

Abstract
Anyone involved in designing or finding molecules in the life sciences over the past few years has witnessed a dramatic change in how we now work due to the COVID-19 pandemic. Computational technologies like artificial intelligence (AI) seemed to become ubiquitous in 2020 and have been increasingly applied as scientists worked from home and were separated from the laboratory and their colleagues. This shift may be more permanent as the future of molecule design across different industries will increasingly require machine learning models for design and optimization of molecules as they become “designed by AI”. AI and machine learning has essentially become a commodity within the pharmaceutical industry. This perspective will briefly describe our personal opinions of how machine learning has evolved and is being applied to model different molecule properties that crosses industries in their utility and ultimately suggests the potential for tight integration of AI into equipment and automated experimental pipelines. It will also describe how many groups have implemented generative models covering different architectures, for de novo design of molecules. We also highlight some of the companies at the forefront of using AI to demonstrate how machine learning has impacted and influenced our work. Finally, we will peer into the future and suggest some of the areas that represent the most interesting technologies that may shape the future of molecule design, highlighting how we can help increase the efficiency of the design-make-test cycle which is currently a major focus across industries.
Keywords: Artificial intelligence, design-make-test, machine learning, molecule design, recurrent neural networks
Introduction
Like most other scientists, those involved in designing or finding molecules for commercial applications in the life sciences (including in human healthcare, animal health, agrochemicals, consumer products and beyond) have witnessed a dramatic change in how we now work due to the COVID-19 pandemic in 2020. During the pandemic, non-essential research was halted, and scientists were sent home in many countries. This led to a noticeable work divide, as those scientists that could use a computer for their research were able to work remotely, while other types of research ceased entirely. What does this tell us about how scientific research will change if this pandemic continues for years to come or if we are faced with other barriers to physical lab access? If scientists must work remotely, could they still do their lab experiments remotely? Perhaps we will see more purpose built “remote-controlled” laboratories that provide this as a service. In the chemical synthesis arena, some groups have already experimented with partial or completely autonomous synthesis [1–6], while in pharmaceutical screening this has been essentially fully automated for decades with minimal human input. Despite these different types of automation, the presence of a scientist for manual lab-work is still needed. However, if we put these elements together, we can automate the complete process and run it remotely such that the design-make-test cycle (Figure 1A, B) is fully autonomous across industries. Would this approach eventually become the norm for R&D labs? Perhaps, in the same way that we see many laboratories have automated liquid dispensers or robots today for repetitive tasks. If so, where does that leave basic research in other scientific domains which are less able to be automated or do not have the financial resources? These groups may be left behind. Automation of the design-make-test cycle in chemistry is a rapidly evolving area that could benefit from its own focused review. In addition, the resource limitations during the pandemic should also make us consider the importance of each experiment and how to do research more frugally if consumables such as pipette tips or other essential items are in short supply. We need to rethink what experiments are the most critical and how we can recycle and reuse data that already exists to ensure experiments are not repeated unnecessarily if the original resource data has yet to be utilized. There is a continually growing and already immense amount of biological data in the public domain. Some of it is readily accessible in databases such as PubChem, ChEMBL etc. [7, 8] or increasingly other repositories like FigShare, GitHub etc., while some of it resides in less accessible areas like publications which may be paywalled or on individual laboratory websites. There is also considerable data that remains inaccessible inside companies for commercial reasons. It is these domains of automation and data accessibility that we and others are interested in addressing so that we can learn from this existing data. What the pandemic also did was accelerate bringing these various aspects of research to a head at the same time to demonstrate the need for tighter integration between research areas and specialties. It has also highlighted how we look at artificial intelligence (AI) and, the area of machine learning as a fundamental technology for molecule design [9–11] which we will explore further herein.
Figure 1.
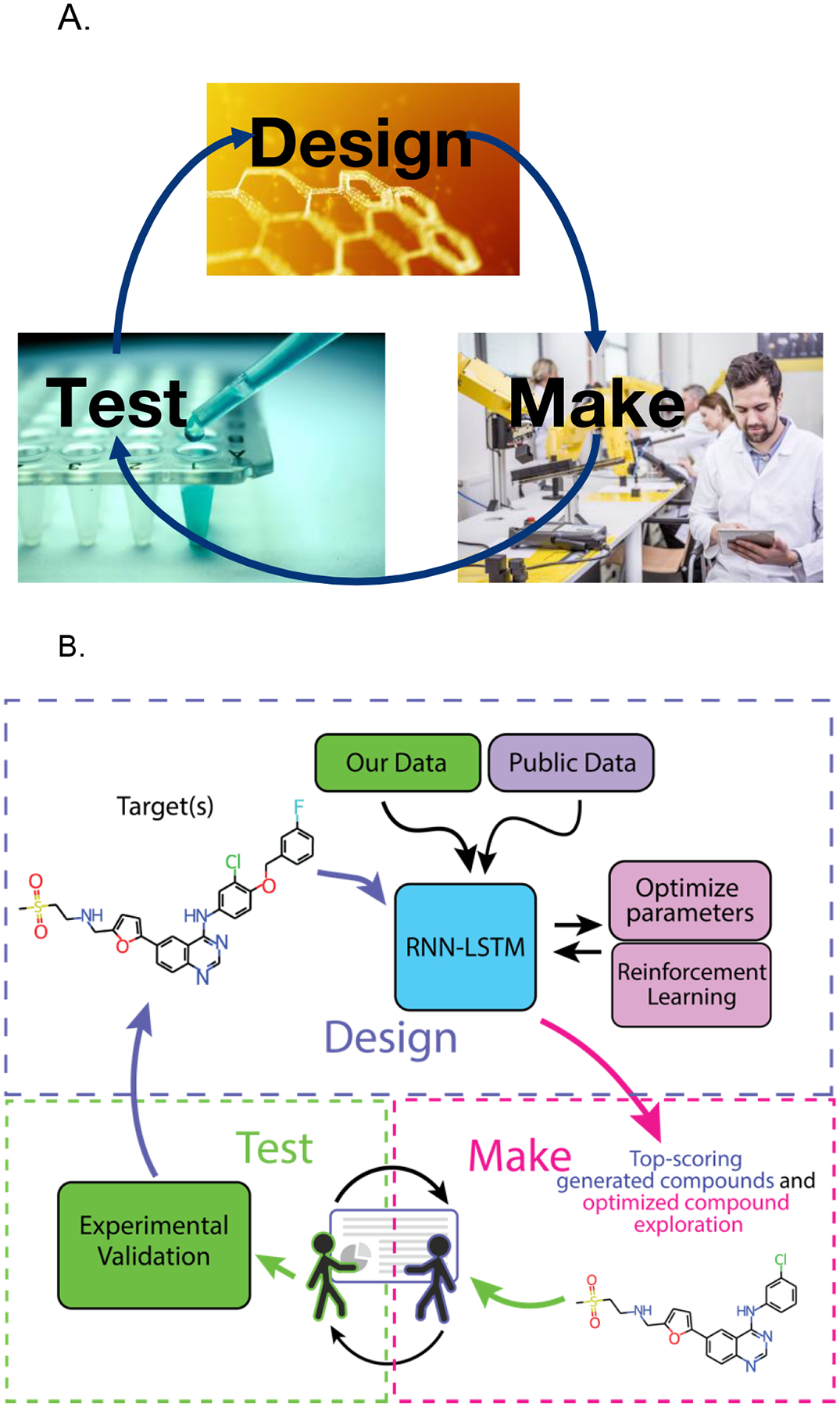
A. The design-make-test cycle. B. A hypothetical example of how a Recurrent Neural Network can be combined with the machine learning models and feedback from scientists to optimize the kinase inhibitor lapatinib.
