

Abstract
CD4+FOXP3+ Tregs are currently explored to develop cell therapies against immune‐mediated disorders, with an increasing focus on antigen receptor‐engineered Tregs. Deciphering their mode of action is necessary to identify the strengths and limits of this approach. Here, we addressed this issue in an autoimmune disease of the CNS, EAE. Following disease induction, autoreactive Tregs upregulated LAG‐3 and CTLA‐4 in LNs, while IL‐10 and amphiregulin (AREG) were increased in CNS Tregs. Using genetic approaches, we demonstrated that IL‐10, CTLA‐4, and LAG‐3 were nonredundantly required for the protective function of antigen receptor‐engineered Tregs against EAE in cell therapy whereas AREG was dispensable. Treg‐derived IL‐10 and CTLA‐4 were both required to suppress acute autoreactive CD4+ T‐cell activation, which correlated with disease control. These molecules also affected the accumulation in the recipients of engineered Tregs themselves, underlying complex roles for these molecules. Noteworthy, despite the persistence of the transferred Tregs and their protective effect, autoreactive T cells eventually accumulated in the spleen of treated mice. In conclusion, this study highlights the remarkable power of antigen receptor‐engineered Tregs to appropriately provide multiple suppressive factors nonredundantly necessary to prevent autoimmune attacks.
Keywords: Autoimmunity, Cell therapy, FOXP3, Gene therapy, Regulatory T cells
Graphical Abstract: Autoreactive Tregs produced by gene therapy are a promising approach for cell therapy of immunological diseases. We found that IL‐10, CTLA‐4, and LAG‐3 were nonredundantly involved in their protective effect in EAE, underlining their unique capacity to appropriately deliver in time and space crucial nonredundant mechanisms of immune regulation.
Introduction
Immune‐mediated inflammatory diseases affect an increasing proportion (5–10%) of the population, and have considerable individual as well as societal costs. The majority of these diseases cannot be cured by available therapies, which mainly consist of immunosuppressive drugs. These drugs do not correct the defects in immune regulation often associated with these chronic disorders. Restoring immune regulation is, thus, an important goal to ensure that these patients do not relapse following the interruption of their immunosuppressive treatment because of the reactivation of remaining pathogenic immune cells.
CD4+ Tregs expressing the transcription factor forkhead box P3 (FoxP3) play a crucial role in the maintenance of immune homeostasis [1], and are intensely explored to develop cellular therapies against immune‐mediated inflammatory diseases including autoimmune disorders, GVH disease, and graft rejection [2]. Clinical trials have already shown the good safety profile of polyclonal Tregs in patients, yet indicated a need for the improvement of their clinical benefit [2, 3, 4, 5, 6].
Antigen‐specific Tregs have shown superior protective efficacy compared to polyclonal Tregs in preclinical models of immune‐mediated diseases, which has driven the field toward the development of antigen‐reactive Tregs for cell‐based therapies. However, the obtention of sufficient numbers of antigen‐reactive Tregs for cell therapy is challenging because these cells are rare and difficult to isolate. This difficulty can be overcome by the engineering of polyclonal Tregs through antigen receptor gene transfer so that they ectopically express a selected antigen receptor, usually a T cell receptor (TCR) or a chimeric antigen receptor [7, 8].
The affinity of the ectopically expressed TCR for the target antigen, and the intensity of TCR signaling are known to influence the suppressive function of Tregs [9, 10, 11]. In a model of CNS autoimmunity, it was found that Tregs genetically engineered to express a high‐affinity autoreactive TCR (TCR isolated from autoreactive Tregs found in CNS autoimmune lesions) almost completely protected recipient mice from disease upon adoptive transfer whereas Tregs expressing a low affinity TCR for the same autoantigen (TCR isolated from autoreactive CNS T‐conventional cells [Tconvs]) had little beneficial effect [7]. The activity of Tregs can also be increased or decreased by modulating TCR signaling [12, 13].
