

Abstract
Medicines and vaccines prescribed to pregnant women often have not had pregnant women or lactating women included in clinical trials and products are often not approved by regulatory agencies for use in pregnant women. As a result, practitioners may need to prescribe medicines and give vaccines to this special population with limited drug efficacy and safety information available. Multiple regulatory guidance documents regarding the development of medications for pregnant and lactating women have been developed to encourage drug development and the investigation of medicines and vaccines used in this population. However, clinical, regulatory, ethical, and drug development challenges are encountered when designing clinical trials that include pregnant women and their fetuses, in which innovative methods and trial designs are essential. This article provides an overview of an industry perspective on maternal–fetal drug development that includes a review of the regulatory landscape for developing medicines for pregnant women and their fetuses, trial designs that include pregnant women, identification of gaps and challenges, and strategies for potential maternal–fetal drug development considerations for the future development of medicines and vaccines for pregnant women. Early involvement and discussion of drug and vaccine products with multiple stakeholders, including therapeutic experts, patients, physicians, and regulators, is encouraged to optimize the development of safe and effective medicines and vaccines for pregnant women and their fetuses.
Keywords: clinical trials, drug development, fetus, maternal, pediatrics, pregnant
During pregnancy, the body undergoes significant anatomical and physiological changes, having profound effects on the pharmacokinetics (PK) of medications. 1 Over‐the‐counter and prescription medicines are often needed during pregnancy. As a result of the lack of clinical, safety, or dosing information, healthcare providers are left using medications off‐label in pregnant women. Therefore, pregnant woman and the fetus are at risk for decreased safety and efficacy. Healthcare practitioners may often prescribe older medications to pregnant patients because of previous experience prescribing the medicine to their patients, or because there is known safety and adverse events information available. Newer drugs may be more effective in this population but are not used as often because the safety profile in pregnant women and their fetuses is unknown. 2 An example of the global impact of off‐label use in this population is shown in aFrench article published in 2020, which was a multicenter, prospective, longitudinal, observational study that included 397 women who gave birth to 400 children, all of whom used at least 1 medication. In this study, the most common on‐label drugs used were acetaminophen, antianemia preparations, analgesics, antibacterial drugs, or acid‐related drugs. Three hundred and twenty‐one (80.9%) pregnant women used at least 1 off‐label drug, 285 (71.8%) pregnant women used at least 1 off‐label high‐risk drug (eg, antithrombotic agents, calcium channel blockers, heparins, or vasoprotectives), and 189 (47.6%) pregnant women used at least 1 contraindicated drug (eg, non‐steroidal anti‐inflammatory drugs). 3 Although this study casts a glance at the potential level of off‐label use of medicines prescribed to pregnant women and the fetus, the final level of off‐label use is unknown. 3 Additionally, whether prescribing on‐ or off‐label medicines or developing new medications or vaccines, the efficacy and safety of the drug in pregnant women and their fetuses might be considered independently, which can cause a conflict at times.
Previously there has been hesitancy in exposing pregnant women and their fetus to investigational drugs because of a concern that it could harm the pregnant woman and potentially the fetus. However, not studying the drug could also lead to inappropriate dosing, insufficient therapeutic response, adverse reactions, and limited efficacy and safety information. Therefore, the objective of this article is to provide an overview of an industry perspective on maternal–fetal drug development by: (1) providing the regulatory landscape for developing medicines for pregnant women and their fetus, (2) designing trials that include pregnant women, (3) identifying potential gaps and challenges, and (4) providing potential strategies for maternal–fetal drug development considerations for the future development of medicines and vaccines for pregnant women and their fetuses.
Maternal–Fetal Drug Development
Regulatory Framework
For decades, pregnant women and children were largely excluded globally from clinical trials. Although in the past 20 years many regulatory agencies have changed or created new regulations and guidance to incorporate this special population in the clinical trials. However, regulations to study medicines in pregnant women and their fetuses remain limited. 4 The United States Congress passed the Best Pharmaceuticals for Children Act (BPCA) in 2002 and the Pediatric Research Equity Act (PREA) in 2003 to help promote the study of medicines in children. 5 , 6 The BPCA is a voluntary program to incentivize the completion of pediatric clinical trials by providing additional marketing exclusivity to drug developers. BPCA utilizes the Food and Drug Administration (FDA)‐issued written requests to describe the pediatric clinical studies to be completed voluntarily for a specific drug. 5 , 7 , 8 , 9 Whereas PREA permits the FDA to require pediatric studies in certain drugs and biological products and request studies to develop the most age‐appropriate formulations. 6 Currently, new medicines developed for conditions that occur in adults and children are now including studies that evaluate the efficacy and safety of the drugs in adults and children of all ages. Also, international collaboration via the Pediatric Cluster teleconference meetings provides a pathway for multiple regulatory agencies to discuss new drug development programs and topics focused on pediatrics. The Pediatric Cluster teleconferences occur monthly and include multiple regulatory agencies, including the FDA, the European Medicines Agency (EMA), Japan's Pharmaceuticals and Medical Device Agency, Health Canada. and Australia's Therapeutic Goods Administration. 10 In addition, the Common Rule of 2018 may aid in the potential inclusion of pregnant women as the rule no longer classifies pregnant women as a vulnerable population under 45 CFR 46.111(a)(3). 4 , 11 However, there are currently no regulatory requirements mandating clinical trial inclusion of pregnant women and their fetuses. The US Health and Human Service regulations apply to the fetus, whereas the pediatric regulations (21 CFR subpart D) are not applicable to the fetus. 12 Although there are limited regulations regarding pregnant and lactating women and fetuses, there are over 7 US guidance documents regarding developing medicines for this special population. 4 , 13 , 14 , 15 , 16 , 17 , 18 , 19
Other initiatives include the United States Congress establishment of the Task Force on Research Specific to Pregnant Women and Lactating Women (PRGLAC) in 2017. The PRGLAC has several representatives, including federal agencies, medical societies, the pharmaceutical industry, and non‐profit organizations. 20 , 21 The task force submitted in the 2018 PRGLAC report to the Department of Health and Human Services guidance regarding the implementation and 15 recommendations for including pregnant and lactating women in clinical research. 20 Additionally, the FDA/University of Maryland Center for Excellence in Regulatory Science and Innovation (M‐CERSI) has held several workshops to advance the understanding and development of medicines in pregnant women and their fetuses. For example, in 2021 the FDA/M‐CERSI held a workshop addressing fetal pharmacology and ways to advance drug development, and in 2022 there was a workshop to engage stakeholders to assess current gaps in advancing pharmacokinetic study evaluation in pregnant women. 22 , 23