The Next Commodity
Commodities are often considered as the basics in life which we happen to take for granted, not only food stuffs, but materials (ores), chemicals and computer memory. When they are in short supply, like the supply chain issues we are seeing during the current pandemic, they can have dramatic effects. While AI is not a new technology to the drug discovery space, in less than a decade, machine learning has been revolutionized with the addition of new architecture such as attention-base models, increased dataset availability, and improved hardware, reducing or removing barriers to machine learning applications [12–14]. In response to this, in recent years we have seen AI and specifically machine learning methods [11, 15, 16] applied in many industries to the point where we would posit it could also now be considered a scientific commodity. Like many other groups, we are interested in applying computational algorithms to drug discovery and over the last decade have noticed how AI has become ubiquitous as it has been applied to many areas of pharmaceutical research. This is by no means new as AI and machine learning or computational approaches in general have been applied in the pharmaceutical industry for many decades. Machine learning has now become a frequent topic of discussion at conferences, with an exploding number of papers describing applications of AI, even crossing over into the popular press. This has implications for the pharmaceutical industry if AI is seen as an essential component of the research and development (R&D) process in the same way that we have synthesis, in vitro and in vivo, clinical testing etc. This may also change the perception of computational approaches as having at least equal importance in the R&D process. This has also made us consider what may be the near and distant future of applying machine learning in drug discovery if it becomes important enough to be now considered a commodity.
Perhaps driving this new-found interest of AI is that over the last few years we have seen companies focusing on AI obtain very significant amounts of funding and sign massive deals with major pharmaceutical companies (Table S1). Several of these companies have used machine learning in different contexts but all have applied their software to drug discovery projects (Table 1). Obviously, not all companies publish on their technologies to the same extent which can lead to a degree of opacity as to how they use AI. It may not be necessary to raise such sums of money in order to compete with this success on an admittedly smaller scale. There has been recognition of the increasing generation and use of open source machine learning and cheminformatics software which has impacted the status quo of commercial cheminformatics software [17] and can be used as a starting point for a new generation of smaller drug discovery companies. For example, it is possible to build on such open-source software to develop machine learning tools and models to assist in drug discovery and toxicology internal projects [18–21] as well as share such technologies with academic collaborators so they can benefit from it. At the same time there are many industries such as consumer product and smaller pharmaceutical companies that do not have such cheminformatics expertise and these efforts could be a useful template for them to license or emulate. To illustrate the potential of this approach of developing and applying machine learning for drug discovery with minimal funding we use our own experiences which have used public data for projects either alone or in conjunction with additional private data (Table S2). While these examples are predominantly for drug discovery they could be extended to other industries or applications. Such models can increasingly leverage public knowledge to enable selection of compounds to test against targets for both rare, neglected and common diseases [22, 23] (Table 1, Table S2). Hence with modest funding it is feasible to perform the computational element of this work and build up wet lab capabilities to facilitate the in vitro work to validate such models. At small drug discovery companies, the pandemic shutdown demonstrated the importance of having machine learning in house and how companies in collaboration with others needed to be more agile in applying such technologies [22, 24, 25]. Machine learning has always provided a way for companies to produce and test new ideas more efficiently, which to some is still seen as evidence of hype. Yet it is widely accepted that prior paradigms such as random high-throughput screening has a success-rate (hit-rate) of 0.01–0.14% [26] and in some cases fails completely. While there are many caveats which must be taken into consideration, we can consider our own predicted vs. verified hit-rates using machine learning, were we often see increases in the success-rate by 10–100 fold, and in some cases even 1000 fold. Again, for illustration purposes, several cases from our own work suggests in vitro hit rates of: 100% (3/3, Ebola) [27], 11% (11/97 Chagas) [28], 25% (1/5, Yellow Fever) [29]. Some of these projects were also validated using in vivo testing (Chagas, 5/97 = 5.2% in vivo hit rate [28]). Combined, these examples demonstrate how companies can use machine learning technologies to create many molecule assets, and that machine learning has proven to be successfully predict molecular hits (Table 1, and Table S2) that a much larger company (not using such approaches) would have only been able to generate with many more employees and a much larger financial investment. While this point is not new, when considered with the advent of generative models, discussed below, it suggests than an accelerated early-stage drug discovery pipeline is just around the corner: Using machine learning models to guide generative models for new molecular IP, we can reasonably expect machine learning to find/generate many more molecules than have been virtually screened in the past. It is likely that while this technology has yet to replace scientists that do this drug discovery research, we would argue it has already augmented those using it with the intelligence of many more experienced scientists. In this context, applying machine learning allows these scientists to identify and generate “inventions” as well as determine which may be worthy of patenting and/or publishing with commercial applications.
