected, were able to disaggregate FFL. Addition of Hsp70/Ydj1 chaperones to wild‐type Hsp104, however, resulted in refolding of nearly 100% of the aggregated luciferase to an enzymatically functional state (Fig. 7C). Interestingly, Hsp104ΔNTD, which lacks the NTD, showed slower initial disaggregation rates compared to those obtained with the wild‐type Hsp104 (Fig. 7C). It is thus possible that, despite having high ATP hydrolysis rates due to the removal of the inhibition element, the lack of the initial binding site for substrates, located in the NTD, results in a less efficient and therefore slower disaggregation of amorphous aggregates. The final yield of luciferase reactivation after 120 min, is however very similar between the two Hsp104 variants (Fig. 7D). Thus, the lack of the NTD initial binding site merely slows down the rate of disaggregation, without significantly affecting its final efficiency.
We then tested the disaggregation rates of Ssa1/Ydj1 in conjunction with the Hsp104DR variant, which lacks the ability to interact with client proteins via its NTD. This mutant displayed significantly reduced disaggregation rates, even compared to Hsp104ΔNTD that lacks the NTD entirely (Fig. 7C). Furthermore, even after 120 min only 67% of luciferase was reactivated (Fig. 7D). The higher decrease in both the rate and yield of disaggregation by Hsp104DR suggest that it is both the initial interaction of the NTD with client proteins, and the release of the NTD‐dependent inhibition of Hsp104 that are required to achieve efficient disaggregation. Moreover, these results indicate that client binding to the NTD enhances the initial rates of the disaggregation, while the release of the NTD inhibition affects both the rate and the final yield.
Functional Hsp104 NTD is required for thermotolerance
Upon exposure to environmental stress, Hsp104 expression is upregulated to broadly counter protein aggregation and promote cell survival, thereby conferring thermotolerance [19, 83]. Following extreme heat shock, at temperatures 50 °C and above, cells expressing Hsp104 are significantly more viable than cells that do not express Hsp104 [19]. This thermotolerance activity is completely dependent on the ability of Hsp104 to prevent the accumulation of protein aggregates within cells by promoting disaggregation.
As we identified that mutations in NTD, that are deficient in substrate binding, lead to impaired Hsp104 disaggregation activity, we hypothesized that Hsp104DR may also display reduced thermotolerance activity in cells.
To test this, we utilized CRISPR to generate Saccharomyces cerevisiae strains endogenously expressing Hsp104DR (with L15D and L96R mutations) or Hsp104ΔNTD (with a deletion of the entire NTD, residues 1–150), and measured their survival compared to strains with wild‐type Hsp104 or Hsp104 deletion (Δhsp104). All variants were generated in a S288C [84] background and under the expression of HSP104 native promoter, fused to mNeonGreen (mNG) [85] at the C‐terminus [86].
The HSP104ΔNTD and HSP104DR strains displayed similar Hsp104 expression patterns to the wild‐type HSP104, with a 4–6‐fold induced expression upon exposure to a 30‐min heat shock at 37 °C (Fig. 8A), as measured by flow cytometry following the fluorescence intensity of mNG fusion. No fluorescence was observed for the Δhsp104 strain that does not express Hsp104.
Fig. 8.
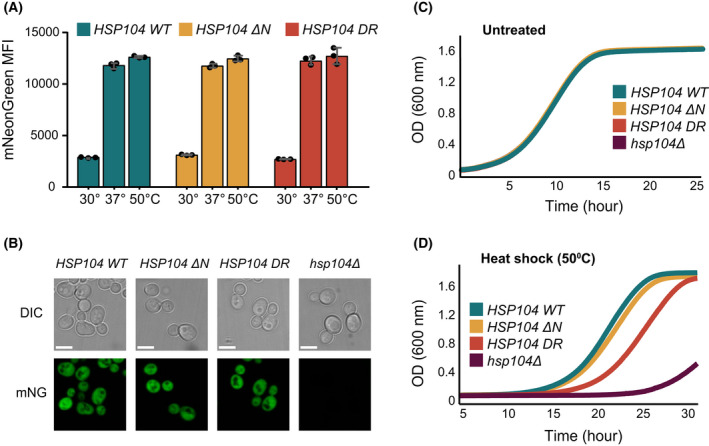
A Functional Hsp104 NTD is required for yeast thermotolerance in vivo. (A) Quantification of Hsp104 protein expression in WT HSP104‐mNG (cyan), HSP104ΔN‐mNG (yellow) and HSP104DR‐mNG (red) yeast strains. Expression was quantified by flow cytometry, measuring mean fluorescence intensity of mNG for untreated cells (kept at 30 °C), cells exposed to a 30 min heat shock at 37 °C, or cells first exposed to 30 min at 37 °C and then another 30 min at 50 °C. No significant changes in Hsp104 expression are observed for the different strains. Data are means ± SEM (n = 3). (B) Light and fluorescence microscopy images of WT HSP104‐mNG, HSP104ΔN‐mNG, HSP104DR‐mNG and Δhsp104 yeast strains following heat‐shock at 37 °C. All cells display normal morphology and Hsp104 WT, Hsp104ΔN, and Hsp104DR variants all show cytosolic localization. No Hsp104 expression was observed in the deletion strain. Scale bars: 5 μm. (C, D) Thermotolerance of Saccharomyces cerevisiae HSP104, HSP104ΔN, HSP104DR and Δhsp104 cells following a 30 min 30 °C (C) or 50 °C (D) treatment. In the absence of heat shock (untreated cells), all strains show similar growth rates at 27 °C, as monitored at OD600. Following heat shock, cells expressing WT Hsp104 and HSP104ΔN show similar recovery rates, while a significant delay in the rate of recovery (one‐way ANOVA, P < 0.05) is observed for Hsp104DR and Δhsp104 cells, as evidenced by an extended lag phase. The experiment was repeated three times with similar results.
