

Abstract
Hepatic fibrosis occurs in response to prolonged tissue injury in the liver, which results in abnormal accumulation of extracellular matrix. Hepatic stellate cells (HSCs) have been suggested to play a major role in liver fibrosis. However, the molecular mechanisms remain incompletely understood. Sirtuin 6 (SIRT6), an NAD+‐dependent deacetylase, has been previously implicated in the regulation of the transforming growth factor β (TGFβ)‐SMAD3 pathway that plays a significant role in liver fibrosis. In this work, we aimed to identify other important players during hepatic fibrogenesis, which are modulated by SIRT6. Yes‐associated protein (YAP) and transcriptional coactivator with PDZ‐binding motif (TAZ or WWTR1), key players in the Hippo pathway, have been implicated in the promotion of hepatic fibrosis. Our data show that HSC‐specific Sirt6 knockout mice are more susceptible to high‐fat‐cholesterol‐cholate diet‐induced hepatic fibrosis than their wildtype counterparts. Our signaling analyses suggest that in addition to the TGFβ‐SMAD3 pathway, YAP and TAZ are also highly activated in the SIRT6‐deficient HSCs. As it is not clear how SIRT6 might regulate YAP and TAZ, we have decided to elucidate the mechanism underlying the regulation of YAP and TAZ by SIRT6 in HSCs. Overexpression or knockdown of SIRT6 corroborates the role of SIRT6 in the negative regulation of YAP and TAZ. Further biochemical analyses reveal that SIRT6 deacetylates YAP and TAZ and reprograms the composition of the TEA domain transcription factor complex to suppress their downstream target genes, particularly those involved in hepatic fibrosis. In conclusion, our data suggest that SIRT6 plays a critical role in the regulation of the Hippo pathway to protect against hepatic fibrosis.
Keywords: deacetylation, fibrosis, hippo, Sirtuin 6, TEAD
Abbreviations
- ACTA2
smooth muscle actin alpha 2
- ANKRD1
ankyrin repeat domain 1
- COL1A1
collagen type 1 alpha 1
- COL3A1
collagen type 3 alpha 1
- CTGF
connective tissue growth factor
- CYR61
cysteine‐rich angiogenic inducer 61
- GFP
green fluorescent protein
- HFCC
high‐fat‐cholesterol‐cholate diet
- HSC
hepatic stellate cell
- IHH
Indian hedgehog
- IP
immunoprecipitation
- KO
knockout
- LRAT
lecithin retinol acyltransferase
- NAFLD
non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- sgRNA
single guide RNA
- SIRT6
Sirtuin 6
- SMAD3
SMAD family member 3
- TAZ
transcriptional co‐activator with a PDZ‐binding motif
- TEAD
TEA domain transcription factor
- TGF‐β1
transforming growth factor β‐1
- VGLL4
vestigial‐like family member 4
- WT
wild type
- WWTR1
WW domain containing transcription regulator 1
- YAP
yes‐associated protein
1. INTRODUCTION
Non‐alcoholic fatty liver disease (NAFLD) is a very common chronic liver disease that begins with simple deposition of fat, known as steatosis, and can progress into an inflammatory and fibrotic manifestation called non‐alcoholic steatohepatitis (NASH), or eventually cirrhosis or liver cancer. 1 , 2 Hepatic fibrosis is a hallmark of the transition from simple steatosis to NASH. 3 Hepatic stellate cells (HSCs) have been shown to play a major role in liver fibrosis by contributing to the majority of the extracellular matrix buildup during liver fibrosis pathogenesis. 4
The evolutionarily conserved Hippo pathway has recently been implicated in liver fibrosis. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 Regulated primarily by a kinase cascade, the major effectors of the Hippo pathway—Yes‐associated protein (YAP) and WW domain containing transcription regulator 1 (WWTR1) or PDZ‐binding motif (TAZ)—shuttle between cytoplasm and nucleus according to their phosphorylation status. Upon stimulation by multiple factors, STE20‐like serine/threonine kinases (MST1/2) and large tumor suppressor kinases 1/2 (LATS1/2) are activated, YAP and TAZ are phosphorylated at Ser127 and Ser89, respectively, and sequestered to the cytoplasm by binding to 14‐3‐3 proteins. In a non‐stimulated state, unphosphorylated YAP/TAZ is localized to the nucleus and exert coactivation of TEA domain transcription factors (TEADs). 18 YAP/TAZ actions in the hepatocytes have been implicated in the development of NASH. 6 , 9 , 15 , 19 Elevated TAZ levels are positively correlated with the expression and secretion of Indian hedgehog (Ihh) ligands, which promote HSC activation and fibrosis. 15 Targeting hepatocyte TAZ using small interfering RNA has been shown to improve hepatic fibrosis in a diet‐induced NASH mouse model. 11 Moreover, YAP is also activated in fibrotic livers of hepatitis C patients, as well as in CCl4‐treated mouse livers. 17 YAP has also been shown to promote HSC activation. 13 , 14 , 17
Sirtuin 6 (SIRT6), an NAD+‐ dependent deacetylase/deacylase, plays a crucial role in the maintenance of hepatic function and health by regulating glucose and lipid metabolism and protecting against the development of steatosis and inflammation. 20 Furthermore, SIRT6 also protects against liver fibrosis by deacetylating key lysine residues on SMAD family members 2 and 3 (SMAD2 and SMAD3). 21 , 22 However, it is unclear whether SIRT6 has a role in the regulation of the Hippo signaling pathway in HSCs. In this study, we explored the relationship between SIRT6 and YAP/TAZ and the related biochemical mechanism in HSCs.
2. MATERIALS AND METHODS
2.1. Animal experiments
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Indiana University School of Medicine in consistence with the guidelines for the care and use of laboratory animals by the National Institutes of Health (NIH). HSC‐ and liver‐specific Sirt6 knockout (HSC‐KO and LKO, respectively) mice were generated using a lecithin retinol acyl‐transferase (Lrat)‐Cre or albumin‐Cre line as previously described. 22 , 23 Four to 6 weeks old Sirt6 KO and wild‐type (WT) mice were housed in ventilated cages at ambient temperature (22 ± 2°C) and humidity (60% ± 5%) with 12:12 h light/dark cycles and fed ad libitum with a high‐fat‐cholesterol‐cholate diet (HFCC; Research Diets D12109C, New Brunswick, NJ) containing 20% calories from protein, 40% calories from fat, 40% calories from carbohydrates, and 1.25% cholesterol and 0.5% sodium cholate by weight. 24 At the end of the experiments, animals were euthanized for tissue and blood collection.
2.2. DNA constructs
Mouse Sirt6, catalytically inactive Sirt6 H133Y, Smad3, vestigial‐like family member 4 (Vgll4), Wwtr1/Taz, and green fluorescent protein (GFP) coding sequences were cloned into a pcDNA3 vector with a FLAG or hemagglutinin (HA) tag. Human YAP1 was a gift from Yosef Shaul (Addgene, Cambridge, MA; plasmid # 18881; http://n2t.net/addgene:18881; RRID:Addgene_18881). Single guide RNAs (sgRNAs) against human YAP1, WWTR1/TAZ, SIRT6, and SMAD3 coding sequences were generated with the assistance of the GPP sgRNA Designer on the Broad Institute website and were cloned into a lentiCRISPRv2 vector as a gift from Dr. Feng Zhang (Addgene, Cambridge, MA; #52961; http://n2t.net/addgene:52961; RRID:Addgene_52961). Multiple human YAP1 and mouse Wwtr1/Taz lysine residues were mutated to arginine residues using a Q5‐site‐directed mutagenesis kit (New England Biolabs, Ipswich, MA). The oligonucleotide sequences were described in Table S1.
2.3. Cell culture
Human HSC line, LX‐2 (MilliporeSigma, Burlington, MA) was cultured in Dulbecco's modified Eagle medium (DMEM, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Thermo Fisher Scientific). Cells were kept in an incubator with 5% CO2 at 37°C. Prior to treatment with transforming growth factor β1 (TGF‐β1, R&D Systems), cells were cultured in serum‐free media overnight, and after 5 ng/ml TGF‐β1 was added the cells were incubated for 24 h. For some experiments, cells were treated with 0.25 or 0.5 μM verteporfin (Cayman Chemical) for 25 h.
