

Abstract
Background
Cellular cholesterol efflux is a key step in reverse cholesterol transport that may impact on atherosclerotic cardiovascular risk. The process may be reliant on the availability of apolipoprotein (apo) B‐100‐containing lipoproteins to accept cholesterol from high‐density lipoprotein. Evolocumab and atorvastatin are known to lower plasma apoB‐100‐containing lipoproteins that could impact on cholesterol efflux capacity (CEC).
Methods
We conducted a 2‐by‐2 factorial trial of the effects of subcutaneous evolocumab (420 mg every 2 weeks) and atorvastatin (80 mg daily) for 8 weeks on CEC in 81 healthy, normolipidaemic men. The capacity of whole plasma and apoB‐depleted plasma, including ATP‐binding cassette transporter A1 (ABCA1)‐mediated and passive diffusion, to efflux cholesterol, was measured.
Results
Evolocumab and atorvastatin independently decreased whole plasma CEC (main effect p < .01 for both). However, there were no significant effects of evolocumab and atorvastatin on apoB‐depleted plasma, ABCA1‐mediated and passive diffusion‐mediated CEC (p > .05 in all). In the three intervention groups combined, the reduction in whole plasma CEC was significantly correlated with the corresponding reduction in plasma apoB‐100 concentration (r = .339, p < .01). In the evolocumab monotherapy group, the reduction in whole plasma CEC was also significantly correlated with the corresponding reduction in plasma lipoprotein(a) concentration (r = .487, p < .05).
Conclusions
In normolipidaemic men, evolocumab and atorvastatin decrease the capacity of whole plasma to efflux cellular cholesterol. These effects may be chiefly owing to a fall in the availability of apoB‐100‐containing lipoproteins. Reduction in circulating lipoprotein(a) may also contribute to the decrease in whole plasma cholesterol efflux with evolocumab monotherapy.
Keywords: cholesterol efflux, cholesterol‐lowering therapies, PCSK9, statin
1. INTRODUCTION
Cholesterol efflux from macrophages, the first step of reverse cholesterol transport (RCT), plays a major role in anti‐atherogenesis. 1 , 2 , 3 Recent observational studies suggest that the capacity of plasma to affect cholesterol efflux is inversely related to atherosclerotic cardiovascular disease (ASCVD), independent of traditional cardiovascular risk factors, including the levels of low‐density lipoprotein (LDL)‐cholesterol and high‐density lipoprotein (HDL)‐cholesterol. 4 , 5
Cholesterol efflux capacity (CEC) is a measure of plasma acceptors to accept cholesterol released from cells through different receptor‐mediated pathways, such as ATP‐binding cassette transporter A1 (ABCA1), ABCG1 and scavenger receptor class B type I (SR‐BI), 6 , 7 , 8 and unspecific passive diffusion. 9 HDL plays a central role in RCT through interaction with cellular receptors, lipid transfer proteins, lipases and apolipoprotein (apo) B‐100‐containing lipoproteins. Such an integrated system enables cholesterol efflux from peripheral cells, particularly macrophages and foam cells, and transport of cholesterol back to the liver, so‐called RCT. 3 RCT is the best‐recognised mechanism by which HDL protects against atherogenesis. 3 However, the precise mechanisms or factors governing cholesterol efflux is complex but may be affected by the latter steps in RCT. These include the availability of apoB‐100‐containing lipoproteins to accept cholesteryl esters from HDL particles, which allows the plasma system to maintain the capacity of HDL to take up free cholesterol in HDL particles constant. 10 We have shown in men with a wide range of body mass index (BMI) that cellular cholesterol efflux was positively associated with the plasma level of apoB‐100 and inversely with plasma HDL‐cholesterol concentration and the clearance of apoB‐100‐containing lipoproteins. 11
Proprotein convertase subtilisin/kexin type 9 (PCSK9), a secretory protease expressed chiefly in the liver, is a key regulator of apoB‐100‐containing lipoprotein metabolism because of its ability to enhance the degradation of the LDL receptor. 12 , 13 PCSK9 inhibition with monoclonal antibodies (mAbs), such as evolocumab and alirocumab, has been consistently shown to profoundly lower plasma concentrations of apoB‐100‐containing lipoproteins, including very‐low‐density lipoprotein (VLDL), intermediate‐density lipoprotein (IDL), LDL and lipoprotein(a) [Lp(a)], with a modest significant increase in HDL‐cholesterol but no significant effect on apoA‐I concentrations. 14 , 15 Statins inhibit 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase, which stimulates LDL receptor activity for hepatic clearance of apoB‐100‐containing lipoproteins, thereby leading to markedly lower concentrations of plasma apoB‐100 and LDL‐cholesterol but do not have a significant impact on HDL‐cholesterol concentration. 14 Recent endpoint trials have demonstrated an improvement in clinical outcomes with PCSK9mAb in high‐risk patients against background statin therapy. 16 , 17 However, patients are still at high residual risk even after a substantial reduction in plasma LDL‐cholesterol concentration with statins and PCSK9 inhibitor treatment. 18 , 19 This suggests that these agents may not be sufficient to correct, or in contrast, to worsen other metabolic cardiovascular risk factors. Given that most cholesterol esters (>70%) are removed from plasma through the catabolism of apoB‐100‐containing lipoproteins, 20 we speculate that the reduction in apoB‐containing lipoprotein with PCSK9mAb and statin may influence the capacity of plasma to affect cholesterol efflux from macrophages, measured as CEC. Since these agents only have modest effects on HDL metabolism, 14 we considered that their predominant effect on CEC would be mediated by changes in apoB‐100‐containing lipoproteins.
We previously reported in a factorial trial that evolocumab and atorvastatin decreased plasma concentration of apoB‐100 in VLDL, IDL and LDL. 14 Evolocumab alone or in combination with atorvastatin also lowered plasma Lp(a) concentration. 21 The primary aim of the present study was to investigate the effects of evolocumab and atorvastatin on ex vivo CEC. The second aim was to explore whether the reduction in plasma apoB‐100‐containing lipoprotein concentration with evolocumab and/or atorvastatin was associated with the corresponding changes in cellular cholesterol efflux ex vivo.