Table 1.
Examples of drug discovery applications of various machine learning to targets and diseases from AI companies.
| Area of research / Disease | Target/property | Outcome | Company | References |
|---|---|---|---|---|
| Canavan disease | aspartate N-acetyltransferase | AtomNet deep neural network for structure-based drug discovery uses a model trained on bioactivity data and protein structures. They scored 10M molecules and 60 were tested in vitro with 5 compounds having low or sub μM activity. | Atomwise | [62] |
| Infectious disease | COVID-19 | Workflow used knowledge graph information from recent literature using machine learning (ML) based extraction to identify baricitinib. This molecule progressed from a clinical trial to emergency FDA approval. | BenevolentAI | [63] |
| Various | Various drug rediscovery examples | de novo generative design benchmarking study used rediscovery of various drugs with different algorithms. | BenevolentAI | [64] |
| Rare disease | Fragile X | Disease-Gene Expression Matching approach to repurposing identified sulindac which rescued the phenotype in the Fmr1 KO mouse. | Healx | [65] |
| Fibrosis | DDR1 kinase | Generative machine learning to discover novel compounds validated in vivo | In silico Medicine | [52] |
| Infectious disease | Antibacterials against E. coli | Machine learning, virtual screeing and in vitro testing | In silico Medicine | [66] |
| Various | Various | Different generative approaches were used and evaluated including entangled conditional adversarial autoencoder, reinforced adversarial neural computer, and Adversarial threshold neural computer. They either purchased compounds similar to those proposed and then tested them in vitro against various kinases or alternatively they synthesized proposed compounds and tested them | In silico Medicine | [67] [68] [69] |
| Various | sEH, ERa and c-KIT | Applied machine learning algorithms (random forest or graph convolutional neural network (GCNN)) to DNA encoded libraries then validated the predictions in vitro. GCNN models had higher hit rates and potencies. | X-Chem | [56] |
| Various | IMPDH, JNK3 etc. | Graph based deep generative model to create linkers for combining two fragments for scaffold hopping and PROTACS using a gated graph neural network incorporating 3D information. Molecules were assessed with a range of 2D and 3D metrics and outperformed a baseline. | ExScientia Ltd | [70] |
| Various | Various | Multiple machine learning approaches applied to searching commercial and proprietary libraries, lead optimization and repurposing. | Collaborations Pharmaceuticals, Inc. | See Table S2 |
It was clear to us that 2020 demonstrated that the pathway from “ideas to molecules to treatments” can be increasingly aided by machine learning algorithms, to the point where they become relatively transparent because they are accepted as part of the drug discovery or design process like other types of tools. While this software is freely available through open-source projects, replacing what was previously only commercially available and used by experts, how they are applied and integrated makes the difference to their likely success or failure. This can also be considered as one definition of what is termed end-to-end machine learning [11]. It is also likely that what will ultimately differentiate such companies in this space from competitors (Table S1) are the curation of the available underlying experimental data and ensuring the quality and validity of the machine learning models that form the basis for each companies differentiating technology. Continual curation of data in larger companies may allow them to capture the decades of drug discovery and toxicology domain knowledge of employees which they have considerably more of compared with newer companies. Knowing what are the ‘pros and cons’ of the different machine learning algorithms is also important, as no single algorithm or resulting model is likely to be the best for all prediction tasks [30] (Table S2, S3). Drug discovery is challenging and not an area to embark upon if you have no concept of what the application domain is. There is still a need for a scientist in the loop for most drug discovery machine learning models, however this does not mean we are far from their autonomous use.
The Future of Molecules Designed by AI
The future of molecule design across multiple industries (pharmaceuticals, agrochemicals, consumer products etc.) will require machine learning models for the design and optimization of molecules and their properties through the complete design-make-test cycle (Figure 1). “Designed by AI” is not the end for machine learning. While machine learning can be used to model and predict most types of data that are generated in the research and development process [11], this is certainly not limited to predicting a bioactivity or toxicity endpoints. Machine learning models may also help at different stages of research to aid in molecule purification, identification or quantification where perhaps a molecule has never been synthesized and no reference data is available. For example, modeling outputs of analytical data such as spectra (MS, FT-IR, UV-Vis [31]) or more complex in vivo data all the way to more abstract predictions, such as potential success of commercialization [32, 33] are possible with machine learning. While learning from known molecule related data is potentially valuable, going beyond what is currently known or state-of-the-art and proposing new molecules to synthesize based on the machine learning models, a physicochemical property, or other data is an area of major interest. There has been substantial activity in recent years with small molecules designed and generated by generative models using many different architectures such as Variational Autoencoder [34], Generative Adversarial Networks [35] and Recurrent Neural Networks [36] (RNN, Figure 1, Table 1)) [36–41] to produce molecules de novo [10, 36, 42–48]. For further detail the reader is pointed to the multiple reviews on this area [49–51]. Prospective testing of the proposed molecules using these methods is generally rare [52] and many prefer to skip the synthesis and find compounds that are structurally similar but commercially available from vendors. When such generative machine learning model derived molecules are eventually synthesized this is usually not done in an automated or tightly integrated fashion but handed over to a contract research organization, collaborator or perhaps left for other researchers to follow up. The application of generative approaches for de novo design of larger molecules is also relatively unexplored (although other approaches have been developed for macrolide library enumeration [53]) and yet there are certainly many large biotech companies focused on biologics whose patents will eventually expire too. One would assume that such companies are also exploring how such machine learning methods could help them design new biologics or optimize their current products [54]. As a test case example for this perspective, we have used a generative long short-term memory (LSTM) algorithm to generate novel peptides with predicted glucagon-like peptide-1 (GLP-1) agonist activity (Figure 2) using publicly available data for the machine learning model. This illustrated that the de novo proposed molecules from the algorithm are in very close structural and predicted bioactivity proximity to known commercial GLP-1 agonists, which would provide some confidence of their utility. Clearly, the ultimate proof of this will require synthesis and testing of these proposed molecules, but this is just one such additional area of use for generative models and there are many therapeutic modalities where they could help us explore chemical and property space. This is scalable such that computationally one could generate many such examples for different targets, diseases, structural scaffolds or molecular entities and then prioritize the targets or diseases to pursue.