2.4. Isolation and culture of mouse primary HSCs
Primary mouse HSCs were isolated from mouse livers using the density gradient centrifugation technique as previously described. 25 Mouse livers were digested using collagenase I and passed through a 70‐μm strainer. HSCs were isolated using Optiprep (MilliporeSigma) gradient. 26 , 27 HSCs were collected and cultured in DMEM containing 10% fetal bovine serum, 10% horse serum, and 1% penicillin/streptomycin antibiotics.
2.5. Histologic analysis
Mouse liver tissue specimens were fixed by formalin and sent for paraffin embedding and sectioning at the Indiana University Histology Core. Tissue sections (5‐μm thick) were deparaffinized in xylene and rehydrated in a graded series of ethanol concentrations. Deparaffinized liver tissue sections were further processed for hematoxylin and eosin (H&E) staining and Sirius Red staining (MilliporeSigma). Liver tissue specimens were also prepared in an embedding medium or O.C.T. Compound (Thermo Fisher Scientific) for cryosections. Oil Red O (MilliporeSigma) staining was performed on these sections using a standard protocol. Images for H&E, Sirius Red, and Oil Red O were captured using a Leica DM750 microscope equipped with an EC3 digital camera and LAS EZ software. Images for immunofluorescence (IF) analysis were obtained using a ZEISS fluorescence microscope with an AxioVision Rel 4.8 software. IF images were quantified from randomly selected fields from each sample using ImageJ 1.52 software (NIH, Bethesda, MD).
2.6. Immunocytochemistry
LX‐2 cells grown on a glass‐bottom dish were fixed with 4% paraformaldehyde for 15 min at room temperature, then washed 3 times with phosphate‐buffered saline (PBS) and incubated overnight with primary antibodies as described (Table S2). After washing, cells were incubated with Alexa Fluor‐conjugated secondary antibodies (Thermo Fisher Scientific, 1:250) for 1.5 h, counterstained, and mounted with 50% glycerol with 4′,6‐diamidino‐2‐phenylindole (DAPI, Thermo Fisher Scientific), and imaged under a fluorescent microscope (Zeiss USA, Thornwood, NY).
2.7. Immunoblotting and immunoprecipitation (IP)
Total protein from tissue lysates was prepared using a lysis buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X‐100, 1.5 mM MgCl2, 1 mM EDTA, 10 mM sodium pyrophosphate, 100 mM sodium fluoride, and freshly added 100 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride, and 1× cOmplete protease inhibitor cocktail (Roche, Indianapolis, IN). Total cellular protein was prepared with lysis buffer containing 1% NP‐40, 20 mM Tris, pH 7.4, 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, and 1× cOmplete protease inhibitor cocktail. Protein samples were resolved by 10% SDS‐PAGE and transferred to nitrocellulose membranes (Bio‐Rad). For immunoblotting, the membrane was blocked with 5% non‐fat dry milk in PBS with 0.1% Tween‐20 for 1 h, followed by incubation with the specific primary antibody at 4°C overnight and with horseradish peroxidase‐conjugated secondary antibody for 2 h at room temperature. Signals were detected by chemiluminescence with Pierce ECL Western Blotting substrate and scanned with a ChemiDoc MP System (Bio‐Rad Laboratories, Hercules, CA, USA). Images were analyzed using ImageJ software for quantitative analysis. For immunoprecipitation experiments, equal amounts of protein lysates were incubated with 1 μg of specific antibodies. For some immunoprecipitation experiments, FLAG beads (MilliporeSigma) were used to pull down overexpressed proteins. After a 16‐h incubation at 4°C, protein A/G plus agarose (Santa Cruz Biotechnology, Dallas, TX) was added and incubated at 4°C for 3 h. Normal rabbit immunoglobulin G (IgG) or normal mouse IgG was used as a negative control. Proteins were analyzed by immunoblotting. Antibodies used in the experiments were described in Table S2.
2.8. Gene expression analysis
Total cellular RNA was extracted with TRI reagent (MilliporeSigma) and cDNA was synthesized from 1 μg of total RNA using a cDNA synthesis kit (Thermo Fisher Scientific) in a final volume of 20 μl. Real‐time PCR was performed using SYBR green Master Mix (Thermo Fisher Scientific) in a Realplex PCR system (Eppendorf North America, Hauppauge, NY). Gene expression was normalized with the expression of the internal control gene peptidylprolyl isomerase A (PPIA) by the ΔΔC t method. The PCR primer sequences were described in Table S1.
2.9. Statistical analysis
All statistical data are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism 9 software from GraphPad (La Jolla, CA). Comparisons between two groups were performed using a two‐tailed unpaired Student t‐test, and comparisons for more than two groups were performed using a one‐way analysis of variance followed by a Tukey post hoc test.
3. RESULTS
3.1. SIRT6 regulates SMAD3‐dependent and ‐independent pathways in hepatic stellate cells
Previously, we have identified SIRT6 as a key epigenetic factor that protects against fibrogenesis by deacetylating SMAD3. 22 To examine the role of SMAD3‐dependent and ‐independent pathways that mediate the SIRT6 effect on HSCs, we first knocked down the SMAD3 gene using CRISPR sgRNAs against the coding sequence of SMAD3 in human HSC LX‐2 cells and treated with 5 ng/ml of TGF‐β1 (Figure 1A). We observed a significant decrease in the expression of COL1A1 and COL3A1 genes, markers of fibrosis, in the SMAD3 knockdown cells as compared to the control sgGFP group, indicating the significance of SMAD3 in fibrogenesis (Figure 1B). To assess whether SMAD3‐independent pathways also mediate the SIRT6 function in HSCs, we simultaneously knocked down the SIRT6 and SMAD3 genes. Both COL1A1 and COL3A1 were significantly downregulated in the SIRT6 and SMAD3 dually deficient HSCs but not to the level in the control sgGFP transfected HSCs (Figure 1B), suggesting that other pathways other than SMAD3 also play a significant role in the downstream of SIRT6. To examine which pathways might be involved in the SIRT6‐regulated hepatic fibrosis in HSCs, we analyzed several key factors that have been implicated in liver fibrosis, 4 including Smad3, platelet‐derived growth factor receptor beta (Pdgf‐β), catenin beta 1 (Ctnnb1), and Yap in WT and Sirt6 HSC‐KO livers. As we previously reported, 22 Smad3 phosphorylation at Ser423/425 was significantly increased (Figure S1A,B). While Pdgfr‐β protein was not significantly changed, Ctnnb1 protein was significantly increased and Yap phosphorylation at Ser127 was significantly decreased (indicating more active) in the Sirt6 HSC‐KO livers as compared to WT livers (Figure S1A,C–E). Connective tissue growth factor (CTGF or CCN2) and cysteine‐rich angiogenic inducer (CYR61 or CCN1), two known target genes in the Hippo pathway, were also induced in the SIRT6‐deficient LX‐2 cells (Figure 1B). As regulation of Ctnnb1 by Sirt6 in renal fibrosis has been reported, 28 , 29 , 30 we decided to further investigate a potential regulation of the Hippo pathway by SIRT6 in HSCs.
FIGURE 1.