2. METHOD
2.1. Subjects and study design
The subjects reported were derived from a substudy of a randomised, double‐blind, placebo‐controlled, intervention trial employing a two‐by‐two factorial design that examined the effect of evolocumab and atorvastatin on apoB metabolism. 14 Full details of study subjects and protocols were published previously. 14 Reporting of the study conforms to broad EQUATOR guidelines. 22 Briefly, we studied healthy, normolipidaemic men aged 18–65 years with fasting plasma LDL‐cholesterol of ≥2.5 and <4.9 mmol/L and triglycerides of <1.7 mmol/L, and BMI of 18–32 kg/m2. None had diabetes; familial hypercholesterolaemia; hypertension; or cardiovascular, renal, hepatic, thyroid, musculoskeletal, psychiatric or other medical disorders; abnormal liver or muscle enzymes; alcohol or substance abuse; nor were taking medications affecting lipid metabolism. All were consuming isocaloric diets and took light‐to‐moderate exercise. The study was approved by a national ethics committee (Bellberry Ltd, Eastwood, South Australia); all subjects provided informed consent.
Eligible subjects were randomised (1:1:1:1) to one of the four treatment groups for 8 weeks—placebo subcutaneous (SC) every two weeks (Q2W) and oral placebo once a day (QD; placebo); placebo SC Q2W and oral atorvastatin 80 mg QD (atorvastatin); SC evolocumab 420 mg Q2W and oral placebo QD (evolocumab); or SC evolocumab 420 mg Q2W and oral atorvastatin 80 mg QD (evolocumab/atorvastatin). Study visits were every 2 weeks for evolocumab dosing, treatment adherence assessment, laboratory testing and safety measurements. Fasting venous blood was collected at baseline and week 8 for laboratory measurements of biochemical analyses and CEC.
2.2. Isolation of apoB‐depleted plasma samples for cholesterol efflux
ApoB‐depleted plasma was obtained after precipitation of apoB‐containing lipoproteins using the polyethylene glycol (PEG) method as previously described. 23 Briefly, plasma samples were treated with PEG solution to precipitate apoB‐containing lipoproteins. The precipitate was removed by high‐speed centrifugation (10,000 rpm, 30 min, 4°C) to obtain the PEG supernatant containing the HDL lipoprotein fraction.
2.3. Measurement of cholesterol efflux capacity
Efflux studies were performed as previously described using J774 macrophages. 23 , 24 Whole plasma, apoB‐depleted plasma, ABCA1‐mediated and passive diffusion‐mediated CEC were determined through the use of specific cell condition models. Briefly, cells were labelled with [1,2‐3H] cholesterol in the presence of an ACAT inhibitor (2 µg/ml, Sandoz 58035; Sigma‐Aldrich, Milano, Italy). J774 cells were treated with 0.3 mM cAMP analogue (cpt‐AMP; Sigma‐Aldrich, Milan, Italy) in 0.2% BSA for 18 h to upregulate ABCA1. The efflux medium was prepared using 2% (v/v) whole plasma (or 2.8% apoB‐depleted plasma) and incubated with cells for 4–6 h. Whole plasma (or apoB‐depleted plasma) CEC was expressed as a percentage of radiolabelled cholesterol released to the medium over the total radioactivity incorporated by cells. Using apoB‐depleted plasma, the ABCA1‐mediated CEC was calculated as the difference between the percentage efflux obtained in cAMP treated cell (i.e. apoB‐depleted plasma CEC) and that obtained in cells not treated with cAPM (i.e. passive diffusion‐mediated CEC). To minimise the intra‐assay variability, every serum sample was run in triplicate and for each, the percentage of efflux was obtained and the average and standard deviation were calculated. A standard pool of human serum from our laboratory (SN1) permitted correction for inter‐assay variability and a second serum standard pool (SN2) was used to determine inter‐assay variability. 25
2.4. Quantification of apoB other analytes
Full details for laboratory methods, including quantification of very‐low‐density (VLDL)‐apoB, intermediate‐density lipoprotein (IDL)‐apoB and LDL‐apoB have been published elsewhere. 14 , 21 Briefly, apoB in the VLDL, IDL and LDL fractions were separated by sequential ultracentrifugation and isolated using the isopropanol method. A modified Lowry method was used to determine the apoB concentration in each fraction. Plasma Lp(a) concentration was measured as particle number in nmol/L by immunoturbidimetry (Denka Seiken Co Ltd, Lp(a) assay, Polymedco). This immunoturbidimetry method has been demonstrated to be insensitive to apo(a) isoform size heterogeneity. 26 All routine lipid and lipoprotein analyses were assayed in serum samples by Medpace Reference Laboratories according to the Centers for Disease Control and Prevention Lipid Standardization Program. Plasma apoE (R&D Systems), lipoprotein lipase (LPL) (Cusabio) and cholesteryl ester transfer protein (CETP) (Cell Biolabs, Inc) concentrations were determined using enzyme‐linked immunoassay. Free PCSK9 concentration was assayed by a quantitative ELISA method (PPD Bioanalytical Lab, Richmond, VA, USA).
2.5. Statistical analyses
Data were analysed using SPSS 26 software (SPSS, Chicago, USA). Data were presented as geometric mean (95% confident interval) unless otherwise indicated. The Shapiro‐Wilk test was used to determine whether variables were normally distributed. Main effects of treatment (i.e. isolated effect of evolocumab treatment irrespective of the effect of atorvastatin treatment) and interactive effects of treatment (i.e. effect of the combination of each treatment) were assessed by maximum‐likelihood random‐effects regression models. The models contained 3‐way interactions of time, evolocumab and atorvastatin. If the 3‐way interaction of evolocumab, atorvastatin and time was not statistically significant, then only the main effects (time‐evolocumab and time‐atorvastatin) were included in the model. 14 Changes in variables with interventions relative to baseline were described as percentages. Associations were examined by Pearson's correlational analyses. Statistical significance was defined at the 5% level using a two‐tailed test.
3. RESULTS
3.1. Baseline clinical and biochemical characteristics
Of 245 subjects screened, 81 completed the study and were randomly assigned to either placebo (n = 20), atorvastatin (n = 22), evolocumab (n = 20) or evolocumab/atorvastatin (n = 19) (see CONSORT flow diagram; Figure S1). Full details of the subject clinical and biochemical characteristics were summarised previously. 14 As shown in Table 1, the 81 eligible subjects were on average 31 years old, nonobese, normotensive, nondiabetic and had overall normal plasma lipid and lipoprotein profiles. There were no significant group differences in any of the variables in Table 1. Adherence to randomised treatments was 100%. The spectrum of an adverse event after treatments was as reported previously. 14
TABLE 1.