Figure 2.
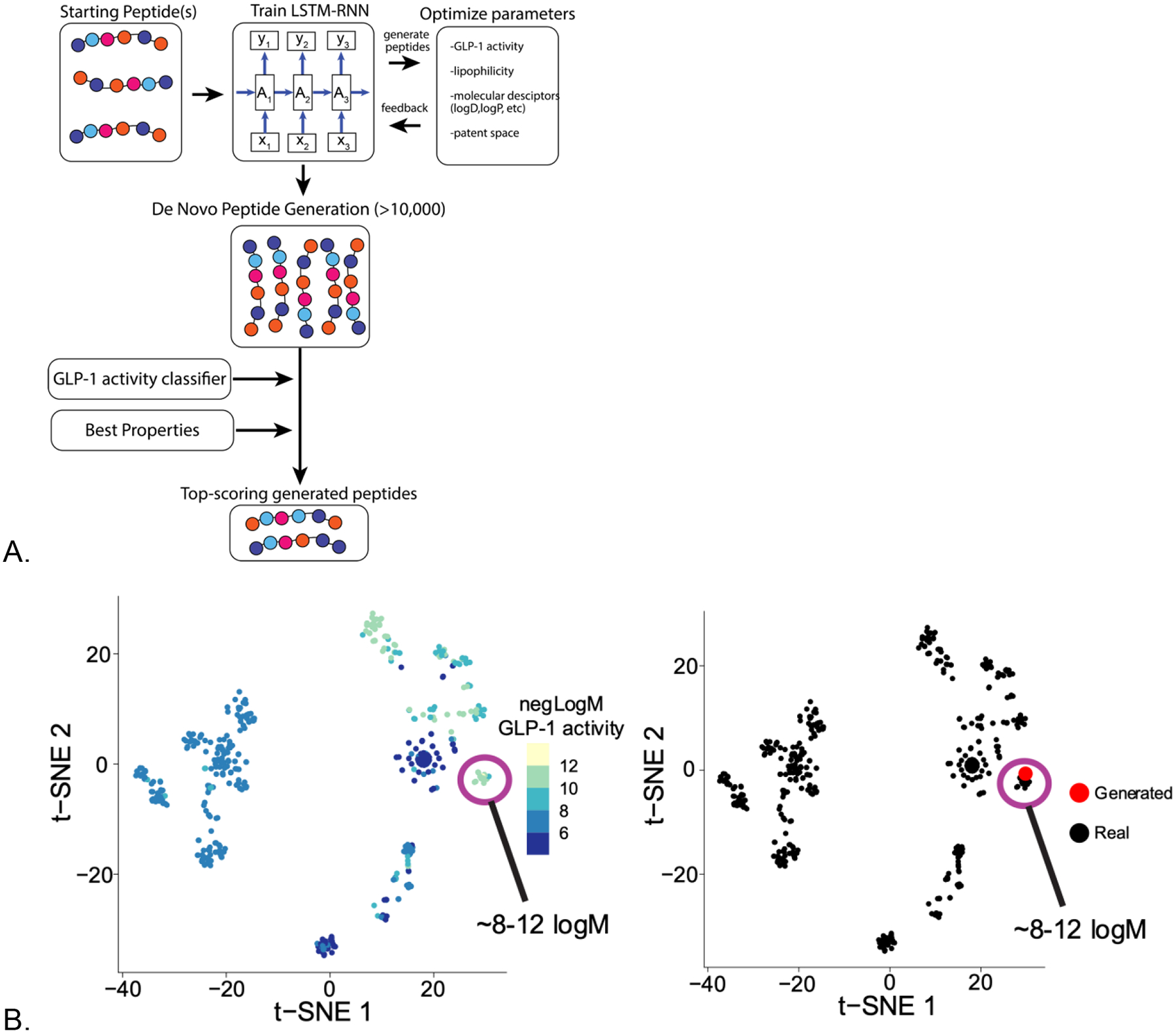
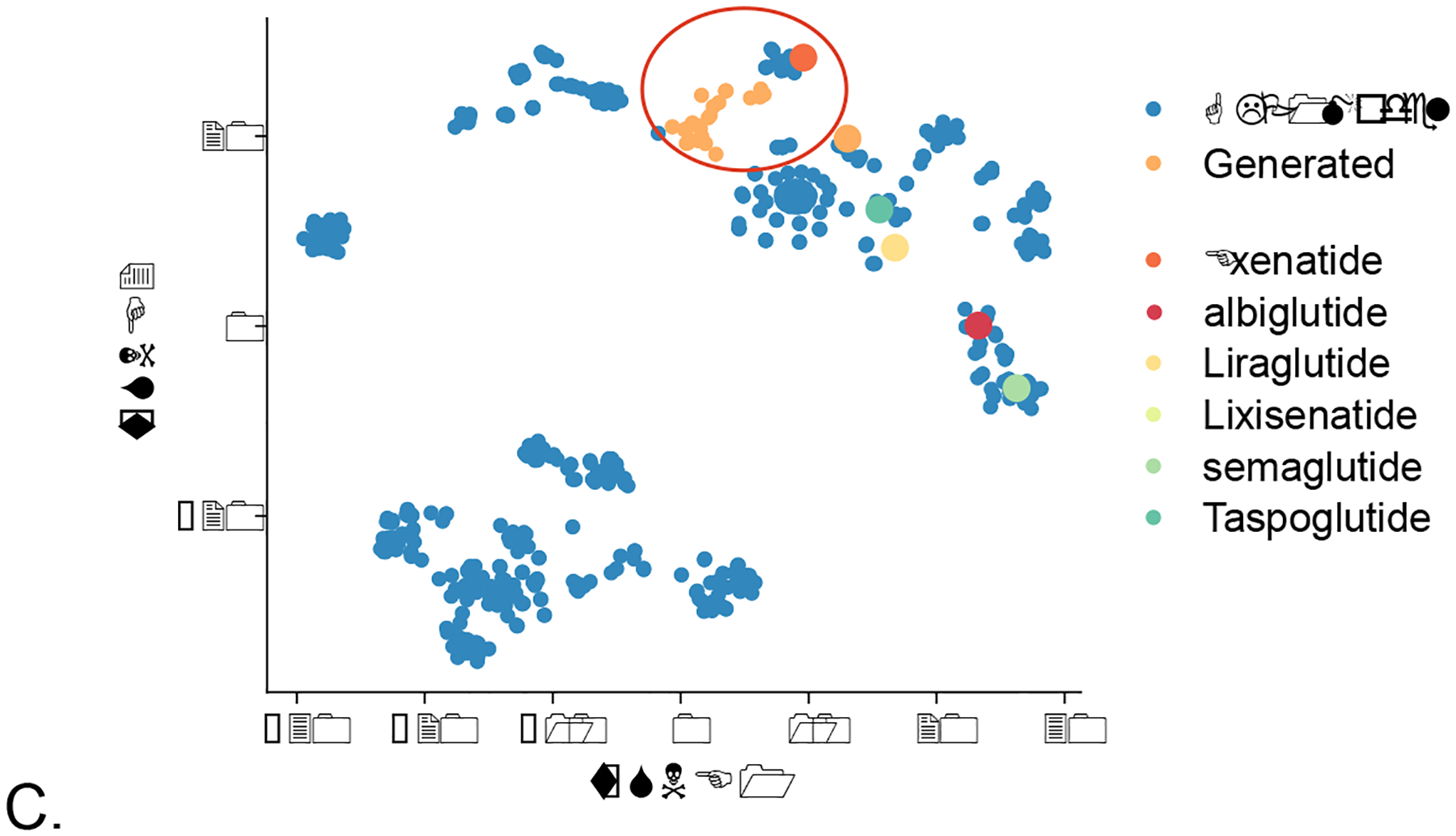
A case study of generative peptide design for GLP-1. A. An RNN-LSTM was trained on a dataset of 1554 antimicrobial peptides and generated peptides were scored with a GLP-1 agonist model generated from data in ChEMBL. B. dimensionality reduction using a t-SNE plot and nearest neighbor distance of generated proposed GLP-1 agonists. C. visualizing de novo generated GLP-1 agonists alongside commercial GLP-1 drugs to illustrate they are close in chemical property space.
Generative approaches are certainly not the only way to produce or optimize molecules and there is a long history of technologies (fragment-based drug discovery [55], structure-based design, computer-aided design etc.). Other technologies such as DNA encoded libraries can rapidly generate billions of potential structures which may need to be scored by machine learning models [56]. A bottleneck for scoring such libraries (or massive numbers of virtual molecules in general) may be in the generation of fingerprints (such as ECFP6) and their storage before processing. One solution is to use the structure encoded as SMILES (or other structural representations such as SELFIES [57]) as the input for modeling using an end-to-end convolution-LSTM model [58]. These types of machine learning algorithms are likely comparable to several others when their statistics are compared, suggesting again that there may be several different machine learning algorithms that can be applied (Table S3). Using convolution-LSTM models for predictions for a billion molecules in a DNA-encoded library, such that calculations take place on the GPU allowing parallel model prediction and pre-processing, produces an approximate 50-fold speed up on prediction generation over models built with ECFP6 fingerprints alone on our in house 10 GPU servers. While this is rather limited example, these types of end-to end machine learning models using SMILES have also been demonstrated in recent comparisons with ECFP6 for prediction of UV-Vis spectra [31] and may be utilized for other types of datasets as well.