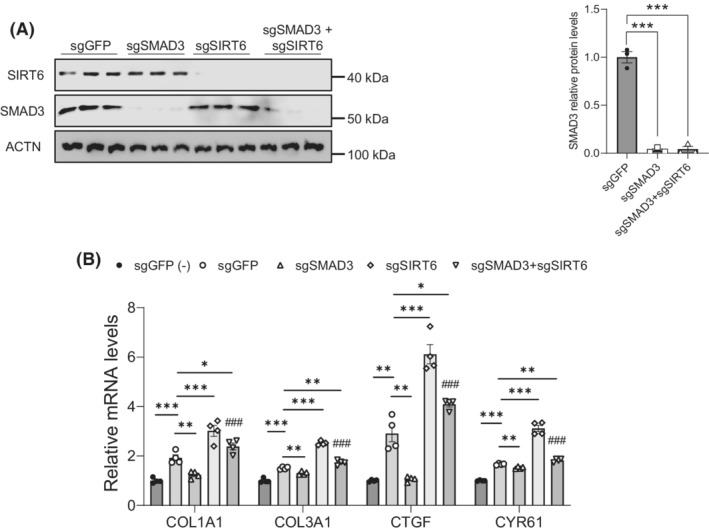
SIRT6 regulates both SMAD3‐dependent and ‐independent pathways in HSCs. (A) Immunoblot and quantification analysis of SIRT6 and SMAD3 in LX‐2 cells transfected with sgGFP, sgSMAD3, sgSIRT6, or sgSMAD3+sgSIRT6 CRISPR/Cas9 plasmids in the presence of 5 ng/ml of TGF‐β1 for 24 h (n = 3). Actinin (ACTN) was used as a loading control. (B) Real‐time PCR analysis of COL1A1, COL3A1, CTGF, and CYR61 in the LX‐2 cells transfected with sgGFP, sgSMAD3, sgSIRT6, or sgSMAD3+sgSIRT6 in the absence (−) or presence of 5 ng/ml of TGF‐β1 for 24 h (n = 4). Data are presented as mean ± SEM. *p < .05, **p < .01, and ***p < .001, and ### p < .001 for sgSMAD3+sgSIRT6 versus sgSMAD3.
3.2. YAP and TAZ drive fibrogenesis in HSCs
Next, we examined the role of the Hippo pathway effectors—YAP and TAZ in HSCs. We knocked down YAP and TAZ in LX‐2 cells using the CRISPR/Cas9 approach. We observed 64% and 85% decreases in YAP protein levels and 70% and 67% decreases in TAZ protein levels in single and double knockdown cells, respectively. Smooth muscle actin alpha 2 (ACTA2) protein levels were also decreased by 55%, 46%, and 66% in YAP, TAZ, and YAP+TAZ knockdown cells, respectively (Figure 2A,B). PCR analysis also showed a decrease of 50%, 49%, and 53% of COL1A1 mRNAs and 24%, 40%, and 57% of COL3A1 mRNAs in the YAP, TAZ, and YAP+TAZ knockdown cells, respectively (Figure 2C). Moreover, we overexpressed YAP, TAZ, or YAP+TAZ in LX‐2 cells, and we observed that ACTA2 protein levels were increased by 222%, 361%, and 233% in YAP, TAZ, or dual overexpression cells, respectively (Figure 2D,E). Next, we assessed lysine acetylation in YAP, TAZ, and VGLL4 in the presence or absence of TGF‐β1. To do this, we overexpressed YAP, TAZ, or VGLL4 in LX‐2 cells and treated them with 5 ng/ml of TGF‐β1. Here, we used SMAD3 as a positive control as we previously reported. 22 Interestingly, YAP, TAZ, SMAD3, and VGLL4 had higher acetylation levels after the TGF‐β1 treatment (Figure 2F).
FIGURE 2.
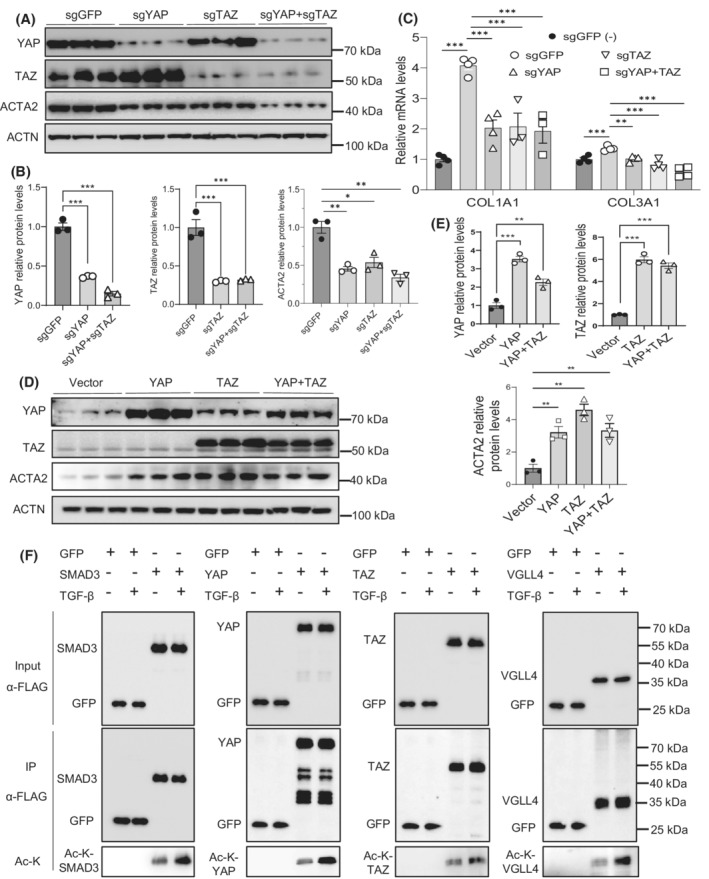
YAP and TAZ activate fibrogenesis in HSCs. (A,B) Immunoblot and quantification analysis of knockdown of YAP, TAZ, or YAP+TAZ and effect on the ACTA2 expression in LX‐2 cells treated with 5 ng/ml of TGF‐β1 for 24 h (n = 3). (C) Real‐time PCR analysis of COL1A1 and COL3A1 mRNAs in LX‐2 cells with the knockdown of YAP, TAZ, or YAP+TAZ by the CRISPR/Cas9 approach (n = 4). (D,E) Immunoblot and quantification analysis of overexpression of YAP, TAZ, or YAP+TAZ and effect on the ACTA2 expression in LX‐2 cells treated with 5 ng/ml of TGF‐β1 for 24 h (n = 3). (F) YAP, TAZ, VGLL4, and SMAD3 acetylation analysis in LX‐2 cells transfected with the indicated constructs in the presence or absence of 5 ng/ml TGF‐β1 for 24 h. Data are presented as mean ± SEM. *p < .05, **p < .01, and ***p < .001.
3.3. YAP and TAZ activities are modulated by SIRT6 in HSCs
To examine whether YAP is regulated by SIRT6, we performed SIRT6 overexpression or knockdown in LX‐2 cells. Our data showed that SIRT6 overexpression increased phosphorylated YAP (Ser127) levels, suggesting that YAP is less active (Figure 3A,B). This was supported by a decrease in the CTGF and CYR61 gene expression (Figure 3C). Moreover, when SIRT6 was knocked down, YAP phosphorylation was barely detectable, indicative of increased YAP activity (Figure 3D,E). As expected, expression of the COL3A1, COL1A1, ACTA2, CTGF, and CYR61 genes was increased after the SIRT6 gene knockdown (Figure 3F). We also observed a 126% increase in p‐YAP levels when SIRT6 was overexpressed and a 31% decrease in p‐YAP after SIRT6 was knocked down in the absence of TGF‐β1 (Figure S2). Moreover, we analyzed hepatic fibrosis and the Hippo pathway in primary HSCs from Sirt6 HSC‐KO mice. We observed an increase in expression of fibrotic genes of Col1a1, Acta2, and Tgfb1 by 206%, 126%, and 437% and the Hippo target genes of Ctgf, Cyr61, and Ankrd1 by 48%, 200%, and 176% in the primary HSCs from the HSC‐KO mice compared to that from the WT mice, respectively (Figure 3G).
FIGURE 3.