The mode of action of antigen receptor‐engineered Tregs remains poorly defined. Their production involves the isolation of peripheral polyclonal Tregs, which are then activated in the presence of anti‐CD3, anti‐CD28, as well as IL‐2 for at least 7 days, and are, during this expansion phase, genetically engineered to ectopically express a single antigen receptor. The impact of such protocol on Treg function postadoptive cell therapy has not been explored extensively. It is, however, essential to elucidate how these modified cells function in vivo to envision their most favorable application and optimize their therapeutic effect in distinct clinical entities.
Nonengineered Tregs can regulate immunity via mechanisms involving CTLA‐4, IL‐10, TGF‐β, IL‐35, CD25, CD39, CD73, perforin, and granzyme B [6, 14]. Some data suggest that Tregs employ different mechanisms to suppress immunity in distinct tissues. Mice with an Il10 deficiency restricted to Tregs displayed an altered immune homeostasis in guts and lungs, with no obvious overt inflammation in other organs [15]. Similarly, the protection afforded by Tregs against kidney damage was IL‐10‐independent in a nephrotoxicity model [16, 17]. Further supporting this notion, upon leaving secondary lymphoid tissues, Tregs remodel their gene expression profile and adapt to their novel niche as they enter peripheral tissues [18, 19]. Tregs from various tissues displayed distinct gene expression programs, and used different factors to promote tissue repair [19, 20].
What mechanisms are used by engineered quasi‐monoclonal Tregs expressing a high‐affinity TCR to protect from different organ‐specific autoimmune disease in cell therapy? Here, we examined the molecular mechanisms involved in the protective function of engineered high‐affinity autoreactive Tregs against CNS autoimmunity. First, we examined the molecular profile of engineered high‐affinity autoreactive Tregs in draining LN after EAE induction, and characterized the transcriptome of CNS Tregs at the peak of EAE. Second, we tested the role of selected factors using genetic approaches. We found that engineered antigen‐reactive Tregs protected mice from EAE using IL‐10, CTLA‐4, and LAG‐3 in a nonredundant manner, whereas their expression of AREG was facultative. Collectively, these data document the remarkable capacity of engineered autoreactive Tregs to appropriately provide different suppressive factors acting in a nonredundant manner to prevent disease progression.
Results
LAG‐3 expression marks TCR‐activated Tregs in draining lymph nodes
Because antigen‐specific Tregs are more efficient than polyclonal Tregs in adoptive cell therapy, we reasoned that they might, upon TCR‐driven activation, upregulate the expression of molecules involved in their suppressive function.
To identify such molecules, we established an experimental system permitting the tracking of engineered antigen‐specific Tregs in recipient mice. Polyclonal Tregs were transduced in vitro with a bicistronic retroviral vector encoding for both a high‐affinity myelin oligodendrocyte glycoprotein (MOG)‐reactive TCR and the marker Thy1.1 that enabled their subsequent identification. These cells were administered into recipient mice that were then immunized with MOG (35‐55). This led to the rapid accumulation of injected Tregs in draining LNs (Fig. 1A‐C). We then measured the expression of molecules controlled in a TCR‐dependent manner on these cells [21]. They displayed the distinctive upregulation of LAG‐3, CD25, CD279 (PD‐1), and CD152 (CTLA‐4), with the strongest differences found on day 3 postimmunization (p.i.) compared to polyclonal (endogenous) Tregs (Fig. 1D). The inhibitory receptor LAG‐3 displayed the strongest upregulation. In contrast, CD83, CD121b (IL‐1R2), and CD200 were expressed at similar levels between transferred antigen‐specific Tregs and endogenous polyclonal Tregs (Supporting information Fig. S1).
Figure 1.