Microscopy images of the four strains showed that, like the wild‐type Hsp104, Hsp104ΔNTD and Hsp104DR are well distributed in the cytosol (Fig. 8B). As before, no fluorescence was observed in Δhsp104 cells that do not express Hsp104‐mNG.
We then compared the survival of the two mutant strains (HSP104ΔNTD and HSP104DR) to cells expressing wild‐type Hsp104. Following a brief pre‐treatment at 37 °C to induce the heat shock response and enhance Hsp104 expression, we exposed the cells to a severe heat shock at 50 °C for 30 min. Following this severe heat shock, the expression levels of Hsp104, Hsp104ΔNTD and Hsp104DR were elevated by ~ 5%, compared to 37 °C (Fig. 8A). The cells growth kinetics was monitored using light scattering for 48 h at 27 °C (Fig. 8C,D). Following outgrowth, the strain expressing Hsp104 lacking the NTD displayed thermotolerance nearly identical to that of wild‐type Hsp104 (15 vs 16 h outgrowth; Fig. 8D). Cells expressing the Hsp104DR variant, however, grew significantly slower than the wild‐type cells and had a significantly higher rate of cell mortality (~ 20 h outgrowth; Fig. 8D).
These observations demonstrate that in the context of full‐length Hsp104 as found in cells, the presence of functional NTD, capable of substrate binding, is required for its function in yeast thermotolerance.
Discussion
Hsp104 protein disaggregases are powerful molecular machines that harness the energy derived from ATP binding and hydrolysis to disaggregate a wide range of protein aggregates and amyloids, as well as to assist in yeast prion propagation. We find that all these activities are mediated by initial binding of the client proteins to the hydrophobic groove found in the NTD of Hsp104. This hydrophobic groove generates a broad‐specificity initial binding platform that contacts the exposed hydrophobic patches in the unfolded, misfolded, amyloid and prion substrates of Hsp104. In addition to its function as the initial substrate recognition platform, we find that the NTD itself has chaperoning activities, which help to protect the exposed hydrophobic regions of its substrates from further misfolding and aggregation, thereby priming them for threading through the Hsp104 central channel.
Moreover, it was recently shown that in the Thermus thermophilus Hsp104 homologue, ClpB, the NTD also serves a regulatory role. Iljina et al. [82] proposed that ClpB NTDs sterically restrict the tilting of the CCDs, which is required for Hsp104 activation. We see that similar inhibition also exists in the eukaryotic Hsp104, as evidenced by the higher ATP hydrolysis rates of Hsp104ΔNTD, which lacks the inhibitory NTD, compared to wild‐type disaggregase. As we further found that this restriction can be lifted upon Hsp104 binding to substrates, it may be that this interaction reduces the mobility of the NTD [66], causing it to adopt a conformation that no longer interferes with CCD motions.
In order to test the importance of this regulatory mechanism, we have generated an Hsp104 mutant that disrupts the hydrophobic groove of the NTD and does not exhibit any substrate binding or standalone NTD chaperone activities. We find that this mutant can no longer be activated by the addition of client proteins and instead remains in a partially‐inhibited state. Interestingly, the behaviour of this mutant in protein disaggregation is very different from the Hsp104 variant in which the NTD has been removed entirely.
Many previous studies showed that the HSP104ΔNTD variant, in which the NTD was completely removed, has disaggregation and thermotolerance activities similar to those of the wild‐type protein [70, 72, 74, 87]. These findings resulted in a general consensus that the NTD is dispensable for disaggregation, prion propagation and thermotolerance. While we likewise observed that HSP104ΔNTD has wild‐type like activities in vitro and in vivo, the deletion of the entire NTD in this construct also removed the inhibition that would normally regulate Hsp104 activity. In contrast, Hsp104DR, which retains the inhibition yet lacks the substrate‐binding sites on the NTD, showed significantly reduced disaggregation activity and impaired thermotolerance.
We believe that these results help to resolve the outstanding question regarding the functional role of the NTD. We find that the NTD, which is an integral part of Hsp104 protein, is indeed required for proper Hsp104 function, facilitating both initial substrate binding to the disaggregase and modulation of its activity. The initial substrate‐binding step to the NTD releases the NTD‐dependent inhibition of Hsp104 activity and primes both the disaggregase and the client protein itself for translocation. Thus, substrate binding to the NTD is required for efficient Hsp104 protein‐disaggregation and prion‐propagation functions. Moreover, a functional NTD is also necessary for thermotolerance and cell survival upon severe stress, highlighting the importance of the Hsp104 NTD both in vitro and in vivo.
Materials and methods
Strains and plasmids
The DNA sequence encoding for the S. cerevisiae Hsp104 NTD (amino acids 1–152) was cloned into pET24a (KanR) plasmid with N‐terminal hexahistidine tag followed by TEV protease cleavage site. Hsp104 and Hsp104 NTD mutants were generated using QuikChange mutagenesis and confirmed by DNA sequencing.