As has happened previously, we can see a time when the machine learning algorithms we take for granted now (such as: deep learning, graph-based methods, LSTM, transformers etc.) will be more widely known and used. We envisage we will also see such models integrated into future generations of laboratory equipment. This will enable such hardware and software combinations to aid in molecule design whilst also proposing and making the molecules based on the computational predictions [1–6]. This would also facilitate tightly integrated “design-make-test” cycles to be repeated until a desired end point was reached (such as a measurable bioactivity, a molecular property, or multiple activities or properties are met). Certainly, the need to integrate these technologies will require working with cheminformatics software and hardware standards to help this come to fruition. These developments when combined may suggest that we are not too far away from the complete design, synthesis and testing in real time guided by AI (if we are not there already by the time this is published!). In the past the larger pharmaceutical companies applied machine learning within relatively small groups and hence it had little impact. In contrast, smaller, newer, pharmaceutical companies are applying machine learning across their companies and are focused on the testing of new molecules as we have highlighted with public or private data (Supplemental references) which is already showing an impact based on the valuation of such companies and deals (Table S1). Machine learning has been broadly applied to tackle hit discovery, lead optimization or beyond (Table 1 and S2). Perhaps the biggest impact of such a technology will be on overall productivity to those industries like animal health or agrochemicals which are facing patent cliffs and have lost their historic connection to pharmaceutical companies as their engine of molecule design and are now requiring new molecules. It has also been widely noted that only a small fraction (a few hundred) of the ~7000 rare diseases have treatments or are even undergoing research [11]. While there are very few applications of AI technologies to rare diseases this could change this dynamic, enabling companies to work on the research and discovery of treatments where there is limited funding, the population may be seen as too small, or there is little return on investment. Similarly, tropical neglected diseases could also benefit, especially as there is growing quantity of in vitro data for these diseases after decades of research, thus providing a valuable starting point for machine learning to aid future drug discovery efforts [59, 60] (Table 1 and Table S2).
