

Abstract
Purpose:
KRAS mutation (MT) is a major oncogenic driver in PDAC. A small subset of PDACs harbor KRAS-wild-type (WT). We aim to characterize the molecular profiles of KRAS-WT PDAC to uncover new pathogenic drivers and offer targeted treatments.
Experimental Design:
Tumor tissue obtained from surgical or biopsy material was subjected to next-generation DNA/RNA sequencing, microsatellite-instability (MSI) and mismatch-repair (MMR) status determination.
Results:
Of the 2,483 patients (male 53.7%, median-age 66 years) studied, 266 tumors (10.7%) were KRAS-WT. The most frequently mutated gene in KRAS-WT-PDAC was TP53 (44.5%), followed by BRAF (13.0%) . Multiple mutations within the DNA-damage-repair (BRCA2, ATM, BAP1, RAD50, FANCE, PALB2), chromatin-remodeling (ARID1A, PBRM1, ARID2, KMT2D, KMT2C, SMARCA4, SETD2), and cell-cycle-control pathways (CDKN2A, CCND1, CCNE1) were detected frequently. There was no statistically-significant difference in PDL1-expression between KRAS-WT (15.8%) and MT (17%) tumors. However, KRAS-WT-PDAC were more likely to be MSI-high (4.7% vs 0.7%; p<0.05), TMB-high (4.5% vs 1%; p<0.05), and exhibit increased infiltration of CD8+ T-cells, NK-cells and myeloid dendritic cells. KRAS-WT-PDACs exhibited gene fusions of BRAF (6.6%), FGFR2 (5.2%), ALK (2.6%), RET (1.3%) and NRG1 (1.3%), as well as amplification of FGF3 (3%), ERBB2 (2.2%), FGFR3 (1.8%), NTRK (1.8%) and MET (1.3%). Real-world evidence reveals a survival advantage of KRAS-WT patients in overall cohorts as well as in patients treated with gemcitabine/nab-paclitaxel or 5FU/oxaliplatin.
Conclusions:
KRAS-WT PDAC represents 10.7% of PDAC and is enriched with targetable alterations, including immuno-oncologic markers. Identification of KRAS-WT patients in clinical practice may expand therapeutic options in a clinically meaningful manner.
Keywords: Pancreatic adenocarcinoma, KRAS, molecular profiling, next generation sequencing
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a challenging disease with overall 5-year survival rate of only 10% (1,2). There is increasing global burden of this disease. The majority of patients with PDAC harbor metastatic disease, present either on initial presentation or subsequently, emphasizing the need for more effective systemic treatments. Progress in drug therapy of PDAC overall has been very limited especially with the use of targeted agents and immunotherapy (3). Outside of germline BRCA mutations (4), there have been very few actionable alterations in PDAC identified thus far. As a result, the adoption of molecular profiling as part of routine clinical practice in this cancer type has been inconsistent.
KRAS (Kirsten rat sarcoma) mutation, a hallmark of PDAC, is an early event in its natural history and is linked to critical aspects of its biology such as inflammation, immune evasion, and altered metabolism (3–6). Mutated KRAS is the major oncogenic driver for PDAC and an attractive treatment target (7,8). To date, no effective treatments have been identified for patients with KRAS mutated PDAC except for the very rare KRAS G12C mutations (9). However, prior studies in patients with PDAC have identified a subset of patients (10–20%) whose tumors harbor no known KRAS mutations at all. These KRAS wild type tumors may have biological differences from KRAS mutated PDACs and may offer treatment opportunities that are unique to this subset. Advances in molecular techniques including whole genome sequencing provide the opportunity to improve our understanding of the molecular biology of PDAC and particularly in tumors that have no KRAS mutations. This may offer a personalized approach in treating patients with PDAC and rationalize the wider use of molecular diagnostics to improve patient outcome. A recent study by Pishvaian (10) et al. reported actionable mutations in 26% of a cohort of 677 patients with PDAC that impacted patient outcome. Their findings demonstrated the potential value matching targeted therapies with molecular profiling in PDAC.
Our hypothesis was that in the absence of KRAS activating mutations, other genomic abnormalities that drive carcinogenesis and are medically targetable will be present in KRAS wild type PDACs. We report on the molecular profiling of PDAC in 2483 tumor samples. The objective of the study was to perform a detailed molecular characterization of KRAS wild type PDAC to better understand the biology of PDAC and to explore therapeutic targets beyond current standard of care that use cytotoxic therapy alone.
Methods
Subjects:
Tumor tissue from patients diagnosed with PDAC was obtained through surgical specimens or tissue obtained by image guided biopsy. Tumors that went through comprehensive molecular profiling testing between 2016 and 2020 at Caris Life Sciences (Phoenix, AZ) were included in the comparative biomarker analyses. In addition, treatment and survival information from insurance data from a large cohort of real-world evidence (RWE) database updated in 2021 were included for survival analysis. Tumors in the biomarker analyses were part of the RWE database.
Next generation sequencing (NGS) of DNA:
NGS was performed on genomic DNA isolated from formalin-fixed paraffin-embedded (FFPE) tumor samples using the NextSeq or NovaSeq platform (Illumina, Inc., San Diego, CA). For NextSeq, a custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). All variants were detected with > 99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of > 500x and an analytic sensitivity of 5%. For NovaSeq, a hybrid pull-down panel of baits designed to enrich for more than 700 clinically relevant genes at high coverage (>500x) and high read-depth was used, along with another panel designed to enrich for an additional >20,000 genes at lower depth (>250x). Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques. Genetic variants identified were interpreted by board-certified molecular geneticists and categorized as ‘pathogenic,’ ‘likely pathogenic,’ ‘variant of unknown significance,’ ‘likely benign,’ or ‘benign,’ according to the American College of Medical Genetics and Genomics (ACMG) standards. When assessing mutation frequencies of individual genes, ‘pathogenic,’ and ‘likely pathogenic’ were counted as mutations while ‘benign’, ‘likely benign’ variants and ‘variants of unknown significance’ were excluded. All variants were detected with greater than 99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of greater than 500 and an analytic sensitivity of 5%.
Tumor mutational burden (TMB):
TMB was measured by counting all non-synonymous missense, nonsense, in-frame insertion/deletion and frameshift mutations found per tumor that had not been previously described as germline alterations in dbSNP151, Genome Aggregation Database (gnomAD) databases or benign variants identified by Caris geneticists. A cutoff point of ≥10 mutations per MB was used (11,12).
MSI/MMR status:
A combination of multiple test platforms was used to determine the MSI, or MMR proficiency status of the tumors, including fragment analysis (FA, Promega, Madison, WI), immunohistochemistry (IHC) (MLH1, M1 antibody; MSH2, G2191129 antibody; MSH6, 44 anti-body; and PMS2, EPR3947 antibody [Ventana Medical Systems, Inc., Tucson, AZ, USA]) and NGS. The three platforms generated highly concordant results as previously reported (13) and in the rare cases of discordant results, the MSI or MMR status of the tumor was determined in the order of IHC, FA and NGS.
RNA sequencing:
All tumors included in the molecular study were also tested using next-generation RNA sequencing (RNA-Seq). FFPE specimens underwent pathology review to diagnose percent tumor content and tumor size; a minimum of 10% of tumor content in the area for microdissection was required to enable enrichment and extraction of tumor-specific RNA. Gene fusion detection was performed on mRNA isolated from formalin-fixed paraffin-embedded tumor samples using the Illumina NovaSeq platform (Illumina, Inc., San Diego, CA) and Agilent SureSelect Human All Exon V7 bait panel (Agilent Technologies, Santa Clara, CA). Qiagen RNA FFPE tissue extraction kit was used for extraction to detect fusions and the RNA quality and quantity was determined using the Agilent TapeStation. Biotinylated RNA baits were hybridized to the synthesized and purified cDNA targets and the bait-target complexes were amplified in a post capture PCR reaction. The resultant libraries were quantified, normalized and the pooled libraries were denatured, diluted, and sequenced; the reference genome used was GRCh37/hg19 and analytical validation of this test demonstrated ≥97% Positive Percent Agreement (PPA), ≥99% Negative Percent Agreement (NPA) and ≥99% Overall Percent Agreement (OPA) with a validated comparator method. Transcripts per million molecules were generated using the Salmon expression pipeline for transcription counting.
Tumor microenvironment characterization and MAPK activation evaluation:
MCP-counter was used for quantification of the abundance of stromal cell populations using transcriptomic data (14), while QuantiSeq (15) was used to quantify the immune cell infiltration. MAPK activation was evaluated by MPAS (MAPK Pathway Activity Score) calculation (16).
PD-L1 expression:
IHC was performed on FFPE sections of glass slides. PD-L1 testing was performed using the SP142 anti-PD-L1 antibody (Spring Biosciences). The staining was regarded as positive if its intensity on the tumor cells was ≥2+ and the percentage of positively stained cells was > 5%. On a semiquantitative scale of 0–3: 0 represented no staining, 1+ weak staining, 2+ moderate staining, and 3+ strong staining.
Statistical methods:
The comparison of molecular alterations between KRAS wild type and mutant tumors was performed using Chi-square, Fisher’s exact or Mann-Whitney test when appropriate. Benjamini-Hochberg method was used to calculate adjusted p values (i.e., q values) and a q<0.05 was regarded as statistically significant to reduce false discovery rate in multiple testing; p<0.05 but q>0.05 was regarded as trending differences. Real-world overall survival (rwOS) information was obtained from insurance claims data and calculated from either tissue collection or first of treatment time to last of contact. Kaplan-Meier estimates were calculated for molecularly defined patient cohorts. In order to explore biomarker differences in cohorts with survivals longer or shorter than the median overall survival, a volcano plot was drawn to display the significance versus fold changes of biomarker alterations and the most significantly different biomarkers were investigated further for their prognostic effects. Significance was determined as p values <0.05.