Additionally, the US regulatory framework, in the form of workshops and guidance available for designing clinical trials for this patient population, continues to expand and simultaneously several other countries and regulatory agencies have also created research initiatives, guidance, and workshops on this important topic. In 2020, for example, the EMA held a workshop to explore the benefit–risk of medicines used during pregnancy and breastfeeding. 24 In 2021, the International Coalition of Medicines Regulatory Authorities (ICRMA) held a pregnancy and lactation workshop that focused on the COVID‐19 medicines available for pregnant women and the severity and clinical outcomes of COVID‐19 in this special population. 25 Also, in the UK, the Medicines and Healthcare products Regulatory Agency (MHRA) developed a 1‐day course on the PK of medicines in pregnancy and the ways that pregnancy can affect plasma drug levels. 26 , 27 In addition to the course, other guidance provided by the EMA for pregnant women and their fetuses include the 2019 “Guideline on Good Pharmacovigilance Practices (GVP) – Product‐ or Population‐Specific Considerations III: Pregnant and Breastfeeding Women” and the 2020 “EMA Regulatory Science to 2025: Strategic Reflection.” 28 , 29
International collaboration efforts continue to increase for maternal and pediatric drug development. One example of international collaboration occurs with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), which is a group that brings multiple regulatory agencies and pharmaceutical industry representatives together to discuss specific technical and scientific topics. Combined international collaboration of multiple group efforts to accelerate maternal drug development led to the May 2022 ICH endorsement of the development of a new ICH Efficacy Guideline on the “Inclusion of Pregnant and Breastfeeding Individuals in Clinical Trials.” 30 Although, these new regulatory guidance and regulations help to encourage future drug development programs to include pregnant women and their fetuses, more can be done in maternal–fetal drug development to provide additional regulatory guidance and incentives to assist with specific funding for studying medications that are off‐patent or used off‐label.
The inclusion of pregnant women in clinical trials can provide information on the use of investigational medicines in this special population, which could lead to obtaining the necessary data to determine whether the medicine or vaccine is safe and effective for use in pregnant women, with that information subsequently added to the drug label. However, often pregnant and lactating women are excluded from clinical trials, especially if there is insufficient information to determine the risk to the fetus or infant. 13 , 31 Therefore, it is vital to study medicines and vaccines that include pregnant women in clinical trials when it is appropriate to include them, but especially in certain situations where potentially the drug or vaccine could be indicated for pregnant women and their fetuses and it is deemed scientifically and ethically applicable to include this patient population.
Impactful and efficient clinical trials that include pregnant women and their fetuses are vital in improving the quality of care as well as increasing the number of available safe and effective medicines and vaccines for this special population. Thus, under the Prescription Drug User Fee Act (PDUFA VII), the FDA has committed to expand the use of the Sentinel network and real‐world evidence for assessing pregnancy safety. 32 Also, the FDA is developing a framework to describe how data from various types of post‐marketing pregnancy safety studies might optimally be used and will conduct 5 demonstration projects to address gaps in knowledge about performance characteristics of different study designs, including pregnancy registries and electronic healthcare data sources. In this expansion, the utilization of real‐world evidence for use in regulatory decision making is also being explored. 32
Regulatory agencies have established patient‐focused drug development guidance and initiatives to incorporate the patient perspective in the drug development and evaluation process. 33 , 34 Partnering with patient advocacy organizations for specific diseases, patients, and advocates for pregnant women, and obtaining their perspectives on the drug development program, is helpful in providing the insight and feedback to better inform the strategy, design, and execution of the study. 34 Patient involvement in a study is needed as their perspectives are extremely valuable. Identification of patient's needs and priorities in a study and their perspective can aid in optimizing the trial protocol designs, improve completion of assessments during the study as well as enhance recruitment and retention of the participant in the study. 35 Therefore, leveraging predictive analysis, strategizing early for the optimal dose and study designs, and obtaining a team of experts to strategize and execute these studies is needed.
Designing Trials That Involve Pregnant Women and Their Fetuses
Advances in medical care have resulted in women who previously would not have survived their diseases now becoming pregnant and needing to continue treatment during their pregnancies. Standard therapies used by pregnant women include drug products such as antibiotics, antidepressants, anti‐epileptic medicines, and anti‐hypertensives, as well as preventative vaccines such as the maternal respiratory syncytial virus (RSV) vaccine. Although the safety profile observed in traditional clinical trials will be a reasonable prediction of what would be observed in a pregnant woman, there are well‐known examples of adverse events in the pregnant women and fetus that are not observed in traditional clinical trials. One example is the antibiotic tetracycline, which if administered after 24 weeks of pregnancy can lead to the permanent staining of a baby's teeth. Likewise, angiotensin‐converting enzyme (ACE) inhibitors, if used during the second and third trimesters of pregnancy for the treatment of hypertension, can cause injury to the infant's kidneys. Lithium carbonate can be used to treat pregnant women with bipolar disorder but is often switched to a similar drug during a portion of the pregnancy when the fetus's heart is developing because Lithium use in pregnancy carries a small risk of fetal heart defects, such as Ebstein's anomaly. Historically, there have been many unfortunate tragedies where pregnant women have used certain medicines that caused significant adverse events to themselves or their unborn child. For example, pregnant women seeking relief from nausea used thalidomide, and some subsequently delivered children with phocomelia.
When a potential drug product or vaccine has been identified for clinical development, a comprehensive plan is outlined listing all planned clinical studies as well as the necessary nonclinical information needed to enroll the appropriate study participants. Important to the well‐being of study participants is knowledge of the PK, safety, and dose translation between species. When the anticipated patient population includes pregnant women, additional nonclinical studies should be considered so that the future clinical studies may be designed to allow investigators to closely monitor both the pregnant woman and the fetus throughout the clinical trial for potential adverse reactions and possible long‐term effects on the mother after the pregnancy and effects on the neonate.