Thinking about the bigger picture, machine learning may also be a means to an end, and that end is the molecule that has a desired activity which is ultimately patented to create intellectual property that a company then monetizes. Machine learning models applied to the continuum of drug discovery could be readily used to develop a pipeline of small or large molecules for future licensing or to serve as a starting point for venture capital investors to found new companies. If multiple companies take a similar approach, then it could create a market around AI-designed molecule assets for their industry segment. At the same time, such AI-based molecule design companies could offer this expertise and capabilities to others to create a new service industry (contract AI organization). This sharing of expertise and cross fertilization of data and technologies across molecule-related industries may be inevitable, blurring the boundaries between chemical industries. Such companies can ultimately improve the design and selection of molecules that avoid likely predictable failures associated with undesirable toxicity which is also an area that crosses industries (e.g human, animal health, agrochemical and consumer products) and which is already possible using the knowledge captured by AI. As a perhaps over-used example, the commercial interest and investment in kinase inhibitors is at an all-time high (US$66.7Bn by the year 2025) but the companies in this area are yet to capitalize on the many afore-mentioned AI technologies like generative de novo approaches to developing compounds that target specific kinases or multiple kinases and avoid others. To date, only a few kinases have been used as examples for generative approaches [52] (Table 1) but they could be used for the hundreds of kinases to identify the most chemically tractable, that would never be possible experimentally. We have yet, to see kinase focused companies using such AI approaches, instead they rely on tried and tested structure-based design and medicinal chemistry. Perhaps we will see them shift to AI approaches as their benefits continue to be described.
Conclusion
In summary, we have highlighted several examples that illustrate how AI applied to molecule design may impact several related industries involved in molecule design including the pharmaceutical industry and others. The increased visibility and awareness of the potential of AI as applied to drug discovery for COVID-19 [61] has been one of the few good things to have come out of the pandemic, even if it has delivered few notable successes to date (Table 1). What will be interesting to see is whether AI technology does indeed increase the long-term productivity and success of the new pharmaceutical companies that are attracting so much recent interest. We are cautiously optimistic that the time for AI in the pharmaceutical industry is here and that it will have a lasting impact. While we do not have all the answers to the questions raised in this perspective, our goal was to illustrate a recent observation that we are currently treating these AI technologies like a commodity. There are clearly still significant challenges and opportunities to applying them, leaving plenty of scope for future research and reviews. There are also ethical issues that have not been addressed, as these generative machine learning technologies are so readily accessible that they could be easily misused without the need for too much underlying knowledge. We look forward to discussing these and other topics with the community of scientists that are involved in this field.
Supplementary Material
Acknowledgments
Dr. Thomas R. Lane, Ana C. Puhl and Maggie A.Z. Hupcey are gratefully acknowledged for their many contributions and discussions.
Funding
This work was supported by NIH funding: R44GM122196-02A1 and 1R41GM131433-01A1 from NIGMS, 3R43AT010585-01S1 from NCCAM and 1R43ES031038-01 from NIEHS (PI – Sean Ekins). “Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number R43ES031038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
Footnotes
Conflicts of interest
S.E. is owner, and F.U. is an employee of Collaborations Pharmaceuticals, Inc.
References
- [1].Ozin G, Siler T. Autonomous chemical synthesis. 2020. https://www.advancedsciencenews.com/autonomous-chemical-synthesis/
- [2].Sanderson K. Automation: Chemistry shoots for the Moon. Nature. 2019;568:577–9. [DOI] [PubMed] [Google Scholar]
- [3].Porwol L, Kowalski DJ, Henson A, Long D-L, Bell NL, Cronin L. An Autonomous Chemical Robot Discovers the Rules of Inorganic Coordination Chemistry without Prior Knowledge. Angew Chem Int Ed Engl. 2020;59:11256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bedard AC, Adamo A, Aroh KC, Russell MG, Bedermann AA, Torosian J, et al. Reconfigurable system for automated optimization of diverse chemical reactions. Science. 2018;361:1220–5. [DOI] [PubMed] [Google Scholar]
- [5].Coley CW, Thomas DA 3rd, Lummiss JAM, Jaworski JN, Breen CP, Schultz V, et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science. 2019;365:eaax1566. [DOI] [PubMed] [Google Scholar]
- [6].Bettenhausen C. AI and robotics come together for synthesis. C&E News. 2020;98. [Google Scholar]
- [7].Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45:D945–D54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang Y, Cheng T, Bryant SH. PubChem BioAssay: A Decade’s Development toward Open High-Throughput Screening Data Sharing. SLAS Discov. 2017;22:655–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Coley CW, Barzilay R, Jaakkola TS, Green WH, Jensen KF. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent Sci. 2017;3:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Blaschke T, Olivecrona M, Engkvist O, Bajorath J, Chen H. Application of Generative Autoencoder in De Novo Molecular Design. Mol Inform. 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ekins S, Puhl AC, Zorn KM, Lane TR, Russo DP, Klein JJ, et al. Exploiting machine learning for end-to-end drug discovery and development. Nat Mater. 2019;18:435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bahdanau D, Cho K, Bengio Y. Neural Machine Translation by Jointly Learning to Align and Translate. 2014;arXiv:1409.0473