Data availability statement:
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. The NGS raw data are owned by Caris Life Sciences and cannot be publicly shared due to the data usage agreement signed by Dr. Philip Philip. Qualified researchers can apply for access to these data by contacting Joanne Xiu (jxiu@carisls.com) and signing a data usage agreement.
Compliance statement:
This study was conducted in accordance with guidelines of the Declaration of Helsinki, Belmont report, and U.S. Common rule. In keeping with 45 CFR 46.101(b)(4), this study was performed utilizing retrospective, deidentified clinical data. Therefore, this study is considered IRB exempt and no patient consent was necessary from the subjects.
Results
Study Population and KRAS status
The study population comprised 2,483 patients, with 53.7% males, and a median age of 66 years (range 22–95 years) (Table 1). Of the total, 266 (10.7%) were KRAS WT. PDAC constituted 92.5% of the analyzed samples with a similar KRAS WT percentage 10.1%. The study also included the additional histological subtypes including mucinous, adenosquamous carcinomas, and sarcomatoid tumors, etc comprising the other 7.5%. Pancreatic acinar cell carcinomas were particularly enriched in KRAS WT (82%). Two of 2 pseudopapillary tumors were KRAS WT. Sex or age were not associated with KRAS wild type status (11% in men and 10% in women). The median age of KRAS WT and MT was 66 and 67 years, respectively. The spectrum of KRAS mutations is listed in Supplemental Table 1. The majority of mutations were G12D (43%), G12V (31%) and G12R (14%). The most common KRAS alteration was the substitution of Glycine at position 12 by Aspartic Acid (G12D) (962/2234 = 43.1%). Replacement of Glycine at position 12 by Valine (G12V; 30.8 %), Arginine (G12R; 14.2%) complete the top three. The potentially targetable G12C mutation was seen in only 1.9% of patients. Seventeen tumors expressed more than one KRAS mutation. Overall, recurring KRAS mutations involved exclusively Glycine 12, glutamine 61 and glycine 13. The quasi-totality of mutations (>99%) were single point mutations with negligible numbers of insertions and deletions.
Table 1:
Tumor characteristics. 1a: tumor histology of analyzed cohort
| Groups | KRAS WT (N) | KRAS WT (%) | KRAS MT (N) | KRAS MT (%) | Total |
|---|---|---|---|---|---|
| Adenocarcinoma, NOS | 233 | 10.1% | 2064 | 89.9% | 2297 |
| Mucinous | 16 | 13.3% | 104 | 86.7% | 120 |
| Squamous/Adenosquamous | 5 | 11.1% | 40 | 88.9% | 45 |
| Acinar | 9 | 81.8% | 2 | 18.2% | 11 |
| Sarcomatoid | 1 | 14.3% | 6 | 85.7% | 7 |
| Pseudopapillary | 2 | 100.0% | 0.0% | 2 | |
| Pleomorphic | 1 | 100.0% | 1 | ||
|
| |||||
| Total | 266 | 10.7% | 2217 | 89.3% | 2483 |
Alterations in RAS WT subgroup
Alterations in the 233 KRAS WT PDAC were analyzed by whole-exome sequencing, whole-transcriptome sequencing and IHC (Figure 1). The most frequently mutated gene was TP53 (44.5%). BRAF, a downstream effector of KRAS, was mutated the second most frequently (13.0%). Unlike melanoma and CRC where V600E mutation constitute the vast majority of BRAF mutations, in the KRAS WT PDAC group approximately a third (37%) of BRAF mutations involved V600 (Class 1), and the RAS-independent mutations (Class 2) were most common (43%); RAS-dependent mutations (Class 3) were the least common (17%) (Figure 1C, Supplemental Table 2). Deletion of a 5 AA stretch (N486_P490del) was the most common RAS-independent BRAF mutation, while D594N was the most common RAS-dependent mutation. Multiple mutations were seen in DNA-damage repair pathway genes (BRCA2: 5.2%, ATM: 4.7%, BAP1: 2.9%, RAD50: 2.3%, FANCE: 2.1%, PALB2: 2.1%), genes involved in chromatin remodeling (ARID1A: 11.6%, PBRM1: 5.6%, ARID2: 3.9%, KMT2D: 3.4%, KMT2C: 3.8%, SMARCA4: 2.2%, SETD2: 1.8%), and cell cycle regulation (CDKN2A: 10.3%, CCND1 amp: 2.6%, CCNE1 amp: 2.3%). KRAS WT PDACs also exhibited copy number amplification of FGF3 (3%), ERBB2 (2.2%), FGFR3 (1.8%), NTRK (1.8%), MET (1.3%). Taken together, over 10% of KRAS WT PDACs exhibited amplifications that may be amenable to known targeted therapies. Consistent with previous reports, we observed targetable fusion events in KRAS WT cohorts, the most prevalent included BRAF, FGFR2, ALK and RET (Supplemental Table 3).
Figure 1:
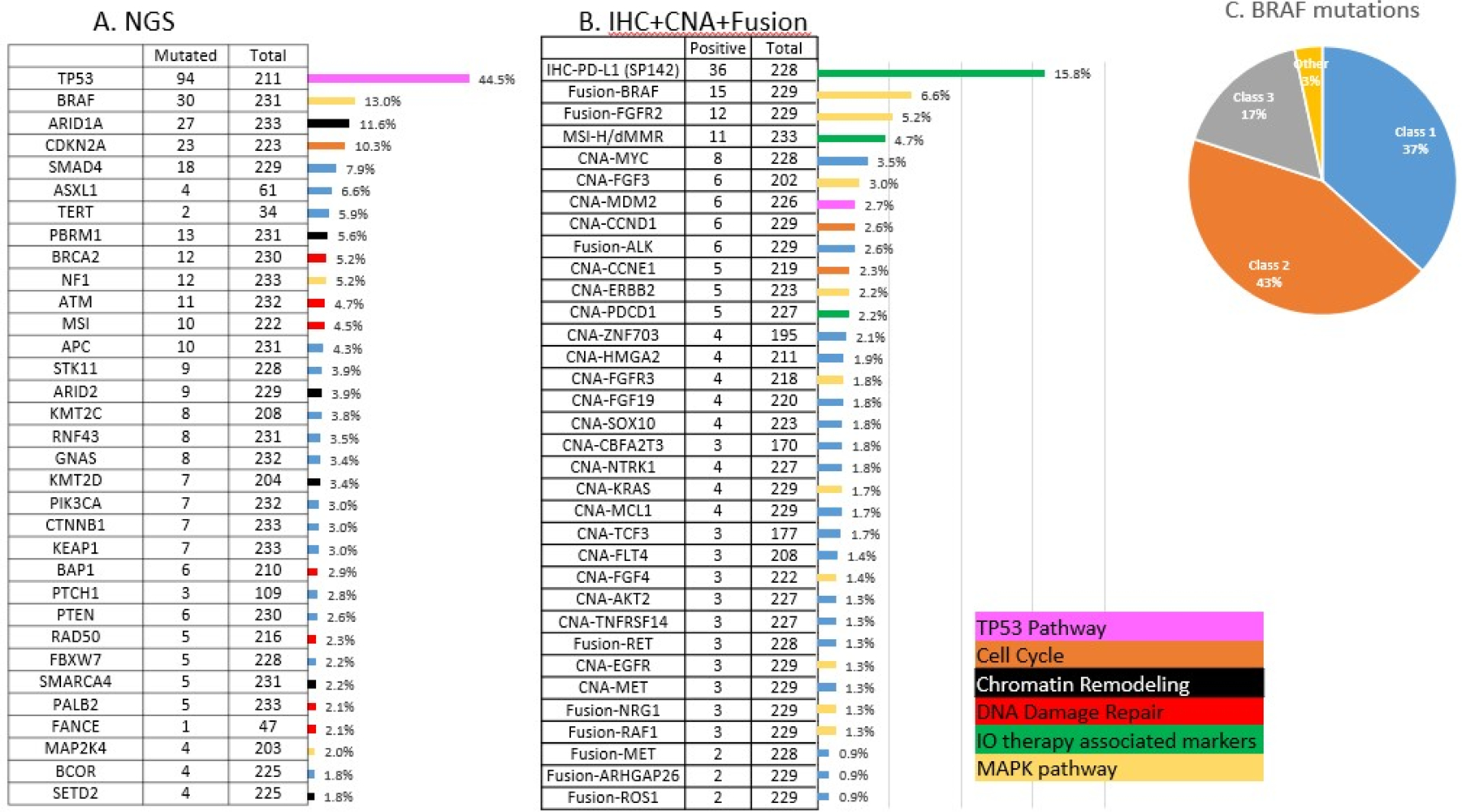
Molecular alterations seen in KRAS-WT tumors. 1A: Mutation rates detected by NGS. 1B: Alteration rates detected by immunohistochemistry, copy number amplification rates detected by NGS and fusion rates detected by RNA sequencing. Bars are color coded according to the oncogenic pathways of each biomarker. 1C: BRAF mutations seen in the cohort categorized into class 1, 2 and 3 based on mechanism of action.
