

ABSTRACT
Early‐onset osteoporosis (EOOP), characterized by low bone mineral density (BMD) and fractures, affects children, premenopausal women and men aged <50 years. EOOP may be secondary to a chronic illness, long‐term medication, nutritional deficiencies, etc. If no such cause is identified, EOOP is regarded primary and may then be related to rare variants in genes playing a pivotal role in bone homeostasis. If the cause remains unknown, EOOP is considered idiopathic. The scope of this review is to guide through clinical and genetic diagnostics of EOOP, summarize the present knowledge on rare monogenic forms of EOOP, and describe how analysis of bone biopsy samples can lead to a better understanding of the disease pathogenesis. The diagnostic pathway of EOOP is often complicated and extensive assessments may be needed to reliably exclude secondary causes. Due to the genetic heterogeneity and overlapping features in the various genetic forms of EOOP and other bone fragility disorders, the genetic diagnosis usually requires the use of next‐generation sequencing to investigate several genes simultaneously. Recent discoveries have elucidated the complexity of disease pathogenesis both regarding genetic architecture and bone tissue‐level pathology. Two rare monogenic forms of EOOP are due to defects in genes partaking in the canonical WNT pathway: LRP5 and WNT1. Variants in the genes encoding plastin‐3 (PLS3) and sphingomyelin synthase 2 (SGMS2) have also been found in children and young adults with skeletal fragility. The molecular mechanisms leading from gene defects to clinical manifestations are often not fully understood. Detailed analysis of patient‐derived transiliac bone biopsies gives valuable information to understand disease pathogenesis, distinguishes EOOP from other bone fragility disorders, and guides in patient management, but is not widely available in clinical settings. Despite the great advances in this field, EOOP remains an insufficiently explored entity and further research is needed to optimize diagnostic and therapeutic approaches. © 2022 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: OSTEOPOROSIS, GENETIC RESEARCH, WNT/β‐CATENIN/LRPS, BONE MODELING AND REMODELING, BONE HISTOMORPHOMETRY
Introduction
Osteoporosis, characterized by deterioration of bone microstructure, low bone mineral density (BMD), and fractures, is a common disease in the elderly population and especially in postmenopausal women.( 1 , 2 ) Because fractures often require hospital care and associate with increased mortality, osteoporosis has considerable human, social, and economic implications worldwide.( 3 , 4 ) On rare occasions, osteoporosis may present already in childhood or early adulthood as a consequence of another condition (eg, chronic diseases of organs such as liver, kidney, lungs and bowel, chronic inflammatory diseases, endocrine diseases including hypogonadism, and neuromuscular disorders), hematological and oncological diseases, long‐term medication (eg, glucocorticoid therapy, cancer treatment), or as a result of nutritional deficiencies (eg, anorexia nervosa, celiac disease, or deficient calcium or vitamin D intake).( 1 , 5 , 6 ) In addition to these secondary forms of osteoporosis, skeletal fragility at an early age can result from a pathogenic variant in a gene playing a pivotal role in bone metabolism.( 7 , 8 ) The diagnosis of early‐onset osteoporosis (EOOP) encompasses all the above‐mentioned forms of osteoporosis presenting in premenopausal women and in men <50 years of age.
Bone undergoes continuous cycles of formation and resorption through bone modeling and bone remodeling.( 9 ) Bone modeling primarily occurs during childhood and adolescence and is responsible for skeletal growth and shaping of the bones in response to mechanical loading and other (eg, endocrine) factors.( 10 ) Upon achievement of peak bone mass by early adulthood, bone mass remains stable in midlife and begins to decline thereafter; this decline is particularly rapid in women during a few years after menopause.( 11 ) Bone remodeling, on the other hand, refers to the continuous renewal of bone tissue by the coupled activity of the three bone cell lineages: the osteoblasts, the osteoclasts, and the osteocytes.( 12 ) The bone extracellular matrix (ECM), which is primarily composed of type I collagen and noncollagenous proteins, plays a pivotal role in bone remodeling by conferring mechanical support and providing growth factors to the bone cells. Impairment of any of these processes can lead to inadequate bone mass accrual and maintenance, predisposing to osteoporosis.( 10 )
Despite extensive research, EOOP remains an inadequately characterized entity. In this review we aim to provide clarity to diagnostic approaches and elucidate the clinical, genetic and bone tissue features of EOOP. We use selected monogenic forms of EOOP to highlight the complex nature of genetics behind EOOP and to underscore the differences in bone tissue characteristics in some genetic forms of EOOP. Comprehensive information on these aspects is needed for patient identification and optimal management.
Definition and Subtypes of EOOP and Path to Diagnosis
The term EOOP refers to osteoporosis occurring in children and young adults. In comparison to postmenopausal osteoporosis, the definition of osteoporosis and intervention threshold in younger individuals are less clear, and the relationship between BMD and fracture risk have not been well established. In growing children, the diagnosis of osteoporosis requires the presence of a significant fracture history and low BMD, as defined by international guidelines (Fig. 1A ).( 13 ) The first criterion is defined as a history of at least two fractures before the age of 10 years or three or more fractures before adulthood.( 7 ) The latter criterion is defined as a BMD Z‐score ≤ −2.0 of the spine or total body. However, the presence of one or more spinal compression fractures in absence of major back trauma constitutes osteoporosis, even if BMD is normal.( 7 , 13 , 14 ) Only fractures resulting from low‐to‐moderate energy events are considered. Furthermore, minor fractures (eg, fingers, toes, nose) are not considered when evaluating fracture history.( 7 ) However, despite these diagnostic criteria, the clinical situation should be considered individually in each child and adolescent with suspected osteoporosis, as anticipated disease course and medications may require osteoporosis management even before the diagnostic criteria are fulfilled.( 6 )
Fig. 1.
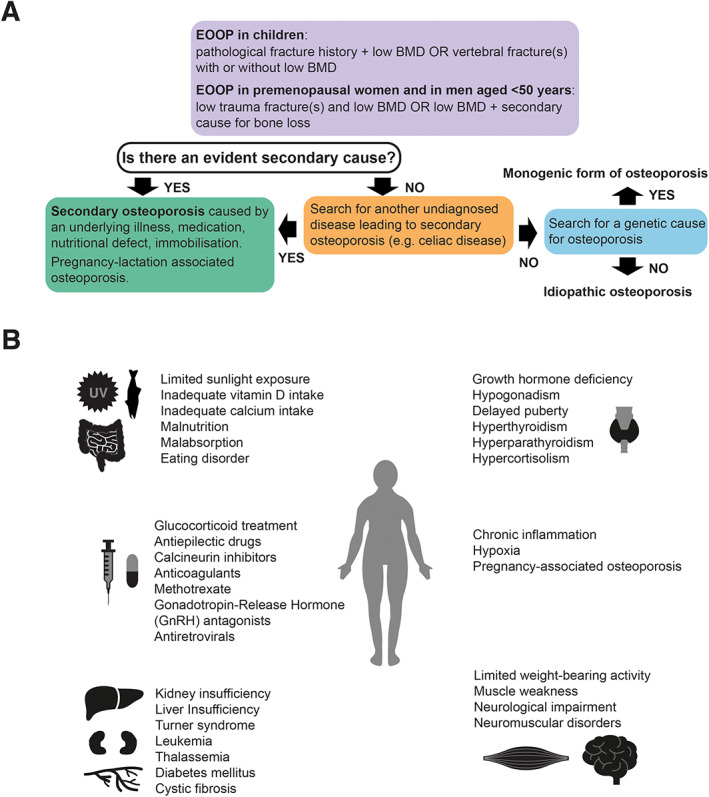
(A) Definition of EOOP and diagnostic path. (B) Summary of the main causes of secondary osteoporosis.
In adults, the diagnosis of EOOP can be given to premenopausal women and males <50 years of with a low BMD, defined as a BMD Z‐score ≤ −2.0 or T‐score ≤ −2.5 at the lumbar spine or femoral neck,( 1 ) when associating with either fragility fractures or an underlying chronic illness (Fig. 1A ). It is noteworthy that routine DXA studies are not performed in this age group but a dual‐energy X‐ray absorptiometry (DXA) scan is usually indicated after two or more fragility fractures (due to low‐to‐moderate energy trauma), after a fracture at an unusual site such as the spine or hip, or in the presence of a chronic illness or medication predisposing to osteoporosis.( 1 , 15 , 16 )
Overall, EOOP is a relatively rare condition, especially when not associated with an underlying chronic disease or other secondary factors (Fig. 1A ).( 1 , 7 ) Exclusion of such secondary causes may require extensive assessments because numerous clinical situations may lead to bone fragility (Fig. 1B ). More extensive lists and descriptions of secondary factors linked to osteoporosis have been described elsewhere.( 15 , 16 , 17 , 18 ) Moreover, EOOP can sometimes manifest with extraskeletal features, including ocular and neurological impairments.
To understand the cause of primary EOOP, genetic testing is recommended, especially if the disease manifests at a very young age or if there is a family history for osteoporosis. In unclear situations, after exclusion of secondary factors and especially if no pathogenic variants in the genes presently linked to bone fragility are identified or a new gene‐disease association is suspected, a bone biopsy may be considered for research purposes. Histology and histomorphometry of transiliac bone biopsy samples could give indication on the type of osteoporosis (eg, low‐turnover versus high‐turnover osteoporosis), unveil a mineralization defect (osteomalacia), or reveal rare causes of osteoporosis like mastocytosis or multiple myeloma. Bone biopsy analysis can also guide in diagnostics and in choosing the therapy, in cases where secondary osteoporosis has been excluded, but this method is not widely used in clinical settings. These aspects are discussed in detail in this review. Despite comprehensive search for secondary causes or genetic defects, the cause of EOOP may remain unknown and such forms are called idiopathic (Fig. 1A ).
Osteogenesis imperfecta (OI) is the best‐known genetic bone fragility disorder and, especially in its mild forms, is sometimes challenging to differentiate from other forms of EOOP. OI is a heterogenous condition mainly characterized by low BMD and frequent fractures, skeletal deformities, and disproportionate short stature.( 19 ) However, a spectrum of other features, including blue sclerae, dentinogenesis imperfecta, hearing loss, scoliosis, joint laxity, and cardiopulmonary impairments are often found in patients with OI. The first clinical classification of OI by Sillence and colleagues( 20 ) in 1979 divided OI into four types based on the severity of clinical features, ranging from mild OI with blue sclerae and a low number of fractures to perinatally lethal forms. Approximately 85% of OI cases are caused by pathogenic variants in the two genes encoding type I collagen, COL1A1 and COL1A2.( 21 ) Collagen type I defects can be either quantitative (low amount but normal quality of type I collagen due to, eg, stop gain mutations) or qualitative (abnormal type I collagen structure, eg, due to a missense mutation). Normally, qualitative defects lead to more severe phenotypes than changes in protein amount because they impair the structure of the collagen fibrils.( 22 ) Recently, the phenotypic and genetic spectrum of OI has considerably expanded and pathogenic variants in at least 16 other genes have been identified.( 23 , 24 ) Most of these genes play a pivotal role in synthesis, posttranslational modification, and processing of type I collagen.( 22 , 23 , 25 , 26 ) This heterogeneity in disease spectrum has complicated the disease classification and has led to challenges in defining each clinical entity in the Online Mendelian Inheritance in Man (OMIM) catalogue. In this catalogue, each disease has a unique phenotype MIM number, but occasionally more than one phenotype MIM number is linked to the same gene. For instance, biallelic variants in WNT1 are linked to “OI” (MIM 615220) whereas monoallelic variants in the same gene are associated with “susceptibility to osteoporosis” (MIM 615221).
In addition to OI, several other rare syndromes featuring bone fragility as a part of a broader phenotype have been identified. For example, osteoporosis‐pseudoglioma syndrome (OPPG, MIM 259770) and spondylo‐ocular syndrome (MIM 605822) are both characterized by severe childhood‐onset skeletal fragility and ocular manifestations.( 23 , 24 ) Further, various mineralization defects, such as hypophosphatasia (HPP) due to pathogenic variants in the gene encoding tissue‐nonspecific alkaline phosphatase (ALPL),( 27 ) or various inherited forms of hypophosphatemia, due to defective renal phosphate handling, lead to skeletal fragility.
Although the clinical classification of OI still largely follows the original Sillence classification,( 28 ) the International Nosology of Genetic Skeletal Disorders( 23 ) groups the various forms of OI and other primary skeletal fragility disorders into one category. Presently, all forms of osteoporosis presenting at an early age are often included under the umbrella term of EOOP. However, because several well‐defined conditions may present with early‐onset skeletal fragility, one should aim at establishing a specific diagnosis whenever possible, instead of using the diagnosis EOOP for all such cases.
In the present review our main focus is not the classical OI and the rare genetic forms directly linked to defective type I collagen, or the various mineralization disorders. Instead, we have chosen to highlight other less well‐known monogenic forms and idiopathic forms of EOOP to underscore the complex genetic and molecular architecture of EOOP, which is also reflected to the tissue‐level manifestations and bone material properties.
Clinical Vignettes
The following two patient descriptions highlight the diagnostic path and challenges in young patients with an unusual fracture history and suspected osteoporosis.
Patient 1
A 36‐year‐old woman was diagnosed with osteoporosis after developing back pain following her third pregnancy. Spinal radiographs indicated vertebral compression fractures in the thoracic spine. She had a height loss of 4 cm. Otherwise the fracture history was negative. She had earlier been diagnosed with atrophic gastritis and received vitamin B12 injections every 2–3 months. She had also undergone thyroidectomy for Hashimoto's thyroiditis and thyroid nodules and received adequate thyroxine replacement therapy.
After the initial diagnosis of osteoporosis based on DXA scanning, she was treated with alendronate until the age of 42 years. Her family history was positive for osteoporosis and fractures, prompting genetic studies for OI. Sequencing of COL1A1 and COL1A2 detected no pathogenic variants, and the cause of osteoporosis remained unknown. At 44 years she was again investigated for osteoporosis to determine the underlying cause. Clinical evaluation showed normal facial features with white sclerae, normal dentition, and absent ligament laxity or skeletal deformities. In addition to height loss, now at 7 cm, she had kyphosis and mild scoliosis. Radiographs showed significant osteopenia in long bones and multiple thoracic vertebral compression fractures in the thoracic spine (Fig. 2A ). DXA showed a BMD Z‐score of −2.9 at the lumbar spine, −1.0 at femoral neck, and −1.7 at the total body. Serum concentrations for calcium, phosphate, parathyroid hormone (PTH), 25‐OH‐vitamin D and thyroid parameters were normal, as were bone turnover markers urine N‐terminal telopeptide (U‐NTX), serum N‐terminal propeptide of type I collagen (S‐P1NP), and serum C‐terminal telopeptide of type I collagen (S‐ICTP). Due to her severe and unexplained spinal osteoporosis and fractures at young age, a transiliac bone biopsy was obtained and confirmed the diagnosis of osteoporosis, showing low bone volume, no osteomalacia, and very low bone turnover.