The DNA sequence encoding for the S. cerevisiae Sup35 NM (amino acids 1–253) was cloned into pJC45 (AmpR) plasmid with N‐terminal hexahistidine tag followed by TEV protease cleavage site [88].
Plasmids encoding for S. cerevisiae Ssa1, Ydj1 and Sse1 were kindly provided by Pierre Goloubinoff (University of Lausanne). Plasmid encoding for Hsp104 was a kind gift from Bernd Bukau (University of Heidelberg).
Endogenous HSP104 variants were generated utilizing CRISPR/Cas9 approach using as a modified S288C yeast strain (Mat a; his3Δ1; ura3Δ0; leu2Δ0:GAL1pr‐NLS‐I‐SCEI‐natNT2; can1Δ::STE2pr‐SpHIS5) with HSP104 fused to C‐terminal mNG background [86]. To achieve endogenous mutations or deletions in HSP104, we used bRA89 plasmid expressing both Cas9 and gRNA under Hygromycin B resistance in yeast, as previously described [89]. First, bRA89 gRNA sequence was replaced with the sequence of guides targeting HSP104 gene (Table S1). The replacement was done using BplI restriction sites (FastDigest BplI, Thermo Fisher Scientific, Massachusetts, United States) followed by ligation (Quick Ligation™ Kit; New England Biolabs, Massachusetts, United States) and bacterial transformation into DH5α. Once verified by sequencing, the modified bRA89 plasmids along with linear fragments (Table S1, synthesized by Twist Bioscience, California, United States) of selected genes containing mutations/deletions were transformed into competent yeast under the selection for NAT and Hygromycin B. SC‐5‐FOA agar plates were employed to drop‐out Cas9 plasmids. The presence of desired mutations/deletion in the loci of interest was verified by Sanger sequencing.
Protein expression and purification
Ssa1 [90], Ydj1 [91], Sse1 [90], PhoA fragments [92], MBP [93], Sup35 NM [88] and tau 2N4R [94] were expressed and purified according to previously published protocols. All proteins were expressed in BL21‐CodonPlus (DE3)‐RIPL (Agilent Technologies, California, United States) cells.
Hsp104 NTD wild‐type and variants were expressed in 1 L LB and grown at 37 °C to OD 600 nm ~ 0.8. Expression was induced by the addition of 1 mm of IPTG and allowed to proceed overnight at 25 °C. Following harvesting, cells were lysed by French Press, and purified on a 5 mL HisTrap HP Ni‐NTA column (GE Healthcare, Illinois, United States). The 6His‐tag was then removed by an overnight cleavage with 2 mg of TEV protease at 4 °C. The cleaved protein was further separated from TEV protease and the uncleaved protein fraction by reverse capture Ni‐NTA. The proteins were further purified on a HiLoad 16/600 Superdex 75 pg gel filtration column equilibrated with 25 mm of HEPES pH 7.4, 300 mm of NaCl and 2 mm of DTT.
C‐terminally tagged Hsp104, Hsp104ΔN and Hsp104 mutants were expressed in 1 L LB and grown at 37 °C to OD 600 nm ~ 0.8. Expression was induced by the addition of 1 mm of IPTG and allowed to proceed overnight at 18 °C. Following harvesting, cells were lysed by French Press, and purified on a 5 mL HisTrap HP Ni‐NTA column (GE Healthcare, Illinois, United States). The purified protein was dialysed into 25 mm of HEPES pH 7.4, 20 mm of NaCl, 10 mm of MgCl2, 1 mm of ATP and 2 mm of DTT. The hexameric proteins were further separated on a HiLoad 16/600 Superdex 200 pg gel filtration column equilibrated with the same buffer.
NMR spectroscopy
All NMR experiments were carried out at 25 °C on 14.1T (600 MHz), 18.8T (800 MHz), or 23.5T (1000 MHz) Bruker spectrometers equipped with triple resonance single (z) or triple (x, y, z) gradient cryoprobes. The experiments were processed with topspin 3.5 (Bruker, Massachusetts, United States) or NMRPipe [95] and analysed with NMRFAM‐SPARKY [96] and ccpn analysis software (version 2.4.2) [97].
Isotopically labelled proteins for NMR were grown in M9 H2O media supplemented with 15NH4Cl (and 13C‐glucose) as the sole nitrogen (and carbon) source.
NMR assignment experiments
Backbone 1H, 15N and 13C resonance assignments were carried out on a sample of 2.4 mm of Hsp104 NTD in 50 mm of HEPES pH 7.4, 50 mm of KCl, 0.02% NaN3 and 10% D2O buffer. Assignments were obtained by recording HNCACB, CBCA(CO)NH, HN(CA)CO, HNCO and HNCA experiments on an 800 MHz Bruker spectrometer.
Reverse labelling was achieved by supplementing the 15N‐based M9 minimal media with 1 g·L−1 14N‐labelled arginine, 0.4 g·L−1 leucine, 0.2 g·L−1 lysine or 0.2 g·L−1 isoleucine amino acids 30 min prior to induction.
Combined, these approaches led to the unambiguous assignment of 95% of non‐proline residues.