Analysis of significant differences in genomic alterations between KRAS WT and MT PDAC revealed associations with alterations in key cancer-related genes. When alterations relevant to immune checkpoint inhibitor therapies were analyzed, KRAS WT PDAC were likelier to be MSI-high/MMR-deficient (4.7% vs 0.7%; p<0.05) and TMB-high (4.5% vs 1%; p<0.05) when compared to KRAS MT tumors (Figure 2). However, there was no statistically significant difference in PDL1 expression between KRAS WT (15.8%) and KRAS MT (17%). Canonical tumor suppressor genes well-known to be involved in tumorigenesis and metastasis including TP53, CDKN2A and SMAD4 mutations were more prevalent in KRAS MT PDAC, however, the vast majority of alterations that were significantly different between WT and MT were seen to be enriched in the WT tumors, including targetable alterations (e.g., BRAF, ALK, ROS1, FGFR2, NRG1, MSI-H, IDH1, RET) (Figure 3). A comprehensive listing of significantly different alterations in KRAS MT and WT PDAC tumors is listed in Supplemental Table 4. An oncoprint of the 233 KRAS WT PDAC tumors detailing immunotherapy-associated markers, gene fusions, mutations and amplifications is shown in Figure 4. Notably, while genomic fusion events are largely absent in KRAS MT tumors, the only exception being a MET fusion (seen in 1 case in KRAS MT cohort), 21% of WT tumors (50 out of 233) were identified to harbor a gene fusion event, involving BRAF (6.6%), FGFR2 (5.2%), ALK (2.6%), RET (1.3%), NRG1 (1.3%), and RAF1 (1.3%) etc. These gene fusion events and other oncogenic alterations demonstrated mutual exclusivity in the majority of cases. As many of genomic alterations identified in KRAS WT PDAC have been associated with activation of MAPK pathway, potentially serving as an alternative oncogenic driver in PDAC in the absence of KRAS mutation, we evaluated the MAPK pathway activity using a previously published 10-gene signature, MAPK Pathway Activity Score (MPAS) (16). KRAS MT PDAC demonstrated a significant elevation of MAPK activity when compared to the KRAS WT cohort (p<0.0001); NF1 mutation, RAF1 fusion, BRAF fusion and mutation all showed higher MPAS when compared to a group of KRAS WT tumors without MAPK pathway mutations (Supplemental Figure 1).
Figure 2:
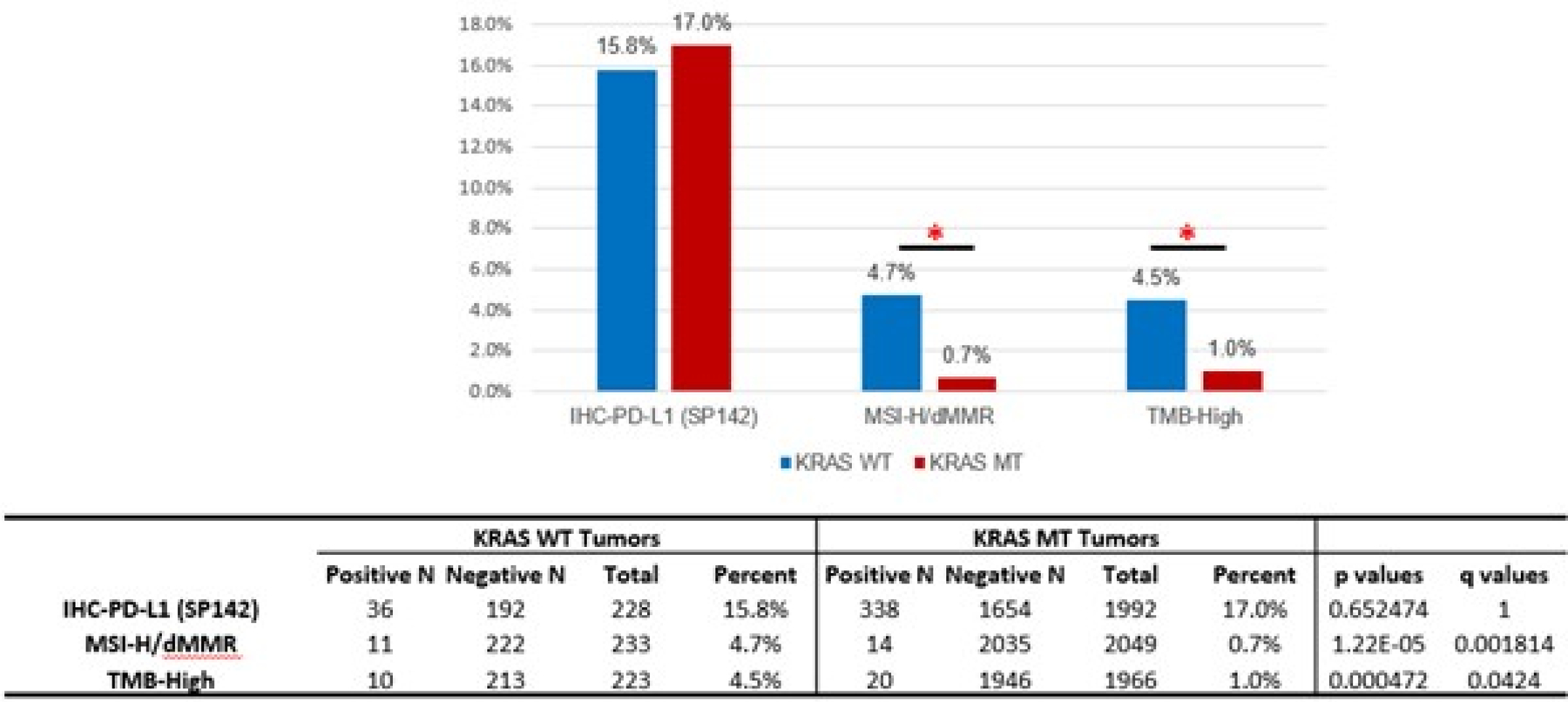
Comparison of immune checkpoint inhibitor-associated biomarkers in KRAS WT and MT tumors. An asterisk indicates a significant difference.
Figure 3:
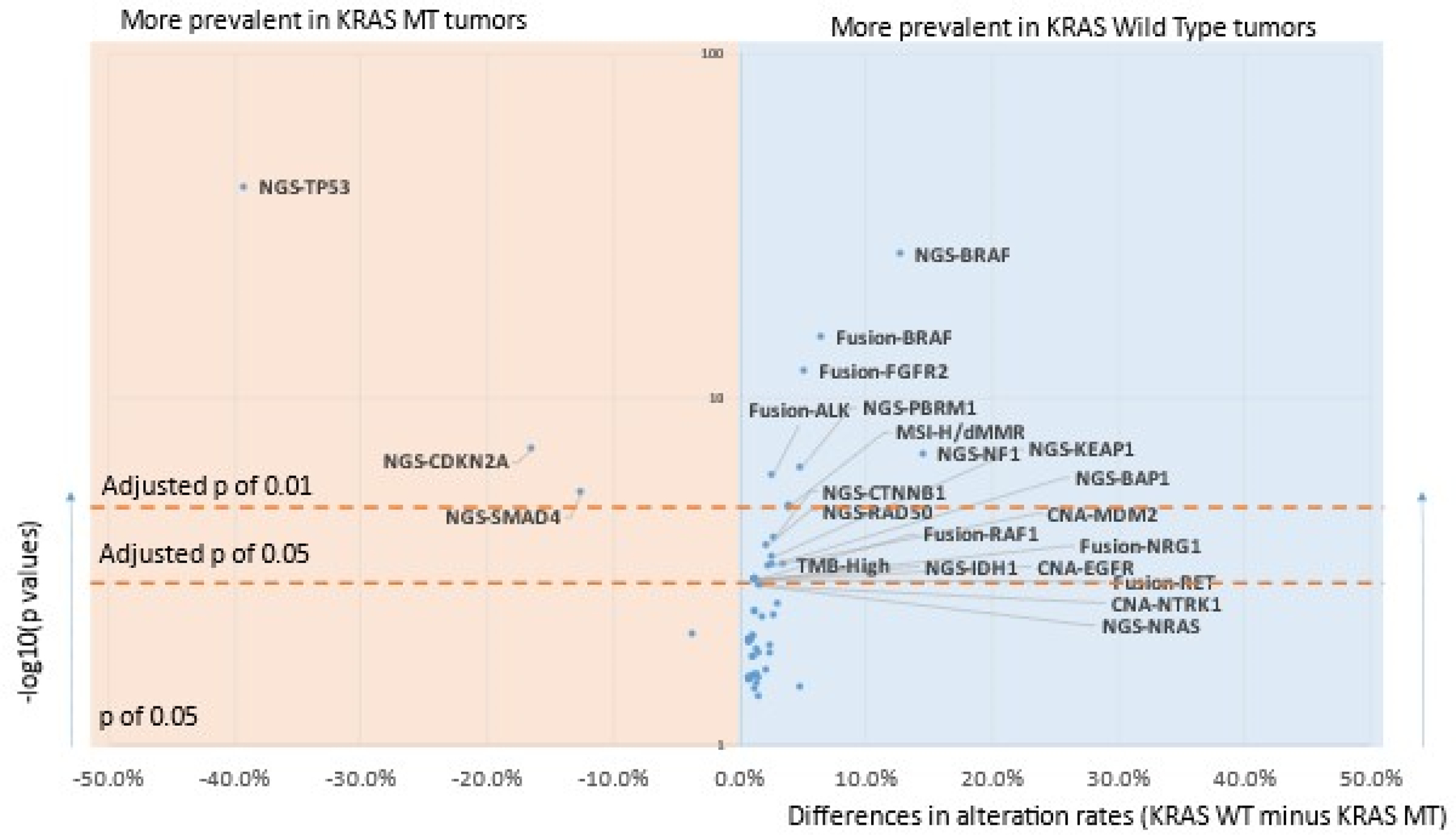
Volcano plot comparing molecular alterations of KRAS MT vs. WT tumors. NGS: Next-Gen Sequencing detected mutations. Only molecular alterations significantly different (adjusted p<0.05) are labeled. Full results can be found in Supplemental table 4.