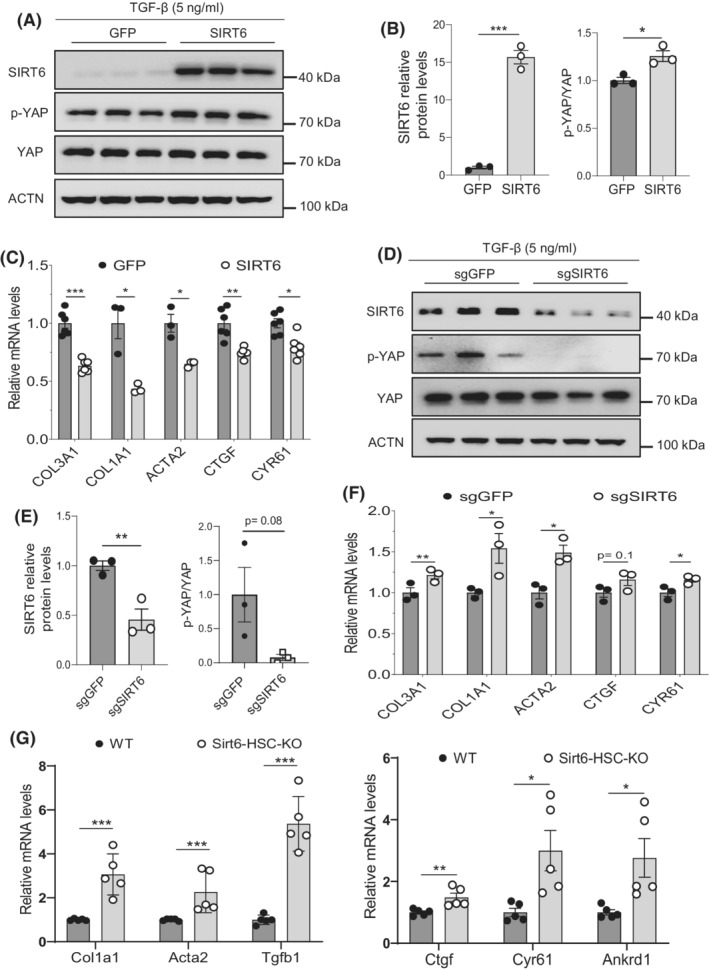
SIRT6 suppresses YAP in HSCs. (A,B) Immunoblot and quantification analysis of YAP and phosphorylated YAP in LX‐2 cells transfected with either control GFP or SIRT6 in the presence of 5 ng/ml TGF‐β1 (n = 3). (C) Real‐time PCR analysis of COL3A1, COL1A1, ACTA2, CTGF, and CYR61 mRNAs in the LX‐2 cells transfected with either control GFP or SIRT6 in the presence of 5 ng/ml TGF‐β1 (n = 6). (D,E) Immunoblot and quantification analysis of YAP and phosphorylated YAP in LX‐2 cells transfected with either sgGFP or sgSIRT6 in the presence of 5 ng/ml TGF‐β1 (n = 3). (F) Real‐time PCR analysis of COL3A1, COL1A1, ACTA2, CTGF, and CYR61 mRNAs in the LX‐2 cells transfected with either sgGFP or sgSIRT6 in the presence of 5 ng/ml TGF‐β1 (n = 3). (G) Real‐time PCR analysis of fibrotic marker genes (Col1a1, Acta2, and Tgfb1) and YAP/TAZ downstream target genes (Ctgf, Cyr61, and Ankrd1) in primary HSCs from WT and Sirt6 HSC‐KO mice (n = 5). Data are presented as mean ± SEM. *p < .05, **p < .01, and ***p < .001.
To corroborate the Hippo pathway involvement in the SIRT6‐regulated HSC activation, we chose a pharmacological approach for inhibition of the YAP coactivation of TEAD transcriptional activity using verteporfin, a well‐validated inhibitor for YAP. 17 To do so, we treated control and SIRT6 knockdown LX‐2 cells with 0.25 or 0.5 μM verteporfin in the presence or absence of 5 ng/ml TGF‐β1. As a readout for fibrosis, COL1A1 gene expression was increased in the SIRT6 knockdown cells in the absence and presence of TGF‐β1 but remarkably decreased by verteporfin, especially at 0.5 μM (Figure S3A). We also analyzed the expression of SMAD7, a downstream target of SMAD3, and found that SMAD7 was highly induced by TGF‐β1 in both control and SIRT6 knockdown cells; however, verteporfin had no significant effect on the SMAD7 gene expression (Figure S3B). To monitor the Hippo pathway activity, we also analyzed two downstream target genes—CTGF and CYR61. Our data showed that CTGF had a much stronger response to TGF‐β1 than CYR61, but the expression of both genes was significantly suppressed by verteporfin as expected (Figure S3C,D).
To further investigate the role of YAP/TAZ in the development of fibrosis, we generated a mouse model deficient in Sirt6 in HSCs as previously described. 22 HSC‐specific Sirt6 knockout was verified by immunofluorescence microscopy and albumin‐Cre‐mediated Sirt6 knockout livers were used as a negative control (Figure 4A). We subjected the mice to 4 or 6 weeks of the HFCC diet to induce NASH. The Sirt6 HSC‐KO mice developed more severe liver fibrosis as observed by Sirius Red staining of liver sections, compared to their WT counterparts (Figure S4). We further corroborated the fibrotic condition by staining collagen type 1 in the liver sections (Figure S4). There was no difference in body weight but liver weight and liver‐to‐body weight ratios were significantly lower in the Sirt6 HSC‐KO mice compared to WT mice (Figure S5).
FIGURE 4.
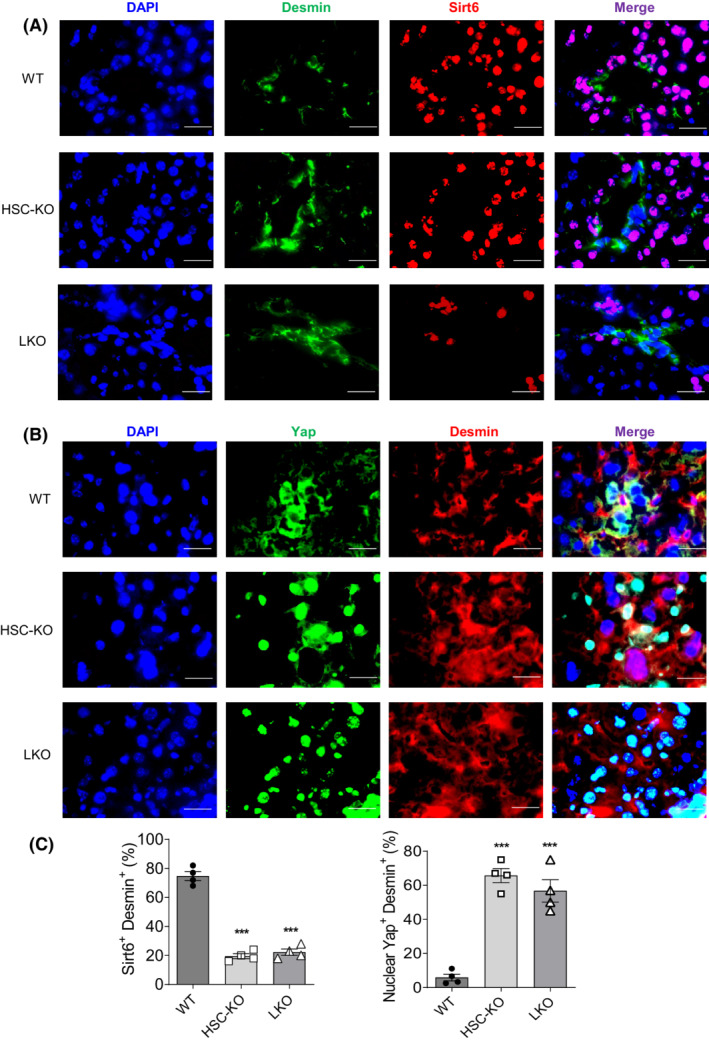
YAP is activated in Sirt6‐deficient HSCs in mouse livers. (A) Confirmation of the Sirt6 gene knockout in HSC‐KO and LKO mouse livers by immunostaining. (B) Immunostaining of Desmin and Yap in liver sections of WT, HSC‐KO, and LKO mice. (C) Quantification analysis of Sirt6+Desmin+ and nuclear Yap+Desmin+ cells in Panels A and B, respectively. Representative images are shown at 630× magnification. Scale bar = 25 μm. Data are presented as mean ± SEM. ***p < .001 versus WT.
Immunofluorescence staining of the liver sections from the HFCC diet fed Sirt6 HSC‐KO mice and LKO mice showed increased nuclear Yap in the desmin‐positive HSCs: 66% and 57% nuclear Yap‐positive HSCs in the HSC‐KO and LKO liver sections, respectively (Figure 4B,C). Next, we verified the role of SIRT6 in the regulation of YAP in LX‐2 cells by overexpression or knockdown of SIRT6 in the presence or absence of TGF‐β1. Upon SIRT6 overexpression, there were fewer nuclear YAP‐positive cells and lower expression of ACTA2, whereas knockdown of SIRT6 led to an increase in nuclear YAP‐positive cells and increased ACTA2 expression compared to controls (Figure 5A–D).