NMR binding experiments
NTD interaction with client proteins was assayed for 200 µm samples of [U‐15N]‐labelled NTD or NTDDR in 50 mm of HEPES pH 7.4, 50 mm of KCl, 1 mm of DTT, 0.02% NaN3 and 10% D2O. The following concentrations of client proteins were used – 400 µm of casein, 400 µm of PhoA22–123, 400 µm of PhoA349–471, 400 µm of α‐lactalbumin, 400 µm of MBP, 450 µm of Sup35 NM, 600 µm of tau 2N4R, 800 µm of GFP and 800 µm of BSA.
1H‐15N HSQC‐TROSY spectra were acquired for each sample and chemical shift differences were calculated from the relation
where ΔδH is the amide proton chemical shift difference and ΔδN is the 15N backbone chemical shift difference.
Chemical shift perturbations between free NTD and NTD‐substrate mixtures that were greater than one standard deviation from the mean were considered significant.
Aggregation prevention
All chaperone activity assays were completed using a 96‐well plate and a BioTek Synergy H1 plate reader.
Calcium‐depleted bovine α‐lactalbumin (300 μm; Sigma‐Aldrich, Missouri, United States) was dissolved in 50 mm of HEPES pH 7.4, 50 mm of KCl buffer and aggregation was initiated by the addition of 2 mm of DTT. The aggregation was monitored at 37 °C by measuring light scattering at 360 nm as a function of time. Hsp104 NTD variants were added to a final concentration of 100 μm. Assays were conducted in triplicate and the mean ± standard deviation is reported.
Human tau 2N4R (10 μm) was pre‐incubated in the presence or absence of NTD (10 μm) or NTDDR (10 μm) for 10 min at 37 °C. All proteins in the assay were buffer exchanged into the assay buffer (50 mm of HEPES pH 7.4, 50 mm of KCl and 2 mm of DTT). Thioflavin T (ThT; Sigma) at a final concentration of 10 μm was added and the aggregation was induced by the addition of a 5 μm freshly prepared heparin salt solution (Sigma). Aggregation reactions were run at 37 °C with continuous shaking (567 r.p.m.) and monitored by ThT fluorescence (excitation = 440 nm, emission = 485 nm, bandwidth), using an area scan mode with a 3 × 3 matrix for each well. Black, flat‐bottom, 96‐well plates (Nunc) sealed with optical adhesive film (Applied Biosystems, Massachusetts, United States) were used. For data processing, baseline curves at the same conditions but without heparin were subtracted from the data. Samples were run in triplicate and the experiments were repeated at least three times with similar results.
The auto‐aggregation of Sup35 NM prion protein was monitored by following the increase of ThT fluorescence at 37 °C in 50 mm of HEPES pH 7.4, 50 mm of KCl buffer. The protein was kept at 4 °C in 8 m urea and diluted 100‐fold into an ice‐cold buffer to a final concentration of 10 μm in the presence or absence of NTD (60 μm) or NTDDR (60 μm). The aggregation assay was performed at 37 °C with continuous shaking (567 r.p.m.) and monitored by ThT fluorescence (excitation = 440 nm, emission = 485 nm, bandwidth), using an area scan mode with a 5 × 5 matrix for each well. Black, flat‐bottom, 96‐well plates (Nunc) sealed with optical adhesive film (Applied Biosystems, Massachusetts, United States) were used.
Co‐sedimentation assay
Preformed Sup35 NM (15 μm) or tau fibres (10 μm) were incubated with NTD (20 μm) or NTDDR (20 μm) for 20 min at 37 °C in 50 mm of HEPES pH 7.4 and 50 mm of KCl. The fibres were separated from the unbound chaperones by centrifugation at 16 900 g for 30 min. The pellets were washed twice, resuspended in 50 μL of buffer with 20% SDS and sonicated for 10 min. Samples were incubated for 5 min at 95 °C and run on a 12% SDS‐PAGE gel. The gels were either blotted with Hsp104 antibody or stained with Coomassie (Bio‐Rad Laboratories, California, United States).
Thermal melts
Thermal melts were performed with a nanoDSF instrument (NanoTemper Technologies GmbH, München, Germany), which was used to monitor the intrinsic fluorescence of NTD and NTD mutants as a function of temperature. Capillaries contained ~ 10 μL of each protein in an NMR buffer. The initial temperature was 20 °C and was set to increase by 1 °C per minute. Fluorescence readings were recorded at 330 and 350 nm, and the melting temperature (T m) was extrapolated.
ATPase activity measurement
The ATPase activities of WT Hsp104 or Hsp104 variants (3 μm) were measured using a NADH‐coupled colorimetric assay [98] by measuring the decrease of NADH absorption at 340 nm. The reactions were carried out in 50 mm of HEPES pH 7.4, 50 mm of KCl, 10 mm of MgCl2 and 10 mm of DTT buffer supplemented with 1.8 mm of phosphoenolpyruvate, 0.5 mm of NADH, 50 μg·mL−1 of pyruvate kinase, 50 μg·mL−1 of lactate dehydrogenase and 3.5 mm of ATP [99]. When indicated, α‐casein (10 μm) or an ATP hydrolysis inactive T204A mutant of Hsc70 (10 μm), was added to the reaction. The changes in absorbance at 340 nm were monitored for 20 min at 25 °C in a 96 wells UV plate (Eppendorf, Hamburg, Germany) using a Synergy H1 plate reader (BioTek).