Clinical and biochemical characteristics of the 81 subjects enrolled at baseline
| Placebo (n = 20) | Atorvastatin (n = 22) | Evolocumab (n = 20) | Evolocumab/Atorvastatin (n = 19) | |
|---|---|---|---|---|
| Age (years) | 32.9 (27.8–39.1) | 29.7 (26.6–33.1) | 32.2 (27.2–38.1) | 30.1 (26.1–34.6) |
| Body mass index (kg/m2) | 24.6 (23.2–26.0) | 24.9 (23.7–26.2) | 24.6 (23.7–25.6) | 25.6 (24.2–27.2) |
| SBP (mmHg) | 127 (124–130) | 123 (119–127) | 124 (120–129) | 124 (118–130) |
| DBP (mmHg) | 78.3 (75.0–81.7) | 76.5 (72.7–80.6) | 74.2 (67.8–81.2) | 76.3 (72.3–80.6) |
| Glucose (mmol/L) | 5.23 (5.06–5.41) | 5.31 (5.16–5.47) | 5.35 (5.11–5.60) | 5.39 (5.24–5.55) |
| Total cholesterol (mmol/L) | 4.57 (4.40–4.76) | 4.69 (4.43–4.97) | 4.52 (4.23–4.83) | 4.60 (4.21–5.02) |
| Triglycerides (mmol/L) | 0.82 (0.75–0.90) | 0.90 (0.79–1.04) | 0.80 (0.67–0.96) | 0.94 (0.77–1.15) |
| HDL‐cholesterol (mmol/L) | 1.17 (1.06–1.30) | 1.22 (1.12–1.33) | 1.11 (0.98–1.25) | 1.14 (1.01–1.28) |
| LDL‐cholesterol (mmol/L) | 3.05 (2.89–3.22) | 3.09 (2.89–3.31) | 3.04 (2.84–3.26) | 2.99 (2.71–3.29) |
| ApoB, g/L | 0.84 (0.79–0.89) | 0.84 (0.79–0.89) | 0.84 (0.79–0.90) | 0.85 (0.78–0.94) |
| VLDL‐apoB, mg/L | 41.7 (34.3–50.7) | 47.0 (37.8–58.5) | 46.9 (34.8–63.2) | 53.0 (40.3–69.5) |
| IDL‐apoB, mg/L | 32.7 (28.4–37.7) | 33.2 (28.5–38.9) | 36.5 (29.2–45.7) | 40.9 (33.5–50.0) |
| LDL‐apoB, mg/L | 465 (411–527) | 431 (378–492) | 447 (369–541) | 456 (391–533) |
| Lipoprotein (a), nmol/L | 23.2 (12.0–45.1) | 11.7 (7.33–18.7) | 19.9 (10.3–38.3) | 20.9 (10.9–40.0) |
Values expressed as geometric mean (95% CI).
Abbreviations: Apo, apolipoprotein; CI, confidence interval; DBP, diastolic blood pressure; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; SBP, systolic blood pressure.
3.2. Treatment effect on fasting plasma lipid, lipoprotein and apolipoprotein
Treatment effects on fasting plasma lipid, lipoprotein and apolipoprotein concentration in 81 subjects were reported previously. 14 Briefly, both evolocumab and atorvastatin independently decreased fasting plasma concentration of total cholesterol, LDL‐cholesterol, total apoB and LDL‐apoB (p < .001 for all); the reduction being significantly greater with combination therapy compared with monotherapy (p < .001 for all). There were significant main effects of evolocumab in raising HDL‐cholesterol (+9%, p < .001) and lowering VLDL‐apoB (−31%, p = .001), IDL‐apoB (−30%, p = .001), apoE (−27%, p < .001) and Lp(a) (−25%, p = .002), and of atorvastatin in lowering triglycerides (−23%, p = .001), VLDL‐apoB (−30%, p = .001), IDL‐apoB (−31%, p = .001) and apoE (−28%, p < .001) concentrations (Table S1). There were also significant effects (p < .001) of atorvastatin in increasing (+41%) and of evolocumab in decreasing (−98%) plasma‐free PCSK9 levels. There were no significant main effects on apoA‐I, LPL or CETP mass concentrations with evolocumab or atorvastatin.
3.3. Treatment effect on cholesterol efflux capacity
Table 2 shows the treatment effect on whole plasma, apoB‐depleted, ABCA1‐mediated and passive diffusion‐mediated CEC in the subjects. As seen, all of the four indices of CEC at baseline were not significantly different among the four groups (ANOVA p > .05 for all). There were also no significant interactions between evolocumab and atorvastatin for any of the variables shown in Table 2. Relative to the nontreatment group, evolocumab and atorvastatin independently decreased whole plasma CEC by −12% and −13% (main effect p < .01 in both, Figure 1). However, there were no significant main effects of evolocumab or atorvastatin on apoB‐depleted CEC, ABCA1‐mediated CEC and passive diffusion‐mediated CEC (p > .05 in all).
TABLE 2.