Figure 4:
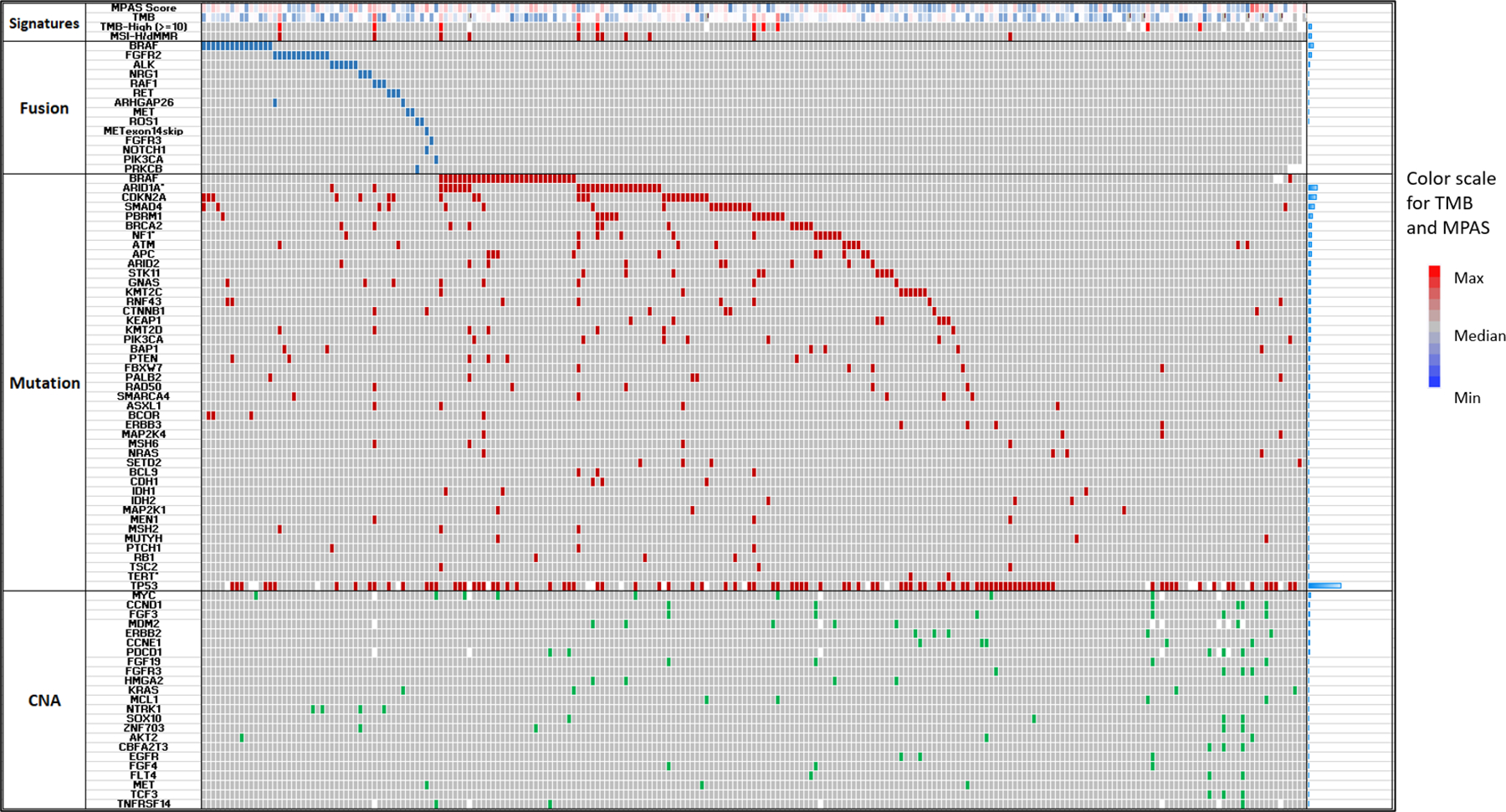
An oncoprint displaying the molecular alteration patten of the 233 PDAC tumors. Each row represents a biomarker of either fusion, mutation or copy number amplification, as well as genomic signatures such as TMB or MSI/MMR. Red, blue and green represents TMB-H, MSI-high/MMR-deficient or mutations detected using DNA-sequencing; green represents copy number amplification detected by DNA sequencing, while navy blue represents fusions detected by RNA Sequencing. Grey represents no alteration detected while blanks represent unavailable data (indeterminate results due to low coverage or noisy signals). Bars on the right represents the prevalence of molecular alterations of each row.
RNA deconvolution analysis was utilized to characterize the immune-microenvironment (Quantiseq, (15)). A statistically significant over-representation of myeloid dendritic cells, NK cells and CD8+ T-cells was observed (Figure 5A) in KRAS WT PDAs. Conversely, KRAS WT PDACs harbored significantly less neutrophils. B-cells were numerically increased in KRAS WT PDAC but this difference did not achieve statistical significance when adjusted for multiple comparison. Estimation of the stromal cell population showed a trend for decreased fibroblasts in the WT tumors and increased endothelial cells in KRAS WT tumors (14) (Figure 5B). As MSI-H/dMMR causes genomic mutations and production of the neoantigens on the cell surface which may induce infiltrations of different cell lymphocyte populations, we further analyzed the immune cell populations in tumors with confirmed MSS/pMMR status and compared tumors with or without KRAS mutation. All significant results as shown in Figure 5 hold true in the MSS cohort, suggesting that the observed immune-microenvironment differences are specific to KRAS mutations and not secondary effect to MSI-H/dMMR.
Figure 5:
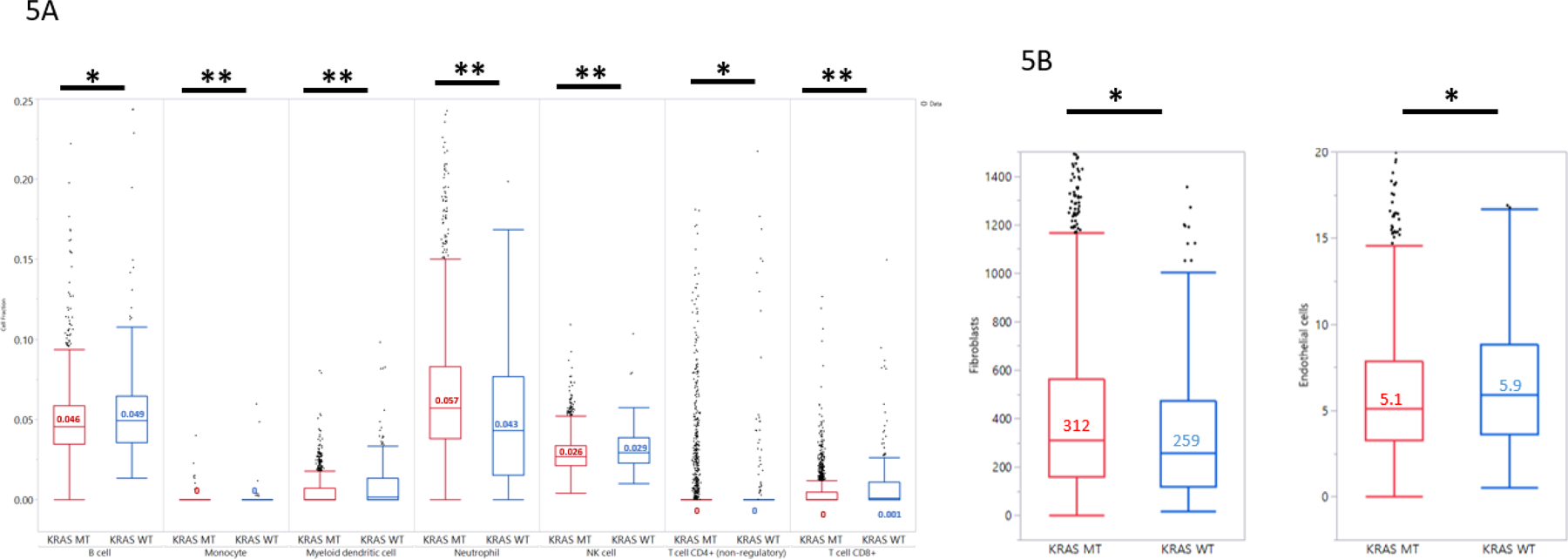
Comparison of Tumor Microenvironment (TME) characteristics in KRAS MT vs. WT tumors. 5A: Lymphocyte cell fractions estimated by RNA sequencing using Quantiseq.5B: Stromal cell populations estimated by RNA sequencing using MCP counter.**: significantly different after correcting for multiple comparison; * trending differences.
Treatment and outcome analysis
Treatment and survival information were available from a total of 5324 pancreatic cancer patients tested as either KRAS WT (N=705) or KRAS MT (N=4619) using NextGen sequencing platforms (NextSeq or Novaseq). KRAS WT patients had a small but statistically significant prolongation of overall survival (calculated from tissue collection to last day of contact) compared to KRAS MT patients (Fig 6A. HR=1.152, p=0.002); the improvement of survival was more prominent when data on only patients with distant metastases profiled were analyzed (Fig 6B. HR=1.259, p<0.0001). In patients treated with 5FU and oxaliplatin (Fig 6C, HR=1.432, p<0.0001) and those treated with gemcitabine and nab-paclitaxel (Fig 6D, HR=1.362, p=0.0003), KRAS WT patients had increased overall survival compared with their KRAS MT counterpart. Within KRAS WT cohort, we explored molecular alterations that are more enriched in patients with longer overall survival by using a volcano plot analysis and observed that TP53 wild type status is associated with longer survival: patients with TP53 mutations had significantly decreased overall survival compared to the wild type (Fig 6E HR=1.456, p<0.0001).