Firefly luciferase disaggregation assay
Firefly luciferase aggregates were generated by unfolding FFL (2 µm) in 8 m urea at 30 °C for 30 min. Then the unfolded FFL was rapidly diluted in ice‐cold buffer (50 mm of HEPES, pH 7.4, 50 mm of KCl, 10 mm of MgCl2 and 10 mm of DTT) to a final concentration of 0.1 µm, and aggregation was allowed to occur on ice for 30 min. The disaggregation reaction was initiated by the addition of 5 μm of indicated Hsp104 variants, 5 μm of Hsc70 and 2.5 μm of Ydj1 and ATP regeneration system to 20 nm of luciferase aggregates. The ATP regeneration system was composed of 50 U Creatine Phosphokinase, 5 mm of ATP and 20 mm of Phosphocreatine. Luminescence was measured at various time points for 2 h, by addition of 50 μm of luciferin reagent (Promega, Wisconsin, United States) to 3 μL of the refolding reaction. Luminescence was measured in a white 96 well (Nunc) plate using a Synergy H1plate reader (BioTek).
Microscopy
Images were acquired using an Olympus IX83 microscope coupled to a Yokogawa CSU‐W1 spinning disc confocal scanner with dual Hamamatsu ORCA‐Flash4.0 V2 sCMOS cameras. All images are of cells grown at 30 °C in yeast extract peptone dextrose (YPD) medium to OD600 0.6 and then incubated at 37 °C to induce Hsp104 expression [19]. Following 30 min recovery at 30 °C, 6 μL of 50‐fold concentrated cells were placed on 22 × 22 mm glass coverslips pretreated with Concanavalin A. All images were acquired at x60 magnification.
Flow cytometry
Cells were allowed to grow to OD600 0.6 at 30 °C, diluted 60‐fold and then incubated for 30 min at 37 °C to induce the expression of Hsp104 chaperone. Cells were then either allowed to grow for 30 min at 30 °C or subjected to a 30 min severe heat shock of 50 °C. The mNG fluorescence intensity of cells was measured for the untreated cells (kept at 30 °C), as well as after the 37 and 50 °C incubations, and corresponds to the level of expressed Hsp104 protein variants C‐terminally fused to mNG. The mNG fluorescence intensity for 70 μL of each sample was quantified by Flow cytometry measurements using an Attune NxT Flow Cytometer (Thermo Fisher Scientific).
Thermotolerance assay
Saccharomyces cerevisiae cultures were grown in a standard rich (YPD; 1% yeast extract, 2% peptone, 2% glucose) medium, supplemented with 100 μg·mL−1 nourseothricin antibiotics, to OD600 0.6 at 30 °C, then diluted 12‐fold to OD600 of 0.05, and pretreated for 30 min at 37 °C to induce heat‐shock protein expression [19]. Following the pre‐treatment, the cells were either subjected to a 30 min heat shock at 50 °C or kept at 25 °C (control). The treated and control cells were grown in 96 well clear plate (Thermo Fisher Scientific), at 27 °C at a final volume of 200 μL. Cultures were kept in suspension by vigorous continuous orbital shaking (548 r.p.m.) and the empty wells in the plate were filled with water to prevent evaporation. The rate of cell growth was monitored every 5 min at OD600 for 48 h, by a Synergy H1 (BioTek) plate reader. The experiment was repeated three times with similar results.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AH, and GZ, and RR planned the experiments; AH, and GZ, and TL performed experiments; AH, GZ and RR analysed the data; AH, GZ, TL and RR wrote the paper.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16441.
Supporting information
Table S1
Acknowledgements
The authors thank Dr. Lihi Radomir for her help with flow cytometry experiments. RR is supported by the European Research Council starting grant (ERC‐2018‐STG 802001), Israel Science Foundation and a research grant from the Blythe Brenden‐Mann New Scientist Fund. This work was supported by the Cooperation Program in Cancer Research of the Deutsches Krebsforschungszentrum (DKFZ) and Israel's Ministry of Science and Technology (MOST).
Anna Harari and Guy Zoltsman contributed equally to this work.
Data availability statement
The data that supports the findings of this study are available from the corresponding authors upon request. NMR chemical shifts for Hsp104 NTD have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) under the accession code 51038.