Fig. 2.
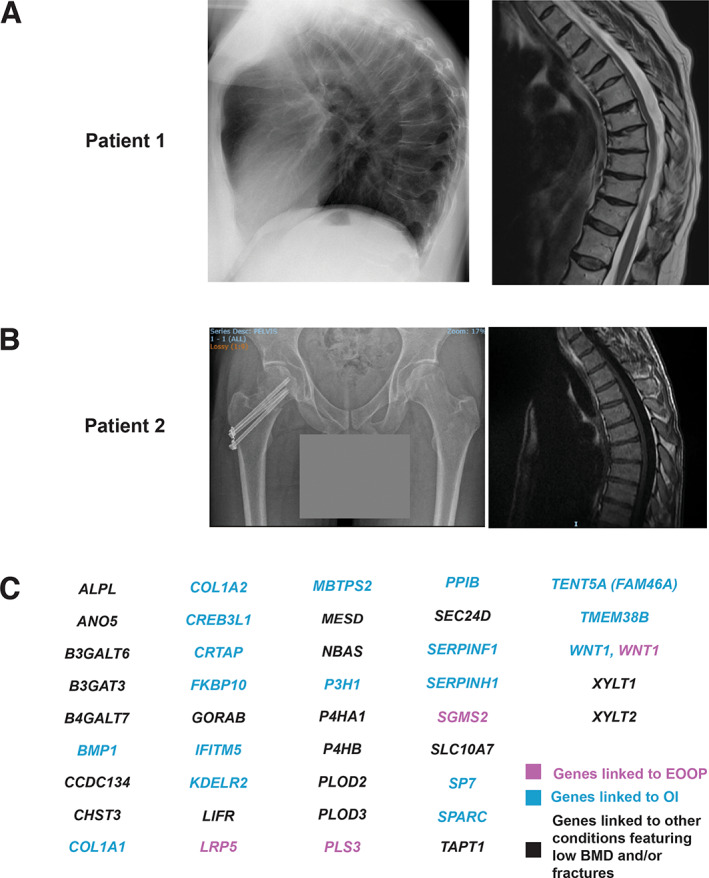
(A) Radiograph of patient 1 at 44 years of age (left) showing generalized osteoporosis with multiple compressions, narrow disk spaces and anterior lipping of the narrow disk spaces; increased thoracic kyphosis. Magnetic resonance imaging (MRI) of the thoracic spine 9 years later (right), at the start of teriparatide treatment, shows progression of the vertebral compression fractures in the thoracic spine. (B) Radiograph of patient 2 (left) showing that the femoral neck fracture was surgically corrected. The pelvis and femurs appear osteopenic. Spinal MRI (right) showing multiple vertebral compression fractures. (C) Gene panel used for patient 2.
The family history was again reviewed in detail and considered indicative of an autosomal dominant inheritance pattern with several male and female relatives, including the patient's mother and aunt, developing EOOP in childhood or early adulthood. Therefore, despite the history of pregnancy and lactation preceding osteoporosis and some chronic illnesse possibly contributing to bone fragility, a monogenic form of EOOP was considered. Linkage analysis followed by targeted sequencing lead to identification of a heterozygous missense mutation p.(Cys218Gly) in the WNT1 gene. WNT1 variants were not yet linked to EOOP at the time of analysis. The same mutation was confirmed by Sanger sequencing in all other affected family members.( 29 , 30 ) Because of the low bone turnover and the earlier, apparently unsuccessful, treatment with bisphosphonates, she was treated with teriparatide for 2 years with a modest increase in bone turnover markers but no significant improvement in BMD or bone quality (Fig. 2A ).( 31 ) Although the biopsy was taken 2 years after bisphosphonate treatment, the previous medication may have contributed to low turnover rate and also to the suboptimal response to teriparatide.
Patient 2
A 27‐year‐old man experienced a hip fracture after a moderate‐energy trauma (Fig. 2B ). He had sustained one previous fracture after a fall during childhood. DXA measurements were indicative of osteoporosis with a lumbar spine BMD T‐score of −3.0; femoral neck BMD showed osteopenia. Spinal radiographs and magnetic resonance imaging (MRI) revealed mild anterior wedging of two vertebral bodies (Fig. 2B ). These findings confirmed the presence of EOOP and prompted further investigations. Secondary causes, such as underlying endocrine or hematological diseases, were excluded (Table 1). The only remarkable finding was a relatively low serum alkaline phosphatase (ALP) of 36 U/L (Table 1). Diagnosis of HPP was thus considered. In addition to low ALP, HPP leads to increased levels of ALP substrates such as serum pyridoxal phosphate (vitamin B6) and urinary phosphoethanolamine.( 32 , 33 ) These substrates were not elevated, making the diagnosis of HPP unlikely but not completely excluded. To clarify the diagnosis, a bone biopsy was taken at the posterior iliac crest. It confirmed osteoporosis with normal mineralization but very low bone turnover. This was consistent with the finding of a low serum ALP despite normal values of the bone turnover markers beta‐isomerized type I collagen C‐terminal telopeptide (β‐CTx) and P1NP.
Table 1.
Laboratory Evaluations for Excluding Secondary Osteoporosis in Patient 2
| Parameter | Result | Reference range |
|---|---|---|
| Calcium | 2.45 mmol/L | 2.20–2.65 mmol/L |
| Phosphate | 1.11 mmol/L | 0.8–1.40 mmol/L |
| Alkaline phosphatase | 36 U/L | <115 U/L |
| Tryptase | 4.8 μg/L | <11.4 μg/L |
| Vitamin B6 | 101 nmol/L | 35–110 nmol/L |
| TSH | 1.83 mU/L | 0.26–4.27 mU/L |
| PTH | 0.8 and 1.2 pmol/L | 0.68–4.40 pmol/L |
| Testosterone | 19.7 nmol/L | 10–30 nmol/L |
| 25‐OH Vitamin D | 120 nmol/L | 50–120 nmol/L |
| b‐CTx | 0.53 μg/L | <1.0 μg/L |
| P1NP | 72 μg/L | 19.4–95.4 μg/L |
| BALP | 11.3 μg/L | <20.1 μg/L |
| Calcium (in urine) | 6.0 mmol/L per 24 hour | 2.5–7.5 mmol/L per 24 hour |
b‐CTx = beta‐isomerized type I collagen C‐telopeptide; BALP = bone‐specific alkaline phosphatase; P1NP = procollagen type I N‐propeptide; PTH = parathyroid hormone; TSH = thyroid stimulating hormone.
These prompted evaluations for genetic causes of his osteoporosis. The family history for osteoporosis was negative. No extraskeletal or OI‐type features were present. A targeted gene panel for 41 known genes linked to OI and other monogenic bone fragility disorders found no likely pathogenic variants nor variants of undetermined significance in these candidate genes (Fig. 2C ), including ALPL. The etiology thus remained unknown and he was diagnosed with idiopathic osteoporosis, in line with the patient′s young age, presence of low BMD and fragility fractures, absent secondary causes and no identified genetic cause,( 1 , 15 , 16 ) and supported by the findings in the bone biopsy.( 34 )
Treatment guidelines for this age group remain unestablished. Modification of lifestyle factors with exercise, healthy nutrition and adequate intake of calcium and vitamin D are advocated and when these measures are insufficient and fracture risk regarded as high, antiresorptive and anabolic drugs can be used. Due to his very low BMD and low bone turnover, teriparatide treatment was initiated and is still ongoing. Given the inherent low bone turnover, need for antiresorptive medication after the planned 2‐year treatment with teriparatide remains to be seen.
Approaches to Genetic Diagnostics in EOOP
Until not so long ago, a tentative diagnosis of bone disorders was based on a detailed clinical characterization and extensive literature review followed by testing for a specific candidate gene.( 35 ) With the exploding number of known disease genes and the possibility for parallel testing of several genes by next generation sequencing (NGS), the field of genetic diagnostics has adopted a strategy that is often called “sequencing first.”( 36 ) In EOOP, a solid clinical and biochemical phenotypic characterization remains crucial for establishing a correct diagnosis. However, genetic testing is recommended, especially if the disease manifests at a very young age or if there is a family history of osteoporosis.
There are several approaches to identify the disease‐specific candidate gene variants. Screening of individual sequences by Sanger sequencing has become the exception and reserved for hotspot variants causing well recognizable disorders; eg, achondroplasia due to the frequent p.(Gly380Arg) variant in FGFR3 ( 37 , 38 ) or Caffey disease due to the recurrent p.(Arg836Cys) change in COL1A1.( 39 ) If no such single candidate gene or variant is known, NGS gene panels can be used (Fig. 3). They can comprise dozens to several hundreds of genes associated with a certain disease group or phenotype.( 40 ) Although there are also amplification‐based gene panels, in most cases the sequences to be analyzed are isolated by enrichment using complementary, sequence‐specific probes (Fig. 3). However, EOOP‐specific genes alone are not sufficient to fill such a gene panel and are usually combined with genes linked to overlapping disorders, such as OI or disorders of bone mineralization (Fig. 2C ),( 23 , 26 ) until the total enriched sequence reaches a reasonable size, usually 50–500 kilobases (kb). Because of the limited size, each gene in such a panel is usually well covered and the results are very reliable. Phenotype‐specific custom‐designed panels are expensive and therefore standardized. Larger gene panels, which are not indication‐specific, have also been developed and may contain, eg, all known genes for monogenic disorders (OMIM genes). Currently, 4532 OMIM genes covering roughly 9 megabases (Mb) are known and require a sequencing output of 1 gigabases (Gb) for a good coverage. Although such panels may be technically unproblematic, determining the relevance of the panel findings for the individual patient may prove challenging. Moreover, all commercial laboratories use different gene panels and these must be repeatedly updated as novel disease genes are discovered. This problem is avoided with whole‐exome sequencing (WES).( 41 ) The exome is a large gene panel (around 45 Mb) enriching all exons of all 20,000 genes known so far (Fig. 3). Differences exist between exome versions; eg, some focus mainly on the coding sequences of verified protein‐coding genes and others comprise additional genes and more noncoding sequences. Exomes have gaps; for example, they only partially cover most of the introns. An advantage of using WES is the possibility of identifying novel gene‐disease associations.
Fig. 3.
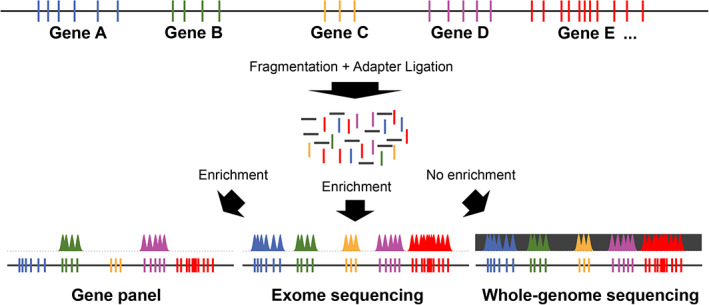
Different NGS‐based diagnostic approaches. A schematic genomic region is depicted containing five genes with different numbers of exons (colored bars). Interspersed noncoding sequences are represented in dark gray. After fragmentation of the entire genomic DNA and ligation of sequencing adapters the enrichment strategy determines which part of the genome is sequenced. Gene panels can be of different sizes, the exome contains all protein‐coding genes plus variable noncoding and putative genes, and the genome most of the coding and noncoding sequences.
Although most disease‐coding variants are exonic or located nearby in upstream or downstream regions, deep intronic variants leading to abnormal splicing can also be disease‐causing. A potential approach to identify noncoding variants influencing either gene expression or splicing is RNA sequencing.( 42 ) However, bone disease genes are often not expressed by readily available leukocytes, thus fibroblasts derived from a skin biopsy are needed as RNA source, limiting the use of this more invasive method in clinical settings due to ethical and patient‐related issues. Therefore, the all‐in‐one solution for coding and noncoding variants as well as structural variants (mostly copy number variants) is whole‐genome sequencing (WGS) (3 Gb) (Fig. 3).( 43 ) Although routine diagnostics has largely been taken over by gene panels and WES, WGS is likely to become the standard within the coming years also in the clinical settings,( 44 ) and especially in research searching novel disease genes.
Sequencing of 36 Mb of exome target sequence reveals around 90,000 single‐nucleotide variants (SNVs), or approximately one SNV in 400 base pairs (bp). This magnitude of variants can only be interpreted with the help of bioinformatics.( 45 ) Software tools allow filtering of variants according to their frequency, predicted or known pathogenicity, inheritance pattern, and phenotype relevance. Only with such information their significance can be determined. However, phenotype information as well as family history are crucial for the interpretation process and an integral factor in the filtering strategy based on the human phenotype ontology (HPO).( 46 , 47 ) Once the genetic cause is identified, it is possible to provide genetic counseling and guide in disease surveillance and management.
Unfortunately, the significance of a single variant often remains uncertain and many variants are determined as “of unknown significance” (VUS). In the clinical settings it is not possible to functionally test each variant and it remains a task for advanced research to establish which variants in specific genes could lead to disease manifestations.( 48 )
Sequencing family trios (index patient and parents) is more powerful than sequencing the sole index case because several variants can be discarded if they do not fit the inheritance pattern. In addition, WES and WGS also offer the possibility of combining the samples into cohort‐wide burden tests using collapsing analysis frameworks.( 49 ) Finally, WES and WGS data can be analyzed retrospectively. Due to the large size of the data, the use of NGS technologies requires high computational capacity and large data storage systems. Storing NGS data not only has high costs but it might also lead to ethical issues due to the presence of sensitive data.
Although the costs of genetic tests have decreased during the recent years, NGS, especially WGS, is still an expensive tool and not all clinics have the possibility to use this method on a routine basis. For this reason, the decision on the approach chosen for investigating a family with EOOP not only depends on the clinical findings but also on the financial resources of the laboratory/clinic.
Rare Genetic Forms of EOOP Highlight Complexity of Disease Pathogenesis
Although the International Skeletal Dysplasia Society (ISDS) Nomenclature includes a large number of monogenic conditions featuring bone fragility,( 23 ) most of the disorders in this category represent various types of OI with defects in type I collagen metabolism or other well‐defined entities with specific extraskeletal manifestations, such as ocular impairments like in spondylo‐ocular syndrome or cutaneous symptoms like in geroderma osteodysplasticum (MIM 231070). Only a handful of other monogenic forms of EOOP have been characterized. We have chosen to focus on these genetic diseases in which low BMD and fractures are the main features. The discussed genetic forms of EOOP include LRP5 and WNT1, involved in the WNT signaling pathway, plastin‐3 (PLS3), and sphingomyelin synthase 2 (SGMS2). These highlight the diversity in molecular pathology in EOOP and describe proteins with very diverse functions that are often not restricted to bone metabolism. These genetic conditions further underscore the importance of genetic testing in arriving at an exact diagnosis, for differentiating between clinically similar skeletal fragilities, and for considering possible extraskeletal symptoms related to the underlying molecular defect. Some of these monogenic forms were identified only recently and their pathomolecular mechanisms remain elusive.