FIGURE 5.
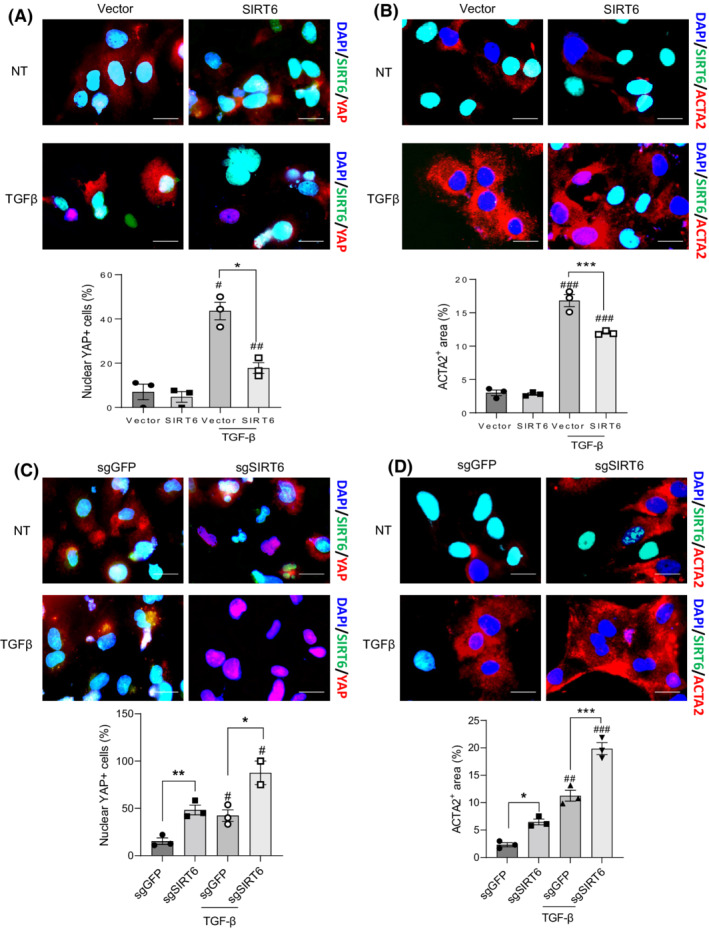
SIRT6 suppresses YAP activation and fibrosis in cultured HSCs. (A,B) Immunostaining and quantification analysis of SIRT6, YAP, and ACTA2 in LX‐2 cells transfected with either vector control or SIRT6 plasmids in the absence (non‐treated, NT) or presence of TGF‐β1. (C,D) Immunostaining and quantification analysis of SIRT6, YAP, and ACTA2 in LX‐2 cells transfected with either sgGFP or sgSIRT6 plasmids in the absence or presence of TGF‐β1. Scale bar = 10 μm. Data are presented as mean ± SEM. # p < .05, ## p < .001, and ### p < .001 for TGF‐β1 treated versus non‐treated, and *p < .05, **p < .01 and ***p < .001 for SIRT6 or sgSIRT6 versus control.
In addition to the modulation of Yap nuclear translocation, we also analyzed the acetylation levels of Yap and Taz in primary HSCs isolated from the Sirt6 HSC‐KO mice and WT mice. Yap or Taz protein was immunoprecipitated using specific antibodies and total acetylation levels were analyzed using acetylated lysine antibodies. Our data showed an increase in Yap and Taz acetylation in the Sirt6‐deficient HSCs as compared to WT HSCs (Figures 6A and S6A,B).
FIGURE 6.
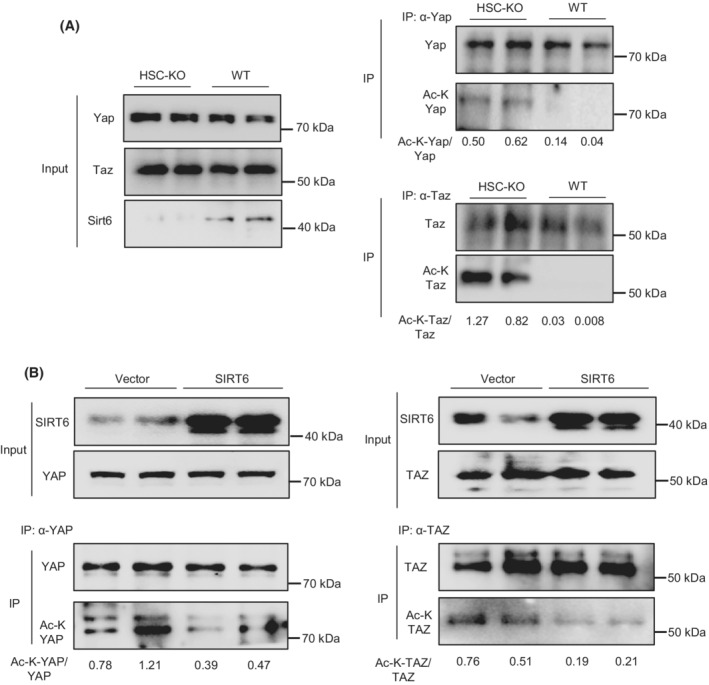
YAP and TAZ acetylation is modulated by SIRT6 in HSCs. (A) Yap and Taz acetylation analysis in primary HSCs from WT and HSC‐KO mice (n = 2). (B) YAP and TAZ acetylation analysis in LX‐2 cells transfected with either vector control or SIRT6 plasmids and treated with TGF‐β1 (n = 2).
Moreover, we overexpressed SIRT6 and analyzed YAP and TAZ acetylation in LX‐2 cells. Our data showed that the acetylation levels in YAP and TAZ were both downregulated by the SIRT6 overexpression (Figure 6B). Additionally, we analyzed Yap and Taz acetylation in the liver of WT and Sirt6‐LKO mice. Our data showed that both Yap and Taz were hyperacetylated and Yap was hypophosphorylated in the LKO livers compared to the WT controls (Figure S7A,B).
3.4. SIRT6 physically interacts with YAP and TAZ
As we observed a decrease in acetylation levels of YAP and TAZ by the SIRT6 overexpression, we hypothesized that SIRT6 might directly interact and deacetylate YAP and TAZ. To test this hypothesis, we co‐transfected Flag‐tagged YAP, TAZ, VGLL4, and SMAD3 along with HA‐tagged SIRT6 into LX‐2 cells with or without 5 ng/ml TGF‐β1 treatment. We pulled down protein complexes using Flag antibodies and analyzed the immunoprecipitated proteins by immunoblotting. Our data showed that SIRT6 indeed interacted with YAP, TAZ, and VGLL4 (SMAD3 served as a positive control), and those interactions were increased under the TGF‐β1 stimulatory conditions (Figure 7A). Next, we examined whether the catalytic activity of SIRT6 is required or not for its interaction with YAP or TAZ by overexpressing WT SIRT6 or catalytically inactive SIRT6 (H133Y) in LX‐2 cells in the presence of 5 ng/ml TGF‐β1. The data showed that the H133Y mutant did not interfere with the SIRT6 interaction with YAP or TAZ (Figure 7B). Moreover, we also verified protein–protein interactions between endogenous YAP or TAZ and SIRT6 in LX‐2 cells treated with TGF‐β1. For this experiment, we used PCNA as a negative control and SMAD3 as a positive control. Our data showed that the endogenous SIRT6 also interacted with YAP or TAZ (Figure 7C).
FIGURE 7.