References
- 1. Kim YE, Hipp MS, Bracher A, Hayer‐Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–55. [DOI] [PubMed] [Google Scholar]
- 2. Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem. 2015;84:435–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nillegoda NB, Wentink AS, Bukau B. Protein disaggregation in multicellular organisms. Trends Biochem Sci. 2018;43:285–300. [DOI] [PubMed] [Google Scholar]
- 4. Zarouchlioti C, Parfitt DA, Li W, Gittings LM, Cheetham ME. DNAJ Proteins in neurodegeneration: essential and protective factors. Philos Trans R Soc Lond B Biol Sci. 2018;373:20160534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tittelmeier J, Nachman E, Nussbaum‐Krammer C. Molecular chaperones: a double‐edged sword in neurodegenerative diseases. Front Aging Neurosci. 2020;12:581374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarparanta J, Jonson PH, Kawan S, Udd B. Neuromuscular diseases due to chaperone mutations: a review and some new results. Int J Mol Sci. 2020;21:1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. [DOI] [PubMed] [Google Scholar]
- 8. Wentink A, Nussbaum‐Krammer C, Bukau B. Modulation of amyloid states by molecular chaperones. Cold Spring Harb Perspect Biol. 2019;11:a033969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. 2019;20:665–80. [DOI] [PubMed] [Google Scholar]
- 10. Ayala Mariscal SM, Kirstein J. J‐domain proteins interaction with neurodegenerative disease‐related proteins. Exp Cell Res. 2021;399:112491. [DOI] [PubMed] [Google Scholar]
- 11. Balchin D, Hayer‐Hartl M, Hartl FU. Recent advances in understanding catalysis of protein folding by molecular chaperones. FEBS Lett. 2020;594:2770–81. [DOI] [PubMed] [Google Scholar]
- 12. Kohler V, Andreasson C. Hsp70‐mediated quality control: should I stay or should I go? Biol Chem. 2020;401:1233–48. [DOI] [PubMed] [Google Scholar]
- 13. Liberek K, Lewandowska A, Zietkiewicz S. Chaperones in control of protein disaggregation. EMBO J. 2008;27:328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doyle SM, Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trends Biochem Sci. 2009;34:40–8. [DOI] [PubMed] [Google Scholar]
- 15. Mokry DZ, Abrahao J, Ramos CH. Disaggregases, molecular chaperones that resolubilize protein aggregates. An Acad Bras Cienc. 2015;87:1273–92. [DOI] [PubMed] [Google Scholar]
- 16. Mogk A, Bukau B, Kampinga HH. Cellular handling of protein aggregates by disaggregation machines. Mol Cell. 2018;69:214–26. [DOI] [PubMed] [Google Scholar]
- 17. Katikaridis P, Bohl V, Mogk A. Resisting the heat: bacterial disaggregases rescue cells from devastating protein aggregation. Front Mol Biosci. 2021;8:681439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. [DOI] [PubMed] [Google Scholar]
- 19. Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248:1112–5. [DOI] [PubMed] [Google Scholar]
- 20. Hodson S, Marshall JJ, Burston SG. Mapping the road to recovery: the ClpB/Hsp104 molecular chaperone. J Struct Biol. 2012;179:161–71. [DOI] [PubMed] [Google Scholar]
- 21. Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29. [DOI] [PubMed] [Google Scholar]
- 22. Puchades C, Sandate CR, Lander GC. The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat Rev Mol Cell Biol. 2020;21:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seraphim TV, Houry WA. AAA+ proteins. Curr Biol. 2020;30:R251–7. [DOI] [PubMed] [Google Scholar]
- 24. Miot M, Reidy M, Doyle SM, Hoskins JR, Johnston DM, Genest O, et al. Species‐specific collaboration of heat shock proteins (Hsp) 70 and 100 in thermotolerance and protein disaggregation. Proc Natl Acad Sci USA. 2011;108:6915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mogk A, Kummer E, Bukau B. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front Mol Biosci. 2015;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zietkiewicz S, Lewandowska A, Stocki P, Liberek K. Hsp70 chaperone machine remodels protein aggregates at the initial step of Hsp70‐Hsp100‐dependent disaggregation. J Biol Chem. 2006;281:7022–9. [DOI] [PubMed] [Google Scholar]
- 27. Lee J, Kim JH, Biter AB, Sielaff B, Lee S, Tsai FT. Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc Natl Acad Sci USA. 2013;110:8513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosenzweig R, Moradi S, Zarrine‐Afsar A, Glover JR, Kay LE. Unraveling the mechanism of protein disaggregation through a ClpB‐DnaK interaction. Science. 2013;339:1080–3. [DOI] [PubMed] [Google Scholar]
- 29. Doyle SM, Shastry S, Kravats AN, Shih YH, Miot M, Hoskins JR, et al. Interplay between E. coli DnaK, ClpB and GrpE during protein disaggregation. J Mol Biol. 2014;427:312–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–65. [DOI] [PubMed] [Google Scholar]
- 31. Chernoff YO, Lindquist SL, Ono B, Inge‐Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion‐like factor [psi+]. Science. 1995;268:880–4. [DOI] [PubMed] [Google Scholar]
- 32. Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae . Science. 1994;264:566–9. [DOI] [PubMed] [Google Scholar]
- 33. Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae . Genetics. 1997;147:507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Satpute‐Krishnan P, Langseth SX, Serio TR. Hsp104‐dependent remodeling of prion complexes mediates protein‐only inheritance. PLoS Biol. 2007;5:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol. 2012;179:152–60. [DOI] [PubMed] [Google Scholar]
- 36. Doyle SM, Hoskins JR, Wickner S. Collaboration between the ClpB AAA+ remodeling protein and the DnaK chaperone system. Proc Natl Acad Sci USA. 2007;104:11138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, et al. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell. 2014;156:170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park YN, Zhao X, Yim YI, Todor H, Ellerbrock R, Reidy M, et al. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot Cell. 2014;13:635–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals. 2008;16:63–74. [DOI] [PubMed] [Google Scholar]