Defects in the WNT signaling pathway
WNT signaling is one of the key pathways governing skeletal health—from early skeletal development to bone mass accrual throughout childhood and to bone mass maintenance and bone metabolism in adulthood.( 50 , 51 , 52 ) The deleterious skeletal consequences of aberrant WNT signaling are exemplified in several human skeletal disorders( 29 , 53 , 54 , 55 , 56 , 57 , 58 , 59 ) and the integral role of this pathway for bone homeostasis has been validated by extensive functional studies in both cell and animal models.( 60 , 61 , 62 , 63 , 64 , 65 , 66 )
The landmark discovery was the identification of biallelic loss‐of‐function mutations in the gene for low‐density lipoprotein (LDL) receptor‐related protein 5 (LRP5) in patients with OPPG.( 53 , 67 ) This disease presents in early childhood with low BMD and multiple peripheral and vertebral fractures, often involving the whole spine. The skeletal features are accompanied by congenital or infancy‐onset blindness (“pseudoglioma”) secondary to defective vascularization. LRP5 was identified as the key transmembrane co‐receptor for WNT ligands and the gatekeeper for the intracellular cascade regulating osteoblast function and bone formation( 52 , 68 , 69 ) (Fig. 4A‐C ). Variants in the LRP5 gene were subsequently linked to several bone mass abnormalities of both low and high bone mass, including phenotypes of EOOP in subjects with monoallelic loss of function (LOF) variants in LRP5 (MIM 166710).( 55 )
Fig. 4.
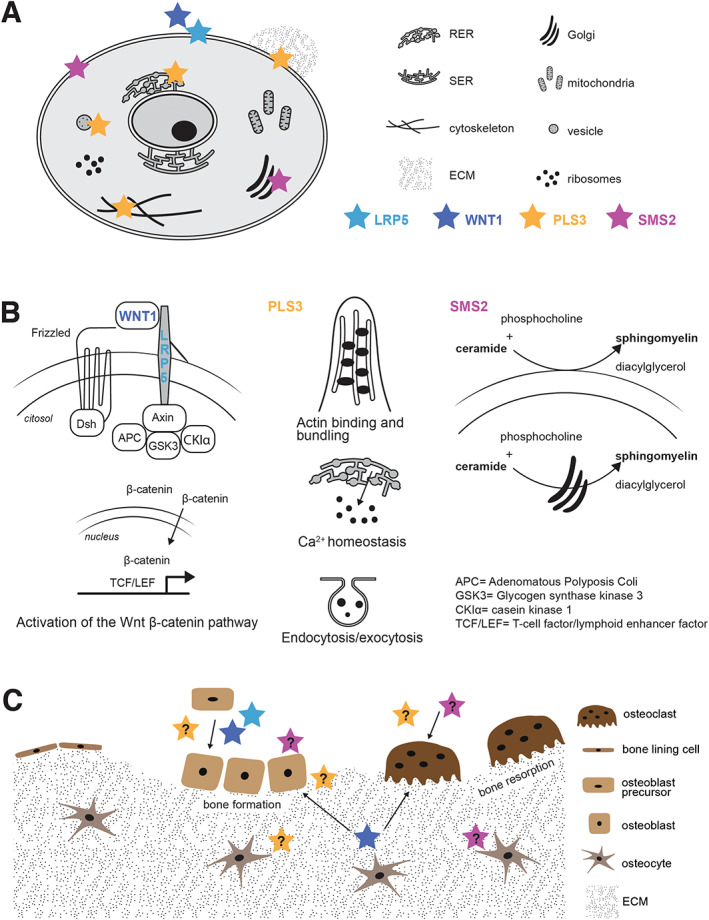
EOOP due to LRP5, WNT1, PLS3, and SMS2 defects. (A) Subcellular localization of the proteins encoded by LRP5, WNT1, PLS3, and SGMS2. (B) Molecular pathways involved in EOOP. (Left) When the WNT1 ligand binds to the receptor seven‐span transmembrane Frizzled and the co‐receptor LRP5, Dishevelled (Dsh) is recruited and the β‐Catenin destruction complex (composed by axin, APC, GSK3, and CK1α) is inactivated thus leading to the accumulation of β‐Catenin in the cytosol. Subsequently, β‐Catenin translocates to the nucleus and activates the transcription of target genes, including those responsible for osteoblast differentiation and function. Monoallelic loss‐of‐function variants in WNT1 and LRP5 lead to osteoporosis due to decrease activation of the canonical WNT signaling pathway. (Center) Plastin‐3, encoded by PLS3, is involved in several molecular mechanisms, including actin binding and bundling, calcium homeostasis, and cell endocytosis and exocytosis. (Right) The sphingomyelin synthase 2 (SMS2), encoded by SGMS2, is involved in sphingomyelin synthesis primarily in the plasma membrane but also in the Golgi apparatus. (C) LRP5 (light blue star), WNT1 (dark blue star), PLS3 (yellow star), and SMS2 (pink star) are involved in bone modeling and remodeling. Although both WNT and LRP5 are known to regulate osteoblast differentiation and function, the role of the other proteins is not fully understood yet. PLS3 might be involved in osteocyte mechanosensing, matrix mineralization, but also osteoblast differentiation and osteoclastogenesis. SMS2 could also partake in matrix mineralization, osteoblast activity, and osteoclastogenesis.
Compared to patients harboring biallelic variants, carriers of LRP5 variants feature a milder phenotype, characterized by low BMD and fractures but without severe eye impairments or with only mild vitreoretinopathy.( 53 , 70 ) Studies have identified monoallelic LRP5 variants in individuals with childhood or early‐adulthood onset symptomatic osteoporosis.( 55 ) In addition, both rare and common LRP5 variants have been associated with childhood bone mass, peak bone mass, and propensity to fractures( 55 , 71 , 72 , 73 ) and, in genomewide association studies (GWASs), with BMD and fractures in the general population.( 72 ) Some larger cohort studies on EOOP have identified various monoallelic LRP5 variants in affected adults, but the true contribution of these variants to disease pathogenesis, without functional evidence, has often remained uncertain.( 74 ) Pathogenic LRP5 variants have also been described in patients developing EOOP during pregnancy and lactation.( 70 ) Another study involving >350 individuals with EOOP, diagnosed based on low BMD, identified LRP5 or LRP6 variants in 8.3% of patients.( 74 ) Individuals carrying rare LRP5 or LRP6 variants had low bone turnover markers, in line with decreased WNT signaling( 74 ) but overall there was significant heterogeneity in skeletal manifestations and response to osteoporosis treatment in those harboring a variant.
Only years after the discovery of LRP5’s role in osteoporosis, WNT1 was identified as a key ligand to the canonical WNT pathway in bone (Fig. 4A,B ). We and others reported on biallelic or monoallelic WNT1 variants in subjects with low BMD and prevalent spinal and peripheral fractures.( 29 , 57 , 58 , 59 ) Monoallelic WNT1 variants cause a milder dominantly inherited phenotype with EOOP, peripheral and vertebral compression fractures, and loss of adult height (MIM 615221) whereas biallelic WNT1 variants result in a much more complicated phenotype reminiscent of OI with severe skeletal fragility, long‐bone deformities, kyphoscoliosis, and short stature (MIM 615220). After the first publications, several cases with biallelic WNT1 mutations have been reported,( 75 , 76 , 77 , 78 ) confirming the severe OI type III‐like clinical phenotype in affected individuals, who often show ptosis, a specific hallmark of this disease.( 75 )
Despite the fact that the phenotype in autosomal recessive WNT1 skeletal fragility is welldescribed, the full spectrum of clinical features associated with monoallelic WNT1 variants is less well characterized, with only less than 20 families reported to date (Table S1). We have described two large unrelated Finnish families with a monoallelic missense variant p.Cys218Gly and consequently EOOP with prevalent peripheral fractures in childhood and vertebral fractures later in adulthood, with otherwise normal growth and development.( 29 ) In our cohort, comprising 25 mutation‐positive individuals aged 11 to 76 years, the age‐of‐onset, fracture susceptibility, and disease progression were greatly variable. This is supported by other reports, describing patients with heterozygous WNT1 variants with normal to osteopenic BMD, no fractures, and normal quality of life, but also individuals with considerable skeletal fragility, hypotonia, joint hypermobility, delayed motor skills, and developmental abnormalities requiring extensive rehabilitation.( 59 , 79 ) Isolated findings include gastrointestinal symptoms, strabismus, and neurological symptoms.( 79 )
Although the significance of WNT1 ligand for bone homeostasis is well‐established, the exact underlying molecular mechanisms remain unclear.( 80 ) WNT1 partakes in the crosstalk between osteoblasts and hematopoietic stem cells to regulate bone cell development, differentiation, and proliferation in the developmental stages, and later, in adult bone, is secreted from osteocytes and activates adjacent osteoblasts and osteocytes in a paracrine manner (Fig. 4C ).( 64 ) Net yield from the activated pathway is anabolic, leading to increased bone formation and decreased bone resorption.( 50 , 64 , 81 ) Loss of Lrp5 in mice did not reduce the bone‐anabolic effect of Wnt1, thus suggesting that this co‐receptor is not needed for Wnt1 function.( 80 ) Aberrant WNT1 signaling in patients seems to primarily reduce cortical bone thickness due to decreased periosteal bone formation, whereas trabecular bone formation and rate of bone resorption are less affected.( 81 ) These are congruent to findings in patients' bone biopsies with low number of bone cells and low bone turnover rate.( 29 , 82 ) In‐depth immunohistochemistry of bone biopsies revealed increased fibroblast growth factor 23 (FGF23) expression, increased marrow adiposity, apoptotic osteocytes in cortical bone, and abnormalities in osteocyte morphology in individuals with prior bisphosphonate treatment.( 83 )
Optimal means for diagnosis and follow‐up of WNT1 patients are unknown. Despite the severely disturbed bone metabolism, serum concentrations of conventional metabolic bone markers are unchanged.( 82 , 84 ) An extensive biomarker survey found significantly elevated serum concentrations of both intact and C‐terminal FGF23 while, interestingly, the two osteocytic inhibitors of WNT signaling, dickkopf‐related protein 1 (DKK1) and sclerostin, were normal.( 84 ) Further, screening of serum samples for a panel of 192 common microRNAs (miRNAs) distinguished a unique miRNA signature in WNT1 mutation‐positive subjects with nine differentially expressed miRNAs.( 85 )
Plastin‐3 related osteoporosis
In 2013, the same year that WNT1 was discovered, a Dutch group reported on an X‐linked form of inherited primary osteoporosis presenting predominantly in males (MIM 300910).( 86 , 87 ) This form of EOOP was caused by hemizygous variants in the X‐chromosomal gene encoding plastin‐3, PLS3. The disease is defined in males by low BMD, multiple peripheral fractures, and especially spinal compression fractures in the thoracic spine since young childhood, leading to progressive kyphosis, severe spinal pathology, and height loss already by early adulthood.( 25 , 88 , 89 , 90 ) Mild extraskeletal features may also be present, such as facial dysmorphism, joint hypermobility, waddling gait, grayish sclerae, hearing loss, and opalescent teeth.( 86 , 87 , 91 , 92 , 93 , 94 , 95 , 96 , 97 , 98 ) Due to its X chromosomal inheritance pattern, males are more severely and earlier affected than females.( 93 )
To date, less than 30 pathogenic LOF PLS3 variants have been identified, mostly frameshift variants( 99 ) (including some partial and whole‐gene deletions( 92 , 94 , 100 ) and an intragenic duplication( 101 )) and nonsense variants,( 99 ) although some missense changes( 99 ) have also been found in individuals with EOOP. No genotype–phenotype correlation is observed and the variants affect both the regulatory domain and the four calponin homology domains of PLS3.( 99 ) Both frameshift variants and missense mutations can lead to severe osteoporosis.( 99 ) Moreover, common PLS3 variants have been associated with osteoporosis in postmenopausal women.( 86 , 102 )
In subjects with PLS3‐related EOOP, basic parameters of calcium homeostasis are normal and bone turnover markers are either normal or slightly reduced,( 84 , 87 , 94 , 103 ) as compared with age‐matched mutation‐negative controls. However, PLS3 mutation‐positive subjects had elevated DKK1 concentrations, suggesting that PLS3 impairment might also alter WNT signaling in bone. Similar to patients with WNT1 osteoporosis, seven miRNAs that are predicted to play a role in bone were found to be significantly upregulated or downregulated in patients with pathogenic PLS3 variants.( 84 , 104 )
Even if PLS3 has an undeniably important role in bone metabolism, the underlying molecular mechanisms, however, remain elusive. Plastins 1–3 (PLS1–PLS3) are a family of proteins that bind to and crosslink actin structures into larger fibers( 105 ) (Fig. 4A,B ). PLS3, the most abundant plastin, has a wide expression range and is highly expressed in adipocytes and endothelial cells but nearly absent in the bone marrow (https://www.proteinatlas.org).( 106 ) In general, PLS3 is more expressed in mesenchymal‐lineage cells than in myeloid‐lineage cells.( 106 ) In contrast, PLS2 is selectively enhanced in bone marrow and lymphoid tissues, especially in monocytes, but has a very low expression in mesenchymal‐lineage cells.( 106 ) Finally, PLS1 is primarily found in intestinal tissues and nearly absent in the bone marrow.( 106 ) PLS3 is an F‐actin binding and bundling protein that is involved in cytoskeletal remodeling (Fig. 4). However, this protein might also have roles in calcium homeostasis,( 107 ) vesicular trafficking, endocytosis, and exocytosis( 99 ) (Fig. 4). Interestingly, other medical conditions not affecting bone are also associated to PLS3 defects.( 99 ) For example, high PLS3 expression is associated with different types of hematological and solid cancers, including acute myeloid leukemia and colon cancer,( 108 , 109 , 110 ) but it is also found to be a protective modifier for spinal muscular atrophy.( 111 )
Although PLS2, which is highly expressed in hematopoietic cell lineages, has a function in osteoclast actin ring formation, movement, and bone resorptive activity,( 100 , 112 , 113 , 114 ) the information on the role of PLS3 in bone is still limited (Fig. 4C ). A recent study by Yorgan and colleagues( 115 ) showed that a Pls3 knockout mouse model features reduced cortical thickness without low BMD and a decreased mineralization capacity by osteoblasts. In contrast, Neugebauer and colleagues( 114 ) reported reduced cortical thickness with osteoporosis due to increased bone resorption in Pls3 knockout mice. However, the effects of PLS3 overexpression or knockout on osteoclasts are very modest.( 114 ) The fact that anti‐resorptive treatment in patients with PLS3‐related osteoporosis increases BMD, suggests that osteoclasts are active in PLS3‐related osteoporosis.( 84 , 87 , 100 ) PLS3 has also been suggested to play a role in osteocytes' mechanosensing and mechanotransduction (Fig. 4C ). Osteocyte shape is dependent on actin filaments and osteocyte processes are rich in actin.( 116 ) Moreover, mechanical forces generated by the ECM are transferred inside the cells through integrins, propagated to filaments of F‐actin and finally transmitted to the nucleus.( 117 ) In chicken, PLS3 is located in the dendrites of the osteocytes, supporting a role in mechanotransduction.( 118 ) Therefore, PLS3 could be especially important for osteocyte function (Fig. 4C ).