Figure 6:
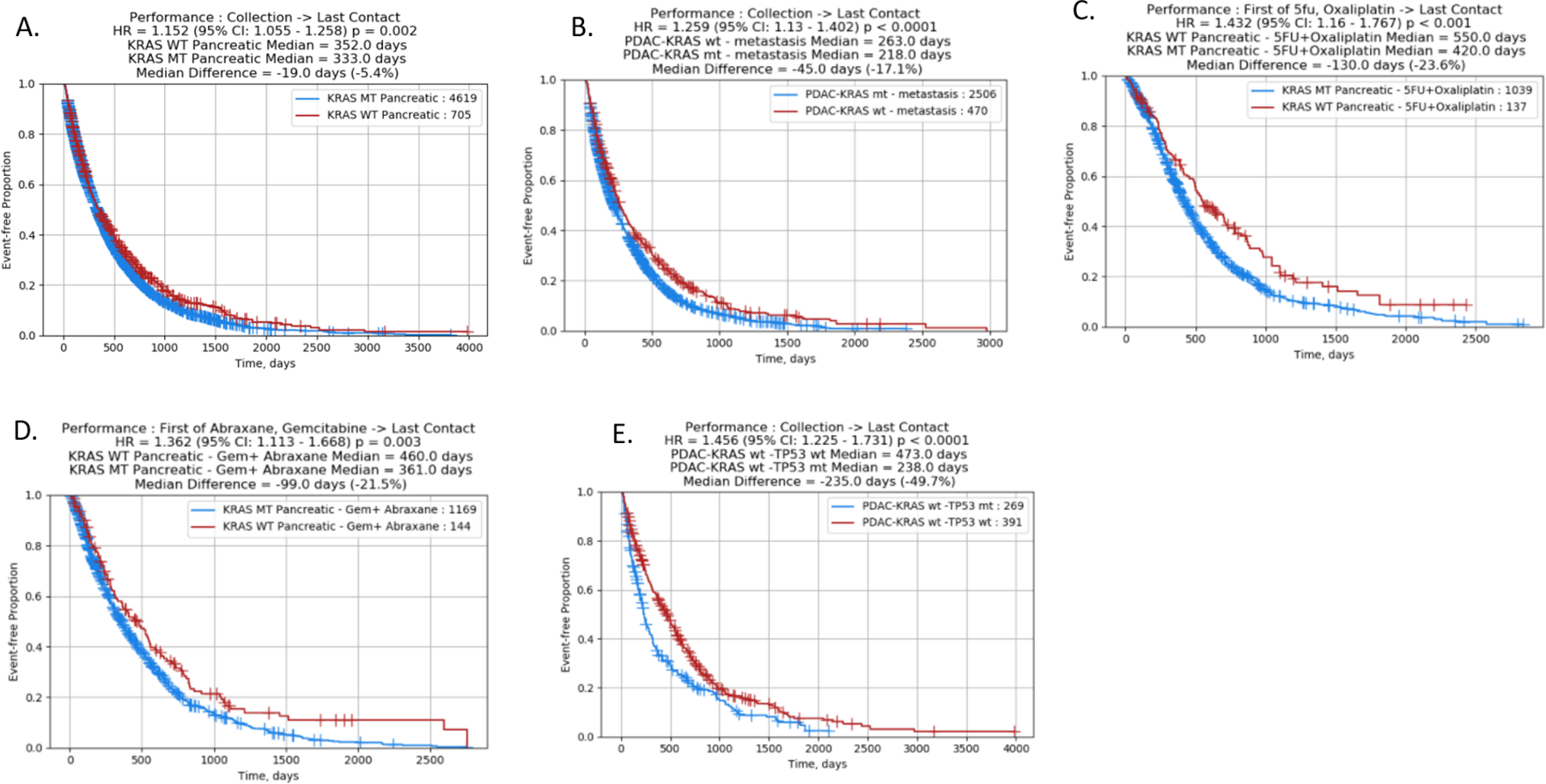
rwOS in KRAS MT and KRAS WT tumors. 6A: overall survival (calculated from tissue collection to last day of contact) of KRAS WT compared to KRAS MT patients; 6B: overall survival of KRAS MT compared to KRAS MT patients with metastatic tumors; 6C: comparison of survival of KRAS WT patients treated with 5FU and oxaliplatin (calculated from start of treatment to last day of contact) with KRAS MT; 6D: comparison of survival of KRAS WT patients treated with gemcitabine and abraxane with KRAS MT; 6E: overall survival (from tissue collection to last day of contact) of KRAS WT patients with or without TP53 mutation.
Discussion:
Routine molecular profiling of PDAC is currently established to identify germline BRCA mutations, and DNA mismatch repair deficiency (10,17). Additionally, certain gene fusions have been described as effective therapeutic targets in PDAC (18–20). However, the utility of comprehensive multigene tumor profiling in patients with pancreatic cancer remains to be established (21). This is largely a result of the low prevalence of actionable mutations in unselected cases as well as the difficulty to obtain adequate tumor tissue, especially in the locally advanced, unresectable setting. Here, we demonstrate enrichment of KRAS WT PDAC for therapeutically targetable molecular alterations as suggested by previous reports (22). To our knowledge, this is the largest study investigating the molecular profiles of PDAC with a focus on molecular characteristics of 233 KRAS WT PDAC. The entire cohort of tumors were interrogated for a broad spectrum of molecular alterations that include mutations, gene amplifications, fusions, changes in gene expression levels as well as protein expression of selected markers such as PD-L1 and MMR proteins. DNA sequencing data of 592 cancer-related genes (NextSeq) and whole exome sequencing were analyzed for mutations while whole transcriptome sequencing was used for gene expression analysis and sensitive fusion detection (23). We are also the first to report on tumor immune infiltration in KRAS WT vs. KRAS MT PDAC. Additionally, our work represents the largest the largest outcome data for PDAC by KRAS status. KRAS WT comprised 10.7% of the study population, and no differences in median age or gender when compared to KRAS MT patients was seen. The frequency of the KRAS WT was within the previously reported range from smaller cohorts of PDAC tumors (24). Fusion events reported here include therapeutically targetable ALK, ROS1, NRG1, BRAF, FGFR2, RET, RAF1, MET (25).
Our data highlight the distinctly different molecular composition of KRAS WT tumors compared with KRAS MT PDAC, suggesting potentially different molecular pathogenesis mechanisms and adding to our understanding of inter-individual tumor heterogeneity in this disease. Despite being the most altered gene in KRAS WT PDAC, TP53 is even more frequently mutated in KRAS MT tumors. Activating mutations of BRAF, a downstream effector of KRAS signaling, were found in 13% of the KRAS WT tumors and were mutually exclusive with KRAS mutations, a difference that was highly statistically significant (p-value 8.19E-27) when compared with KRAS MT tumors. While class 1 BRAF mutations, including the frequent V600E mutation can be effectively targeted using BRAF inhibitors with or without MEK inhibitors for numerous cancer types, class 2 and class 3 BRAF mutants have also become targetable. As reported in the PancSeq study (26), the in-frame BRAF deletions, considered as a class 2 mutant, confer MAPK sensitivity in preclinical models and were treated with MEK inhibitors accordingly with promising clinical activity. Class 3 mutants have been reported to rely on additional MAPK activation signals and would be more effectively targeted in combination with inhibitors of receptor tyrosine kinases. (27–29). Similarly, gene fusions involving BRAF were the most prevalent fusions in KRAS WT compared to KRAS MT (6.6% vs. 0%) with statistical significance (p<0.001); these fusions are known to dimerize and present elevated kinase activity, highlighting the importance of the MAPK kinases pathway in PDAC biology. Targeting BRAF fusions clinically with MEK inhibitor or BRAF inhibitors has been reported: A case series of 2 melanoma tumor harboring BRAF fusions treated with trametinib showed a 90% reduction in extracranial metastases in one patient with PPFIBP2-BRAF fusion, and improvement in symptoms with slight disease progression in imaging in another patient with a KIAA1549-BRAF fusion (30). Another study examining BRAF fusions across solid tumors reported a Spitzoid melanoma harboring a ZKSCAN1-BRAF fusion responded to trametinib treatment and the lung metastasis was rendered resectable after the treatment; as well as response of a malignant spindle cell tumor of the chest wall harboring a KIAA1549-BRAF fusion to combination therapy with sorafenib, bevacizumab and temsirolimus (31, 32). In our study, the fraction of KRAS WT tumors harboring BRAF alterations was substantially higher than reported in previous studies, possibly reflecting differences in analytical methods and highlight the advantage of fusion detection using RNA-based assays (22). Reliable detection of these alterations is critical from a clinical perspective since they represent meaningful therapeutic target (23).
In agreement with previous reports from our group and others, NRG1 fusion events were seen exclusively in KRAS WT PDAC cohort (N=3). Targeting the protein product of these gene fusions using monoclonal antibodies including seribantumab has recently been shown to result in clinical responses in a subset of patients (33). Recently, zenocutuzumab (MCLA-128), a bispecific humanized IgG1 monoclonal antibody, showed an ORR of 40% and DCR of 90% in a cohort of 10 pancreatic cancer patients. All 9 patients who had disease control had a >50% decline in their CA19–9. Duration of response is pending (34). In addition, FGFR and IDH alterations were exclusively seen in KRAS WT disease, potentially representing meaningful therapeutic targets as seen in biliary tract cancers (35,36). So far, FGFR inhibitors have been tested in combination with chemotherapy in unselected metastatic PDAC populations as with the addition of dovutinib to gemcitabine and capecitabine and BGJ398 to mFOLFIRINOX (37).