Effect of the interventions on whole plasma CEC, apoB‐depleted CEC, ABCA1‐mediated CEC and passive diffusion
| Placebo | ATV | EVO | ATV + EVO | Main effect | Interaction | ||
|---|---|---|---|---|---|---|---|
| (n = 20) | (n = 22) | (n = 20) | (n = 19) | p‐value | p‐value | ||
| ATV | EVO | ATV vs EVO | |||||
| Whole plasma CEC (%) | |||||||
| Baseline | 18.1 (16.5–19.9) | 20.9 (19.0–23.0) | 19.5 (17.3–22.0) | 19.2 (16.9–21.7) | .003 | .001 | .67 |
| Week 8 | 17.2 (15.6–19.1) | 16.3 (14.5–18.3) | 15.4 (13.6–17.4) | 13.5 (12.1–15.2) | |||
| ApoB‐depleted CEC (%) | |||||||
| Baseline | 13.6 (12.8–14.4) | 13.9 (13.0–14.8) | 14.1 (12.7–15.7) | 13.6 (12.5–14.9) | .486 | .133 | .146 |
| Week 8 | 15.0 (14.2–15.8) | 15.3 (14.3–16.4) | 15.4 (14.2–16.6) | 14.3 (13.3–15.4) | |||
| ABCA1‐mediated CEC (%) | |||||||
| Baseline | 2.5 (1.8–3.6) | 2.9 (2.3–3.8) | 2.6 (1.8–3.6) | 2.2 (1.6–3.0) | .594 | .689 | .732 |
| Week 8 | 4.0 (3.4−4.7) | 4.5 (3.8–5.3) | 4.0 (3.4–4.7) | 3.7 (2.8–4.8) | |||
| Passive diffusion (%) | |||||||
| Baseline | 10.6 (10.0–11.2) | 10.6 (10.0–11.2) | 11.1 (10.0–12.3) | 11.1 (10.4–11.8) | .180 | .295 | .231 |
| Week 8 | 10.7 (10.0–11.4) | 10.7 (10.1–11.3) | 11.0 (10.4–11.5) | 10.2 (9.8–10.7) | |||
Data presented as geometric mean (95% CI). Bold values denote statistically significance of main effect of treatments compared with the placebo group using maximum‐likelihood random‐effects regression models.
Abbreviations: ABC, ATP‐binding cassette transporter; Apo, apolipoprotein; ATV, atorvastatin; CEC, cholesterol efflux capacity; EVO, evolocumab.
FIGURE 1.
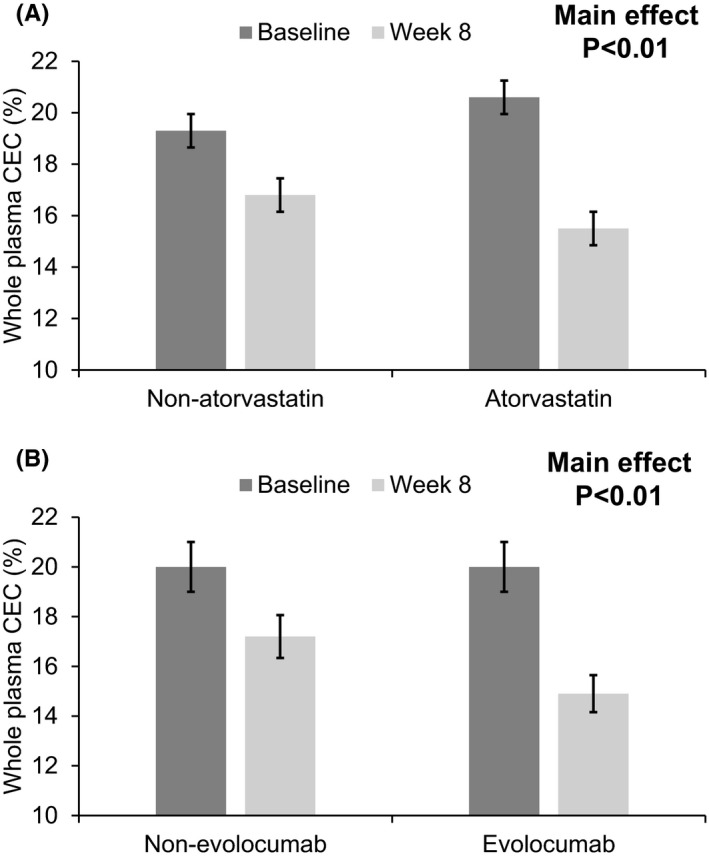
Main effects of atorvastatin (A) and evolocumab (B) on whole plasma cholesterol efflux capacity (CEC)
At baseline, the capacity of whole plasma to effect cholesterol efflux ex vivo was significantly and positively correlated with plasma apoB (r = .240; p = .031), but not with VLDL‐apoB (r = −.034; p = .762), IDL‐apoB (r = −.062; p = .584), LDL‐apoB (r = −.062; p = .465) and Lp(a) concentration (r = .047; p = .680). Plasma HDL‐cholesterol concentrations were also not significantly associated (p > .05 for all) with whole plasma (r = .042), apoB‐depleted plasma (r = .004), ABCA1‐mediated (r = −.114) and passive diffusion‐mediated CEC (r = .107).
In the three intervention groups combined, the percentage reduction in whole plasma CEC was significantly correlated with the corresponding reduction in plasma LDL‐cholesterol (r = .359, p < .05; Figure 2A), LDL‐apoB concentrations (r = .264, p < .05; Figure 2B) and apoB (r = .339, p < .01; Figure 2C). However, there was no significant association (p > .05 for all) between the changes in whole plasma CEC and plasma concentrations of VLDL‐apoB (r = .008), IDL‐apoB (r = .126) and Lp(a) concentration (r = −.041). Using data from our previous report, 14 the reduction in whole plasma CEC was also significantly correlated with the corresponding increase in the fractional catabolic rate (FCR) of LDL‐apoB‐100 (r = −.327, p < .05).
FIGURE 2.