PLS3 has also been implicated in bone matrix mineralization. Transiliac bone biopsy samples in children and adults with PLS3 mutations show low bone turnover, often associated with increased osteoid( 89 , 93 ) and high cortical osteocyte apoptosis,( 83 , 94 ) uneven mineralization pattern in childhood, and a more uniform increase in mineralization in adults.( 31 , 94 ) During the mineralization process, matrix vesicles are formed by budding from the mineralizing cell microvilli. These contain, as a structural core, a dense bundle of cross‐linked actin microfilaments. In a mineralizing osteocyte‐like cell line PLS3 was present both in the budding matrix vesicles and in the apical microvilli from which the vesicles were formed.( 119 ) It can be hypothesized that defective PLS3 function disturbs this matrix vesicle‐mediated mineralization process, leading to defective mineralization that can be seen in patient‐derived bone biopsies. Nevertheless, the number of analyzed patients as well as the number of controls is limited. Moreover, PLS3‐osteoporosis arises during childhood, but the so far investigated bone biopsies are taken at an adult age. For this reason, some bone surfaces have already disappeared, thus this may imply a risk for potential bias when performing histomorphometric analyses.
Defects in sphingomyelin synthase 2
In 2019, we reported a novel disease gene underlying a rare skeletal disorder, “osteoporosis with calvarial doughnut lesions” (OP‐CDL; MIM 126550). OP‐CDL was clinically described already in 1990s,( 120 ) followed by several reports of similarly affected patients, but the genetic cause remained unknown until years later. Our study confirmed that monoallelic mutations in SGMS2, encoding sphingomyelin synthase 2 (SMS2), are responsible for OP‐CDL.
Sphingomyelin is a major lipid of the plasma membrane and is enriched in microdomains, lipid rafts, of the plasma membrane that are critical for signal transduction. SMS2 is an enzyme that resides primarily in the plasma membrane,( 121 , 122 ) where it synthesizes sphingomyelin from ceramide and phosphatidylcholine (Fig. 4A,B ).
The disease manifests as childhood‐onset osteoporosis with low BMD and frequent fractures. However, a much more severe skeletal dysplasia was also associated with the same gene. To date, three different SGMS2 variants have been identified and the disease severity varies depending on the underlying variant.( 123 , 124 ) Although the recurrent nonsense variant p.Arg50* associates with a milder phenotype featuring EOOP, the missense variants p.Ile62Ser and p.Met64Arg result in a more severe phenotype of spondylometaphyseal dysplasia with multiple spinal and peripheral fractures, scoliosis, and severe short stature.
Regardless of the SGMS2 variant, patients also exhibit a peculiar cranial feature of multiple sclerotic, doughnut‐shaped skull lesions. Although the radiographically observed lesions seem to be confined to the skull, radiographs portray similar uneven mineralization in long bones with alternating areas of increased density and osteopenic appearance.( 123 ) Furthermore, bone biopsies from affected patients show gross disturbance and inconsistency in bone matrix mineralization.( 123 , 125 ) Our detailed analyses of transiliac bone biopsies from two SGMS2 variant‐positive adult males with EOOP depicted an overall decrease in bone volume with disorganized arrangement of collagenous fibrils (woven bone appearance) and disruption in organization and continuity of the osteocyte–canalicular network and osteocyte orientation.( 125 ) Moreover, we showed that eroded surface to bone surface ratio (ES/BS) was highly elevated in both patients, while the osteoclast surface to bone surface ratio (Oc.S/BS) showed variable outcomes.( 125 ) Although another study also revealed reduced cortical volumetric BMD and thickness in another two patients with OP‐CDL,( 124 ) the number of osteoclasts and the extent of eroded surface in the iliac crest were slightly reduced in these patients.( 124 ) Patient‐derived osteoclasts, induced form CD14+ cells, did neither differentiate nor resorb differently.( 123 ) Similar to WNT1 and PLS3‐related osteoporosis, markers of bone metabolism are normal but circulating levels of alkaline phosphatase tend to be elevated.( 123 )
Several features of this form of EOOP remain unclear but some hypotheses can be drawn from existing data. The cranial lesions, which seem to be confined to the calvarial bones, are an interesting feature of the disease. These lesions seem to be absent in childhood and appear during adulthood. They also seem to grow in size and become more lytic‐appearing with increasing age. Interestingly, the sclerotic/lytic calvarial lesions share some resemblance with calvarial lesions observed in tumor metastasis.( 126 , 127 ) Such bone lesions are triggered by a vicious cycle, which to a large extent is driven by growth factors and inflammatory cytokines.( 128 ) In fact, SMS2 has been linked to inflammatory responses and total suppression or inhibition of SMS2 reduces the inflammatory response in several specific conditions.( 129 , 130 , 131 ) Furthermore, osteoclasts originate from hematopoietic stem cells like immune cells such as macrophages and monocytes,( 9 ) and considering the tight interplay between immune cells and osteoclasts,( 132 ) there might be a link between the patients' sclerotic lesions and osteoclast function. Inhibition or knockdown of SMS2 attenuates the permeability of endothelial cells in vitro and in vivo.( 129 , 133 ) One can hypothesize that this could potentially cause reduced oxygen tension contributing to osteolysis. While a study by Yan and colleagues( 134 ) identified SGMS2 as a regulatory gene of late embryonic craniofacial development in mice, the restriction of lytic lesions to the skull bones in OP‐CDL still remains unclear.
Although osteoblasts have been reported to be negatively affected by SMS2 impairment, the information is still sparse. Matsumoto and colleagues( 135 ) reported that sphingomyelin synthase 1, but not SMS2, is involved in bone formation by osteoblasts in mice. Moreover, Yoshikawa and colleagues( 136 ) reported that Sgms2 knockdown in primary murine osteoblasts reduces the expression of the nuclear factor κB (NF‐κB) ligand and increases the expression of osteoprotegerin, thus decreasing osteoclastogenesis. However, these studies do not reflect the situation in humans because SGMS2 deficiency leads to osteoporosis, patchy mineralization, cortical trabecularization, and calvarial osteolytic/doughnut lesions.( 123 , 124 , 125 )
Differential diagnosis between various monogenic forms of EOOP
Despite differences in pathophysiology, it is evident that the described forms of monogenic osteoporosis have substantial overlap and may have greatly varying clinical presentations regarding age of onset, disease severity, and treatment response. Further variability is introduced by different variants in the same gene, as seen in SGMS2‐related pathology. Taking into account further changes introduced by aging, lifestyle factors, and comorbidities, the clinical presentation can be easily obscured and unclear even to an experienced clinician.
With increasing clinical data from newly diagnosed patients, subtle trends in skeletal characteristics are observed, especially regarding sites of fracture, bone deformities, spinal involvement, and cranial abnormalities. PLS3 variants seem to have a major impact on the spine, resulting in vertebral compressions and long bone fractures already at a young age. Vertebral compression fractures lead to thoracic kyphosis and loss of adult height. Similar vertebral complications are seen in WNT1 osteoporosis, although usually only later in adulthood and not in childhood.( 137 , 138 )
Although the severe form of OP‐CDL manifests as severe long bone and spinal changes, patients with the nonsense variant p.Arg50* tend to have milder vertebral changes but suffer from frequent long‐bone fractures. These typically present in early childhood and subside by adulthood. Peripheral fractures are also common in patients with WNT1 osteoporosis; they often occur in childhood but become even more prevalent in adulthood.
Second to differences in skeletal characteristics are the presence and array of possible extraskeletal features. The affected genes are often ubiquitously expressed, regulate a wide range of developmental and cellular processes, and may therefore predispose to other organ manifestations. WNT1, for one, has a key role in the central nervous system and in patients harboring WNT1 mutations neurological manifestations such as severe cerebellar hypoplasia, epilepsy, cerebellar, pontine, and mesencephalic tectum hypoplasia, severe global developmental delay, and gross brain atrophy have been reported.( 58 , 139 ) Although these are usually diagnosed in patients with biallelic WNT1 variants, it is possible that even patients with a monoallelic WNT1 variant could manifest milder neurological features.
Neurologic features are also a central feature in OP‐CDL.( 123 ) Alongside skeletal manifestations, SGMS2 mutation‐positive subjects may present with recurrent and transient facial, trochlear, and oculomotor nerve palsies that are isolated and spontaneously remitting. Other recorded features include hearing loss, sensory neuropathy with progressive ataxia, and gross developmental delay. Despite the skull lesions, the symptoms are not secondary to cranial sclerosis and extensive clinical evaluations or brain imaging studies have found no structural changes, suggesting that the changes in nerve cell function arise directly from abnormal neuronal sphingomyelin metabolism.
Bone Biopsy as a Tool to Explore Bone Tissue Characteristics in EOOP
Beyond bone mass and geometry, alterations affecting bone material properties at different levels of hierarchy can reduce the capacity of bone to dissipate the energy of an impact without fracturing.( 140 ) Transiliac bone biopsy samples offer a unique and microscopic look into changes in bone material properties and can be obtained for diagnostic purposes to evaluate histology and histomorphometry in unclear pathological conditions.( 141 , 142 ) In “classical” OI cases with genetically confirmed type I collagen defects, bone biopsy is usually not indicated as there are treatment guidelines. In other forms of primary EOOP the analysis of bone biopsy may provide important insight into pathogenesis and guide also in disease management. Double tetracycline‐labeled bone biopsies may be indicated when the cause of the osteoporosis or fractures is unclear, when the presentation is unusual or fractures continue despite therapy. Biopsies help to differentiate between osteomalacia and osteoporosis and between high and low bone turnover states.( 142 ) Transiliac crest bone biopsies are also useful to evaluate treatment effects (Table 2). This tool may not be readily available in clinical practice but has been widely used in research settings.
Table 2.
Overview of bone tissue characteristics in transiliac bone biopsy samples and effect of treatment for OI type I and EOOP due to LRP5, WNT1, PLS3, and SGMS2 defects. The results have been compared to reference data from healthy children and adolescents. 141 , 145 , 161
| OI type I | LRP5 | WNT1 | PLS3 | SGMS2 | |||||
|---|---|---|---|---|---|---|---|---|---|
| n=19 145 , 159 , 160 | n=1 197 | n=1 31 , 83 | n=2 87 | n=1 94 | n=1 195 | n=2 31 , 83 | n=2 123 | n=1 124 | |
|
Age (years, y) Gender (male, m; female, f) |
2.2‐14.1y 13m/6f |
6y/m | 14y/m |
4y/m 8y/m |
9.7y/m | 12y/m |
9y/m 13y/m |
12 y/f 15 y/m |
5y/f* |
| Histology and histomorphometry : 141 | |||||||||
| Cortical width | ↓ | ↑ | ↔ | ↓ | ↓ | ↓ | ↓ | ↔, ↔ | ↔ |
| Trabecular bone volume/tissue volume (%) | ↓ | ↓ | ↔ | ↓ | ↓ | ↓ | ↓ | ↔, ↓ | n.a |
| Osteoid thickness (μm) | ↔ |
↔ Tb ↔ Ct |
↔ | ↓ | ↑ | ↓ | ↔ | ↔, ↑ | ↔ |
| Osteoid surface/bone surface (%) | ↑ |
↓ Tb ↔ Ct |
↔ | ↓ | ↑ | ↓ | ↔ | ↔, ↑ | ↔ |
| Osteoblast surface/bone surface (%) | ↑ | n.a | ↔ | ↔ | ↔ | ↓ | ↔ | ↓, ↔ | ↑ |
| Mineralizing surface/bone surface (%) | ↑ |
↓ Tb ↔ Ct |
↔ | ↔ | ↓ | ↓ | ↔ | ↔; n.a | ↑ |
| Mineral apposition rate (μm/d) | ↓ |
↓ Tb ↔ Ct |
n.a | ↔ | ↑ | ↓ | n.a | ↔; n.a | ↓ |
| Bone formation rate/bone surface (μm3/μm2/year) | ↔ |
↓ Tb ↔ Ct |
n.a | ↔ | ↓ | ↓ | ↔ | ↔; n.a | ↔ |
| Bone formation rate/osteoblast surface (%/year) | ↓ | n.a | n.a | n.a | n.a | n.a | n.a | n.a | ↔ |
| Mineralization lag time (days) | ↔ | n.a | n.a | n.a | ↑ | ↓ | n.a | ↑, n.a | ↑ |
| Osteoclast surface/bone surface (%) | ↔ | n.a | ↔ | ↔ | ↔ | ↓↓ | n.a | ↔, ↔ | ↔ |
| Eroded surface (%) | ↔ | ↔ | ↔ | n.a | ↓ | ↓ | n.a | ↔, ↑ | ↔ |
| Lamellar (LB)/Woven bone (WB) | LB | n.a | LB | n.a | n.a | LB | LB | WB | n.a |
| Bone mineralization density distribution by quantitative backscattered electron microscopy: 143, 161 | |||||||||
| Mean calcium concentration (CaMean, weight % Ca) | ↑ |
↔ Tb ↑ Ct |
↓ | ↔ | ↓ | ↑ | ↔, ↓ Tb ↔, ↔Ct | ↓ | n.a |
| Most frequent calcium concentration (CaPeak, weight % Ca) | ↑ |
↔ Tb ↑ Ct |
↓ | ↔ | ↓ | ↑ |
↔, ↓ Tb ↔, ↓ Ct |
↓, ↓ Tb; ↑, ↓ Ct; |
n.a |
| Heterogeneity in mineralization (CaWidth, Δ weight % Ca) | ↓ |
↑ Tb ↔ Ct |
↑ | ↔ | ↑ | ↔ | ↑ |
↑, ↑ Tb ↑, ↑ Ct |
n.a |
| Percentage of lowly mineralized bone areas (CaLow, % bone area) | ↔ |
↑ Tb ↓ Ct |
↑ | ↔ | ↑ | ↓ |
↔, ↑ Tb ↔, ↔ Ct |
↑, ↑ Tb; ↑, ↑ Ct; |
n.a |
| Percentage of highly mineralized bone areas (CaHigh, % bone areas) | ↑ |
↑ Tb ↑ Ct |
↓ Tb ↔ Ct |
↔ | ↓ | ↑ | ↔ |
↔, ↔ Tb; ↑, ↔ Ct; |
n.a |
| Osteocyte lacunae sections (OLS) characteristics by quantitative backscattered electron microscopy: 145 | |||||||||
| Porosity (%) | ↑ | n.a | ↑ | n.a | n.a | n.a | n.a | ↑ | n.a |
| Density (number/mm2) | ↑ | n.a | ↓ | ↑ | n.a | n.a | n.a | ↑ | n.a |
| Area (μm2) |
↓ Tb ↔ Ct |
n.a | ↑ | n.a | n.a | n.a | n.a | ↑ | n.a |
Tb: trabecular bone, Ct: cortical bone; ↑: increased; ↓: decreased; ↔: within normal limits
*Histomorphometry outcomes were obtained from cortical bone (trabecular bone was not available) and reference values are from. 196
The residual bone sample block can be used for further analyses such as quantitative backscattered electron imaging (qBEI) to gain information on mineralized bone volume, the degree of mineralization of the ECM and two‐dimensional (2D) osteocyte lacunae sections (OLS) characteristics (Table 2).( 143 , 144 , 145 ) This technique utilizes the linear correlation between the intensity of the backscattered signals and the calcium content of the ECM.( 143 , 146 , 147 , 148 , 149 , 150 ) Other methods to analyze bone tissue properties at the microscale and nanoscale, such as small‐angle X‐ray scattering to determine size and orientation of the mineral particles,( 151 , 152 , 153 , 154 , 155 ) and vibrational spectroscopy (Fourier‐transform infrared or Raman)( 156 , 157 ) to investigate the structure of the organic matrix or a mechanical investigation using, eg, nanoindentation,( 158 ) are reported elsewhere. For many of these methods, reference values for healthy individuals (children and adults) are available.( 141 , 143 , 145 , 152 , 159 , 160 , 161 , 162 , 163 , 164 )
To elucidate the variability of bone tissue features in different forms of EOOP and to underscore the considerable amount of information obtained from bone biopsy with different analytic methods, we discuss here bone material properties based on the evaluation of transiliac bone biopsy samples in two representative pediatric patients. One patient has EOOP due to a monoallelic WNT1 variant and the other child has typical OI due to a COL1A1 variant leading to a quantitative defect in type I collagen. Although the net result of these variants in both cases is increased bone fragility, the tissue pathology differs considerably. As shown in Fig. 5, qBEI microscopy of bone biopsy samples indicated a reduction of 40% in trabecular mass in the patient with OI( 160 ) compared to a healthy girl,( 161 ) whereas this parameter was within lower normal limits in the child with EOOP due to a WNT1 variant( 31 ) (Fig. 5A ). In OI, the low trabecular bone volume is associated with abnormal bone remodeling; bone histomorphometric analyses have shown that bone turnover is increased in OI due to elevated numbers of osteoblasts and osteoclasts, whereas ECM production at the single cell level is markedly reduced. This metabolic imbalance leads to poor increase in trabecular thickness and increased resorption of the bone ECM.( 159 , 165 ) Consistently, the nearly normal trabecular bone volume in the child with a WNT1 variant is in line with normal indices of bone formation and resorption found in this and other children with WNT1 variants.( 31 , 166 )
Fig. 5.