Additional targetable alterations (ALK, RET and MET) were also more prevalent in our KRAS WT PDAC cohort. A previously reported case series (18) of 4 patients with PDAC exhibiting ALK-fusions treated with ALK-inhibitors with promising results. In particular, one patient with metastatic PDAC exhibiting and exon 13 EML4–exon 20 ALK translocation treated sequentially with crizotinib, ceritinib and alectinib remained alive for 52 months after diagnosis. Another case report (38) described a patient with locally advanced PDAC with STRN-ALK rearrangement initially resistant mFOLFIRINOX with sustained response to a combination of IMRT and crizotinib that led to an avoidance of surgery. Interestingly, six out of 7 patients with ALK translocations were reported in patients under the age of 50 (39). Preliminary data from the basket phase I/II ARROW trial showed partial responses with 2 patients with pancreatic cancer with RET-fusions treated with pralsetinib with a duration of response of 5.5 months and 7.4 months in each (40). Pralsetinib has been recently approved for thyroid and NSCLC with RET fusions. Of note, RET fusions have been reported in a subset of pancreatic acinar cell carcinomas (41). Additionally, one patient with a novel MET fusion was reported to have a complete response that lasted over 12 months.
Using MPAS, a RNA signature previously reported to evaluate activation of MAPK pathway, we show that numerous alterations seen in the KRAS WT PDAC are associated with significant MAPK activation with some reaching MPAS scores comparable to the KRAS mutant PDAC cohort (16). While targeting this pathway with an anti-EGFR or anti-IGFR strategy failed in the past when treatment was tested without appropriate selection strategies (42, 43), such strategies should be reconsidered in KRAS WT PDAC especially with MAPK activation profile in PDAC cells lacking the KRAS function (44).
Findings of this study also suggest that immunotherapy with checkpoint inhibitors that has shown no benefit in unselected PDAC patients thus far (45) may have potential activity in the KRAS WT population based on the finding of a higher frequency of MSI-high/MMR-deficient and TMB-high tumors. Treatment of such tumors with pembrolizumab can result in objective responses as demonstrated by a subset analysis of the KEYNOTE-158 study which showed an ORR of 18% in pancreatic tumors, lagging most other MSI-High tumors (46). These findings agree with prior observations of an association between KRAS WT and immune response biomarkers (22,47) and recent data on the role of KRAS MT on immune evasion in pre-clinical models (48).
Increasing evidence from our data as well as data from others suggest that there are systematic differences between KRAS MT and WT tumors at the genomic level that result in altered transcriptomic profiles and are expected to drive differences in clinical behavior of the two disease subtypes. In agreement with this, our real-world evidence confirms prolonged survival of patients with KRAS WT tumors which is consistent with previously reported smaller series (49). Molecular features that potentially lead to improved survival include higher frequency of MSI-high/MMR-deficient and associated increased lymphocyte infiltration. In addition, it is conceivable that the slightly prolonged overall survival observed in the platinum-treated patients compared to gemcitabine-nab-pacltiaxel treated patients may be a result of higher rate of DDR genes mutations, including RAD50 and PALB2 in the KRAS WT cohort. Our results adds to the current literature which provides inconsistent information on the prognostic or predictive impact of KRAS in PDAC (49,50). One weakness of our study is that there was no breakdown of the study population based on the stage of the disease at presentation. However, with the typically advanced nature of PDAC at diagnosis it is unlikely that disease stage will have a significant impact on its molecular profile. This study included tumor tissue from primary or metastatic sites. Given that KRAS mutations are very early events in PDAC oncogenesis, it is highly unlikely a difference would be seen if studied separately as was supported recent reports (51,52), despite the recent study potentially challenging the notion (53). To further study activation of oncogenic pathways in KRAS WT tumors is an important consideration because genetic models of PDAC are mostly focused on the KRAS MT genotype despite a growing evidence of a complex pattern of PDAC evolution based on single-cell sequencing of precursor lesions (54). Another challenge of tissue-based study in pancreatic cancer is the paucity of material obtained through standard diagnostic procedures such as using endoscopic ultrasound. The adoption of liquid biopsies will help address this issue in the future.
In conclusion
In conclusion, patients with PDAC with KRAS WT status represent a distinct subgroup who may benefit from comprehensive molecular profiling to improve their treatment outcomes. In this subgroup of PDACs with an estimated incidence of over 5,000 patients per year in the USA, early identification of targetable mutations will optimize treatment planning at a personalized level. Collectively, multigene profiling, including determination of the KRAS status as part of initial diagnostic workup, should be considered in the routine management of PDAC (Figure 7). Future studies will focus on identification of predictive transcriptomic signatures in the KRAS WT population. This is in particular relevant for a large range of drugs targeting non-oncogene dependencies compared to mutated genes (55).
Figure 7:
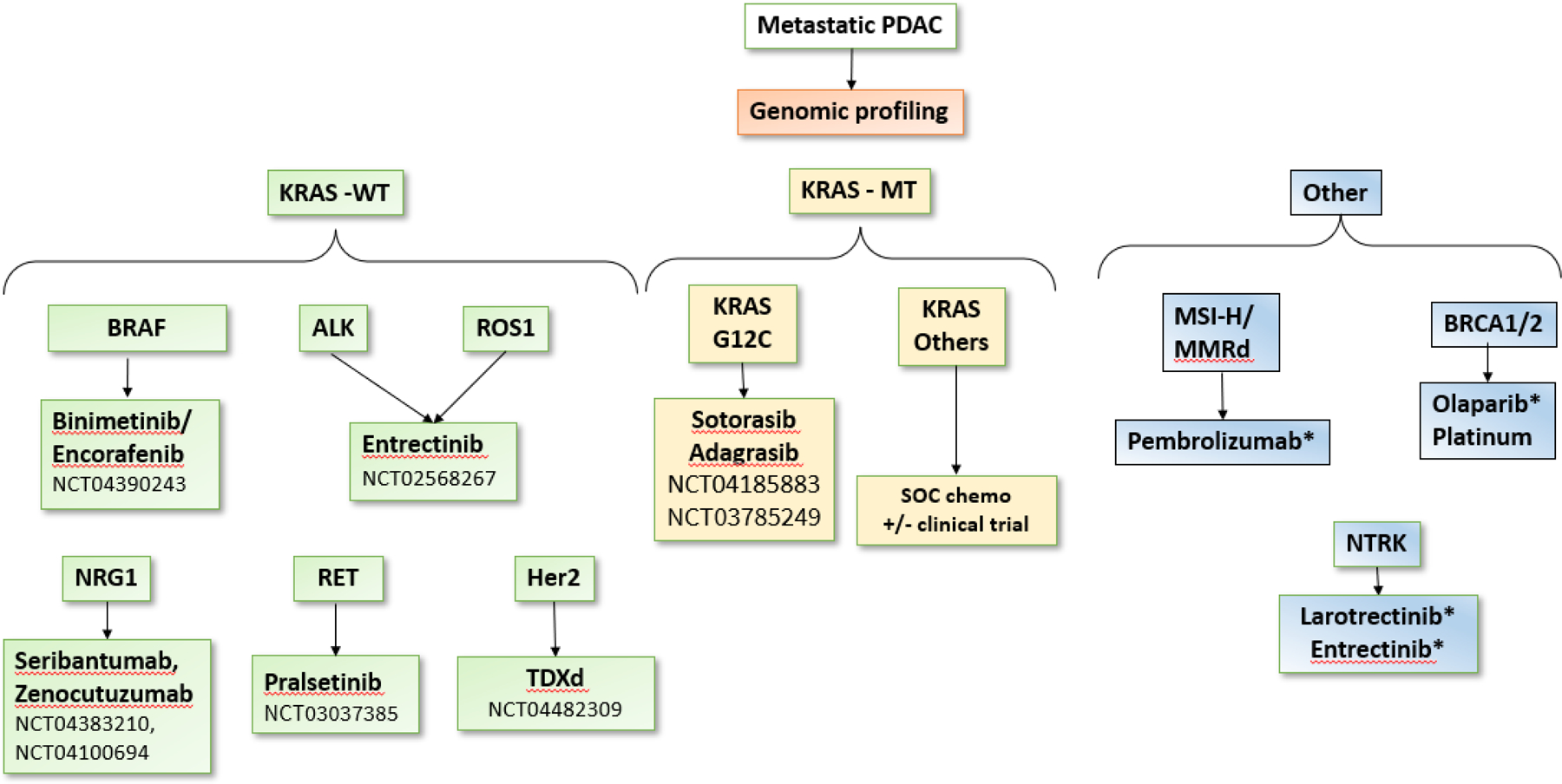
Genomic profiling of advanced pancreatic adenocarcinoma to determine targetable molecular abnormalities. Drugs with an * indicate FDA-approved agents for treatment in pancreatic adenocarcinoma. KRAS WT tumors are enriched with several targetable mutations when compared to KRAS MT tumors. Currently, olaparib approval by the FDA is limited to treating patients with germline BRCA1/2 mutations only. WT: wild type. MT: mutant. PDAC: pancreatic ductal adenocarcinoma. SOC: standard of care.
Supplementary Material
Supplemental Figure 1: MAPK Pathway Activity Score (MPAS) in KRAS WT pancreatic tumor molecular groups. Double asterisks suggest statistical significant differences (adjusted p<0.05); single asterisks suggest trending (p<0.05); green asterisks: when compared to ‘Other KRAS Wild type’, red asterisks: when compared to KRAS mutant cohort.