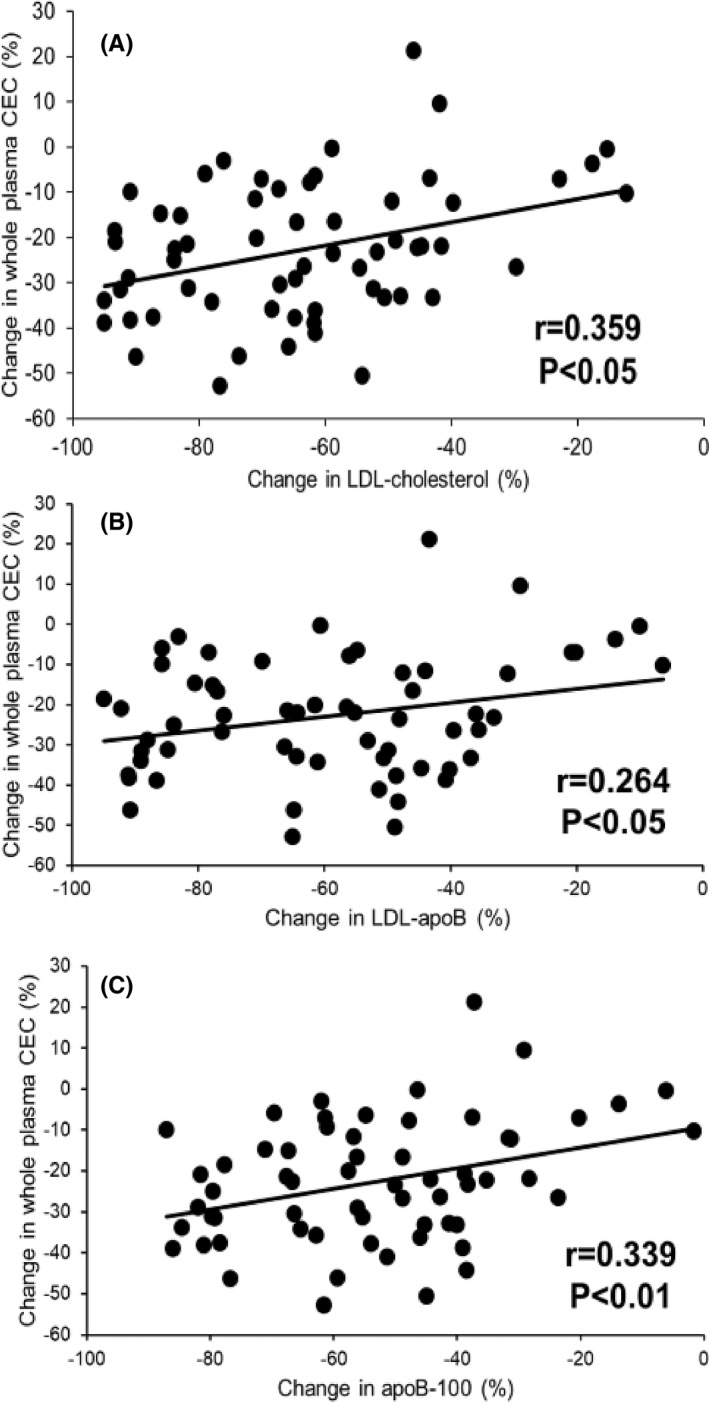
Correlation between change in whole plasma cholesterol efflux capacity (CEC) and change in plasma concentrations of LDL‐cholesterol (A), LDL‐apoB‐100 (B) and apoB‐100 (C) in three intervention groups
In the evolocumab monotherapy group, the reduction in whole plasma CEC was significantly correlated with the corresponding reduction in plasma Lp(a) concentration (r = .487, p < .05; Figure 3); this association remained significant (p < .05 for all) after adjusting for the corresponding changes in plasma LDL‐cholesterol (r = .508), LDL‐apoB (r = .540) or apoB (r = .534). However, the decrease in whole plasma CEC was not significantly correlated with the corresponding reduction in plasma Lp(a) concentration in patients with combination treatment.
FIGURE 3.
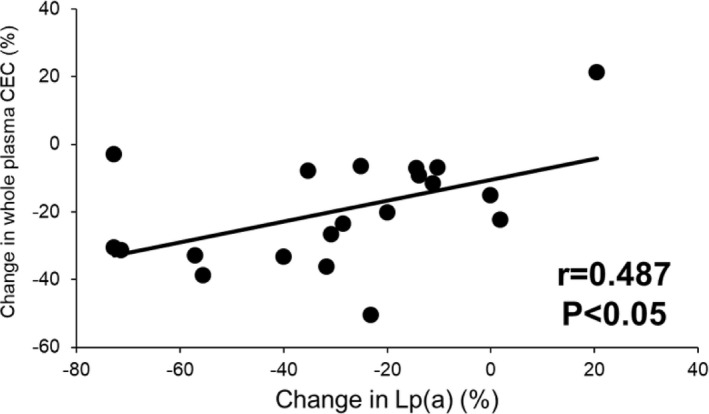
Correlation between change in whole plasma cholesterol efflux capacity (CEC) and change in Lp(a) concentration in subjects receiving evolocumab monotherapy
4. DISCUSSION
Our principal finding was that evolocumab and atorvastatin lowered the capacity of whole plasma to affect cholesterol efflux ex vivo in healthy normolipidaemic subjects. The reduction in whole plasma cholesterol capacity may be mediated by their effects in lowering plasma apoB‐100‐containing lipoprotein concentrations, including LDL and/or Lp(a) particles. However, evolocumab and atorvastatin did not show a significant effect on cholesterol efflux to apoB‐depleted plasma mediated by both ABCA1 and passive diffusion pathways.
4.1. Previous cellular cholesterol efflux studies with statins and PCSK9 inhibitors
The effects of statins on cellular cholesterol efflux have been previously examined, but with divergent results showing increased, decreased or no change in the capacity of plasma to efflux cholesterol ex vivo. 27 , 28 , 29 , 30 , 31 , 32 , 33 We have previously reported that rosuvastatin treatment resulted in a significant reduction of whole plasma CEC, but not apoB‐depleted plasma CEC in overweight subjects. 29 The discrepant findings of the aforementioned studies might be accounted for by differences in study design (controlled vs uncontrolled observations), the methods to measure cellular cholesterol efflux (whole plasma vs apoB‐depleted plasma), the type and dose of statin employed (atorvastatin vs rosuvastatin). Only one study has reported on the effect of PCSK9 monoclonal antibodies on CEC. 34 Lappegård et al found that treatment with evolocumab had no effect on whole plasma CEC in 3 patients with heterozygous familial hypercholesterolemia. 34 However, the results needed to be interpreted with caution because of the very small sample size. We have extended previous studies by investigating the comparative effect of evolocumab and atorvastatin on cellular cholesterol efflux capacity under normal physiological conditions.