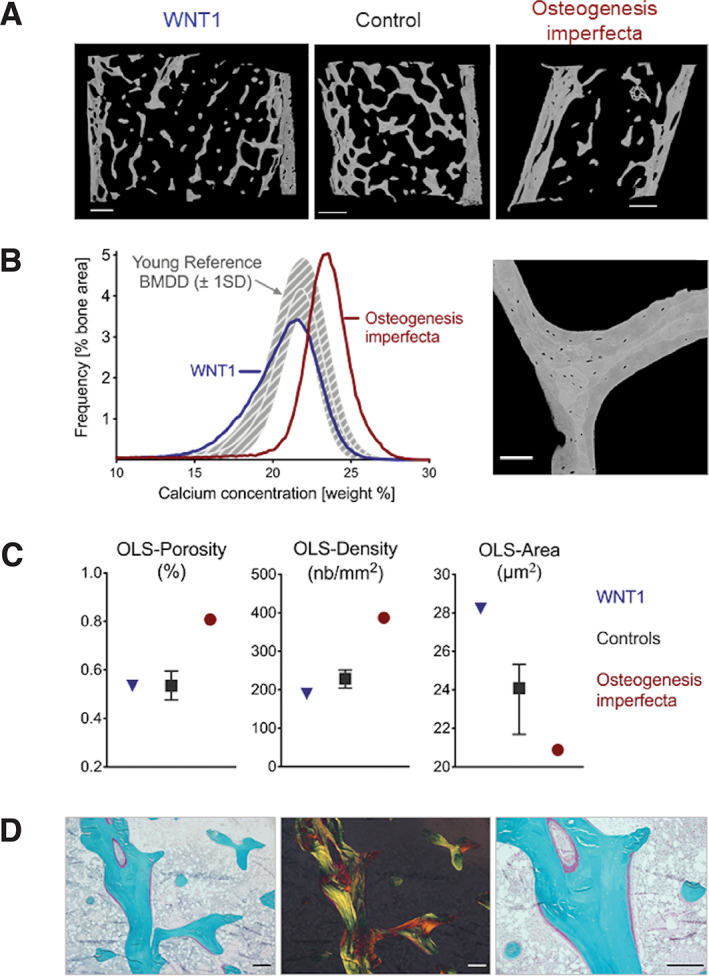
(A) qBEI of selected bone biopsy samples: from a patient with a monoallelic WNT1 variant (left; reproduced with permission from Elsevier( 31 )), from a healthy control (middle; reproduced with permission from Springer( 160 )) and from a patient with OI due to a COL1A1 variant (right; reproduced with permission from Springer( 160 )). The inner and outer cortex and the trabecular compartment in between can be visualized. The trabecular bone volume is strikingly reduced in OI bone. The different gray scales relate to different calcium content of the ECM (Scale bar = 1 mm). (B) (Left) Corresponding BMDD curves: these represent frequency distributions of gray (calcium) levels and can be described by five parameters: the average and the most frequent calcium concentration (CaMean and CaPeak), the proportion of lowly and highly mineralized ECM (CaLow and CaHigh, respectively on the left and right side of the histogram) and the heterogeneity in mineralization, mirrored by the width of the histogram at half maximum (CaWidth).( 143 ) Compared to the reference BMDD from healthy children (gray band), the BMDD from the child with OI is shifted to the right, towards higher matrix mineralization, while the BMDD curve in the patient with WNT1 variant is shifted to the left, towards lower mineralization.( 31 , 160 , 161 ) (Right) qBEI of a trabecular feature at high magnification (0.88 μm/pixel) to assess the osteocyte lacunae sections (OLS). Scale bar = 100 μm, published under a creative common attribution license.( 145 ) (C) Results from the OLS‐analysis. Compared to healthy references( 145 ), bone of a patient with OI shows a large OLS‐density and a reduced OLS‐area, while the opposite is true for bone from WNT1 patients. The decrease in density and increase in area leads to an unremarkable OLS‐porosity for WNT1 bone. (D) Thin section of bone from the patient with the WNT1 variant stained with Goldner trichrome visualized with bright field microscopy (left and right) as well as under polarized light (middle). Under bright field large osteoid seams on the bone surface can be observed (red: osteoid, green: mineralized bone). Under polarized light a rather normal lamellar organization of bone ECM is found (Scale bars = 250 μm). BMDD = bone mineralization density distribution; OLS‐porosity = percentage of total OLS area per bone area); OLS‐density = number of OLS per bone area; qBEI = quantitative backscattered electron microscopy imaging.
The qBEI microscopy image can be translated into a gray‐level histogram corresponding to a bone mineralization density distribution (BMDD) curve (Fig. 5B ). This distribution arises from the fact that bone ECM is not homogenously mineralized, but rather consists of a mosaic of bone packets with variable mineral content depending on local tissue age. Younger bone packets have a lower mineral content than older ones and changes in bone turnover, as well as disturbances in the mineralization process, will have a profound impact on the shape of the BMDD.( 167 , 168 ) In healthy children and adolescents, the BMDD for trabecular bone shows little variation with age, sex, ethnicity, and skeletal site.( 161 , 169 , 170 , 171 ) Any deviation from this physiological window is potentially deleterious and contributes to bone fragility; hypermineralization stiffens the bone material and makes it more brittle, whereas hypomineralization softens it and makes it weaker.
BMDD in the patient with the COL1A1 variant reveals a pronounced hypermineralization (BMDD curve shifted to higher mineralization) corresponding to high‐turnover OI, whereas a BMDD shift to lower mineralization indicative of low‐turnover EOOP is found in the patient harboring the WNT1 variant (Fig. 5B ). Normally, low to normal bone remodeling rates lead to normal or increased mineralization and high bone turnover rates to lower mineralization.( 143 , 168 ) The fact that exactly the opposite is the case in OI and WNT1 osteoporosis, indicates that in both cases, not only the turnover rate but also the mineralization kinetics are disturbed.
Because mineral homeostasis, mechanosensing, and bone remodeling are largely controlled by osteocytes,( 172 , 173 , 174 ) the quantitative assessment of osteocyte parameters such as size and density might provide important insights into material abnormalities. The fingerprint of dead osteocytes are highly mineralized lacunae. This phenomenon, known as “micropetrosis,” is believed to hamper mechanosensitivity and the repair of bone microdamage, contributing to bone fragility.( 175 , 176 ) Hypermineralized osteocyte lacunae are easily identifiable on qBEI as bright dots. So far, no increase in micropetrosis has been found in healthy children or in pediatric patients with EOOP.( 169 , 177 ) Figure 5C shows that OLS‐porosity is high in the child with OI whereas it appears within normal range in the child with the WNT1 variant. Patients with a decreased OLS‐density and an increased OLS‐area may still show normal OLS‐porosity, and conversely, osteocyte dysfunction might be associated with abnormal OLS‐density and/or abnormal OLS‐size.( 31 , 83 ) The OLS analysis of bone sections is a 2D method, whereas a full three‐dimensional (3D) assessment of osteocyte lacunar volume and shape can be achieved using X‐ray tomography.( 178 )
Goldner trichrome staining (Fig. 5D ) allows to distinguish mineralized ECM (stained in green) from osteoid (stained in red) and evaluate structural parameters as well as osteoblast and osteoclast activity.( 141 ) In addition, Goldner‐stained histological sections can be viewed under polarized light to assess collagen fibril orientation. Such analyses allow to distinguish primary disordered woven bone from remodeled lamellar bone. Woven bone is less mature, more cellular, and more prone to fracture.( 179 ) Woven bone formation is characteristic for severe forms of OI and less frequent in the rather mild OI type I,( 180 ) but present also in OP‐CDL.( 123 ) However, even lamellar bone can be of reduced quality if less or defective collagen is secreted, lamellae and collagen fibrils are thinner, and crosslink formation altered.( 19 , 21 , 157 , 159 , 180 )
As discussed in this article, bone biopsy is a valuable tool that provides a large amount of tissue‐level information about the pathology leading to bone fragility. However, this tool may not be readily available in clinical practice due to its invasive nature, large patient volume, and required expertise for biopsy analysis. Bone biopsies are widely used in research settings and these findings can hopefully in the future be translated to general management guidelines in the specified monogenic forms of EOOP, as already is the case with OI. Another downside is the incompleteness of normative data. Although such data are available for several parameters for healthy children and for classical OI, it remains to be established for other monogenic forms across age groups.
Treatment Strategies in Monogenic Forms of EOOP
Due to the paucity of clinical studies in young patients with osteoporosis, there are presently no specific evidence‐based guidelines for treating EOOP. We provide here a concise summary of treatment strategies, focusing especially on the rare monogenic forms. For more extensive information, including management of nongenetic forms, the readers are advised to use other recent reviews specifically focusing on osteoporosis management in young patients and premenopausal women.( 17 , 18 , 181 , 182 )
As a first line of treatment, patients with EOOP should be given adequate calcium and vitamin D supplementation, if dietary evaluation or biochemistry shows insufficiency.( 183 , 184 ) Furthermore, a healthy lifestyle, including good diet, regular exercise, no alcohol consumption, and no smoking, has been shown to improve BMD in both young healthy subjects and patients with EOOP.( 185 , 186 , 187 ) An increase in BMD is also seen when secondary causes of EOOP are treated (eg, gluten‐free diet for celiac disease).( 16 ) When secondary EOOP is excluded but fragility fractures continue occurring, the use of antiresorptive and anabolic drugs should be considered.( 1 ) Their effectiveness in children and young adults with EOOP has not been thoroughly studied due to the rarity of the condition and the fact that only small studies investigating effects on BMD but not on fractures have been carried out. This means that in each situation the potential benefits and side effects of medication will have to be weighed against the severity of bone fragility, also keeping in mind potential harms in women of childbearing age.
Prior studies have primarily focused on evaluating use of antiresorptive and anabolic drugs in collagen type I–related OI. In general, the use of bisphosphonates, such as pamidronate, alendronate, and zoledronate, to reduce osteoclast activity, increases BMD in patients with OI whereas its effect on fractures is less certain.( 188 , 189 ) Clinical studies concerning denosumab, a monoclonal antibody inhibiting osteoclast activation, remain scarce and further investigation is needed to evaluate its full potential.( 190 ) Teriparatide treatment leads to increased BMD and prevents spinal compression fractures in OI patients,( 191 , 192 ) particularly in patients with mild OI compared to patients with severe forms.( 191 ) Preclinical studies on two other anabolic drugs, sclerostin antibodies and transforming growth factor β (TGF‐β) inhibitors to promote bone formation, resulted in increased BMD in different mice models of OI.( 193 , 194 ) Clinical studies for testing anabolic molecules in OI patients are ongoing (clinical trials BPS‐804 and NCT03064074).
However, as discussed earlier, results from studies involving patients with OI cannot be directly extrapolated to treatment approaches in EOOP as the bone tissue characteristics and bone turnover differ greatly. Using bisphosphonates in low bone turnover states may not be beneficial and hence personalized approaches need to be adopted when treating patients with monogenic forms of EOOP. However, although osteoporosis linked to LRP5 and WNT1 variants associates with decreased WNT signaling and decreased osteoblast activity and function, for EOOP due to PLS3 and SGMS2 variants the underlying pathomolecular mechanisms are still incompletely understood. Teriparatide treatment showed accelerated bone turnover in an adult male patient with the LRP5 polymorphism p.Arg1036Gln( 74 ) and in three adult male patients with the WNT1 missense variant p.Cys218Gly.( 30 ) Osteoporosis medication in nine patients with WNT1‐related EOOP did not ameliorate their spinal pathology.( 137 ) Considering the underlying defects, sclerostin antibodies might potentially be beneficial for these patients but clinical studies in this regard are not yet available.