{kind=link}
Table 1b:
patient gender and age
| KRAS WT | KRAS WT | KRAS MT | KMRAS MT | ||
|---|---|---|---|---|---|
| Gender | (N) | (%) | (N) | (%) | Total |
|
| |||||
| Female | 115 | 10.0% | 1034 | 90.0% | 1149 |
| Male | 151 | 11.3% | 1183 | 88.7% | 1334 |
| KRAS WT | KRAS MT | ||||
|
| |||||
| Age-Median | 66 | 67 | |||
| Age Range | 33–92 | 26–95 | |||
Statement of translational relevance:
Pancreatic Ductal adenocarcinoma (PDAC) carries a dismal prognosis. KRAS mutation is considered a main oncogenic driver in the vast majority of PDACs. Current treatment of metastatic PDAC consists of combination cytotoxic agents. So far, targeted and immunotherapy have failed in PDAC all-comers. In this manuscript, we characterize the molecular profiles of KRAS-wild type (WT) PDACs to uncover molecular drivers that offer targeted treatment opportunities. We show that KRAS WT pancreatic adenocarcinomas represent about 10% of all PDACs and are enriched in alterations that can be targeted with targeted therapies that have been FDA-approved in other organ systems. BRAF mutations and mutations in DNA-damage repair pathway were present. KRAS-WT tumors were also more likely to MSI-high opening up the doors for checkpoint inhibitors. Real-world evidence data showed that KRAS-WT tumors exhibited an overall survival advantage. Our work establishes the importance of incorporating molecular characterization of KRAS-WT tumors into routine clinical practice
Footnotes
Conflict of Interests:
Philip A. Philip:
Honoraria - Array BioPharma; AstraZeneca; Bayer; Blueprint Medicines; Celgene; Ipsen; Merck; syncore; TriSalus Life Sciences
Consulting or Advisory Role - Celgene; Daiichi Sankyo; Ipsen; Merck; syncore; Taiho Pharmaceutical; TriSalus Life Sciences
Speakers’ Bureau - Bayer; Celgene; Incyte; Ipsen; Novartis
Research Funding - Advanced Accelerator Applications (Inst); ASLAN Pharmaceuticals (Inst); Bayer (Inst); boston biomedical (Inst); Caris Life Sciences (Inst); Caris Life Sciences (Inst); Genentech (Inst); halozyme (Inst); Immunomedics (Inst); incyte (Inst); incyte (Inst); Karyopharm Therapeutics (Inst); Lilly (Inst); Merck (Inst); merus (Inst); Momenta Pharmaceuticals (Inst); novartis (Inst); Plexxikon (Inst); QED Therapeutics (Inst); QED Therapeutics (Inst); Regeneron (Inst); Taiho Pharmaceutical (Inst); Taiho Pharmaceutical (Inst); TYME (Inst); tyme (Inst)
Travel, Accommodations, Expenses - Abbvie; celgene; Rafael Pharmaceuticals (OPTIONAL) Uncompensated Relationships - Caris MPI; Rafael Pharmaceuticals
Ibrahim Azar: no COI.
Joanne Xiu: Employment - Caris Life Sciences
Michael J. Hall: Research Funding - Ambry Genetics; AstraZeneca; Caris Life Sciences; Foundation Medicine; InVitae; Myriad Genetics
Patents, Royalties, Other Intellectual Property - I share a patent with several Fox Chase investigators for a novel method to investigate hereditary CRC genes (Inst)
Travel, Accommodations, Expenses - AstraZeneca; Caris Life Sciences; Foundation Medicine; Myriad Genetics
Other Relationship - Caris Life Sciences; Foundation Medicine; Invitae; Myriad Genetics
Andrew Eugene Hendifar:
Consulting or Advisory Role - Abbvie; Celgene; Ipsen; Novartis; Perthera
Research Funding - Ipsen
Travel, Accommodations, Expenses - Halozyme
Emil Lou
Honoraria - Boston Scientific; Daiichi Sankyo/UCB Japan (Inst); GlaxoSmithKline; Novocure
Consulting or Advisory Role - Boston Scientific; Novocure; Novocure; Novocure; Novocure
Research Funding - Novocure
Travel, Accommodations, Expenses - GlaxoSmithKline
(OPTIONAL) Uncompensated Relationships - Caris Life Sciences; Minnetronix Medical; NomoCan
Jimmy J. Hwang:
Consulting or Advisory Role - Amgen; Amgen; Amgen; Amgen; Bayer; Bayer; Bayer; Bayer; Boehringer Ingelheim; Boehringer Ingelheim; Boehringer Ingelheim; Boehringer Ingelheim; Bristol-Myers Squibb; Bristol-Myers Squibb; Bristol-Myers Squibb; Bristol-Myers Squibb; Caris Centers of Excellence; Caris Centers of Excellence; Caris Centers of Excellence; Caris Centers of Excellence; Eisai; Eisai; Eisai; Eisai; Genentech/Roche; Genentech/Roche; Genentech/Roche; Genentech/Roche; Ipsen; Ipsen; Ipsen; Ipsen;
Lilly; Lilly; Lilly; Lilly; Taiho Pharmaceutical; Taiho Pharmaceutical; Taiho Pharmaceutical; Taiho Pharmaceutical
Speakers’ Bureau - Amgen; Amgen; Amgen; Amgen; Bristol-Myers Squibb; Bristol-Myers Squibb; Bristol-Myers Squibb; Bristol-Myers Squibb; Celgene; Celgene; Celgene; Celgene; Genentech/Roche; Genentech/Roche; Genentech/Roche; Genentech/Roche; Ipsen; Ipsen; Ipsen; Ipsen
Research Funding - Boehringer Ingelheim (Inst); Boehringer Ingelheim (Inst); Boehringer Ingelheim (Inst); Boehringer Ingelheim (Inst); Caris Centers of Excellence (Inst); Caris Centers of Excellence (Inst); Caris Centers of Excellence (Inst); Caris Centers of Excellence (Inst)
Jun Gong:
Honoraria - Amgen; Astellas Pharma; Clinical Congress Consultants; Elsevier; Exelixis; QED Therapeutics
Consulting or Advisory Role - Amgen; Astellas Pharma; Clinical Congress Consultants; Elsevier; Exelixis; QED Therapeutics
Rebecca Feldman: Employment - Caris Life Sciences
Michelle Ellis: Employment - Caris Life Sciences
Phil Stafford: Employment - Caris Life Sciences
David Spetzler: Employment - Caris Life Sciences
Moh’d M. Khushman:
Stock and Other Ownership Interests - Aprea therapeutics; Blueprint Medicines; Daiichi Sankyo; Global Blood Therapeutics; Guardant Health; Halozyme
Speakers’ Bureau – AstraZeneca
Davendra Sohal:
Honoraria - Foundation Medicine
Consulting or Advisory Role - Ability Pharma; Perthera
Speakers’ Bureau - Incyte
Research Funding - Amgen (Inst); Apexigen (Inst); Bristol-Myers Squibb (Inst); Celgene (Inst); Genentech (Inst); Incyte (Inst); Rafael Pharmaceuticals (Inst)
A. Craig Lockhart:
Research Funding - Astellas Pharma (Inst); Bristol-Myers Squibb (Inst); Merck (Inst); Sarah Cannon Research Institute (Inst)
Benjamin A. Weinberg:
Honoraria - Onclive; Rafael Pharmaceuticals; Tempus
Consulting or Advisory Role - Bayer
Speakers’ Bureau - Bayer; HalioDx; Lilly; Sirtex Medical; Taiho Pharmaceutical
Research Funding - Abbvie (Inst); Ipsen (Inst); Isofol Medical (Inst); Novartis (Inst)
Expert Testimony - AstraZeneca
Travel, Accommodations, Expenses - Boehringer Ingelheim; Caris Life Sciences
Wafik S. El-Deiry:
Stock and Other Ownership Interests - Oncoceutics; p53-Therapeutics
Consulting or Advisory Role - Boehringer Ingelheim
Research Funding - Bayer; D&D Pharmatech; D&D Pharmatech; D&D Pharmatech; Loxo; Morphotek; Tarmeta Biosciences
Patents, Royalties, Other Intellectual Property - Patent on TIC10 (ONC201); Patents pending on the use of small molecules to target mutant p53
Other Relationship - Caris Life Sciences
John Marshall:
Employment - Caris Life Sciences; Indivumed
Honoraria - Amgen; Bayer/Onyx; Caris Life Sciences; Merck; Taiho Pharmaceutical
Consulting or Advisory Role - Amgen; Bayer/Onyx; Caris Life Sciences; Celgene; Genentech/Roche; Taiho Pharmaceutical
Speakers’ Bureau - Amgen; Bayer/Onyx; Merck; Taiho Pharmaceutical
Anthony F. Shields:
Consulting or Advisory Role - Caris Life Sciences; ImaginAb
Speakers’ Bureau - Caris Life Sciences
Research Funding - Alkermes; Astellas Pharma; AstraZeneca; Bayer; Boehringer Ingelheim; Boston Biomedical; Caris Life Sciences; Daiichi Sankyo; Eisai; Esperas Pharma; Esperas Pharma; Exelixis; Five Prime Therapeutics; H3 Biomedicine; Halozyme; Hutchison China Meditech; ImaginAb; Incyte; Inovio Pharmaceuticals; Jiangsu Alphamab Biopharmaceuticals; Lexicon; LSK BioPharma; MSK Pharma; Nouscom; Plexxikon; Repertoire Immune Medicines; Seattle Genetics; Shanghai HaiHe Pharmaceutical; Taiho Pharmaceutical; Telix Pharmaceuticals; Xencor
Travel, Accommodations, Expenses - Caris Life Sciences; GE Healthcare; ImaginAb; Inovio Pharmaceuticals; TransTarget
W. Michael Korn:
Employment - Caris Life Sciences
Leadership - Caris Life Sciences
Consulting or Advisory Role - Merck Sharp & Dohme
Travel, Accommodations, Expenses - Merck Sharp & Dohme
References
- 1.Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet Lond Engl 2020. Jun 27;395(10242):2008–20. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021. Jan;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 3.Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018. Feb 1;554(7690):62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med 2019. Jul 25;381(4):317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 2014. Feb;39(2):91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storz P, Crawford HC. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020. Jun;158(8):2072–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016. Mar 3;531(7592):47–52. [DOI] [PubMed] [Google Scholar]
- 8.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2020. Mar;17(3):153–68. [DOI] [PubMed] [Google Scholar]
- 9.Luo J KRAS mutation in pancreatic cancer. Semin Oncol 2021. Feb 23;S0093-7754(21)00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020. Apr;21(4):508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 2020. Oct;21(10):1353–65. [DOI] [PubMed] [Google Scholar]