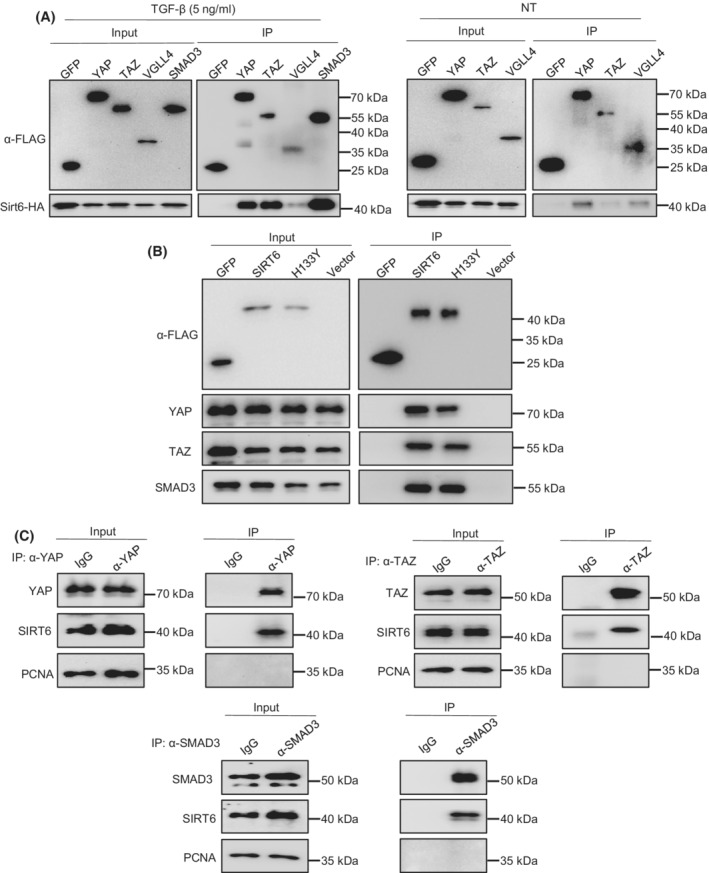
SIRT6 interacts with YAP, TAZ, and VGLL4 in HSCs. (A) Co‐IP analysis of interactions between Flag‐tagged YAP, TAZ, VGLL4, or SMAD3 and HA‐tagged SIRT6 in LX‐2 cells treated with or without 5 ng/ml TGF‐β1. SMAD3 was used as a positive control. (B) Co‐IP analysis of interactions between Flag‐tagged WT‐SIRT6 or mutant SIRT6 (H133Y) and YAP or TAZ in LX‐2 cells treated with 5 ng/ml TGF‐β1. (C) Co‐IP analysis of interactions between endogenous YAP or TAZ and SIRT6 in LX‐2 cells treated with TGF‐β1.
3.5. SIRT6 deacetylates YAP and TAZ and alters the TEAD protein complex composition
To assess whether SIRT6 directly regulates YAP or TAZ acetylation, we overexpressed WT or mutant SIRT6 (H133Y) in the presence of TGF‐β1. Our data showed that overexpression of WT SIRT6 but not mutant SIRT6 decreased YAP and TAZ acetylation levels (Figure 8A,B). SMAD3 was used as a positive control (Figure 8C). These data suggest that the catalytic activity of SIRT6 is required for the deacetylation of YAP and TAZ. As YAP and TAZ normally coactivate TEAD transcription factors, we examined whether SIRT6 alters the TEAD protein complex formation. We performed Co‐IP analysis using TEAD1 antibody in vector control or SIRT6 transfected LX‐2 cells in the presence of TGF‐β1. Our data showed that SIRT6 overexpression remarkably reduced the TEAD1‐YAP interaction and moderately decreased the TEAD1‐TAZ interaction while the TEAD1‐VGLL4 interaction was dramatically increased (Figure 8D). This suggests that SIRT6 suppresses the interaction of TEAD1 with coactivators YAP/TAZ and promotes the interaction with corepressor VGLL4.
FIGURE 8.
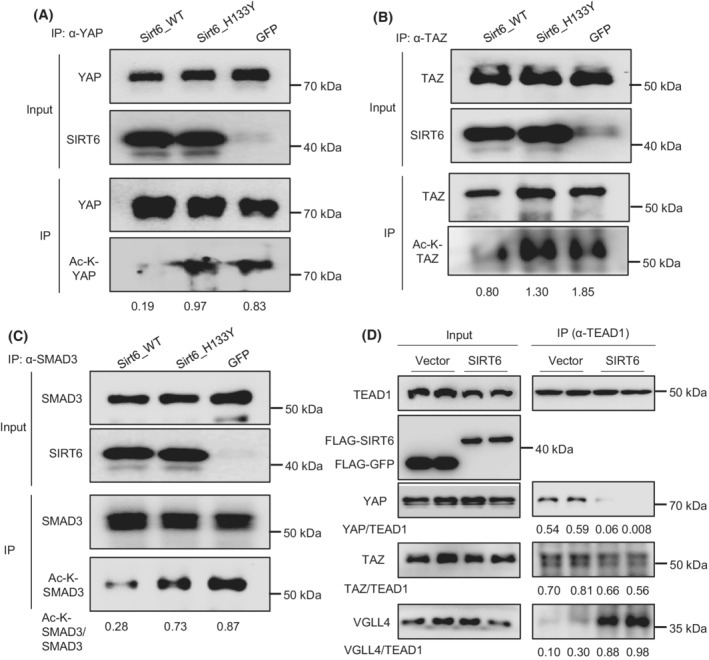
SIRT6 deacetylates YAP and TAZ and alters the TEAD protein complex composition in HSCs. (A–C) YAP, TAZ, and SMAD3 acetylation analysis in LX‐2 cells transfected with either GFP, WT Sirt6, or mutant Sirt6 (H133Y) in the presence of 5 ng/ml TGF‐β1. (D) Co‐IP analysis of interactions between TEAD1 and YAP, TAZ, or VGLL4 in LX‐2 cells transfected with either vector control or SIRT6 plasmids in the presence of 5 ng/ml TGF‐β1.
To further investigate the deacetylation of YAP by SIRT6, we first checked several lysine residues that have been previously reported to be acetylated (Figure 9A). 31 Using site‐directed mutagenesis, we generated single, double, or triple mutations of lysine (K) to arginine (R) in human YAP1. Next, we transfected these mutants into LX‐2 cells and analyzed three downstream target genes including CYR61, CTGF, and ankyrin repeat domain 1 (ANKRD1). Our data showed that the K102R mutant had the largest negative effect on the YAP1 activity, followed by K90R, K440R, K97R, and K76R (Figure 9B). The relative contribution of these lysine residues to the overall acetylation levels was analyzed by immunoblotting (Figure 9C). According to the YAP and TAZ amino acid sequence alignments for the conserved lysine residues, 32 we examined two lysine residues in the mouse Taz TEAD‐binding domain, and our data showed that K39R exerted a more negative effect than K54R on the Taz activity and acetylation levels (Figure 10A–C).
FIGURE 9.
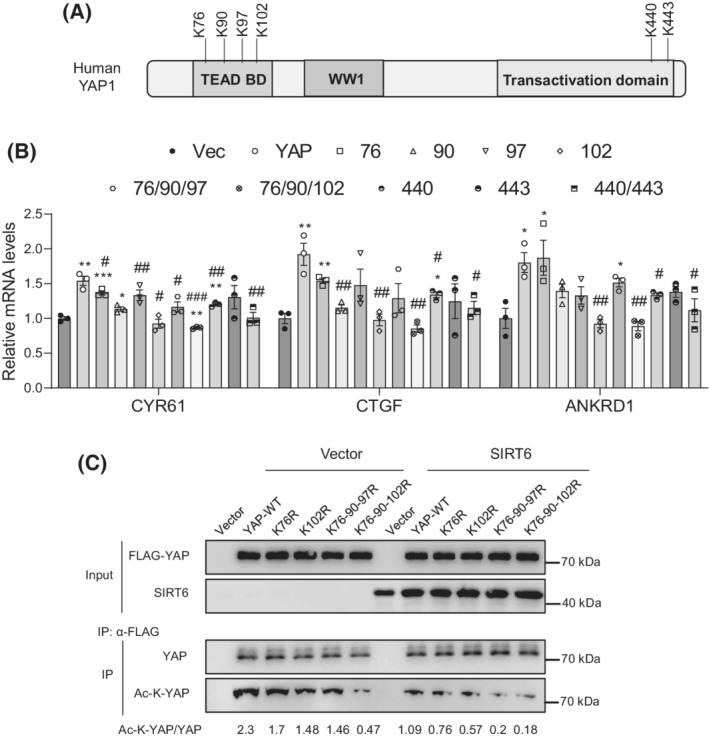
SIRT6 deacetylates YAP1 at multiple lysine residues. (A) A diagram of human YAP1 domain structure and several known acetylated lysine residues. (B) Real‐time PCR analysis of CYR61, CTGF, and ANKRD1 mRNAs in the LX‐2 cells transfected with either WT or mutant human YAP1 plasmids in the presence of 5 ng/ml TGF‐β1 (n = 3). (C) YAP1 acetylation analysis in LX‐2 cells transfected with WT or mutant human YAP1 plasmids together with vector or SIRT6 plasmids in the presence of 5 ng/ml TGF‐β1. Data are presented as mean ± SEM. *p < .05, **p < .01, ***p < .001 versus vector control, and # p < .05, ## p < .001, ### p < .001 versus WT YAP.