As part of the investigational new drug (IND) application, study reports of nonclinical studies and exploratory studies, including PK in at least 2 species, both single and multiple dose, toxicology at multiples of anticipated human exposure, genotoxicity studies, safety pharmacology, efficacy studies in appropriate non‐human models, if available, toxicokinetics, and reproductive/toxicology, including an assessment of teratology and female fertility, are needed. 36 Data obtained from nonclinical studies or animal studies provide valuable information, but is this enough? For example, depending on the drug product, nonclinical studies or animal studies can predict whether a particular drug poses a high risk of birth defects or whether the medicine is excreted in human milk, but still, in certain situations, these data can be limited. 2 Therefore, in special populations (eg, pregnant or lactating women) there may need to be additional nonclinical assessments and sufficient nonclinical information to support proceeding to clinical studies. Additional guidance on conducting nonclinical safety studies to support clinical studies in special populations can be found in the ICH guidelines for industry: S5 (R3) Guideline on Reproductive Toxicology: Detection of Toxicity to Reproduction for Human Pharmaceuticals; S11 Nonclinical Safety Testing in Support of Development of Pediatric Pharmaceuticals; and M3 (R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. 37 , 38 , 39 , 40
Animal studies mainly focus on potential drug effects and possible adverse effects on the reproductive outcome and birth defects, which are often determined by administering the animals a higher dose of the drug. However, selecting the appropriate nonclinical study or animal model as well as trimester to administer the medication is essential to leverage the data appropriately, interpret the findings, and determine the applicability to pregnant women and their fetuses. Animal models have been used to help predict pharmacokinetic pharmacodynamic (PKPD) modeling in pregnancy and fetal drug development but limitations exist, arising from differences in placenta structure, hemodynamics, and other differences among species. 41 , 42
Phase 1 clinical research focuses on the safety and dose finding in a small number of healthy adult volunteers and occasionally in participants diagnosed with the condition or disease for which the compound is being investigated. The purpose of phase 2 is to study the efficacy of the drug and adverse reactions in a larger but relatively small number of participants diagnosed with the condition or disease. Phase 2 studies are usually well controlled and the participants are closely monitored. 43 In phase 3, the number of participants is larger than Phase 2 with the selected disease or condition studied and expands to controlled and uncontrolled trials. After preliminary evidence of drug effectiveness is obtained in phase 2 studies, then phase 3 studies are intended to gather additional information on the safety and effectiveness of the drug. 43
Before 2012, depending upon the anticipated indication, clinical trials included mainly adults but with some participants from special populations (eg, the pediatric population, from birth to 18 years; pregnant women; and the geriatric population, aged 65 years and older). However, after 2012 when the Food and Drug Administration Safety and Innovation Act (FDASIA) made BPCA and PREA permanent, the pediatric population was routinely included in industry‐sponsored clinical studies, but pregnant women and their fetuses often remained understudied. 5 , 6 , 44 PK data is usually collected in industry‐sponsored studies through observational trials or other surveillance opportunities. Occasionally a participant may become pregnant during a study in which there will be continued monitoring of these study participants including the outcome of their pregnancies and the condition of the newborns. Additionally, if the participant is lactating, then an evaluation of the excretion of the drug or its metabolites into human milk should be investigated as well as the infant monitored for potential effects of the drug. 37
With the challenges of including pregnant women, or those who may become pregnant in clinical trials, unconventional trial design options can be considered when developing products for pregnant women and their fetuses, which include but are not limited to longitudinal design, opportunistic study design, staggered trial design, embedded trial design, and adaptive trial design. Prior to selecting the design and conducting the clinical trial it is important to have estimates of PKPD data prior to launching a clinical trial evaluation. 45 In the 2004 FDA Pharmacokinetics in Pregnancy guidance, regulators suggest that PK studies should be conducted before pregnancy, to establish a baseline for comparison, and then continue throughout each trimester. However, there are challenges associated with enrolling participants pre‐pregnancy and so the regulators recommend an alternative approach, obtaining PKPD assessments in the second and third trimester and comparing the pregnancy data with the postpartum data. 45
Adaptive design is a clinical trial design that permits prospectively planned modification in 1 or more aspects of the trial design, and has been used in uncertain cases with regard to safety and efficacy. 46 This type of design is used in smaller patient populations and can be expanded to a broader population based on interim safety data. These types of trial designs have been used in other special populations, such as pediatrics, because it permits more flexibility in the study to allow additional enrollment based on previous data. Additional regulatory guidance has been established regarding adaptive designs and has greatly contributed to the increase in the use and understanding of adaptive designs in clinical trials. 31 , 46 When designing clinical trials for special populations, such as pregnant women or pediatric participants, it is important to obtain sufficient information in clinical trials about medical product efficacy and safety in participants from racial and ethnic minority backgrounds. 31 , 47 Also, selecting the most suitable endpoint to capture the information needed to determine the efficacy and safety of the drug in this unique population is necessary. Including clinical or surrogate endpoints that evaluate both the pregnant woman and the fetus is essential.
Impactful and high‐quality clinical trial designs that obtain the necessary data is critical. However, early in the planning stages it is also essential to consider and incorporate in the development of the drug or vaccine novel methods, designs, and tools for medicines intended for both pregnant women and their fetuses. Additionally, the consideration of how the medication may affect the pregnant individual and how it affects the fetus can vary, and utilizing tools to predict potential effects is needed. Population PK (popPK) and physiologically based PK (PBPK) modeling are pharmacometrics tools that have been used for many years in pharmacological studies involving pregnant and lactating women. 21 For example, an estimate of the PK of oseltamivir in pregnant women diagnosed with influenza was determined by utilizing popPK modeling. 21 To investigate the extent to which a drug transfers across the placenta, the incorporation of PBPK models can in general predict the PK in pregnant women but is limited in predicting the PK in the fetus. 48 Often there is a lack of information regarding the PK of drugs in pregnant women, and anticipated exposure for the dose and dosing regimens are extrapolated from other populations, such as non‐pregnant women, to pregnant women. The extrapolation of information from non‐pregnant to pregnant women could result in a potential risk of supratherapeutic or subtherapeutic dose exposures, leading to the pregnant women and/or their fetuses experiencing toxic or ineffective drug effects. 49 However, there are some situations where PBPK pregnancy models are able to incorporate data collected during pregnancy in women and nonclinical models to build a foundation that will in turn result in a more robust assessment of exposure throughout pregnancy and guide optimal sampling in confirmatory studies. Models may be included in regulatory submissions to provide additional information on time‐dependent factors at any stage of pregnancy, support appropriate dosing and optimal study designs. 50
The optimization of digital tools, modeling and simulation, and the use of real‐world evidence and real‐world data lead to a better understanding of the use of the medicine, decreases the exposure of a drug to pregnant women and their fetuses by reducing the potential sample size of participants needed to be in a clinical trial, and can contribute to the determination of an appropriate dose and design for studies in pregnant women and their fetus.