- 12.Merino DM, McShane LM, Fabrizio D, Funari V, Chen S-J, White JR, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer 2020. Mar;8(1):e000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 2018. Mar;7(3):746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 2019. May 24;11(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagle M-C, Kirouac D, Klijn C, Liu B, Mahajan S, Junttila M, et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis Oncol 2018;2(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamgar M, Dyson G, Shields AF, Tesfaye AA, Philip PA, Al Hallak MN. Comprehensive genomic profiling in metastatic pancreatic adenocarcinoma, correlation with early death versus prolonged survival. J Clin Oncol 2019. Feb 1;37(4_suppl):282–282. [Google Scholar]
- 18.Singhi AD, Ali SM, Lacy J, Hendifar A, Nguyen K, Koo J, et al. Identification of Targetable ALK Rearrangements in Pancreatic Ductal Adenocarcinoma. J Natl Compr Cancer Netw JNCCN 2017. May;15(5):555–62. [DOI] [PubMed] [Google Scholar]
- 19.Sohal DPS, Kennedy EB, Cinar P, Conroy T, Copur MS, Crane CH, et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J Clin Oncol Off J Am Soc Clin Oncol 2020. Aug 5;JCO2001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones MR, Williamson LM, Topham JT, Lee MKC, Goytain A, Ho J, et al. NRG1 Gene Fusions Are Recurrent, Clinically Actionable Gene Rearrangements in KRAS Wild-Type Pancreatic Ductal Adenocarcinoma. Clin Cancer Res Off J Am Assoc Cancer Res 2019. Aug 1;25(15):4674–81. [DOI] [PubMed] [Google Scholar]
- 21.Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol Off J Eur Soc Med Oncol 2020. Nov;31(11):1491–505. [DOI] [PubMed] [Google Scholar]
- 22.Luchini C, Paolino G, Mattiolo P, Piredda ML, Cavaliere A, Gaule M, et al. KRAS wild-type pancreatic ductal adenocarcinoma: molecular pathology and therapeutic opportunities. J Exp Clin Cancer Res CR 2020. Oct 28;39(1):227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin Cancer Res Off J Am Assoc Cancer Res 2019. Aug 1;25(15):4712–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin W-C, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015. Apr 9;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fusco MJ, Saeed-Vafa D, Carballido EM, Boyle TA, Malafa M, Blue KL, et al. Identification of Targetable Gene Fusions and Structural Rearrangements to Foster Precision Medicine in KRAS Wild-Type Pancreatic Cancer. JCO Precis Oncol 2021;5:PO.20.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov 2018. Sep;8(9):1096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 2018. Jun;37(24):3183–99. [DOI] [PubMed] [Google Scholar]
- 28.Owsley J, Stein MK, Porter J, In GK, Salem M, O’Day S, et al. Prevalence of class I-III BRAF mutations among 114,662 cancer patients in a large genomic database. Exp Biol Med Maywood NJ 2021. Jan;246(1):31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017. Aug 10;548(7666):234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menzies AM, Yeh I, Botton T, Bastian BC, Scolyer RA, Long GV. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment Cell Melanoma Res 2015. Sep;28(5):607–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016. Feb 15;138(4):881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hendifar A, Blais EM, Wolpin B, Subbiah V, Collisson E, Singh I, et al. Retrospective Case Series Analysis of RAF Family Alterations in Pancreatic Cancer: Real-World Outcomes From Targeted and Standard Therapies. JCO Precis Oncol 2021. Aug 25;5:PO.20.00494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odintsov I, Lui AJW, Sisso WJ, Gladstone E, Liu Z, Delasos L, et al. The Anti-HER3 mAb Seribantumab Effectively Inhibits Growth of Patient-Derived and Isogenic Cell Line and Xenograft Models with Oncogenic NRG1 Fusions. Clin Cancer Res Off J Am Assoc Cancer Res 2021. Jun 1;27(11):3154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schram AM, Drilon AE, Macarulla T, O’Reilly EM, ROdon J, et al. A phase II basket study of MCLA-128, a bispecific antibody targeting the HER3 pathway, in NRG1 fusion-positive advanced solid tumors. Journal of Clinical Oncology 38, no. 15_suppl [Google Scholar]
- 35.Abou-Alfa GK, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020. Jun;21(6):796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020. May;21(5):671–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma WW, Xie H, Fetterly G, Pitzonka L, Whitworth A, LeVea C, et al. A Phase Ib Study of the FGFR/VEGFR Inhibitor Dovitinib With Gemcitabine and Capecitabine in Advanced Solid Tumor and Pancreatic Cancer Patients. Am J Clin Oncol 2019. Feb;42(2):184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuli R Anaplastic Lymphoma Kinase Rearrangement and Response to Crizotinib in Pancreatic Ductal Adenocarcinoma. JCO Precis Oncol 2017. Aug 17; [DOI] [PubMed] [Google Scholar]
- 39.Gower A, Golestany B, Gong J, Singhi AD, Hendifar AE. Novel ALK Fusion, PPFIBP1-ALK, in Pancreatic Ductal Adenocarcinoma Responsive to Alectinib and Lorlatinib. JCO Precis Oncol 2020;4:PO.19.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subbiah V Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. Journal of Clinical Oncology 38, no 15_suppl. 2020. May 25; [Google Scholar]
- 41.Chou A, Brown IS, Kumarasinghe MP, Perren A, Riley D, Kim Y, et al. RET gene rearrangements occur in a subset of pancreatic acinar cell carcinomas. Mod Pathol Off J U S Can Acad Pathol Inc 2020. Apr;33(4):657–64. [DOI] [PubMed] [Google Scholar]
- 42.Philip PA, Benedetti J, Corless CL, Wong R, O’Reilly EM, Flynn PJ, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol Off J Am Soc Clin Oncol 2010. Aug 1;28(22):3605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Philip PA, Goldman B, Ramanathan RK, Lenz H-J, Lowy AM, Whitehead RP, et al. Dual blockade of epidermal growth factor receptor and insulin-like growth factor receptor-1 signaling in metastatic pancreatic cancer: phase Ib and randomized phase II trial of gemcitabine, erlotinib, and cixutumumab versus gemcitabine plus erlotinib (SWOG S0727). Cancer 2014. Oct 1;120(19):2980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muzumdar MD, Chen P-Y, Dorans KJ, Chung KM, Bhutkar A, Hong E, et al. Survival of pancreatic cancer cells lacking KRAS function. Nat Commun 2017. Oct 23;8(1):1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Reilly EM, Oh D-Y, Dhani N, Renouf DJ, Lee MA, Sun W, et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 2019. Oct 1;5(10):1431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marabelle A Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. Journal of Clinical Oncology 38, no 1. 2020. Jan 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grant RC, Denroche R, Jang GH, Nowak KM, Zhang A, Borgida A, et al. Clinical and genomic characterisation of mismatch repair deficient pancreatic adenocarcinoma. Gut 2020. Sep 15;gutjnl-2020-320730. [DOI] [PubMed] [Google Scholar]
- 48.Ischenko I, D’Amico S, Rao M, Li J, Hayman MJ, Powers S, et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun 2021. Mar 5;12(1):1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Windon AL, Loaiza-Bonilla A, Jensen CE, Randall M, Morrissette JJD, Shroff SG. A KRAS wild type mutational status confers a survival advantage in pancreatic ductal adenocarcinoma. J Gastrointest Oncol 2018. Feb;9(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haas M, Ormanns S, Baechmann S, Remold A, Kruger S, Westphalen CB, et al. Extended RAS analysis and correlation with overall survival in advanced pancreatic cancer. Br J Cancer 2017. May 23;116(11):1462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashimoto D, Arima K, Yokoyama N, Chikamoto A, Taki K, Inoue R, et al. Heterogeneity of KRAS Mutations in Pancreatic Ductal Adenocarcinoma. Pancreas 2016. Sep;45(8):1111–4. [DOI] [PubMed] [Google Scholar]
- 52.Bauer TM, Dhir T, Strickland A, Thomsett H, Goetz AB, Cannaday S, et al. Genetic Drivers of Pancreatic Cancer Are Identical Between the Primary Tumor and a Secondary Lesion in a Long-Term (>5 Years) Survivor After a Whipple Procedure. J Pancreat Cancer 2018;4(1):81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakamoto H, Attiyeh MA, Gerold JM, Makohon-Moore AP, Hayashi A, Hong J, et al. The Evolutionary Origins of Recurrent Pancreatic Cancer. Cancer Discov 2020. Jun;10(6):792–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuboki Y, Fischer CG, Beleva Guthrie V, Huang W, Yu J, Chianchiano P, et al. Single-cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. J Pathol 2019. Mar;247(3):347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hahn WC, Bader JS, Braun TP, Califano A, Clemons PA, Druker BJ, et al. An expanded universe of cancer targets. Cell 2021. Mar 4;184(5):1142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: MAPK Pathway Activity Score (MPAS) in KRAS WT pancreatic tumor molecular groups. Double asterisks suggest statistical significant differences (adjusted p<0.05); single asterisks suggest trending (p<0.05); green asterisks: when compared to ‘Other KRAS Wild type’, red asterisks: when compared to KRAS mutant cohort.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. The NGS raw data are owned by Caris Life Sciences and cannot be publicly shared due to the data usage agreement signed by Dr. Philip Philip. Qualified researchers can apply for access to these data by contacting Joanne Xiu (jxiu@carisls.com) and signing a data usage agreement.