FIGURE 10.
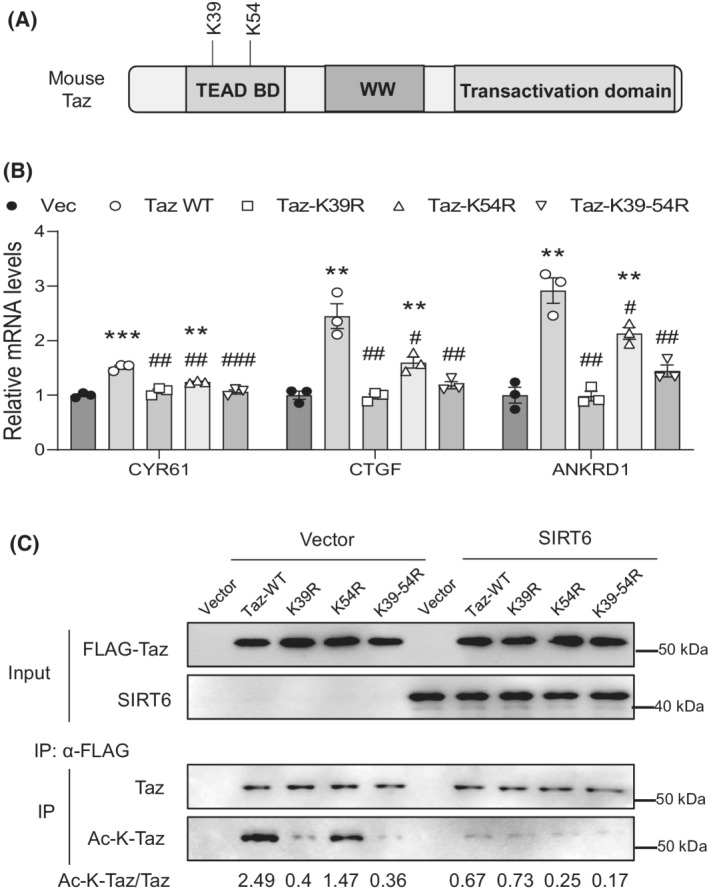
SIRT6 deacetylates Taz at two lysine residues in the TEAD‐binding domain. (A) A diagram of mouse Taz protein domain structure and two conserved lysine residues. (B) Real‐time PCR analysis of CYR61, CTGF, and ANKRD1 mRNAs in the LX‐2 cells transfected with either WT or mutant mouse Taz plasmids in the presence of 5 ng/ml TGF‐β1 (n = 3). (C) Taz acetylation analysis in LX‐2 cells transfected with either WT or mutant mouse Taz plasmids together with vector or SIRT6 plasmids in the presence of 5 ng/ml TGF‐β1. Data are presented as mean ± SEM. *p < .05, **p < .01, ***p < .001 versus vector control, and # p < .05, ## p < .001, ### p < .001 versus WT Taz.
4. DISCUSSION
In this work, we have uncovered a critical regulatory pathway in which SIRT6 suppresses the Hippo downstream effectors including YAP, TAZ, and TEAD. YAP and TAZ have been implicated in the development of hepatic fibrosis. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 In a chemically induced fibrosis mouse model and human fibrotic livers, hepatic YAP is activated. 17 Chemical inhibition of YAP using verteporfin has been shown to suppress the HSC activation in mice. 17 , 33 Interestingly, overexpression of YAP in hepatocytes also induces hepatic inflammation and fibrosis. 9 YAP can be activated by free fatty acids in hepatocytes, and this can be reversed by inhibition of p38δ MAPK. 6 YAP can be activated by Hedgehog signaling, and both drive the HSC transdifferentiation and proliferation through the regulation of glutaminolysis. 13 TAZ protein levels are elevated in the animal and human NASH livers. 9 , 15 , 19 TAZ can be activated by elevated cholesterol levels in the liver through a PKA‐Ca2+‐RHOA pathway. 8 Hepatocyte TAZ has been shown to induce expression of Indian Hedgehog ligands that promote activation of HSCs and hepatic fibrosis. 15
Based on amino acid sequence alignments of YAP and TAZ across different species including human, mouse, and zebra fish, K76 of YAP and K39 of TAZ are the only conserved lysine residue in the TEAD binding domain between YAP and TAZ proteins. 32 Our data suggest that acetylation at K39 in TAZ but not K76 in YAP plays a more important role in the regulation of their respective protein activity. However, K102 of YAP, which aligns with G59 of TAZ, has the most significant role in the regulation of the YAP activity among the six lysine residues analyzed in this study. In addition, K443 of YAP in the transactivation domain also plays a significant role in the regulation of the YAP activity.
As SIRT6 is an NAD+‐dependent deacetylase, our data suggest that the catalytic activity of SIRT6 is required to regulate the acetylation levels and activities of YAP and TAZ in HSCs. Our site‐directed mutagenesis data support the significance of deacetylation of YAP and TAZ at multiple lysine residues, especially K102 of YAP and K39 of TAZ. Previous studies have reported that SIRT1 also modulates YAP acetylation. 31 , 34 Hata et al. have reported that in response to the SN2 alkylating agents such as methyl methanesulfonate (MMS), YAP1 acetylation at the two conserved C‐terminal distal lysine residues K494/K497 (corresponding to K440 and K443 in our study) are increased in HEK293T and HeLa cells by CBP/p300, which can be decreased by SIRT1. It is interesting to note that our K→R mutations of those two residues lead to reduced YAP coactivator activity in response to the TGF‐β1 treatment whereas Hata et al. have shown that the double 494/497K→R YAP mutant leads to increased luciferase reporter activities in response to MMS. In another report, Mao et al. have shown that YAP2 can be acetylated by CBP/p300 and deacetylated by SIRT1 in HEK293T and HepG2 cells. The authors have also used aggregated K→R mutations to suggest that four lysine residues in the TEAD binding domain—K76/K90/K97K102 and two C‐terminal distal lysine residues K494/K497 can be both deacetylated by SIRT1 but the four lysine residues in the TEAD binding domain play a more significant role in the coactivation of TEAD transcription factors. In contrast, our study has pinpointed that the deacetylation of K102 in YAP1 by SIRT6 has the strongest negative effect on the YAP1 coactivation activity in HSCs. We believe that besides the differences in cell type and signaling context, specific lysine deacetylation and protein complex dynamics also significantly contribute to the overall outcome of the YAP function. When SIRT6 is abundant and active, YAP and TAZ are hypoacetylated and less engaged with TEAD transcription factors in HSCs, and this promotes a quiescent state of HSCs. Under pathogenic conditions such as NAFLD, either SIRT6 amount or activity may be significantly decreased, and this leads to YAP/TAZ hyperacetylation, active engagement with TEAD transcription factors, exclusion of VGLL4 from the TEAD protein complex, and subsequently HSC activation (Figure 11).
FIGURE 11.
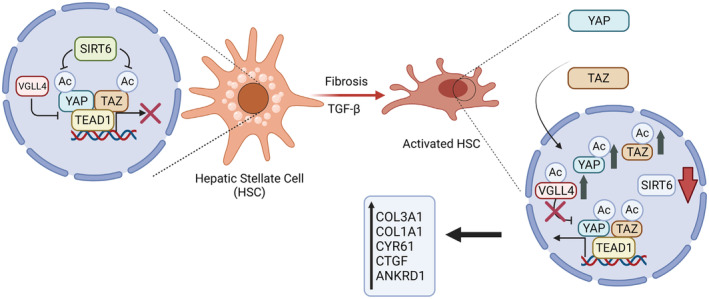
A working model for the role of SIRT6 in the regulation of YAP/TAZ and hepatic fibrogenesis.When SIRT6 is sufficiently active, YAP and TAZ are suppressed in HSCs. This promotes a quiescent state of HSCs. Under pathogenic conditions, SIRT6 activity is markedly reduced, and this leads to highly acetylated YAP and TAZ that promote HSC activation and fibrogenesis.