Challenges and Opportunities
The complexities and challenges of maternal and fetal trials are similar to those of pediatric and neonatal studies. Prior to regulatory mandates to study medicines in children, few children participated in clinical studies. However, with the implementation of regulatory mandates, investigators gained experience and the pharmaceutical industry was able to develop efficient pediatric clinical programs. With the increase in studies including pregnant women, there is recognition that PK, safety, and efficacy data are needed throughout pregnancy. Therefore, development of new pregnancy regulatory guidances, scientific workshops, and exploration of novel methods and tools such as modeling and simulation is being conducted to obtain the pregnancy information. As shown in Figure 1, there are over 380,000 adult (18 to 64 years) studies available and an increasing number of pediatric studies, but the maternal–fetal studies available remain limited. The limited number of maternal–fetal studies may cause difficulty for interested participants to join the study and challenges for the investigators to be part of the study and gain experience in this specialized area.
Figure 1.
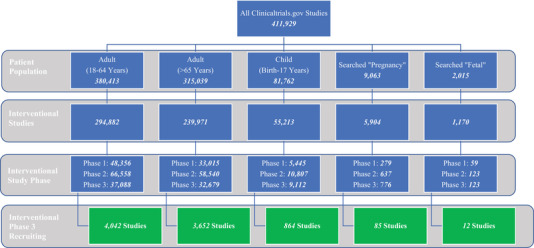
Number of studies for each patient population listed on ClinicalTrials.gov.
Note: National Institute of Health. 51 https://www.clinicaltrials.gov/
The safety profile of a drug at the time of regulatory approval is often incomplete for pregnant women, due to limited sample size, short duration, and inclusion/exclusion criteria. As pregnancy is frequently an exclusionary criterion, there are limited data on the efficacy and safety of available medicines in this population. Post‐marketing surveillance offers an opportunity to gather information in populations not included in the original drug development experience. However, despite knowing that there is a growing expectation on the part of healthcare providers and patients for safety and efficacy information on the use of medicines and vaccines, researchers must continue to carefully consider potential risks and benefits prior to the inclusion of pregnant women and their fetuses in the drug development process.
Other compounds may have existing information on pharmacology, PK, and PD, which can be leveraged in a new drug development program for medicines intended for pregnant women and their fetuses by permitting new programs to have improved study design, reduction in the number of studies needed, and the reduction in the amount of time required to complete the studies.
Some additional challenges in conducting studies in this special population arise when discussing a clinical trial with a pregnant woman, where ethical and safety concerns are usually paramount for the mother and the fetus. 52 Enrollment in these studies can be complicated and difficult unless there is known benefit to both the mother and the fetus. One of the complications for conducting maternal drug or vaccine studies and enrolling pregnant women and their fetuses in a study is the hesitancy to participate . Hesitancy from clinicians and researchers in exposing pregnant women and their fetuses to investigational drugs is mainly associated with ethical concerns regarding potential drug‐related adverse reactions. 53 Thus, investigators and clinical trial units may be hesitant to run these studies because of the unknown safety profile for the pregnant woman and fetus and the questionable benefits. However, the hesitancy of clinicians, researchers, and pregnant women to be part of a clinical study can also result in pregnant women continuing to be prescribed medicines off‐label, where the potential adverse reaction of the drug is unknown for the pregnant woman and fetus, including the long‐term effects on the pregnant individual and the child.
Informed consent could be a potential challenge as both mother and fetus must be considered. Conducting trials that include pregnant women and their fetus may include consenting questions such as the number of consents needed for the participants (eg, pregnant woman and fetus), number of consents needed for each phase of the study, and the country requirements for who is eligible to provide consent for the study. The potential consents required could include but are not limited to a parental consent (maternal and/or maternal and paternal consent) and a separate consent for the fetus, as well as a new consent once the infant is born (eg, for safety and development assessments). 54 Additionally, the clinical trials consent requirements could vary by the type of study, age of participant(s), and country.
Some therapeutics such as vaccines and neonatal‐specific medications may be more straightforward to study. Still, other drugs that would rarely be used in the pregnant woman and fetus could be more complex to study. For example, there could be a medicine or vaccine that is intended for non‐pregnant women but might be used in a pregnant woman, so it is critical to understand whether there are unexpected potential adverse effects to the pregnant woman and the fetus even though the medicine or vaccine is intended mainly for the women and not for the fetus. Additionally, there could be a medicine or vaccine such as the maternal RSV vaccines that focus on the fetus and the development of antibodies that will cross the placenta and provide protection to the infant after birth. Additional complexity in maternal–fetal drug development, which differs from traditional drug development includes: determining the appropriate endpoint for both participants, the pregnant woman and the fetus in the clinical trial; the selection of long‐term outcomes to measure, such as growth and development parameters; and determining the duration of follow‐up for the pregnant woman, the fetus, and eventually the infant.
Pregnant women diagnosed with medical complexities may be under additional stress, leading to reduced interest in clinical trial participation. Potential investigators are likely to be concerned that eligible pregnant women are not likely to participate. Difficult‐to‐enroll studies are challenging for the industry to execute as they could take years to recruit, leading to the use of medicines off‐label before scientific data can be obtained and potential delay in the drug being approved for this special patient population. For medicines approved in adults or adults and children, but not approved for use in pregnant women and used off‐label, it creates increased difficulty to enroll pregnant women into a trial with a drug that is already available on the market. Industry and investigators have also experienced similar types of challenges in running pediatric and neonatal trials. However, for maternal–fetal studies there is an additional complexity because the patient population is limited to only a 9‐month period, and both the woman and the fetus are changing throughout the duration of the pregnancy.