4.2. Effect of atorvastatin on cholesterol efflux capacity
Inhibition of de novo cholesterol synthesis by statins is well recognised to upregulate LDL receptor activity, thereby increasing hepatic removal of apoB‐100‐containing lipoproteins. 35 , 36 Statins are also known to increase transcription of micro‐RNA33 and to decrease transcription of liver X receptor, leading to down‐regulation of the expression of ABCA1 and ABCG1 for cholesterol efflux. 37 We confirmed this in an earlier report by showing that atorvastatin treatment decreased the plasma concentrations of VLDL, IDL and LDL in these patients. 14 We have also previously demonstrated that the capacity of whole plasma to affect cholesterol efflux ex vivo was significantly and positively correlated with plasma apoB‐100 concentration. 11 This supports the notion that the availability of apoB‐100‐containing lipoproteins plays a key role in the latter steps in RCT to accept cholesterol cholesteryl esters from HDL particles and maintain the capacity of HDL to pass cholesterol it acquired from macrophages to cholesterol pools within apoB‐100‐containing lipoproteins. 10 In the present study, the lowering effects of apoB‐100‐containing lipoprotein particles with atorvastatin would be expected to reduce the availability of these lipoproteins to accept cholesterol cholesteryl esters from HDL particles. Accordingly, we found that atorvastatin significantly and independently decreased whole plasma CEC, but not apoB‐depleted plasma CEC (i.e. both ABCA1 and passive diffusion‐mediated pathways). In the pooled analysis from three intervention groups, we also observed that the reduction in whole plasma CEC was significantly associated with the corresponding reductions in plasma LDL‐cholesterol, LDL‐apoB and total apoB concentrations. These observations were further supported by the significant association between the reduction in whole plasma CEC and the corresponding increase in the FCR of LDL‐apoB‐100. The FCR of LDL‐apoB‐100 determines the plasma pool size of apoB in the circulation. It was therefore not unexpected that the reduction in total apoB concentration and the corresponding changes in whole plasma CEC was inversely correlated with the increase in the FCR of LDL‐apoB‐100. Taken together, this finding reinforces the potentially greater role of the total availability of apoB particles in influencing cholesterol efflux in RCT.
4.3. Effect of evolocumab on cholesterol efflux capacity
The role of PCSK9 inhibition in the upregulation of LDLR activity is also well recognised. 14 , 15 Like atorvastatin, the capacity of whole plasma to affect cholesterol efflux ex vivo would be expected to be decreased with evolocumab because this agent decreases the plasma concentration of apoB‐100‐containing lipoproteins by enhanced LDL receptor‐mediated clearance pathway. Accordingly, we found that evolocumab significantly and independently decreased whole plasma CEC, but not apoB‐depleted plasma CEC and other indices of CEC (i.e. ABCA1 and passive diffusion‐mediated CEC).
Cell‐based data also show that PCSK9 may downregulate ABCA1 protein expression and subsequent ABCA1‐mediated cholesterol efflux capacity from peripheral cells in the initial stage of RCT. 24 However, the experimental conditions from these observations do not reflect those of our study. In the present study, we found opposing effects on plasma‐free PCSK9 levels of evolocumab and atorvastatin. Given that whole plasma CEC decreased with both treatments, our data do not support a direct for PCSK9 on cellular cholesterol efflux.
Given the role of HDL in RCT as an initial acceptor of free cholesterol from peripheral cells, the increase in plasma HDL‐cholesterol concentration with evolocumab would be expected to enhance cholesterol efflux from peripheral cells to HDL particles. However, such increase in plasma HDL‐cholesterol was only modest and therefore unlikely to impact on the predominant effect on cholesterol efflux mediated by the reduction in apoB‐100‐containing lipoproteins with evolocumab. Consistent with this notion, we found that whole plasma CEC at baseline was only significantly associated with plasma apoB, but not HDL‐cholesterol. It is also possible that the capacity of HDL to store cholesterol released from cells is limited. To maintain its capacity to affect cholesterol efflux, free cholesterol from HDL needs to be transferred to larger pools of lipoproteins, such as apoB‐100‐containing VLDL and LDL particles. Hence, the association between plasma HDL‐cholesterol concentration and cholesterol efflux might have been diminished and confounded by the simultaneous presence of apoB‐100‐containing lipoproteins. Measurement of HDL kinetics may further help to clarify the precise contributory role of the HDL system in effectuating CEC.
We have previously demonstrated that treatment with evolocumab monotherapy or in combination with atorvastatin, but not atorvastatin monotherapy, resulted in a significant reduction in plasma Lp(a) concentration. 21 Like LDL particles, Lp(a) may act a role as an acceptor of cholesteryl esters from HDL particles. 10 Experimental data also suggest that Lp(a) can directly promote cholesterol efflux from HepG2 cells to HDL through upregulation of ABCA1. 38 , 39 Consistent with it, we found that the reduction in whole plasma CEC with evolocumab monotherapy was significantly associated with the corresponding reduction in plasma Lp(a) concentration (Figure 3), independent of LDL‐cholesterol, LDL‐apoB or apoB concentrations. However, the lack of a significant association between the changes in whole plasma CEC and plasma Lp(a) concentration in patients with the combined treatment (i.e. evolocumab plus atorvastatin) might seem paradoxical. The reason for this observation remains unclear. As discussed earlier, experimental data have suggested that statins can exert a direct effect on cellular cholesterol efflux by regulating the expression of ABCA1 expression in human macrophages. 40 Hence, it is possible that the association between the reduction in whole plasma CEC and plasma Lp(a) with evolocumab might have been diminished in the background setting of atorvastatin treatment. 40 , 41 However, this speculation remains to be investigated.
4.4. Study limitations
Our study has limitations. We only studied healthy, nonobese, insulin sensitive, overall normolipidaemic men. Whether our findings apply to women, patients with obesity, type 2 diabetes, dyslipidaemia or FH remains to be tested. We cannot strictly exclude the possibility that our sample size was not sufficient to demonstrate that evolocumab or atorvastatin might have had a small but significant impact on the capacity of cholesterol efflux to apoB‐depleted plasma or other indices of cholesterol efflux. The association between CEC and plasma concentrations of apoB‐containing lipoproteins [such as LDL‐apoB and Lp(a)] with the two agents were also only moderate and require further confirmation in larger sample size, particularly in patients with elevated LDL and/or Lp(a). Cholesterol efflux was measured ex vivo reflects only the earlier stage of RCT in which the second major part (delivering cholesterol to the liver) is lacking.
4.5. Clinical implications
Cholesterol efflux capacity is a measure of reverse cholesterol transport that is inversely related to the risk of ASCVD, independent of traditional classic risk factors. 2 , 4 Targeting cholesterol efflux may therefore be an important therapeutic approach to preventing and treating ASCVD. 3 , 5 Clinical benefits of statins and PCSK9 inhibitors in the prevention of ASCVD have been established by several landmark clinical trials. 16 , 17 , 42 The benefits of these agents are not only related to their effects on LDL but also involve several anti‐atherosclerotic mechanisms that could involve RCT.