Concerning PLS3‐related EOOP, three adult patients (two males and one female) with the same splicing PLS3 variant responded well to teriparatide treatment but the outcome on fracture risk has yet to be determined.( 30 ) Moreover, bisphosphonate treatment in patients with pathogenic PLS3 variants seems to increase BMD, improve vertebral shaping, and prevent new fractures.( 86 , 93 , 100 , 101 ) Finally, the use of bisphosphonates in three children with SGMS2 osteoporosis prevented further long‐bone fractures and improved back pain.( 123 )
Long‐term effects of osteoporosis treatment in patients with LRP5, WNT1, PLS3, and SGMS2 variants have yet to be evaluated. Moreover, clinical trials comparing EOOP patients and sex‐matched controls need to be carried out to better investigate the effects of osteoporosis treatment and evaluate the safety of the use of these drugs in young patients. Finally, due to the diversity of mechanisms involved in EOOP, the variable and often inadequate response to the available osteoporosis treatments suggests that new personalized treatment approaches need to be identified and tested in larger patient cohorts. In order to identify novel treatment strategies, further studies on the molecular pathology of EOOP are needed.
Concluding Remarks
EOOP remains a rare but important clinical entity that poses challenges in diagnostic approaches and management. Several causative factors and mechanisms have been identified. Although the clinical features of EOOP overlap with other skeletal fragility syndromes, there are also critical differences relating to the underlying causes, clinical and bone‐tissue characteristics, and choice of treatment. A thorough clinical investigation combined with family history and tailored biochemical tests are the first steps toward a diagnosis. When secondary causes of bone fragility are excluded, primary EOOP due to a constitutive genetic defect in a gene playing a pivotal role in bone metabolism should be considered. The broad use of NGS has sped up the identification of disease‐causing variants and has also led to the identification of several novel genes linked to bone fragility. The pathomolecular mechanisms responsible for these recently identified forms of EOOP remain to be fully elucidated. Patient‐derived bone biopsies provide a possibility to understand what the genetic defects cause at the tissue level, giving detailed information about bone structure, turnover, and mineralization. In the absence of large clinical studies on osteoporosis in young subjects, treatment guidelines for EOOP are lacking. Further research and novel personalized treatment strategies are required to optimize patient management.
Author Contributions
Alice Costantini: Conceptualization; data curation; funding acquisition; project administration; writing – original draft; writing – review and editing. Riikka E. Mäkitie: Conceptualization; data curation; writing – original draft; writing – review and editing. Markus A. Hartmann: Conceptualization; data curation; formal analysis; funding acquisition; writing – original draft; writing – review and editing. Nadja Fratzl‐Zelman: Conceptualization; data curation; funding acquisition; methodology; writing – original draft; writing – review and editing. M. Carola Zillikens: Conceptualization; data curation; investigation; writing – original draft; writing – review and editing. Uwe Kornak: Conceptualization; data curation; methodology; writing – original draft; writing – review and editing. Kent Søe: Conceptualization; data curation; writing – original draft; writing – review and editing. Outi Mäkitie: Conceptualization; data curation; funding acquisition; project administration; supervision; writing – original draft; writing – review and editing.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4668.
Supporting information
Table S1 WNT1 variants linked to autosomal dominant EOOP.
Acknowledgments
This study was financially supported by the Stockholm County Council, the Swedish Research Council (2018‐02645; 2020‐00587), the Konung Gustaf V:s och Drottning Victorias Frimurarestiftelsethe, the Academy of Finland (277843), the Novo Nordisk Foundation (21322), the Folkhälsan Research Foundation, and the Sigrid Jusélius Foundation. Financial support from the Research funds of the Austrian workers compensation board (AUVA) and the Austrian Social Health Insurance Fund (OEGK) is algratefully acknowledged. We thank Stéphane Blouin for performing the OLS‐analysis in the patient with the WNT1 variant, as well as Petra Keplinger, Sonja Lueger and Phaedra Messmer from the bone material laboratory at the Ludwig Boltzmann Institute of Osteology for careful sample preparation and qBEI measurements.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1. Makitie O, Zillikens MC. Early‐onset osteoporosis. Calcif Tissue Int. 2022;110(5):546‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kanis JA, Gluer CC. An update on the diagnosis and assessment of osteoporosis with densitometry. Committee of Scientific Advisors, International Osteoporosis Foundation. Osteoporos Int. 2000;11(3):192‐202. [DOI] [PubMed] [Google Scholar]
- 3. Lorentzon M, Cummings SR. Osteoporosis: the evolution of a diagnosis. J Intern Med. 2015;277(6):650‐661. [DOI] [PubMed] [Google Scholar]
- 4. Harvey N, Dennison E, Cooper C. Osteoporosis: impact on health and economics. Nat Rev Rheumatol. 2010;6(2):99‐105. [DOI] [PubMed] [Google Scholar]
- 5. Shaw NJ. Management of osteoporosis in children. Eur J Endocrinol. 2008;159(Suppl 1):S33‐S39. [DOI] [PubMed] [Google Scholar]
- 6. Ward LM, Weber DR, Munns CF, Hogler W, Zemel BS. A contemporary view of the definition and diagnosis of osteoporosis in children and adolescents. J Clin Endocrinol Metab. 2020;105(5):e2088‐e2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Makitie O. Causes, mechanisms and management of paediatric osteoporosis. Nat Rev Rheumatol. 2013;9(8):465‐475. [DOI] [PubMed] [Google Scholar]
- 8. Rivadeneira F, Makitie O. Osteoporosis and bone mass disorders: from gene pathways to treatments. Trends Endocrinol Metab. 2016;27(5):262‐281. [DOI] [PubMed] [Google Scholar]
- 9. Florencio‐Silva R, Sasso GRD, Sasso‐Cerri E, Simoes MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed Res Int. 2015;2015:421746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Langdahl B, Ferrari S, Dempster DW. Bone modeling and remodeling: potential as therapeutic targets for the treatment of osteoporosis. Ther Adv Musculoskelet Dis. 2016;8(6):225‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farr JN, Khosla S. Skeletal changes through the lifespan—from growth to senescence. Nat Rev Endocrinol. 2015;11(9):513‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci. 2006;1092:385‐396. [DOI] [PubMed] [Google Scholar]
- 13. Lewiecki EM, Gordon CM, Baim S, et al. International Society for Clinical Densitometry 2007 adult and pediatric official positions. Bone. 2008;43(6):1115‐1121. [DOI] [PubMed] [Google Scholar]
- 14. Bishop N, Arundel P, Clark E, et al. Fracture prediction and the definition of osteoporosis in children and adolescents: the ISCD 2013 Pediatric Official Positions. J Clin Densitom. 2014;17(2):275‐280. [DOI] [PubMed] [Google Scholar]
- 15. Ferrari S, Bianchi ML, Eisman JA, et al. Osteoporosis in young adults: pathophysiology, diagnosis, and management. Osteoporos Int. 2012;23(12):2735‐2748. [DOI] [PubMed] [Google Scholar]
- 16. Pepe J, Body JJ, Hadji P, et al. Osteoporosis in premenopausal women: a clinical narrative review by the ECTS and the IOF. J Clin Endocrinol Metab. 2020;105(8):dgaa306. [DOI] [PubMed] [Google Scholar]
- 17. Lewiecki EM. Evaluating patients for secondary causes of osteoporosis. Curr Osteoporos Rep. 2022;20(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 18. Cohen A. Premenopausal osteoporosis. Endocrinol Metab Clin North Am. 2017;46(1):117‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016;387(10028):1657‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16(2):101‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marini JC, Forlino A, Bachinger HP, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. [DOI] [PubMed] [Google Scholar]
- 22. Besio R, Chow CW, Tonelli F, Marini JC, Forlino A. Bone biology: insights from osteogenesis imperfecta and related rare fragility syndromes. FEBS J. 2019;286(15):3033‐3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mortier GR, Cohn DH, Cormier‐Daire V, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179(12):2393‐2419. [DOI] [PubMed] [Google Scholar]
- 24. Ghatan S, Costantini A, Li R, et al. The polygenic and monogenic basis of Paediatric fractures. Curr Osteoporos Rep. 2021;19(5):481‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Makitie RE, Costantini A, Kampe A, Alm JJ, Makitie O. New insights into monogenic causes of osteoporosis. Front Endocrinol (Lausanne). 2019;10:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jovanovic M, Guterman‐Ram G, Marini JC. Osteogenesis imperfecta: mechanisms and signaling pathways connecting classical and rare OI types. Endocr Rev. 2022;43(1):61‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weiss MJ, Cole DE, Ray K, et al. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc Natl Acad Sci U S A. 1988;85(20):7666‐7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014;164A(6):1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laine CM, Joeng KS, Campeau PM, et al. WNT1 mutations in early‐onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368(19):1809‐1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valimaki VV, Makitie O, Pereira R, et al. Teriparatide treatment in patients with WNT1 or PLS3 mutation‐related early‐onset osteoporosis: a pilot study. J Clin Endocrinol Metab. 2017;102(2):535‐544. [DOI] [PubMed] [Google Scholar]
- 31. Fratzl‐Zelman N, Wesseling‐Perry K, Makitie RE, et al. Bone material properties and response to teriparatide in osteoporosis due to WNT1 and PLS3 mutations. Bone. 2021;146:115900. [DOI] [PubMed] [Google Scholar]
- 32. Bianchi ML, Bishop NJ, Guanabens N, et al. Hypophosphatasia in adolescents and adults: overview of diagnosis and treatment. Osteoporos Int. 2020;31(8):1445‐1460. [DOI] [PubMed] [Google Scholar]
- 33. Whyte MP. Hypophosphatasia—aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12(4):233‐246. [DOI] [PubMed] [Google Scholar]
- 34. Ostertag A, Cohen‐Solal M, Audran M, et al. Vertebral fractures are associated with increased cortical porosity in iliac crest bone biopsy of men with idiopathic osteoporosis. Bone. 2009;44(3):413‐147. [DOI] [PubMed] [Google Scholar]
- 35. Byers PH, Krakow D, Nunes ME, Pepin M, American College of Medical Genetics. Genetic evaluation of suspected osteogenesis imperfecta (OI). Genet Med. 2006;8(6):383‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next‐generation sequencing: single‐gene, gene panel, or exome/genome sequencing. Genet Med. 2015;17(6):444‐451. [DOI] [PubMed] [Google Scholar]
- 37. Shiang R, Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78(2):335‐342. [DOI] [PubMed] [Google Scholar]
- 38. Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370(9582):162‐172. [DOI] [PubMed] [Google Scholar]
- 39. Glorieux FH. Caffey disease: an unlikely collagenopathy. J Clin Invest. 2005;115(5):1142‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adams DR, Eng CM. Next‐generation sequencing to diagnose suspected genetic disorders. N Engl J Med. 2019;380(2):201. [DOI] [PubMed] [Google Scholar]
- 41. Yang Y, Muzny DM, Reid JG, et al. Clinical whole‐exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369(16):1502‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kremer LS, Bader DM, Mertes C, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Turro E, Astle WJ, Megy K, et al. Whole‐genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583(7814):96‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xuan J, Yu Y, Qing T, Guo L, Shi L. Next‐generation sequencing in the clinic: promises and challenges. Cancer Lett. 2013;340(2):284‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smedley D, Jacobsen JO, Jager M, et al. Next‐generation diagnostics and disease‐gene discovery with the Exomiser. Nat Protoc. 2015;10(12):2004‐2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zemojtel T, Kohler S, Mackenroth L, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease‐associated genome. Sci Transl Med. 2014;6(252):252ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kohler S, Gargano M, Matentzoglu N, et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021;49(D1):D1207‐D1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Formosa MM, Bergen DJM, Gregson CL, et al. A roadmap to gene discoveries and novel therapies in monogenic low and high bone mass disorders. Front Endocrinol (Lausanne). 2021;12:709711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cohen A, Hostyk J, Baugh EH, et al. Whole exome sequencing reveals potentially pathogenic variants in a small subset of premenopausal women with idiopathic osteoporosis. Bone. 2022;154:116253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179‐192. [DOI] [PubMed] [Google Scholar]
- 51. Canalis E. Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol. 2013;9(10):575‐583. [DOI] [PubMed] [Google Scholar]
- 52. Johnson ML, Harnish K, Nusse R, Van Hul W. LRP5 and Wnt signaling: a union made for bone. J Bone Miner Res. 2004;19(11):1749‐1757. [DOI] [PubMed] [Google Scholar]
- 53. Gong Y, Slee RB, Fukai N, et al. LDL receptor‐related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513‐523. [DOI] [PubMed] [Google Scholar]
- 54. Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL‐receptor–related protein 5. N Engl J Med. 2002;346(20):1513‐1521. [DOI] [PubMed] [Google Scholar]
- 55. Hartikka H, Makitie O, Mannikko M, et al. Heterozygous mutations in the LDL receptor‐related protein 5 (LRP5) gene are associated with primary osteoporosis in children. J Bone Miner Res. 2005;20(5):783‐789. [DOI] [PubMed] [Google Scholar]
- 56. Korvala J, Loija M, Makitie O, et al. Rare variations in WNT3A and DKK1 may predispose carriers to primary osteoporosis. Eur J Med Genet. 2012;55(10):515‐519. [DOI] [PubMed] [Google Scholar]
- 57. Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013;50(5):345‐348. [DOI] [PubMed] [Google Scholar]