If a healthcare provider or pregnant woman was interested in participating in a clinical trial, there could be significant difficulty navigating where to locate an open clinical trial that includes pregnant women in the trial. One of the options to learn about potential clinical trials conducted in the USA and around the world that may include pregnant women is to review ClinicalTrials.gov, which is a searchable registry and results database that provides information about the purpose of the trial, who is eligible to participate, locations, and contact information to obtain more information about the study. However, to determine potential studies that include pregnant women it is a challenge, as currently in ClinicalTrials.gov there is only an option to select “Adult (18–64 years),” “Adult (>65 years),” and “Child (birth–17 years),” but no option to select “pregnant” or “fetal.” Also, currently there is no option to select whether the study excludes pregnant women. Thus, a healthcare provider or a patient that is interested in locating a clinical trial for pregnant women would have to search for “pregnant” as the condition or disease and then review all the potential trials that would be applicable. Currently there is a large discrepancy in the number of studies available for adults, pediatrics, and adults aged >65 years versus the number of studies for pregnant women and their fetuses (Figure 1). Therefore, it could be advantageous if ClinicalTrials.gov or similar platforms to search for clinical trials included an advanced search option to select “pregnancy or maternal” to enable easier and quicker access for the healthcare provider and the potential participant to locate clinical trials for medicines and vaccines where pregnant women and/or their fetuses are eligible to participate in the study. Although there is a significant difference between the studies available for adults and pediatrics in comparison with pregnant women and their fetuses (Figure 1), the number of studies captured in the figure include studies for drug research, vitamins, procedures, and behavioral research. Additional applicable regulation and guidance documents for industry for developing medicines and vaccines in pregnant women and their fetuses, registries, and incentives to conduct research and develop medicines and vaccines in this area, as well as an increased awareness of potential clinical trials available to this special population are needed to help narrow the gap and discrepancy between the number of studies conducted and the medicines and vaccines approved across patient populations.
In September 2018, Moore et al published a summary of the estimated costs from 2015 through 2016 of pivotal trials required to show scientific evidence for FDA approval. The cost ranged from $5 million to over $345 million per trial, with a mean study cost of $13.5 million. 55 Most of these trials in this study were conducted in adults. Clinical trial cost is generally higher when studying special populations such as maternal‐fetal or pediatric studies.
Pediatric studies, for example, require extra time and additional steps such as obtaining consent from both parents and assent from the patient, and often having the consent discussion multiple times as parents are not always together. Studies in pediatric populations routinely require more sites and investigators to run trials in multiple countries, compared with studies including adults only. Individual sites are at times only able to enroll low numbers of patients, adding to costs because of the need to obtain additional global sites to recruit the required number of participants for the study. Additional funding would be needed for the recruitment and retention of pregnant women and their fetuses into a study. However, if it is a long‐term safety study more funding would be needed to evaluate the mother as well as the fetus over a longer period of time as the child grows. As BPCA incentives have helped move pediatric trials forward, similar incentives and opportunities would help support the additional cost of drug development and potential expansion of a priority list of therapeutic areas or medicines to be studied for pregnant women. 5 , 56
Considerations and Strategies for Maternal–Fetal Drug Development Programs
Traditionally, the drug safety and efficacy data in pregnant women and fetuses have been obtained from post‐marketing surveillance, but are limited. However, through federal resources and a focus on the gap in knowledge on maternal–fetal drug development, in the early 2000s the National Institute of Child Health and Human Development (NICHD) established a collaborative multicenter research network known as the Obstetric–Fetal Pharmacology Research Units (OPRUs). The mission of the ORPUs was to improve the safety and efficacy of medications used in pregnant and lactating women. In 2015, the network name was changed to the Obstetric–Fetal Pharmacology Research Centers (OPRCs). The ORPCs identified commonly used medicines in pregnancy and lactation and investigators conducted PKPD studies of these medications. As a result of this work, several studies from the OPRCs have been published on medicines commonly used in pregnancy and during lactation. 21 In addition to consider utilizing the ORPCs, there are several other considerations that need to be potentially incorporated when conducting maternal–fetal clinical research, such as thoughtful design strategy and execution and the involvement of multiple functional areas and experts throughout the drug development process. Depending on the drug or vaccine development program the considerations and strategies could fluctuate between categories when designing trials for pregnant women and their fetus (Figure 2). The incorporation of trial considerations and elements that have been successful in previous maternal–fetal development programs is encouraged as it may optimize the maternal–fetal strategy and execution of the new drug or vaccine development program. There are several potential design elements and strategies to consider when developing a maternal–fetal drug or vaccine program which include building a core study team internally of appropriate experts representing multiple functional lines, such as regulatory, clinical, safety, trial operations, clinical pharmacology and pharmacometrics expertise, as well as engaging externally with therapeutically aligned experts and research networks.
Figure 2.
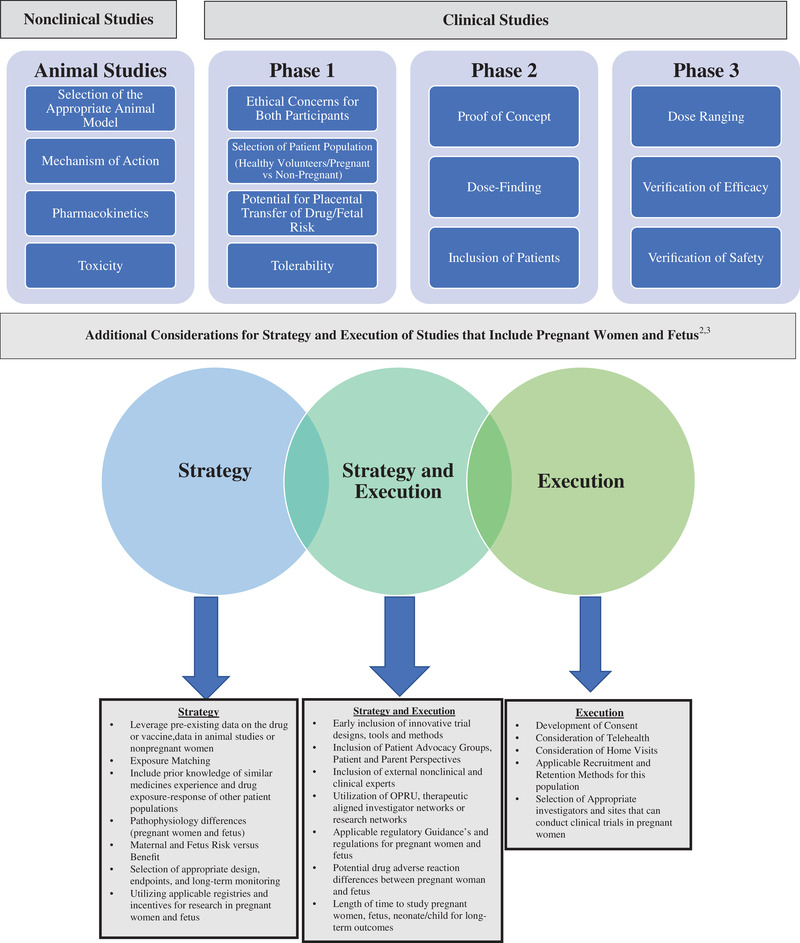
Drug development considerations and strategies when designing studies that include pregnant women and fetuses. Sources: Roset Bahmanyar et al 52 and Green et al. 4