Our findings suggest that the capacity of plasma to affect cholesterol efflux is reduced by both evolocumab and atorvastatin; this is in part, associated with a reduction in the acceptor capacity of apoB‐100‐containing lipoproteins, including LDL and/or Lp(a). The clinical significance of reduced cholesterol efflux observed in vitro after evolocumab and/or atorvastatin treatment is uncertain; it may appear counterintuitive given the documented ASCVD benefits of lowering elevated plasma levels of apoB‐100‐containing lipoproteins. 16 , 17 , 42 , 43 The anti‐atherogenic benefits of lowering apoB‐100‐containing lipoproteins with evolocumab and atorvastatin may override the negative impact on their effect on cholesterol efflux in RCT, reflecting a feedback mechanism in response to the reduction in vivo flux of cholesterol into the artery wall from LDL and VLDL. It is also noteworthy that the net flux of cholesterol to the liver for faecal excretion in the later stages of RCT may be enhanced due to the upregulation of LDL receptor activity by both evolocumab and atorvastatin treatments. 44 However, this speculation needs further testing with detailed HDL kinetic and cholesterol balance studies.
5. CONCLUSION
Given that the capacity of cholesterol efflux was decreased by both evolocumab and atorvastatin, further investigation should also explore the incremental effect of peroxisome proliferator‐activated receptor (PPAR) alpha agonists or selective PPAR γ modulators (SPARMs) added to a statin or PCSK9 inhibitor. 45 , 46 Such an approach may be useful for addressing the high residual risk of ASCVD demonstrated with statins and PCSK9 inhibitors in clinical outcome trials in secondary prevention. 16 , 17 , 42
CONFLICT OF INTEREST
GFW: reports funding or honoraria from Amgen, Pfizer, Esperion, Arrowhead, Regenron, Sanofi, AstraZeneca, Novartis. The remaining authors declare no conflict of interest.
AUTHOR CONTRIBUTION
QY analysed the data and wrote the manuscript. GFW and DC led and designed the study, analysed the data and edited the manuscript. JP supervised data collection and edited the manuscript. AR and EF collaborated with measuring cholesterol efflux capacity and edited the manuscript. All authors contributed to the drafting and editing of the review, and subsequent revisions.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
JP is supported by an Investigator Grant from the National Health and Medical Research Council (NHMRC). This study was funded by Amgen Inc. Open access publishing facilitated by The University of Western Australia, as part of the Wiley ‐ The University of Western Australia agreement via the Council of Australian University Librarians. [Correction added on 29 May 2022, after first online publication: CAUL funding statement has been added.]
Ying Q, Ronca A, Chan DC, Pang J, Favari E, Watts GF. Effect of a PCSK9 inhibitor and a statin on cholesterol efflux capacity: A limitation of current cholesterol‐lowering treatments? Eur J Clin Invest. 2022;52:e13766. doi: 10.1111/eci.13766
REFERENCES
- 1. Rothblat GH, de la Llera‐Moya M, Atger V, Kellner‐Weibel G, Williams DL, Phillips MC. Cell cholesterol efflux: integration of old and new observations provides new insights. J Lipid Res. 1999;40:781‐796. [PubMed] [Google Scholar]
- 2. Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905‐1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adorni MP, Ronda N, Bernini F, Zimetti F. High density lipoprotein cholesterol efflux capacity and atherosclerosis in cardiovascular disease: pathophysiological aspects and pharmacological perspectives. Cells. 2021;10(3):574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khera AV, Cuchel M, de la Llera‐Moya M, et al. Cholesterol efflux capacity, high‐density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rohatgi A, Grundy SM. Cholesterol efflux capacity as a therapeutic target: rationale and clinical implications. J Am Coll Cardiol. 2015;66:2211‐2213. [DOI] [PubMed] [Google Scholar]
- 6. Wang N, Silver DL, Thiele C, Tall AR. ATP‐binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J Biol Chem. 2001;276:23742‐23747. [DOI] [PubMed] [Google Scholar]
- 7. Kennedy MA, Barrera GC, Nakamura K, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121‐131. [DOI] [PubMed] [Google Scholar]
- 8. Jian B, de la Llera‐Moya M, Ji Y, et al. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599‐5606. [DOI] [PubMed] [Google Scholar]
- 9. Mendez AJ. Cholesterol efflux mediated by apolipoproteins is an active cellular process distinct from efflux mediated by passive diffusion. J Lipid Res. 1997;38:1807‐1821. [PubMed] [Google Scholar]
- 10. Hoang A, Drew BG, Low H, et al. Mechanism of cholesterol efflux in humans after infusion of reconstituted high‐density lipoprotein. Eur Heart J. 2012;33:657‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan DC, Hoang A, Barrett PH, et al. Apolipoprotein B‐100 and apoA‐II kinetics as determinants of cellular cholesterol efflux. J Clin Endocrinol Metab. 2012;97:E1658‐E1666. [DOI] [PubMed] [Google Scholar]
- 12. Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis‐regulated convertase 1 (NARC‐1): Liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100:928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114:1022‐1036. [DOI] [PubMed] [Google Scholar]
- 14. Watts GF, Chan DC, Dent R, et al. Factorial effects of evolocumab and atorvastatin on lipoprotein metabolism. Circulation. 2017;135:338‐351. [DOI] [PubMed] [Google Scholar]
- 15. Reyes‐Soffer G, Pavlyha M, Ngai C, et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. 2017;135:352‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713‐1722. [DOI] [PubMed] [Google Scholar]
- 17. Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097‐2107. [DOI] [PubMed] [Google Scholar]
- 18. Reith C, Armitage J. Management of residual risk after statin therapy. Atherosclerosis. 2016;245:161‐170. [DOI] [PubMed] [Google Scholar]