- [13].Luong M-T, Pham H, Manning CD. Effective Approaches to Attention-based Neural Machine Translation. arXiv. 2015;arXiv:1508.04025. [Google Scholar]
- [14].Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, HGomez AN, et al. Attention Is All You Need. ArXiv. 2017;1706.03762. [Google Scholar]
- [15].Hernandez D The ways AI is transforming drug development. Wall Street Journal 2017. [Google Scholar]
- [16].Anon. Special report: The return of the machinery question. The Economist; 2016. [Google Scholar]
- [17].Gupta RR, Gifford EM, Liston T, Waller CL, Hohman M, Bunin BA, et al. Using open source computational tools for predicting human metabolic stability and additional absorption, distribution, metabolism, excretion, and toxicity properties. Drug Metab Dispos. 2010;38:2083–90. [DOI] [PubMed] [Google Scholar]
- [18].Zorn KM, Lane TR, Russo DP, Clark AM, Makarov V, Ekins S. Multiple Machine Learning Comparisons of HIV Cell-based and Reverse Transcriptase Data Sets. Mol Pharm. 2019;16:1620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Minerali E, Foil DH, Zorn KM, Lane TR, Ekins S. Comparing Machine Learning Algorithms for Predicting Drug-Induced Liver Injury (DILI). Mol Pharm. 2020;17:2628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Minerali E, Foil DH, Zorn KM, Ekins S. Evaluation of Assay Central® Machine Learning Models for Rat Acute Oral Toxicity Prediction. ACS Sustain Chem Eng. 2020;8:16020–7. [Google Scholar]
- [21].Lane TR, Foil DH, Minerali E, Urbina F, Zorn KM, Ekins S. A Very Large-Scale Bioactivity Comparison of Deep Learning and Multiple Machine Learning Algorithms for Drug Discovery. Mol Pharm. 2020;18:403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Klein JJ, Baker N, Foil DH, Zorn KM, Urbina F, Puhl AC, et al. Using Bibliometric Analysis and Machine Learning to Identify Compounds binding to Sialidase-1. ACS Omega. 2021;6:3186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Anderson E, Havener TM, Zorn KM, Foil DH, Lane TR, Capuzzi SJ, et al. Synergistic drug combinations and machine learning for drug repurposing in chordoma. Sci Rep. 2020;10:12982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vignaux P, Minerali E, Foil DH, Puhl AC, Ekins S. Machine Learning for Discovery of GSK3β Inhibitors. ACS Omega. 2020;5:26551–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vignaux PA, Minerali E, Lane TR, Foil DH, Madrid PB, Puhl AC, et al. The Antiviral Drug Tilorone Is a Potent and Selective Inhibitor of Acetylcholinesterase. Chem Res Toxicol. 2021;34:1296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhu T, Cao S, Su PC, Patel R, Shah D, Chokshi HB, et al. Hit identification and optimization in virtual screening: practical recommendations based on a critical literature analysis. J Med Chem. 2013;56:6560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ekins S, Freundlich JS, Clark AM, Anantpadma M, Davey RA, Madrid P. Machine learning models identify molecules active against the Ebola virus in vitro. F1000Res. 2015;4:1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ekins S, de Siqueira-Neto JL, McCall LI, Sarker M, Yadav M, Ponder EL, et al. Machine Learning Models and Pathway Genome Data Base for Trypanosoma cruzi Drug Discovery. PLoS Negl Trop Dis. 2015;9:e0003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gawriljuk VO, Foil DH, Puhl AC, Zorn KM, Lane TR, Riabova O, et al. Development of Machine Learning Models and the Discovery of a New Antiviral Compound against Yellow Fever Virus. J Chem Inf Model. 2021;61:3804–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shalev-Shwartz S, Ben-David S. Understanding Machine Learning: From Theory to Algorithms. USA: Cambridge University Press; 2014. [Google Scholar]
- [31].Urbina F, Batra K, Luebke KJ, White JD, Matsiev D, Olson LL, et al. UV-adVISor: Attention-Based Recurrent Neural Networks to Predict UV-Vis Spectra. Anal Chem. 2021;93:16076–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lo AW, Siah KW, Wong CH. Machine Learning with Statistical Imputation for Predicting Drug Approvals. Harvard Data Science Review. 2019;1:1. [Google Scholar]
- [33].Siah KW, Kelley NW, Ballerstedt S, Holzhauer B, Lyu T, Mettler D, et al. Predicting drug approvals: The Novartis data science and artificial intelligence challenge. Patterns (N Y). 2021;2:100312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gomez-Bombarelli R, Wei JN, Duvenaud D, Hernandez-Lobato JM, Sanchez-Lengeling B, Sheberla D, et al. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent Sci. 2018;4:268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Prykhodko O, Johansson SV, Kotsias PC, Arus-Pous J, Bjerrum EJ, Engkvist O, et al. A de novo molecular generation method using latent vector based generative adversarial network. J Cheminform. 2019;11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Segler MHS, Kogej T, Tyrchan C, Waller MP. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent Sci. 2018;4:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gupta A, Muller AT, Huisman BJH, Fuchs JA, Schneider P, Schneider G. Erratum: Generative Recurrent Networks for De Novo Drug Design. Mol Inform. 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bjerrum EJ, Threlfall R. Molecular Generation with Recurrent Neural Networks (RNNs). arXiv. 2017;1705.04612. [Google Scholar]
- [39].Domenico A, Nicola G, Daniela T, Fulvio C, Nicola A, Orazio N. De Novo Drug Design of Targeted Chemical Libraries Based on Artificial Intelligence and Pair-Based Multiobjective Optimization. J Chem Inf Model. 2020;60:4582–93. [DOI] [PubMed] [Google Scholar]