Collaboration with the research networks and initiatives is advantageous when building a new drug or vaccine development plan. The connection with these specialized groups can help to understand and integrate experience obtained from past clinical studies for the therapeutic area or drug class that is being developed. For example, for many years pregnant and lactating women with HIV have participated in PK studies. However, many of the women with HIV are also breastfeeding their newborns and so it would be important to gather the previous experience of the PK studies and understand the extent that the newborn is exposed to the maternal drugs in the breastmilk. 57 Another example related to maternal–fetal health initiatives includes the WHO department of Essential Medicines and Health Products, United Nations Population Fund, and United Nations International Children's Emergency Fund groups, who created a priority list of medicines for women and children that encompasses medications based on the global burden of disease and evidence that the efficacy and safety of the drugs could prevent or treat major illnesses that cause maternal and pediatric mortality and morbidity. 58 Therefore, connecting with research initiatives such as the Innovative Medicines Initiative ConcePTION or networks such as the World Health Organization (WHO) and the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) can lead to sharing knowledge and creating new paths to advance drug development for pregnant women. The ConcePTION group focuses on improving the monitoring and communication of the safety of medicines used in pregnancy and breastfeeding, 59 whereas WHO/IMPAACT is a group with a mission to decrease the incidence of HIV and HIV‐associated infections in pregnant women and children. WHO/IMPAACT produced a document titled “Research for informed choices: Accelerating the study of new drugs for HIV in pregnant and breastfeeding women” that described a new framework for pregnant women with HIV to be included earlier in clinical trials. 57 , 60 Additionally, WHO/IMPAACT provided techniques to accelerate studies in pregnancy, such as conducting reproductive toxicity studies during early pre‐clinical stages of drug development and permitting women enrolled in a study to decide whether they want to continue in the study if they become pregnant. 57 , 60 Therefore, leveraging prior data on the drug or vaccine as well any data obtained from available animal studies or non‐pregnant individuals can be beneficial when determining study design endpoints or possible quantitative or innovative tools to incorporate into the development program.
In creating a maternal–fetal development strategy plan it is important to utilize regulatory guidance, established registries, research incentives, and clinical research networks to ultimately lead to improved dosing, safety, and efficacy of medicine and vaccines intended for pregnant women and their fetuses, as shown in Figure 2. 59 Regulators encourage sponsors with maternal–fetal development programs to have early meetings with the intended regulatory agencies to discuss the program and when and how to include pregnant women in the development plan. 13
Conclusion
Leveraging existing data, experts in their field (eg, bioethics, neonatology, OBGYN/maternal health, and perinatology), digital tools, and previous experience with medicine and vaccine studies in pregnant women is essential to creating an informed clinical trial design and development program. It is imperative that multiple stakeholders be involved early in the clinical trial design for medicines and vaccines intended for pregnant women and their fetuses. Potential stakeholders include patient, industry, academia, and regulatory representatives. It is vital to harness the patient voice and use patient experts to explore the current field, the challenges, and the potential actional solutions.
Regulatory guidance, pregnancy research initiatives, and regulatory cluster teleconference meetings focused on medicines commonly used or that might be used in pregnant women and their fetuses would be beneficial. Through collaboration with multiple stakeholders, continued research, education, and awareness about the potential effects of medicines and vaccines on the pregnant individual and fetus, initiatives to develop medicines and vaccines in this special population, registries, workshops, and consortiums are all needed to understand and address the gaps in this specialized area. The integration of novel study designs and innovative strategies are vital to accelerating drug development and increasing the number of safe and effective medicines and vaccines available to pregnant women and their fetuses.
Conflicts of Interest
The authors are employees of Pfizer and shareholders. The authors received no financial support for the research, authorship, and/or publication of this article. The opinions expressed in this article are those of the authors and should not be interpreted as the position of Pfizer.
Data Sharing
Data sharing is not applicable to this article as no new data were created or analyses conducted in this study.
Funding
The authors received no financial support for this article's research, authorship, and/or publication.
Author Contributions
Conception and design of the article: E.D. and J.B. Drafting or revising the article: E.D. and J.B. Research: E.D. and J.B. Data analysis: E.D. and J.B.
Acknowledgments
The authors would like to thank Andrew Emmett, Mary Alice Hiatt, Joan Korth‐Bradley, Maria Maddalena Lino, Iona Munjal, and Charles Thompson for their contributions.
References
- 1. Pariente G, Leibson T, Carls A, Adams‐Webber T, Ito S, Koren G. Pregnancy‐associated changes in pharmacokinetics: a systematic review. PLoS Med. 2016;13(11):e1002160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meadows M. U.S. Food and Drug Administration: FDA Consumer Magazine: Pregnancy and the Drug Dilemma. https://www.perinatology.com/Archive/FDA%20CAT.htm. Published 2001. Accessed August 25, 2022.
- 3. Laroche ML, Blin A, Coubret A, Grau M, Roux B, Aubard Y. Off‐label prescribing during pregnancy in France: the NeHaVi cohort. Int J Clin Pharmacol Ther. 2020;58(4):198‐208. [DOI] [PubMed] [Google Scholar]
- 4. Green DJ, Park K, Bhatt‐Mehta V, Snyder D, Burckart GJ. Regulatory considerations for the mother, fetus and neonate in fetal pharmacology modeling. Front Pediatr. 2021;9:698611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. U.S. Food and Drug Administration: Best Pharmaceuticals for Children Act (BPCA). https://www.fda.gov/drugs/development-resources/best-pharmaceuticals-children-act-bpca#:~:text=Best%20Pharmaceuticals%20for%20Children%20Act%20%28BPCA%29%20BPCA%20provides,Written%20Request%20issued%20by%20FDA.%20Written%20Requests%20Issued. Accessed August 7, 2022.
- 6. U.S. Food and Drug Administration: Pediatric Research Equity Act (PREA). https://www.fda.gov/drugs/development‐resources/pediatric‐research‐equity‐act‐prea. Accessed August 7,2022.
- 7. U.S. Food and Drug Administration. Approved Active Moieties to which FDA has issued a Written Request for Pediatric Studies under Section 505A of the Federal Food, Drug, and Cosmetic Act. https://www.fda.gov/drugs/development-resources/written-requests-issued. Accessed August 7, 2022.