- 40. Jackrel ME, Shorter J. Protein‐remodeling factors as potential therapeutics for neurodegenerative disease. Front Neurosci. 2017;11:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. March ZM, Mack KL, Shorter J. AAA+ protein‐based technologies to counter neurodegenerative disease. Biophys J. 2019;116:1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. March ZM, Sweeney K, Kim H, Yan X, Castellano LM, Jackrel ME, et al. Therapeutic genetic variation revealed in diverse Hsp104 homologs. eLife. 2020;9:e57457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lo Bianco C, Shorter J, Regulier E, Lashuel H, Iwatsubo T, Lindquist S, et al. Hsp104 antagonizes alpha‐synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest. 2008;118:3087–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman‐Nick M, Neuhaus‐Follini A, et al. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell. 2012;151:778–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cushman‐Nick M, Bonini NM, Shorter J. Hsp104 suppresses polyglutamine‐induced degeneration post onset in a drosophila MJD/SCA3 model. PLoS Genet. 2013;9:e1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jackrel ME, Shorter J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease‐linked proteins. Dis Model Mech. 2014;7:1175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, et al. Nuclear‐import receptors reverse aberrant phase transitions of RNA‐binding proteins with prion‐like domains. Cell. 2018;173:677–92.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tariq A, Lin J, Noll MM, Torrente MP, Mack KL, Murillo OH, et al. Potentiating Hsp104 activity via phosphomimetic mutations in the middle domain. FEMS Yeast Res. 2018;18:foy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jia C, Ma X, Liu Z, Gu J, Zhang X, Li D, et al. Different heat shock proteins bind alpha‐synuclein with distinct mechanisms and synergistically prevent its amyloid aggregation. Front Neurosci. 2019;13:1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tariq A, Lin J, Jackrel ME, Hesketh CD, Carman PJ, Mack KL, et al. Mining disaggregase sequence space to safely counter TDP‐43. FUS, and alpha‐synuclein proteotoxicity. Cell Rep. 2019;28:2080–95.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kushnirov VV, Dergalev AA, Alexandrov AI. Amyloid fragmentation and disaggregation in yeast and animals. Biomolecules. 2021;11:1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wayne NJ, Dembny KE, Pease T, Saba F, Zhao X, Masison DC, et al. Huntingtin polyglutamine fragments are a substrate for Hsp104 in Saccharomyces cerevisiae . Mol Cell Biol. 2021;41:e0012221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yokom AL, Gates SN, Jackrel ME, Mack KL, Su M, Shorter J, et al. Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat Struct Mol Biol. 2016;23:830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, Kendsersky NM, et al. Ratchet‐like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 2017;357:273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Deville C, Carroni M, Franke KB, Topf M, Bukau B, Mogk A, et al. Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci Adv. 2017;3:e1701726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang X, Zhang S, Zhang L, Lu J, Zhao C, Luo F, et al. Heat shock protein 104 (HSP104) chaperones soluble Tau via a mechanism distinct from its disaggregase activity. J Biol Chem. 2019;294:4956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deville C, Franke K, Mogk A, Bukau B, Saibil HR. Two‐step activation mechanism of the ClpB disaggregase for sequential substrate threading by the main ATPase motor. Cell Rep. 2019;27:3433–46.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rizo AN, Lin J, Gates SN, Tse E, Bart SM, Castellano LM, et al. Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat Commun. 2019;10:2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, et al. The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115:229–40. [DOI] [PubMed] [Google Scholar]
- 60. Seyffer F, Kummer E, Oguchi Y, Winkler J, Kumar M, Zahn R, et al. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol. 2012;19:1347–55. [DOI] [PubMed] [Google Scholar]
- 61. Kummer E, Szlachcic A, Franke KB, Ungelenk S, Bukau B, Mogk A. Bacterial and yeast AAA+ disaggregases ClpB and Hsp104 operate through conserved mechanism involving cooperation with Hsp70. J Mol Biol. 2016;428:4378–91. [DOI] [PubMed] [Google Scholar]
- 62. Wendler P, Ciniawsky S, Kock M, Kube S. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta. 2012;1823:2–14. [DOI] [PubMed] [Google Scholar]
- 63. Biter AB, Lee J, Sung N, Tsai FT, Lee S. Functional analysis of conserved cis‐ and trans‐elements in the Hsp104 protein disaggregating machine. J Struct Biol. 2012;179:172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hou Z, Chen D, Ryder BD, Joachimiak LA. Biophysical properties of a tau seed. Sci Rep. 2021;11:13602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee S, Roh SH, Lee J, Sung N, Liu J, Tsai FTF. Cryo‐EM structures of the Hsp104 protein disaggregase captured in the ATP conformation. Cell Rep. 2019;26:29–36.