In summary, our data have shown a critical role of SIRT6 in the regulation of the Hippo pathway during hepatic fibrogenesis. SIRT6 interacts with and deacetylates YAP and TAZ at multiple conserved lysine residues. In addition, SIRT6 also reprograms the TEAD protein complex by removing the YAP and TAZ coactivators and recruiting the VGLL4 corepressor. In doing so, SIRT6 suppresses HSC activation and liver fibrosis. These findings support the therapeutic potential of SIRT6 in the prevention and treatment of hepatic fibrosis.
AUTHOR CONTRIBUTIONS
Kushan Chowdhury was responsible for the acquisition of data, analysis, and interpretation of data, drafting, and writing of the manuscript. Menghao Huang was responsible for the acquisition of data, analysis, and interpretation of data, and drafting of the manuscript. Hyeong‐Geug Kim was responsible for the acquisition of data. X. Charlie Dong was responsible for the study concept and design, analysis, and interpretation of data, drafting of the manuscript, statistical analysis, obtaining funding, and study supervision.
DISCLOSURES
The authors declare no conflicts of interest.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1
Table S2
ACKNOWLEDGMENTS
The authors want to thank Dr. Heather Francis, Dr. Hongxia Ren, and Dr. Ronald C. Wek at Indiana University School of Medicine for their comments and suggestions. This work was supported in part by the following funding sources: R01DK121925 (X.C.D.), R01DK120689 (X.C.D.), R01AA028506 (X.C.D), and R21AG072288 (X.C.D). The sponsors had no role in the study design and data collection, analysis, or interpretation.
Chowdhury K, Huang M, Kim H‐G, Dong XC. Sirtuin 6 protects against hepatic fibrogenesis by suppressing the YAP and TAZ function. The FASEB Journal. 2022;36:e22529. doi: 10.1096/fj.202200522R
Kushan Chowdhury and Menghao Huang contributed equally to this work.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol. 2016;11:451‐496. [DOI] [PubMed] [Google Scholar]
- 2. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537‐2564. [DOI] [PubMed] [Google Scholar]
- 3. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology. 2020;158:1913‐1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397‐411. [DOI] [PubMed] [Google Scholar]
- 5. Kim CL, Choi SH, Mo JS. Role of the hippo pathway in fibrosis and cancer. Cell. 2019;8:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salloum S, Jeyarajan AJ, Kruger AJ, et al. Fatty acids activate the transcriptional coactivator YAP1 to promote liver fibrosis via p38 mitogen‐activated protein kinase. Cell Mol Gastroenterol Hepatol. 2021;12:1297‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dai Y, Hao P, Sun Z, et al. Liver knockout YAP gene improved insulin resistance‐induced hepatic fibrosis. J Endocrinol. 2021;249:149‐161. [DOI] [PubMed] [Google Scholar]
- 8. Wang X, Cai B, Yang X, et al. Cholesterol stabilizes TAZ in hepatocytes to promote experimental non‐alcoholic steatohepatitis. Cell Metab. 2020;31:969‐986.e967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mooring M, Fowl BH, Lum SZC, et al. Hepatocyte stress increases expression of yes‐associated protein and transcriptional coactivator with PDZ‐binding motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology. 2020;71:1813‐1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Y, Tu K, Liu D, et al. p300 acetyltransferase is a cytoplasm‐to‐nucleus shuttle for SMAD2/3 and TAZ nuclear transport in transforming growth factor beta‐stimulated hepatic stellate cells. Hepatology. 2019;70:1409‐1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X, Sommerfeld MR, Jahn‐Hofmann K, et al. A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol Commun. 2019;3:1221‐1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y, Lu T, Zhang C, et al. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia‐reperfusion injury. J Hepatol. 2019;71:719‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du K, Hyun J, Premont RT, et al. Hedgehog‐YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154:1465‐1479.e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang K, Chang Y, Shi Z, et al. Omega‐3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci Rep. 2016;6:30029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Zheng Z, Caviglia JM, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2016;24:848‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin K, Pritchett J, Llewellyn J, et al. PAK proteins and YAP‐1 signalling downstream of integrin beta‐1 in myofibroblasts promote liver fibrosis. Nat Commun. 2016;7:12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mannaerts I, Leite SB, Verhulst S, et al. The hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679‐688. [DOI] [PubMed] [Google Scholar]
- 18. Ma S, Meng Z, Chen R, Guan KL. The hippo pathway: biology and pathophysiology. Annu Rev Biochem. 2019;88:577‐604. [DOI] [PubMed] [Google Scholar]
- 19. Khajehahmadi Z, Mohagheghi S, Nikeghbalian S, et al. Downregulation of hedgehog ligands in human simple steatosis may protect against nonalcoholic steatohepatitis: is TAZ a crucial regulator? IUBMB Life. 2019;71:1382‐1390. [DOI] [PubMed] [Google Scholar]
- 20. Dong XC, Chowdhury K, Huang M, Kim HG. Signal transduction and molecular regulation in fatty liver disease. Antioxid Redox Signal. 2021;35:689‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang J, Li Y, Liu Q, et al. Sirt6 alleviated liver fibrosis by deacetylating conserved lysine 54 on Smad2 in hepatic stellate cells. Hepatology. 2021;73:1140‐1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhong X, Huang M, Kim HG, et al. SIRT6 protects against liver fibrosis by deacetylation and suppression of SMAD3 in hepatic stellate cells. Cell Mol Gastroenterol Hepatol. 2020;10:341‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang M, Kim HG, Zhong X, et al. Sestrin 3 protects against diet‐induced nonalcoholic steatohepatitis in mice through suppression of transforming growth factor beta signal transduction. Hepatology. 2020;71:76‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High‐yield and high‐purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015;10:305‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aparicio‐Vergara M, Tencerova M, Morgantini C, Barreby E, Aouadi M. Isolation of Kupffer cells and hepatocytes from a single mouse liver. Methods Mol Biol. 2017;1639:161‐171. [DOI] [PubMed] [Google Scholar]
- 27. Chang W, Yang M, Song L, et al. Isolation and culture of hepatic stellate cells from mouse liver. Acta Biochim Biophys Sin (Shanghai). 2014;46:291‐298. [DOI] [PubMed] [Google Scholar]
- 28. Jin J, Li W, Wang T, Park BH, Park SK, Kang KP. Loss of proximal tubular Sirtuin 6 aggravates unilateral ureteral obstruction‐induced tubulointerstitial inflammation and fibrosis by regulation of beta‐catenin acetylation. Cell. 2022;11:1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu L, Wu Y, Wang P, et al. PSC‐MSC‐derived exosomes protect against kidney fibrosis in vivo and in vitro through the SIRT6/beta‐catenin signaling pathway. Int J Stem Cells. 2021;14:310‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai J, Liu Z, Huang X, et al. The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking beta‐catenin target gene expression. Kidney Int. 2020;97:106‐118. [DOI] [PubMed] [Google Scholar]
- 31. Hata S, Hirayama J, Kajiho H, et al. A novel acetylation cycle of transcription co‐activator yes‐associated protein that is downstream of hippo pathway is triggered in response to SN2 alkylating agents. J Biol Chem. 2012;287:22089‐22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reggiani F, Gobbi G, Ciarrocchi A, Sancisi V. YAP and TAZ are not identical twins. Trends Biochem Sci. 2021;46:154‐168. [DOI] [PubMed] [Google Scholar]
- 33. Yu HX, Yao Y, Bu FT, et al. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol Immunol. 2019;107:29‐40. [DOI] [PubMed] [Google Scholar]
- 34. Mao B, Hu F, Cheng J, et al. SIRT1 regulates YAP2‐mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene. 2014;33:1468‐1474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1
Table S2
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.