- 8. U.S. Food and Drug Administration: Qualifying for Pediatric Exclusivity Under Section 505A of the Federal Food, Drug, and Cosmetic Act: Frequently Asked Questions on Pediatric Exclusivity (505A). https://www.fda.gov/drugs/development‐resources/qualifying‐pediatric‐exclusivity‐under‐section‐505a‐federal‐food‐drug‐and‐cosmetic‐act‐frequently. Accessed August 7, 2022.
- 9. U.S. Food and Drug Administration: Guidance for Industry Qualifying for Pediatric Exclusivity Under Section 505A of the Federal Food, Drug, and Cosmetic Act. https://www.fda.gov/media/72029/download. Accessed June 20, 2022.
- 10. U.S. Food and Drug Administration ‐ International Collaboration/Pediatric Cluster. https://www.fda.gov/science‐research/pediatrics/international‐collaboration‐pediatric‐cluster. Accessed August 7, 2022.
- 11. National Archives: Code of Federal Regulations ‐ 45 CFR 46.111(a)(3). https://www.ecfr.gov/current/title‐45/subtitle‐A/subchapter‐A/part‐46. Accessed August 7, 2022.
- 12. National Archives: Code of Federal Regulations ‐ 21 CFR Subpart D. https://www.ecfr.gov/current/title‐21/chapter‐I/subchapter‐A/part‐50/subpart‐D. Accessed August 7, 2022.
- 13. U.S. Food and Drug Administration: Pregnant Women: Scientific and Ethical Considerations for Inclusion in Clinical Trials Guidance for Industry. https://www.fda.gov/media/112195/download. Accessed August 7, 2022.
- 14. U.S. Food and Drug Administration: Reviewer Guidance: Evaluating the Risks of Drug Exposure in Human Pregnancies. https://www.fda.gov/media/71368/download. Accessed August 7, 2022.
- 15. U.S. Food and Drug Administration: Clinical Pharmacology Considerations for Neonatal Studies for Drugs and Biological Products. https://www.fda.gov/media/129532/download. Accessed August 7, 2022.
- 16. U.S. Food and Drug Administration: Postapproval Pregnancy Safety Studies Guidance for Industry. https://www.fda.gov/media/124746/download. Accessed August 7, 2022.
- 17. U.S. Food and Drug Administration: Guidance for Industry: Nonclinical Safety Evaluation of the Immunotoxic Potential of Drugs and Biologics. https://www.fda.gov/media/135312/download.Accessed August 7, 2022.
- 18. U.S. Food and Drug Administration: Safety Testing of Drug Metabolites Guidance for Industry. https://www.fda.gov/media/72279/download. Accessed August 7, 2022.
- 19. U.S. Food and Drug Administration: Pregnancy, Lactation, and Reproductive Potential: Labeling for Human Prescription Drug and Biological Products‐Content and Format. https://www.fda.gov/media/90160/download. Accessed August 7, 2022.
- 20. Task Force on Research Specific To Pregnant Women and Lactating Women: Report Implementation Plan. https://www.nichd.nih.gov/sites/default/files/inline‐files/PRGLAC_Implement_Plan_083120.pdf. Accessed August 7, 2022.
- 21. Ren Z, Bremer AA, Pawlyk AC. Drug development research in pregnant and lactating women. Am J Obstet Gynecol. 2021;225(1):33‐42. [DOI] [PubMed] [Google Scholar]
- 22. U.S. Food and Drug Administration/M‐CERSI Workshop: Pharmacokinetic Evaluation in Pregnancy; May 16–17, 2022. https://www.fda.gov/drugs/news‐events‐human‐drugs/pharmacokinetic‐evaluation‐pregnancy‐virtual‐public‐workshop‐05162022. Accessed August 7, 2022.
- 23. U.S. Food and Drug Administration/M‐CERSI Workshop: Fetal Pharmacology and Therapeutics; October 21–22, 2022. https://www.fda.gov/drugs/news‐events‐human‐drugs/fetal‐pharmacology‐and‐therapeutics‐october‐21‐22‐2021‐10212021‐10222021. Accessed August 7, 2022.
- 24. European Medicines Agency: Works on Benefit‐Risk of Medicines used During Pregnancy and Breastfeeding; September 9, 2022. https://www.ema.europa.eu/en/events/workshop‐benefit‐risk‐medicines‐used‐during‐pregnancy‐breastfeeding. Accessed August 7, 2022.
- 25. International Coalition of Medicines Regulatory Authorities (ICRMA): Pregnancy and Lactation Workshop https://icmra.info/drupal/en/covid‐19/9february2021. Accessed August 7, 2022.
- 26. Medicines and Healthcare products Regulatory Agency: Use of Medicines in Pregnancy and Breastfeeding Guidance. https://www.gov.uk/guidance/use‐of‐medicines‐in‐pregnancy‐and‐breastfeeding. Accessed August 7, 2022.
- 27. Medicines and Healthcare products Regulatory Agency: MHRA Course on Pharmacokinetics of Medicines in Pregnancy. https://medregs.blog.gov.uk/2020/06/22/mhra‐course‐on‐pharmacokinetics‐of‐medicines‐in‐pregnancy‐understanding‐how‐pregnancy‐affects‐plasma‐drug‐levels/. Accessed August 7, 2022.
- 28. European Medicines Agency: Guideline on Good Pharmacovigilance Practice (GVP) ‐ Product or Population Specific Consideration for Pregnant and Breastfeeding Women. https://www.ema.europa.eu/en/documents/scientific‐guideline/draft‐guideline‐good‐pharmacovigilance‐practices‐product‐population‐specific‐considerations‐iii_en.pdf. Accessed August 7, 2022.
- 29. Nooney J, Thor S, de Vries C, et al. Assuring access to safe medicines in pregnancy and breastfeeding. Clin Pharmacol Ther. 2021;110(4):941‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). https://www.ich.org/pressrelease/press‐release‐ich‐assembly‐hybrid‐meeting‐athens‐may‐2022. Accessed August 7, 2022.
- 31. U.S. Food and Drug Administration: Enhancing the Diversity of Clinical Trial Populations ‐ Eligibility Criteria, Enrollment Practices, and Trial Designs Guidance for Industry. 2020; https://www.fda.gov/media/127712/download. Accessed August 7, 2022.
- 32. U.S. Food and Drug Administration: PDUFA Reauthorization Performance Goals and Procedures Fiscal Yearrs 2023 through 2027. https://www.fda.gov/media/151712/download. Accessed August 7, 2022.