- [40].Winter R, Montanari F, Steffen A, Briem H, Noe F, Clevert DA. Efficient multi-objective molecular optimization in a continuous latent space. Chem Sci. 2019;10:8016–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Maziarka L, Pocha A, Kaczmarczyk J, Rataj K, Danel T, Warchol M. Mol-CycleGAN: a generative model for molecular optimization. J Cheminform. 2020;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Olivecrona M, Blaschke T, Engkvist O, Chen H. Molecular de-novo design through deep reinforcement learning. Journal of Cheminformatics. 2017;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Krenn M, Häse F, Nigam A, Friederich P, Aspuru-Guzik A. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Machine Learning: Science and Technology. 2020;1:045024. [Google Scholar]
- [44].Jin W, Barzilay R, Jaakola T. Junction Tree Variational Autoencoder for Molecular Graph Generation. arXiv 2019. https://arxiv.org/pdf/1802.04364.pdf [Google Scholar]
- [45].Hochreiter S, Schmidhuber J. Long Short-Term Memory. Neural Computation. 1997;9:1735–80. [DOI] [PubMed] [Google Scholar]
- [46].Sanchez-Lengeling B, Outeiral C, Guimaraes GL, Aspuru-Guzik A. Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). ChemRxiv 2017. https://chemrxiv.org/engage/chemrxiv/article-details/60c73d91702a9beea7189bc2 [Google Scholar]
- [47].Winter R, Montanari F, Steffen A, Briem H, Noé F, Clevert D-A. Efficient multi-objective molecular optimization in a continuous latent space. Chemical Science. 2019;10:8016–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gao K, Nguyen DD, Tu M, Wei G-W. Generative Network Complex for the Automated Generation of Drug-like Molecules. Journal of Chemical Information and Modeling. 2020;60:5682–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Meyers J, Fabian B, Brown N. De novo molecular design and generative models. Drug Discov Today. 2021;26:2707–15. [DOI] [PubMed] [Google Scholar]
- [50].Bhisetti G, Fang C. Artificial Intelligence-Enabled De Novo Design of Novel Compounds that Are Synthesizable. Methods Mol Biol. 2022;2390:409–19. [DOI] [PubMed] [Google Scholar]
- [51].Palazzesi F, Pozzan A. Deep Learning Applied to Ligand-Based De Novo Drug Design. Methods Mol Biol. 2022;2390:273–99. [DOI] [PubMed] [Google Scholar]
- [52].Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019;37:1038–40. [DOI] [PubMed] [Google Scholar]
- [53].Zin PPK, Williams G, Fourches D. SIME: synthetic insight-based macrolide enumerator to generate the V1B library of 1 billion macrolides. J Cheminform. 2020;12:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Castillo-Hair SM, Seelig G. Machine Learning for Designing Next-Generation mRNA Therapeutics. Acc Chem Res. 2022;55:24–34. [DOI] [PubMed] [Google Scholar]
- [55].de Esch IJP, Erlanson DA, Jahnke W, Johnson CN, Walsh L. Fragment-to-Lead Medicinal Chemistry Publications in 2020. J Med Chem. 2022;65:84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].McCloskey K, Sigel EA, Kearnes S, Xue L, Tian X, Moccia D, et al. Machine Learning on DNA-Encoded Libraries: A New Paradigm for Hit Finding. J Med Chem. 2020;63:8857–66. [DOI] [PubMed] [Google Scholar]
- [57].Nigam A, Pollice R, Krenn M, Gomes GDP, Aspuru-Guzik A. Beyond generative models: superfast traversal, optimization, novelty, exploration and discovery (STONED) algorithm for molecules using SELFIES. Chem Sci. 2021;12:7079–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Shi X, Chen Z, Wang H, Yeung D-Y, Wong W, Woo W. Convolutional LSTM Network: A Machine Learning Approach for Precipitation Nowcasting. arXiv. 2015;1506.04214. [Google Scholar]
- [59].Zorn KM, Sun S, McConnon CL, Ma K, Chen EK, Foil DH, et al. A Machine Learning Strategy for Drug Discovery Identifies Anti-Schistosomal Small Molecules. ACS Infect Dis. 2021;7:406–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hernandez HW, Soeung M, Zorn KM, Ashoura N, Mottin M, Andrade CH, et al. High Throughput and Computational Repurposing for Neglected Diseases. Pharm Res. 2018;36:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Muratov EN, Amaro R, Andrade CH, Brown N, Ekins S, Fourches D, et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem Soc Rev. 2021;50:9121–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Stecula A, Hussain MS, Viola RE. Discovery of Novel Inhibitors of a Critical Brain Enzyme Using a Homology Model and a Deep Convolutional Neural Network. J Med Chem. 2020;63:8867–75. [DOI] [PubMed] [Google Scholar]
- [63].Smith DP, Oechsle O, Rawling MJ, Savory E, Lacoste AMB, Richardson PJ. Expert-Augmented Computational Drug Repurposing Identified Baricitinib as a Treatment for COVID-19. Front Pharmacol. 2021;12:709856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Brown N, Fiscato M, Segler MHS, Vaucher AC. GuacaMol: Benchmarking Models for de Novo Molecular Design. J Chem Inf Model. 2019;59:1096–108. [DOI] [PubMed] [Google Scholar]
- [65].Tranfaglia MR, Thibodeaux C, Mason DJ, Brown D, Roberts I, Smith R, et al. Repurposing available drugs for neurodevelopmental disorders: The fragile X experience. Neuropharmacology. 2019;147:74–86. [DOI] [PubMed] [Google Scholar]
- [66].Ivanenkov YA, Zhavoronkov A, Yamidanov RS, Osterman IA, Sergiev PV, Aladinskiy VA, et al. Identification of Novel Antibacterials Using Machine Learning Techniques. Front Pharmacol. 2019;10:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Polykovskiy D, Zhebrak A, Vetrov D, Ivanenkov Y, Aladinskiy V, Mamoshina P, et al. Entangled Conditional Adversarial Autoencoder for de Novo Drug Discovery. Mol Pharm. 2018;15:4398–405. [DOI] [PubMed] [Google Scholar]
- [68].Putin E, Asadulaev A, Ivanenkov Y, Aladinskiy V, Sanchez-Lengeling B, Aspuru-Guzik A, et al. Reinforced Adversarial Neural Computer for de Novo Molecular Design. J Chem Inf Model. 2018;58:1194–204. [DOI] [PubMed] [Google Scholar]
- [69].Putin E, Asadulaev A, Vanhaelen Q, Ivanenkov Y, Aladinskaya AV, Aliper A, et al. Adversarial Threshold Neural Computer for Molecular de Novo Design. Mol Pharm. 2018;15:4386–97. [DOI] [PubMed] [Google Scholar]
- [70].Imrie F, Bradley AR, van der Schaar M, Deane CM. Deep Generative Models for 3D Linker Design. J Chem Inf Model. 2020;60:1983–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.