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rosenzweig R, Farber P, Velyvis A, Rennella E, Latham MP, Kay LE. ClpB N‐terminal domain plays a regulatory role in protein disaggregation. Proc Natl Acad Sci USA. 2015;112:E6872‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee J, Sung N, Mercado JM, Hryc CF, Chang C, Lee S, et al. Overlapping and specific functions of the Hsp104 N domain define its role in protein disaggregation. Sci Rep. 2017;7:11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sweeny EA, Tariq A, Gurpinar E, Go MS, Sochor MA, Kan ZY, et al. Structural and mechanistic insights into Hsp104 function revealed by synchrotron X‐ray footprinting. J Biol Chem. 2020;295:1517–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang P, Li J, Weaver C, Lucius A, Sha B. Crystal structures of Hsp104 N‐terminal domains from Saccharomyces cerevisiae and Candida albicans suggest the mechanism for the function of Hsp104 in dissolving prions. Acta Crystallogr D Struct Biol. 2017;73:365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hung GC, Masison DC. N‐terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics. 2006;173:611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lum R, Niggemann M, Glover JR. Peptide and protein binding in the axial channel of Hsp104. Insights into the mechanism of protein unfolding. J Biol Chem. 2008;283:30139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sielaff B, Tsai FT. The M‐domain controls Hsp104 protein remodeling activity in an Hsp70/Hsp40‐dependent manner. J Mol Biol. 2010;402:30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sweeny EA, Jackrel ME, Go MS, Sochor MA, Razzo BM, DeSantis ME, et al. The Hsp104 N‐terminal domain enables disaggregase plasticity and potentiation. Mol Cell. 2015;57:836–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tipton KA, Verges KJ, Weissman JS. In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Mol Cell. 2008;32:584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reidy M, Sharma R, Masison DC. Schizosaccharomyces pombe disaggregation machinery chaperones support Saccharomyces cerevisiae growth and prion propagation. Eukaryot Cell. 2013;12:739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schlieker C, Weibezahn J, Patzelt H, Tessarz P, Strub C, Zeth K, et al. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11:607–15. [DOI] [PubMed] [Google Scholar]
- 77. Romanova NV, Chernoff YO. Hsp104 and prion propagation. Protein Pept Lett. 2009;16:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kelly MJ, Krieger C, Ball LJ, Yu Y, Richter G, Schmieder P, et al. Application of amino acid type‐specific 1H‐ and 14N‐labeling in a 2H‐, 15N‐labeled background to a 47 kDa homodimer: potential for NMR structure determination of large proteins. J Biomol NMR. 1999;14:79–83. [DOI] [PubMed] [Google Scholar]
- 79. Sweeny EA, Shorter J. Mechanistic and structural insights into the prion‐disaggregase activity of Hsp104. J Mol Biol. 2016;428:1870–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Biancalana M, Koide S. Molecular mechanism of thioflavin‐T binding to amyloid fibrils. Biochim Biophys Acta. 2010;1804:1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Klosowska A, Chamera T, Liberek K. Adenosine diphosphate restricts the protein remodeling activity of the Hsp104 chaperone to Hsp70 assisted disaggregation. eLife. 2016;5:e15159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Iljina M, Mazal H, Goloubinoff P, Riven I, Haran G. Entropic inhibition: how the activity of a AAA+ machine is modulated by its substrate‐binding domain. ACS Chem Biol. 2021;16:775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Parsell DA, Kowal AS, Singer MA, Lindquist S. Protein disaggregation mediated by heat‐shock protein Hsp104. Nature. 1994;372:475–8. [DOI] [PubMed] [Google Scholar]
- 84. Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR‐mediated gene disruption and other applications. Yeast. 1998;14:115–32. [DOI] [PubMed] [Google Scholar]
- 85. Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum . Nat Methods. 2013;10:407–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Meurer M, Duan Y, Sass E, Kats I, Herbst K, Buchmuller BC, et al. Genome‐wide C‐SWAT library for high‐throughput yeast genome tagging. Nat Methods. 2018;15:598–600. [DOI] [PubMed] [Google Scholar]
- 87. Johnston DM, Miot M, Hoskins JR, Wickner S, Doyle SM. Substrate discrimination by ClpB and Hsp104. Front Mol Biosci. 2017;4:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Helsen CW, Glover JR. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104‐kDa heat shock protein (Hsp104). J Biol Chem. 2012;287:542–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Anand R, Beach A, Li K, Haber J. Rad51‐mediated double‐strand break repair and mismatch correction of divergent substrates. Nature. 2017;544:377–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kumar V, Peter JJ, Sagar A, Ray A, Jha MP, Rebeaud ME, et al. Interdomain communication suppressing high intrinsic ATPase activity of Sse1 is essential for its co‐disaggregase activity with Ssa1. FEBS J. 2020;287:671–94. [DOI] [PubMed] [Google Scholar]
- 91. Jiang Y, Rossi P, Kalodimos CG. Structural basis for client recognition and activity of Hsp40 chaperones. Science. 2019;365:1313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Saio T, Guan X, Rossi P, Economou A, Kalodimos CG. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science. 2014;344:1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gardner KH, Zhang XC, Gehring K, Kay LE. Solution NMR studies of a 42 KDa Escherichia coli maltose binding protein beta‐cyclodextrin complex: chemical shift assignments and analysis. J Am Chem Soc. 1998;120:11738–48. [Google Scholar]
- 94. Irwin R, Faust O, Petrovic I, Wolf SG, Hofmann H, Rosenzweig R. Hsp40s play complementary roles in the prevention of tau amyloid formation. eLife. 2021;10:e69601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. [DOI] [PubMed] [Google Scholar]
- 96. Goddard TD, Knelle DG. SPARKY 3. University of California, San Francisco; 2000. [Google Scholar]
- 97. Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–96. [DOI] [PubMed] [Google Scholar]
- 98. Norby JG. Coupled assay of Na+, K+‐ATPase activity. Methods Enzymol. 1988;156:116–9. [DOI] [PubMed] [Google Scholar]
- 99. Stiggall DL, Galante YM, Hatefi Y. Preparation and properties of complex V. Methods Enzymol. 1979;55:308–15, 819–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that supports the findings of this study are available from the corresponding authors upon request. NMR chemical shifts for Hsp104 NTD have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) under the accession code 51038.