- 33. U.S. Food and Drug Administration: CDER Patient‐Focused Drug Development. https://www.fda.gov/drugs/development‐approval‐process‐drugs/cder‐patient‐focused‐drug‐development. Accessed August 7, 2022.
- 34. U.S. Food and Drug Administration: Patient‐Focused Drug Development: Selecting, Developing, or Modifying Fit‐for Purpose Clinical Outcome Assessments Guidance https://www.fda.gov/media/159500/download. Accessed August 7, 2022.
- 35. Hoos A, Anderson J, Boutin M, et al. Partnering with patients in the development and lifecycle of medicines: a call for action. Ther Innov Regul Sci. 2015;49(6):929‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andrade EL, Bento AF, Cavalli J, et al. Non‐clinical studies in the process of new drug development ‐ Part II: good laboratory practice, metabolism, pharmacokinetics, safety and dose translation to clinical studies. Braz J Med Biol Res. 2016;49(12):e5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. U.S. Food and Drug Administration: E8 (R1) General Considerations for Clinical Studies Guidance for Industry.https://www.fda.gov/media/157560/download. Accessed August 7, 2022.
- 38. U.S. Food and Drug Administration: S5(R3) Detection of Reproductive and Developmental Toxicity for Human Pharmaceuticals Guidance for Industry. https://www.fda.gov/media/148475/download. Accessed August 7, 2022.
- 39. U.S. Food and Drug Administration: S11 NonClinical Safety Testing in Support of Development of Pediatric Pharmaceuticals Guidance for Industry. https://www.fda.gov/media/148478/download. Accessed August 7, 2022.
- 40. U.S. Food and Drug Administration: M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. https://www.fda.gov/media/71542/download. Accessed August 7, 2022. [PubMed]
- 41. Gingrich J, Filipovic D, Conolly R, Bhattacharya S, Veiga‐Lopez A. Pregnancy‐specific physiologically‐based toxicokinetic models for bisphenol A and bisphenol S. Environ Int. 2021;147:106301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emond C, Raymer JH, Studabaker WB, Garner CE, Birnbaum LS. A physiologically based pharmacokinetic model for developmental exposure to BDE‐47 in rats. Toxicol Appl Pharmacol. 2010;242(3):290‐298. [DOI] [PubMed] [Google Scholar]
- 43. U.S. Food and Drug Administration: Code of Federal Regulations ‐ 21 CFR 312.21. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=312.21. Accessed August 7, 2022.
- 44. U.S. Food and Drug Administration: Food and Drug Administration Safety and Innovation Act (FDASIA). https://www.fda.gov/drugs/development‐resources/reviews‐pediatric‐studies‐conducted‐under‐bpca‐and‐prea‐2012‐present. Accessed August 7, 2022.
- 45. U.S Food and Drug Administration: Pharmacokinetics in Pregnancy ‐ Study Design, Data Analysis, and Impact on Dosing and Labeling Guidance. https://www.fda.gov/media/71353/download#:~:text=PK%20studies%20in%20pregnant%20women,regulations%20(21%20CFR%20312.2). Accessed August 7, 2022.
- 46. U.S. Food and Drug Administration: Adaptive Designs for Clinical Trials of Drugs and Biologics Guidance for Industry. 2019; https://www.fda.gov/media/78495/download. Accessed August 7, 2022.
- 47. U.S. Food and Drug Administration: Diversity Plans to Improve Enrollment of Participants from Underrepresented Racial and Ethnic Populations in Clinical Trials Guidance for Industry. 2022; https://www.fda.gov/media/157635/download. Accessed August 7, 2022.
- 48. Liu XI, Green DJ, van den Anker JN, et al. Mechanistic modeling of placental drug transfer in humans: how do differences in maternal/fetal fraction of unbound drug and placental influx/efflux transfer rates affect fetal pharmacokinetics? Front Pediatr. 2021;9:723006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dallmann A, Pfister M, van den Anker J, Eissing T. Physiologically based pharmacokinetic modeling in pregnancy: a systematic review of published models. Clin Pharmacol Ther. 2018;104(6):1110‐1124. [DOI] [PubMed] [Google Scholar]
- 50. CERTARA: Dosing for Two: How Pharmacometrics Supports Drug Safety in Pregnancy. https://www.certara.com/blog/dosing‐for‐two‐how‐pharmacometrics‐supports‐drug‐safety‐in‐pregnancy/. Accessed August 7, 2022.
- 51. National Institute of Health . US National Library of Medicine. https://clinicaltrials.gov/. Accessed August 25, 2022.
- 52. Roset Bahmanyar E, Out HJ, van Duin M. Women and babies are dying from inertia: a collaborative framework for obstetrical drug development is urgently needed. Am J Obstet Gynecol. 2021;225(1):43‐50. [DOI] [PubMed] [Google Scholar]
- 53. McKiever M, Frey H, Costantine MM. Challenges in conducting clinical research studies in pregnant women. J Pharmacokinet Pharmacodyn. 2020;47(4):287‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Johansson M, Hermeren G, Sahlin NE. Paternal consent in prenatal research: ethical aspects. Med Health Care Philos. 2020;23(2):325‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moore TJ, Zhang H, Anderson G, Alexander GC. Estimated costs of pivotal trials for novel therapeutic agents approved by the US Food and Drug Administration, 2015–2016. JAMA Intern Med. 2018;178(11):1451‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. National Archives: Federal Register ‐ Best Pharmaceuticals for Children Act (BPCA) Priority List of Needs in Pediatric Therapeutics. https://www.federalregister.gov/documents/2019/04/23/2019‐08167/best‐pharmaceuticals‐for‐children‐act‐bpca‐priority‐list‐of‐needs‐in‐pediatric‐therapeutics. Accessed August 7, 2022.
- 57. Eke AC, Olagunju A, Momper J, et al. Optimizing pharmacology studies in pregnant and lactating women using lessons from HIV: a consensus statement. Clin Pharmacol Ther. 2021;110(1):36‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hill S, Yang A, Bero L. Priority medicines for maternal and child health: a global survey of national essential medicines lists. PloS one. 2012;7(5):e38055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Innovative Medicines Initiative: ConcePTION. https://www.imi‐conception.eu/. Accessed August 7, 2022.
- 60. World Health Organization(WHO)/International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT): Research for Informed Choices ‐ Accelerating the Study of New Drugs for HIV in Pregnant and Breastfeeding Women. https://www.impaactnetwork.org/sites/default/files/inline‐files/PW_Call_to_Action_final.pdf. Accessed August 7, 2022.