- 58. Pyott SM, Tran TT, Leistritz DF, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. 2013;92(4):590‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Keupp K, Beleggia F, Kayserili H, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92(4):565‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cui Y, Niziolek PJ, MacDonald BT, et al. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17(6):684‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yadav VK, Ryu JH, Suda N, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135(5):825‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Riddle RC, Diegel CR, Leslie JM, et al. Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS One. 2013;8(5):e63323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Joeng KS, Lee YC, Jiang MM, et al. The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet. 2014;23(15):4035‐4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Joeng KS, Lee YC, Lim J, et al. Osteocyte‐specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest. 2017;127(7):2678‐2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Qiu W, Andersen TE, Bollerslev J, Mandrup S, Abdallah BM, Kassem M. Patients with high bone mass phenotype exhibit enhanced osteoblast differentiation and inhibition of adipogenesis of human mesenchymal stem cells. J Bone Miner Res. 2007;22(11):1720‐1731. [DOI] [PubMed] [Google Scholar]
- 66. Yorgan TA, Peters S, Jeschke A, et al. The anti‐osteoanabolic function of sclerostin is blunted in mice carrying a high bone mass mutation of Lrp5. J Bone Miner Res. 2015;30(7):1175‐1183. [DOI] [PubMed] [Google Scholar]
- 67. Van Wesenbeeck L, Cleiren E, Gram J, et al. Six novel missense mutations in the LDL receptor‐related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003;72:763‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moon R, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through b‐Catenin. Science. 2002;296(5573):1644‐1646. [DOI] [PubMed] [Google Scholar]
- 69. Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280(20):19883‐19887. [DOI] [PubMed] [Google Scholar]
- 70. Campos‐Obando N, Oei L, Hoefsloot LH, et al. Osteoporotic vertebral fractures during pregnancy: be aware of a potential underlying genetic cause. J Clin Endocrinol Metab. 2014;99(4):1107‐1111. [DOI] [PubMed] [Google Scholar]
- 71. Saarinen A, Mayranpaa MK, Lehesjoki AE, Makitie O. Low‐density lipoprotein receptor‐related protein 5 (LRP5) variation in fracture prone children. Bone. 2010;46(4):940‐945. [DOI] [PubMed] [Google Scholar]
- 72. van Meurs JB, Trikalinos TA, Ralston SH, et al. Large‐scale analysis of association between LRP5 and LRP6 variants and osteoporosis. JAMA. 2008;299(11):1277‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ferrari SL, Deutsch S, Baudoin C, et al. LRP5 gene polymorphisms and idiopathic osteoporosis in men. Bone. 2005;37(6):770‐775. [DOI] [PubMed] [Google Scholar]
- 74. Sturznickel J, Rolvien T, Delsmann A, et al. Clinical phenotype and relevance of LRP5 and LRP6 variants in patients with early‐onset osteoporosis (EOOP). J Bone Miner Res. 2021;36(2):271‐282. [DOI] [PubMed] [Google Scholar]
- 75. Nampoothiri S, Guillemyn B, Elcioglu N, et al. Ptosis as a unique hallmark for autosomal recessive WNT1‐associated osteogenesis imperfecta. Am J Med Genet A. 2019;179(6):908‐914. [DOI] [PubMed] [Google Scholar]
- 76. Hayat A, Hussain S, Bilal M, et al. Biallelic variants in four genes underlying recessive osteogenesis imperfecta. Eur J Med Genet. 2020;63(8):103954. [DOI] [PubMed] [Google Scholar]
- 77. Kausar M, Siddiqi S, Yaqoob M, et al. Novel mutation G324C in WNT1 mapped in a large Pakistani family with severe recessively inherited osteogenesis imperfecta. J Biomed Sci. 2018;25(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Umair M, Alhaddad B, Rafique A, et al. Exome sequencing reveals a novel homozygous splice site variant in the WNT1 gene underlying osteogenesis imperfecta type 3. Pediatr Res. 2017;82(5):753‐758. [DOI] [PubMed] [Google Scholar]
- 79. Alhamdi S, Lee YC, Chowdhury S, et al. Heterozygous WNT1 variant causing a variable bone phenotype. Am J Med Genet A. 2018;176(11):2419‐2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Luther J, Yorgan TA, Rolvien T, et al. Wnt1 is an Lrp5‐independent bone‐anabolic Wnt ligand. Sci Transl Med. 2018;10(466):eaau7137. [DOI] [PubMed] [Google Scholar]
- 81. Wang F, Rummukainen P, Heino TJ, Kiviranta R. Osteoblastic Wnt1 regulates periosteal bone formation in adult mice. Bone. 2021;143:115754. [DOI] [PubMed] [Google Scholar]
- 82. Makitie RE, Haanpaa M, Valta H, et al. Skeletal characteristics of WNT1 osteoporosis in children and young adults. J Bone Miner Res. 2016;31(9):1734‐1742. [DOI] [PubMed] [Google Scholar]
- 83. Wesseling‐Perry K, Makitie RE, Valimaki VV, et al. Osteocyte protein expression is altered in low‐turnover osteoporosis caused by mutations in WNT1 and PLS3. J Clin Endocrinol Metab. 2017;102(7):2340‐2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Makitie RE, Kampe A, Costantini A, Alm JJ, Magnusson P, Makitie O. Biomarkers in WNT1 and PLS3 osteoporosis: altered concentrations of DKK1 and FGF23. J Bone Miner Res. 2020;35(5):901‐912. [DOI] [PubMed] [Google Scholar]
- 85. Makitie RE, Hackl M, Niinimaki R, Kakko S, Grillari J, Makitie O. Altered microRNA profile in osteoporosis caused by impaired WNT signaling. J Clin Endocrinol Metab. 2018;103(5):1985‐1996. [DOI] [PubMed] [Google Scholar]
- 86. Van Dijk FS, Zillikens MC, Micha D, et al. PLS3 mutations in X‐linked osteoporosis with fractures. N Engl J Med. 2013;369(16):1529‐1536. [DOI] [PubMed] [Google Scholar]
- 87. Fahiminiya S, Majewski J, Al‐Jallad H, et al. Osteoporosis caused by mutations in PLS3: clinical and bone tissue characteristics. J Bone Miner Res. 2014;29(8):1805‐1814. [DOI] [PubMed] [Google Scholar]
- 88. Kampe AJ, Makitie RE, Makitie O. New genetic forms of childhood‐onset primary osteoporosis. Horm Res Paediatr. 2015;84(6):361‐369. [DOI] [PubMed] [Google Scholar]
- 89. Laine CM, Wessman M, Toiviainen‐Salo S, et al. A novel splice mutation in PLS3 causes X‐linked early onset low‐turnover osteoporosis. J Bone Miner Res. 2015;30(3):510‐518. [DOI] [PubMed] [Google Scholar]
- 90. Makitie RE, Kampe AJ, Taylan F, Makitie O. Recent discoveries in monogenic disorders of childhood bone fragility. Curr Osteoporos Rep. 2017;15(4):303‐310. [DOI] [PubMed] [Google Scholar]
- 91. Costantini A, Krallis P, Kampe A, et al. A novel frameshift deletion in PLS3 causing severe primary osteoporosis. J Hum Genet. 2018;63(8):923‐926. [DOI] [PubMed] [Google Scholar]
- 92. Kannu P, Mahjoub A, Babul‐Hirji R, Carter MT, Harrington J. PLS3 mutations in X‐linked osteoporosis: clinical and bone characteristics of two novel mutations. Horm Res Paediatr. 2017;88(3‐4):298‐304. [DOI] [PubMed] [Google Scholar]
- 93. Kampe AJ, Costantini A, Makitie RE, et al. PLS3 sequencing in childhood‐onset primary osteoporosis identifies two novel disease‐causing variants. Osteoporos Int. 2017;28(10):3023‐3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kampe AJ, Costantini A, Levy‐Shraga Y, et al. PLS3 deletions lead to severe spinal osteoporosis and disturbed bone matrix mineralization. J Bone Miner Res. 2017;32(12):2394‐2404. [DOI] [PubMed] [Google Scholar]
- 95. Nishi E, Masuda K, Arakawa M, et al. Exome sequencing‐based identification of mutations in non‐syndromic genes among individuals with apparently syndromic features. Am J Med Genet A. 2016;170(11):2889‐2894. [DOI] [PubMed] [Google Scholar]
- 96. Hu J, Li LJ, Zheng WB, et al. A novel mutation in PLS3 causes extremely rare X‐linked osteogenesis imperfecta. Mol Genet Genomic Med. 2020;8(12):e1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cao YJ, Zhang H, Zhang ZL. Novel mutations in the Wnt1, Tmem38b, P4hb, and Pls3 genes in four unrelated Chinese families with osteogenesis imperfecta. Endocr Pract. 2019;25(3):230‐241. [DOI] [PubMed] [Google Scholar]
- 98. Chen T, Wu H, Zhang C, et al. Clinical, genetics, and bioinformatic characterization of mutations affecting an essential region of PLS3 in patients with BMND18. Int J Endocrinol. 2018;2018:8953217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wolff L, Strathmann EA, Muller I, et al. Plastin 3 in health and disease: a matter of balance. Cell Mol Life Sci. 2021;78(13):5275‐5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lv F, Ma M, Liu W, et al. A novel large fragment deletion in PLS3 causes rare X‐linked early‐onset osteoporosis and response to zoledronic acid. Osteoporos Int. 2017;28(9):2691‐2700. [DOI] [PubMed] [Google Scholar]
- 101. Costantini A, Skarp S, Kampe A, et al. Rare copy number variants in array‐based comparative genomic hybridization in early‐onset skeletal fragility. Front Endocrinol (Lausanne). 2018;9:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shao C, Wang YW, He JW, Fu WZ, Wang C, Zhang ZL. Genetic variants in the PLS3 gene are associated with osteoporotic fractures in postmenopausal Chinese women. Acta Pharmacol Sin. 2019;40(9):1212‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Balasubramanian M, Cartwright A, Smith K, Arundel P, Bishop NJ. Copy number variants in association with type 1 collagenopathy: atypical osteogenesis imperfecta. Am J Med Genet A. 2016;170A(2):476‐481. [DOI] [PubMed] [Google Scholar]
- 104. Makitie RE, Hackl M, Weigl M, et al. Unique, gender‐dependent serum microRNA profile in PLS3 gene‐related osteoporosis. J Bone Miner Res. 2020;35(10):1962‐1973. [DOI] [PubMed] [Google Scholar]
- 105. Lin CS, Park T, Chen ZP, Leavitt J. Human plastin genes. Comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J Biol Chem. 1993;268(4):2781‐2792. [PubMed] [Google Scholar]
- 106. Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue‐based map of the human proteome. Science. 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 107. Schwebach CL, Kudryashova E, Kudryashov DS. Plastin 3 in X‐linked osteoporosis: imbalance of ca(2+)‐dependent regulation is equivalent to protein loss. Front Cell Dev Biol. 2020;8:635783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Velthaus A, Cornils K, Hennigs JK, et al. The actin binding protein Plastin‐3 is involved in the pathogenesis of acute myeloid leukemia. Cancers (Basel). 2019;11(11):1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Szkandera J, Winder T, Stotz M, et al. A common gene variant in PLS3 predicts colon cancer recurrence in women. Tumour Biol. 2013;34(4):2183‐2188. [DOI] [PubMed] [Google Scholar]
- 110. Ning Y, Gerger A, Zhang W, et al. Plastin polymorphisms predict gender‐ and stage‐specific colon cancer recurrence after adjuvant chemotherapy. Mol Cancer Ther. 2014;13(2):528‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Oprea GE, Kroeber S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chellaiah MA. L‐plastin phosphorylation: possible regulation by a TNFR1 signaling cascade in osteoclasts. Cells. 2021;10(9):2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ma T, Sadashivaiah K, Madayiputhiya N, Chellaiah MA. Regulation of sealing ring formation by L‐plastin and cortactin in osteoclasts. J Biol Chem. 2010;285(39):29911‐29924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Neugebauer J, Heilig J, Hosseinibarkooie S, et al. Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum Mol Genet. 2018;27(24):4249‐4262. [DOI] [PubMed] [Google Scholar]
- 115. Yorgan TA, Sari H, Rolvien T, et al. Mice lacking plastin‐3 display a specific defect of cortical bone acquisition. Bone. 2020;130:115062. [DOI] [PubMed] [Google Scholar]
- 116. Tanaka‐Kamioka K, Kamioka H, Ris H, Lim SS. Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin‐rich projections. J Bone Miner Res. 1998;13(10):1555‐1568. [DOI] [PubMed] [Google Scholar]
- 117. Wang N, Tytell JD, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009;10(1):75‐82. [DOI] [PubMed] [Google Scholar]
- 118. Geiger B. A 130K protein from chicken gizzard: its localization at the termini of microfilament bundles in cultured chicken cells. Cell. 1979;18(1):193‐205. [DOI] [PubMed] [Google Scholar]
- 119. Thouverey C, Malinowska A, Balcerzak M, et al. Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast‐like cells. J Proteomics. 2011;74(7):1123‐1134. [DOI] [PubMed] [Google Scholar]
- 120. Stock JL, Coderre JA, Overdorf JH, Fitzpatrick LA, Shapiro JR. Calvarial doughnut lesions associated with high‐turnover osteoporosis presenting in childhood. J Clin Densitom. 1999;2(1):45‐53. [DOI] [PubMed] [Google Scholar]
- 121. Li Z, Zhang H, Liu J, et al. Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol Cell Biol. 2011;31(20):4205‐4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Taniguchi M, Okazaki T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration‐from cell and animal models to human disorders. Biochim Biophys Acta. 2014;1841(5):692‐703. [DOI] [PubMed] [Google Scholar]
- 123. Pekkinen M, Terhal PA, Botto LD, et al. Osteoporosis and skeletal dysplasia caused by pathogenic variants in SGMS2. JCI Insight. 2019;4(7):e126180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Robinson ME, Bardai G, Veilleux LN, Glorieux FH, Rauch F. Musculoskeletal phenotype in two unrelated individuals with a recurrent nonsense variant in SGMS2. Bone. 2020;134:115261. [DOI] [PubMed] [Google Scholar]
- 125. Makitie RE, Blouin S, Valimaki VV, et al. Abnormal bone tissue organization and osteocyte Lacunocanalicular network in early‐onset osteoporosis due to SGMS2 mutations. JBMR Plus. 2021;5(11):e10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Messiou C, Cook G, deSouza NM. Imaging metastatic bone disease from carcinoma of the prostate. Br J Cancer. 2009;101(8):1225‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ciray I, Astrom G, Andreasson I, et al. Evaluation of new sclerotic bone metastases in breast cancer patients during treatment. Acta Radiol. 2000;41(2):178‐182. [DOI] [PubMed] [Google Scholar]
- 128. Suva LJ, Washam C, Nicholas RW, Griffin RJ. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7(4):208‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gowda S, Yeang C, Wadgaonkar S, et al. Sphingomyelin synthase 2 (SMS2) deficiency attenuates LPS‐induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L430‐L440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ohnishi T, Hashizume C, Taniguchi M, et al. Sphingomyelin synthase 2 deficiency inhibits the induction of murine colitis‐associated colon cancer. FASEB J. 2017;31(9):3816‐3830. [DOI] [PubMed] [Google Scholar]