- 19. Tall AR, Thomas DG, Gonzalez‐Cabodevilla AG, Goldberg IJ. Addressing dyslipidemic risk beyond LDL‐cholesterol. J Clin Invest. 2022;132:e148559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594‐1607. [DOI] [PubMed] [Google Scholar]
- 21. Watts GF, Chan DC, Somaratne R, et al. Controlled study of the effect of proprotein convertase subtilisin‐kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur Heart J. 2018;39:2577‐2585. [DOI] [PubMed] [Google Scholar]
- 22. Simera I, Moher D, Hoey J, Schulz KF, Altman DG. A catalogue of reporting guidelines for health research. Eur J Clin Invest. 2010;40:35‐53. [DOI] [PubMed] [Google Scholar]
- 23. Ronda N, Favari E, Borghi MO, et al. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. 2014;73:609‐615. [DOI] [PubMed] [Google Scholar]
- 24. Adorni MP, Cipollari E, Favari E, et al. Inhibitory effect of PCSK9 on Abca1 protein expression and cholesterol efflux in macrophages. Atherosclerosis. 2017;256:1‐6. [DOI] [PubMed] [Google Scholar]
- 25. Vigna GB, Satta E, Bernini F, et al. Flow‐mediated dilation, carotid wall thickness and HDL function in subjects with hyperalphalipoproteinemia. Nutr Metab Cardiovasc Dis. 2014;24:777‐783. [DOI] [PubMed] [Google Scholar]
- 26. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57:526‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chamaria S, Johnson KW, Vengrenyuk Y, et al. Intracoronary imaging, cholesterol efflux, and transcriptomics after intensive statin treatment in diabetes. Sci Rep. 2017;7:7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kini AS, Vengrenyuk Y, Shameer K, et al. Intracoronary imaging, cholesterol efflux, and transcriptomes after intensive statin treatment: the YELLOW II study. J Am Coll Cardiol. 2017;69:628‐640. [DOI] [PubMed] [Google Scholar]
- 29. Sviridov D, Hoang A, Ooi E, Watts G, Barrett PH, Nestel P. Indices of reverse cholesterol transport in subjects with metabolic syndrome after treatment with rosuvastatin. Atherosclerosis. 2008;197:732‐739. [DOI] [PubMed] [Google Scholar]
- 30. Nicholls SJ, Ruotolo G, Brewer HB, et al. Cholesterol efflux capacity and pre‐beta‐1 HDL concentrations are increased in dyslipidemic patients treated with evacetrapib. J Am Coll Cardiol. 2015;66:2201‐2210. [DOI] [PubMed] [Google Scholar]
- 31. Muñoz‐Hernandez L, Ortiz‐Bautista RJ, Brito‐Córdova G, et al. Cholesterol efflux capacity of large, small and total HDL particles is unaltered by atorvastatin in patients with type 2 diabetes. Atherosclerosis. 2018;277:72‐79. [DOI] [PubMed] [Google Scholar]
- 32. Soran H, Liu Y, Adam S, et al. A comparison of the effects of low‐ and high‐dose atorvastatin on lipoprotein metabolism and inflammatory cytokines in type 2 diabetes: results from the protection against nephropathy in diabetes with atorvastatin (PANDA) randomized trial. J Clin Lipidol. 2018;12:44‐55. [DOI] [PubMed] [Google Scholar]
- 33. Kralova Lesna I, Adamkova V, Pagacova L. Effect of rosuvastatin treatment on cholesterol efflux from human macrophages. Neuro Endocrinol Lett. 2011;32(suppl 2):24‐28. [PubMed] [Google Scholar]
- 34. Lappegård KT, Kjellmo CA, Ljunggren S, et al. Lipoprotein apheresis affects lipoprotein particle subclasses more efficiently compared to the PCSK9 inhibitor evolocumab, a pilot study. Transfus Apher Sci. 2018;57:91‐96. [DOI] [PubMed] [Google Scholar]
- 35. Bilheimer DW, Grundy SM, Brown MS, Goldstein JL. Mevinolin and colestipol stimulate receptor‐mediated clearance of low density lipoprotein from plasma in familial hypercholesterolemia heterozygotes. Proc Natl Acad Sci U S A. 1983;80:4124‐4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grundy SM. Statins: definitive translational research. Mol Med. 2014;20(suppl 1):S20‐S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Niesor EJ, Schwartz GG, Perez A, et al. Statin‐induced decrease in ATP‐binding cassette transporter A1 expression via microRNA33 induction may counteract cholesterol efflux to high‐density lipoprotein. Cardiovasc Drugs Ther. 2015;29:7‐14. [DOI] [PubMed] [Google Scholar]
- 38. Sharma M, Von Zychlinski‐Kleffmann A, Porteous CM, Jones GT, Williams MJ, McCormick SP. Lipoprotein (a) upregulates ABCA1 in liver cells via scavenger receptor‐B1 through its oxidized phospholipids. J Lipid Res. 2015;56:1318‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yeang C, Tsimikas S. HDL‐C, ABCA1‐mediated cholesterol efflux, and lipoprotein(a): insights into a potential novel physiologic role of lipoprotein(a). J Lipid Res. 2015;56:1241‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong J, Quinn CM, Gelissen IC, Jessup W, Brown AJ. The effect of statins on ABCA1 and ABCG1 expression in human macrophages is influenced by cellular cholesterol levels and extent of differentiation. Atherosclerosis. 2008;196:180‐189. [DOI] [PubMed] [Google Scholar]
- 41. Tsimikas S, Gordts P, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41:2275‐2284. [DOI] [PubMed] [Google Scholar]
- 42. Borén J, Chapman MJ, Krauss RM, et al. Low‐density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41:2313‐2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B particles and cardiovascular disease: a narrative review. JAMA Cardiol. 2019;4:1287‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cedó L, Metso J, Santos D, et al. LDL receptor regulates the reverse transport of macrophage‐derived unesterified cholesterol via concerted action of the HDL‐LDL axis: Insight from mouse models. Circ Res. 2020;127:778‐792. [DOI] [PubMed] [Google Scholar]
- 45. Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator‐activated receptor agonists. Diabetes. 2005;54:2460‐2470. [DOI] [PubMed] [Google Scholar]
- 46. Fruchart JC, Hermans MP, Fruchart‐Najib J, Kodama T. Selective peroxisome proliferator‐activated receptor alpha modulators (SPPARMα) in the metabolic syndrome: is pemafibrate light at the end of the tunnel? Curr Atheroscler Rep. 2021;23:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material