- 131. Sakamoto H, Yoshida T, Sanaki T, et al. Possible roles of long‐chain sphingomyelines and sphingomyelin synthase 2 in mouse macrophage inflammatory response. Biochem Biophys Res Commun. 2017;482(2):202‐207. [DOI] [PubMed] [Google Scholar]
- 132. Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Cross talk between the bone and immune systems: osteoclasts function as antigen‐presenting cells and activate CD4+ and CD8+ T cells. Blood. 2010;116(2):210‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Anjum F, Joshi K, Grinkina N, Gowda S, Cutaia M, Wadgaonkar R. Role of sphingomyelin synthesis in pulmonary endothelial cell cytoskeletal activation and endotoxin‐induced lung injury. Am J Respir Cell Mol Biol. 2012;47(1):94‐103. [DOI] [PubMed] [Google Scholar]
- 134. Yan F, Jia P, Yoshioka H, Suzuki A, Iwata J, Zhao Z. A developmental stage‐specific network approach for studying dynamic co‐regulation of transcription factors and microRNAs during craniofacial development. Development. 2020;147(24):dev192948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Matsumoto G, Hashizume C, Watanabe K, Taniguchi M, Okazaki T. Deficiency of sphingomyelin synthase 1 but not sphingomyelin synthase 2 reduces bone formation due to impaired osteoblast differentiation. Mol Med. 2019;25(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yoshikawa Y, Yoshizawa T, Domae E, et al. Knockdown of sphingomyelin synthase 2 inhibits osteoclastogenesis by decreasing RANKL expression in mouse primary osteoblasts. Biomed Res. 2019;40(5):189‐196. [DOI] [PubMed] [Google Scholar]
- 137. Makitie RE, Niinimaki T, Nieminen MT, Schalin‐Jantti C, Niinimaki J, Makitie O. Impaired WNT signaling and the spine‐heterozygous WNT1 mutation causes severe age‐related spinal pathology. Bone. 2017;101:3‐9. [DOI] [PubMed] [Google Scholar]
- 138. Makitie RE, Niinimaki T, Suo‐Palosaari M, et al. PLS3 mutations cause severe age and sex‐related spinal pathology. Front Endocrinol (Lausanne). 2020;11:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Faqeih E, Shaheen R, Alkuraya FS. WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. J Med Genet. 2013;50(7):491‐492. [DOI] [PubMed] [Google Scholar]
- 140. Wagermaier W, Klaushofer K, Fratzl P. Fragility of bone material controlled by internal interfaces. Calcif Tissue Int. 2015;97(3):201‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Glorieux FH, Travers R, Taylor A, et al. Normative data for iliac bone histomorphometry in growing children. Bone. 2000;26(2):103‐109. [DOI] [PubMed] [Google Scholar]
- 142. Rauch F. Watching bone cells at work: what we can see from bone biopsies. Pediatr Nephrol. 2006;21(4):457‐462. [DOI] [PubMed] [Google Scholar]
- 143. Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone. 2008;42(3):456‐466. [DOI] [PubMed] [Google Scholar]
- 144. Blouin S, Fratzl‐Zelman N, Glorieux FH, et al. Hypermineralization and high osteocyte lacunar density in osteogenesis imperfecta type V bone indicate exuberant primary bone formation. J Bone Miner Res. 2017;32(9):1884‐1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mahr M, Blouin S, Behanova M, et al. Increased osteocyte lacunae density in the Hypermineralized bone matrix of children with osteogenesis imperfecta type I. Int J Mol Sci. 2021;22(9):4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bloebaum RD, Skedros JG, Vajda EG, Bachus KN, Constantz BR. Determining mineral content variations in bone using backscattered electron imaging. Bone. 1997;20(5):485‐490. [DOI] [PubMed] [Google Scholar]
- 147. Boyde A, Elliott JC, Jones SJ. Stereology and histogram analysis of backscattered electron images: age changes in bone. Bone. 1993;14(3):205‐210. [DOI] [PubMed] [Google Scholar]
- 148. Boivin G, Meunier PJ. The degree of mineralization of bone tissue measured by computerized quantitative contact microradiography. Calcif Tissue Int. 2002;70(6):503‐511. [DOI] [PubMed] [Google Scholar]
- 149. Roschger P, Fratzl P, Eschberger J, Klaushofer K. Validation of quantitative backscattered electron imaging for the measurement of mineral density distribution in human bone biopsies. Bone. 1998;23(4):319‐326. [DOI] [PubMed] [Google Scholar]
- 150. Hartmann MA, Blouin S, Misof BM, et al. Quantitative backscattered electron imaging of bone using a thermionic or a field emission electron source. Calcif Tissue Int. 2021;109(2):190‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Fratzl‐Zelman N, Valenta A, Roschger P, et al. Decreased bone turnover and deterioration of bone structure in two cases of pycnodysostosis. J Clin Endocrinol Metab. 2004;89(4):1538‐1547. [DOI] [PubMed] [Google Scholar]
- 152. Fratzl‐Zelman N, Schmidt I, Roschger P, et al. Mineral particle size in children with osteogenesis imperfecta type I is not increased independently of specific collagen mutations. Bone. 2014;60:122‐128. [DOI] [PubMed] [Google Scholar]
- 153. Fratzl‐Zelman N, Schmidt I, Roschger P, et al. Unique micro‐ and nano‐scale mineralization pattern of human osteogenesis imperfecta type VI bone. Bone. 2015;73:233‐241. [DOI] [PubMed] [Google Scholar]
- 154. Pabisch S, Wagermaier W, Zander T, Li C, Fratzl P. Imaging the nanostructure of bone and dentin through small‐ and wide‐angle X‐ray scattering. Methods Enzymol. 2013;532:391‐413. [DOI] [PubMed] [Google Scholar]
- 155. Fratzl P, Gupta HS, Roschger P, Klaushofer K. Bone nanostructure and its relevance for mechanical performance. Dis Treat Nanotechnol. 2010;2:345‐360. [Google Scholar]
- 156. Paschalis EP, Gamsjaeger S, Klaushofer K. Vibrational spectroscopic techniques to assess bone quality. Osteoporos Int. 2017;28(8):2275‐2291. [DOI] [PubMed] [Google Scholar]
- 157. Paschalis EP, Gamsjaeger S, Fratzl‐Zelman N, et al. Evidence for a role for Nanoporosity and Pyridinoline content in human mild osteogenesis imperfecta. J Bone Miner Res. 2016;31(5):1050‐1059. [DOI] [PubMed] [Google Scholar]
- 158. Weber M, Roschger P, Fratzl‐Zelman N, et al. Pamidronate does not adversely affect bone intrinsic material properties in children with osteogenesis imperfecta. Bone. 2006;39(3):616‐622. [DOI] [PubMed] [Google Scholar]
- 159. Rauch F, Travers R, Parfitt AM, Glorieux FH. Static and dynamic bone histomorphometry in children with osteogenesis imperfecta. Bone. 2000;26(6):581‐589. [DOI] [PubMed] [Google Scholar]
- 160. Roschger P, Fratzl‐Zelman N, Misof BM, Glorieux FH, Klaushofer K, Rauch F. Evidence that abnormal high bone mineralization in growing children with osteogenesis imperfecta is not associated with specific collagen mutations. Calcif Tissue Int. 2008;82(4):263‐270. [DOI] [PubMed] [Google Scholar]
- 161. Fratzl‐Zelman N, Roschger P, Misof BM, et al. Normative data on mineralization density distribution in iliac bone biopsies of children, adolescents and young adults. Bone. 2009;44(6):1043‐1048. [DOI] [PubMed] [Google Scholar]
- 162. Gamsjaeger S, Hofstetter B, Fratzl‐Zelman N, et al. Pediatric reference Raman data for material characteristics of iliac trabecular bone. Bone. 2014;69:89‐97. [DOI] [PubMed] [Google Scholar]
- 163. Rehman MT, Hoyland JA, Denton J, Freemont AJ. Age related histomorphometric changes in bone in normal British men and women. J Clin Pathol. 1994;47(6):529‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Rinnerthaler S, Roschger P, Jakob HF, Nader A, Klaushofer K, Fratzl P. Scanning small angle X‐ray scattering analysis of human bone sections. Calcif Tissue Int. 1999;64(5):422‐429. [DOI] [PubMed] [Google Scholar]
- 165. Rolvien T, Sturznickel J, Schmidt FN, et al. Comparison of bone microarchitecture between adult osteogenesis imperfecta and early‐onset osteoporosis. Calcif Tissue Int. 2018;103(5):512‐521. [DOI] [PubMed] [Google Scholar]
- 166. Palomo T, Al‐Jallad H, Moffatt P, et al. Skeletal characteristics associated with homozygous and heterozygous WNT1 mutations. Bone. 2014;67:63‐70. [DOI] [PubMed] [Google Scholar]
- 167. Boivin G, Farlay D, Bala Y, Doublier A, Meunier PJ, Delmas PD. Influence of remodeling on the mineralization of bone tissue. Osteoporos Int. 2009;20(6):1023‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ruffoni D, Fratzl P, Roschger P, Klaushofer K, Weinkamer R. The bone mineralization density distribution as a fingerprint of the mineralization process. Bone. 2007;40(5):1308‐1319. [DOI] [PubMed] [Google Scholar]
- 169. Jandl NM, von Kroge S, Sturznickel J, et al. Large osteocyte lacunae in iliac crest infantile bone are not associated with impaired mineral distribution or signs of osteocytic osteolysis. Bone. 2020;135:115324. [DOI] [PubMed] [Google Scholar]
- 170. Roschger P, Gupta HS, Berzlanovich A, et al. Constant mineralization density distribution in cancellous human bone. Bone. 2003;32(3):316‐323. [DOI] [PubMed] [Google Scholar]
- 171. Koehne T, Vettorazzi E, Kusters N, et al. Trends in trabecular architecture and bone mineral density distribution in 152 individuals aged 30‐90 years. Bone. 2014;66:31‐38. [DOI] [PubMed] [Google Scholar]
- 172. Wang K, Ren Y, Lin S, et al. Osteocytes but not osteoblasts directly build mineralized bone structures. Int J Biol Sci. 2021;17(10):2430‐2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Ayoubi M, van Tol AF, Weinkamer R, et al. 3D interrelationship between osteocyte network and forming mineral during human bone remodeling. Adv Healthc Mater. 2021;10(12):e2100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Robling AG, Bonewald LF. The osteocyte: new insights. Annu Rev Physiol. 2020;82:485‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Frost HM. Micropetrosis. J Bone Joint Surg Am. 1960;42‐A:144‐150. [PubMed] [Google Scholar]
- 176. Milovanovic P, Busse B. Phenomenon of osteocyte lacunar mineralization: indicator of former osteocyte death and a novel marker of impaired bone quality? Endocr Connect. 2020;9(4):R70‐R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Jones SJ, Glorieux FH, Travers R, Boyde A. The microscopic structure of bone in normal children and patients with osteogenesis imperfecta: a survey using backscattered electron imaging. Calcif Tissue Int. 1999;64(1):8‐17. [DOI] [PubMed] [Google Scholar]
- 178. Goff E, Buccino F, Bregoli C, et al. Large‐scale quantification of human osteocyte lacunar morphological biomarkers as assessed by ultra‐high‐resolution desktop micro‐computed tomography. Bone. 2021;152:116094. [DOI] [PubMed] [Google Scholar]
- 179. Razi H, Predan J, Fischer FD, Kolednik O, Fratzl P. Damage tolerance of lamellar bone. Bone. 2020;130:115102. [DOI] [PubMed] [Google Scholar]
- 180. Shapiro F, Maguire K, Swami S, et al. Histopathology of osteogenesis imperfecta bone. Supramolecular assessment of cells and matrices in the context of woven and lamellar bone formation using light, polarization and ultrastructural microscopy. Bone Rep. 2021;14:100734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Shane E, Shiau S, Recker RR, et al. Denosumab after Teriparatide in premenopausal women with idiopathic osteoporosis. J Clin Endocrinol Metab. 2022;107(4):e1528‐e1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Cohen A, Shiau S, Nair N, et al. Effect of teriparatide on bone remodeling and density in premenopausal idiopathic osteoporosis: a phase II trial. J Clin Endocrinol Metab. 2020;105(10):e3540‐e3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Islam MZ, Shamim AA, Viljakainen HT, et al. Effect of vitamin D, calcium and multiple micronutrient supplementation on vitamin D and bone status in Bangladeshi premenopausal garment factory workers with hypovitaminosis D: a double‐blinded, randomised, placebo‐controlled 1‐year intervention. Br J Nutr. 2010;104(2):241‐247. [DOI] [PubMed] [Google Scholar]
- 184. Peris P, Monegal A, Martinez MA, Moll C, Pons F, Guanabens N. Bone mineral density evolution in young premenopausal women with idiopathic osteoporosis. Clin Rheumatol. 2007;26(6):958‐961. [DOI] [PubMed] [Google Scholar]
- 185. Zhu K, Prince RL. Lifestyle and osteoporosis. Curr Osteoporos Rep. 2015;13(1):52‐59. [DOI] [PubMed] [Google Scholar]
- 186. Lorentzon M, Mellstrom D, Ohlsson C. Association of amount of physical activity with cortical bone size and trabecular volumetric BMD in young adult men: the GOOD study. J Bone Miner Res. 2005;20(11):1936‐1943. [DOI] [PubMed] [Google Scholar]
- 187. Roddam AW, Neale R, Appleby P, Allen NE, Tipper S, Key TJ. Association between plasma 25‐hydroxyvitamin D levels and fracture risk: the EPIC‐Oxford study. Am J Epidemiol. 2007;166(11):1327‐1336. [DOI] [PubMed] [Google Scholar]
- 188. Ralston SH, Gaston MS. Management of osteogenesis imperfecta. Front Endocrinol (Lausanne). 2019;10:924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2016;10:CD005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Li G, Jin Y, Levine MAH, Hoyer‐Kuhn H, Ward L, Adachi JD. Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. Acta Paediatr. 2018;107(3):534‐537. [DOI] [PubMed] [Google Scholar]
- 191. Orwoll ES, Shapiro J, Veith S, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124(2):491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Gatti D, Rossini M, Viapiana O, et al. Teriparatide treatment in adult patients with osteogenesis imperfecta type I. Calcif Tissue Int. 2013;93(5):448‐452. [DOI] [PubMed] [Google Scholar]
- 193. Roschger A, Roschger P, Keplingter P, et al. Effect of sclerostin antibody treatment in a mouse model of severe osteogenesis imperfecta. Bone. 2014;66:182‐188. [DOI] [PubMed] [Google Scholar]
- 194. Grafe I, Alexander S, Yang T, et al. Sclerostin antibody treatment improves the bone phenotype of Crtap(−/−) mice, a model of recessive osteogenesis imperfecta. J Bone Miner Res. 2016;31(5):1030‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Balasubramanian M, Fratzl‐Zelman N, O'Sullivan R, et al. Novel PLS3 variants in X‐linked osteoporosis: Exploring bone material properties. Am J Med Genet A. 2018;176(7):1578‐86. [DOI] [PubMed] [Google Scholar]
- 196. Rauch F, Travers R, Glorieux FH. Cellular activity on the seven surfaces of iliac bone: a histomorphometric study in children and adolescents. J Bone Miner Res. 2006;21(4):513‐9. [DOI] [PubMed] [Google Scholar]
- 197. Fahiminiya S, Majewski J, Roughley P, et al. Whole‐exome sequencing reveals a heterozygous LRP5 mutation in a 6‐year‐old boy with vertebral compression fractures and low trabecular bone density. Bone. 2013;57(1):41‐6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 WNT1 variants linked to autosomal dominant EOOP.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.