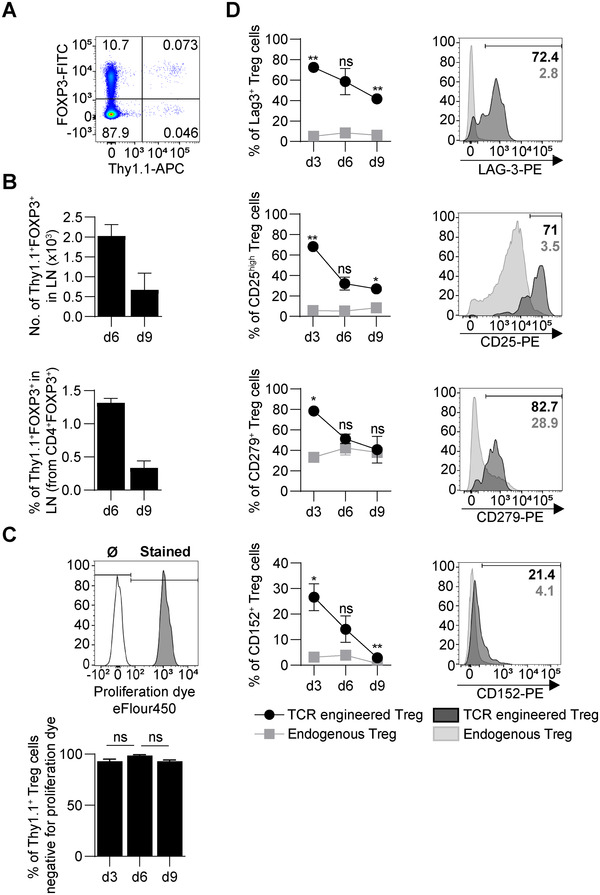
Engineered antigen‐specific Tregs distinctively upregulate surface expression of LAG‐3, CD25, PD‐1, and CTLA‐4. Polyclonal Treg cells were isolated from C57BL/6 mice and transduced with high‐avidity MOG‐reactive TCR. A total of 1 × 106 engineered Treg cells were injected into C57BL/6 recipient mice one day before immunization with MOG (35‐55) peptide. On days 3, 6, and 9 p.i. cells from draining LN (inguinal and popliteal) were harvested and stained for selected intracellular and surface markers. In each experiment, three mice were sacrificed on the selected day, and cells from all LN were pooled. Data are pooled from two experiments. Graphs show mean ± SEM. (A) Representative flow cytometry plot showing the percentages of Foxp3+Thy1.1+ engineered Treg (0.073%) within the CD4+ population in LN on day 3 p.i. Endogenous Tregs are Foxp3+Thy1.1–. (B) Absolute number (top) and percentage (bottom) of engineered Treg (CD4+Foxp3+Thy1.1+) in draining LN on days 6 and 9 p.i. (C) Engineered MOG‐specific Treg cells were labeled with proliferation dye. Gray histogram shows stained Foxp3+Thy1.1+ cells. A total of 100% of the cells were stained just before injection. The white histogram shows a control sample that was not stained (Ø). On 3, 6, and 9 days p.i., the percentage of engineered Treg cells negative for proliferation dye was determined (bottom). (D) Expression of LAG‐3, CD25, CD279 (PD‐1), CD152 (CTLA‐4) on the cell surface of engineered (dark) and endogenous Treg (light grey) cells. Representative flow cytometry plots show the percentage of cells with high expression of selected surface markers on day 3 p.i. **p < 0.01, *p < 0.05, ns–not significant.
We conclude that engineered TCR‐activated Tregs distinctively upregulate the expression of LAG‐3, CD25, CD279, and CD152 in draining LNs. These molecules are known to have suppressive functions, and might thus, contribute to the protective effect of engineered cells.
LAG‐3 is involved in the protective function of autoreactive Tregs in EAE
LAG‐3 is an immune regulatory molecule with complex roles, being involved in the suppression of immunity [22], the suppressive function of Tregs [22], yet also limiting the regulatory impact of the latter in some contexts [23]. We, thus, directly tested how LAG‐3 modulated the protective function of engineered MOG‐reactive Tregs in EAE.
Lag3‐deficient as well as control Tregs were engineered with a high‐affinity MOG‐reactive TCR and then compared for their protective function in EAE (Fig. 2A). WT engineered Tregs completely protected recipient mice from disease, while their Lag3‐deficient counterpart only achieved a partial protection compared to WT Tregs (Fig. 2A). Thus, LAG‐3 intrinsically contributes moderately to the protective function of engineered autoreactive Tregs against CNS autoimmunity.
Figure 2.
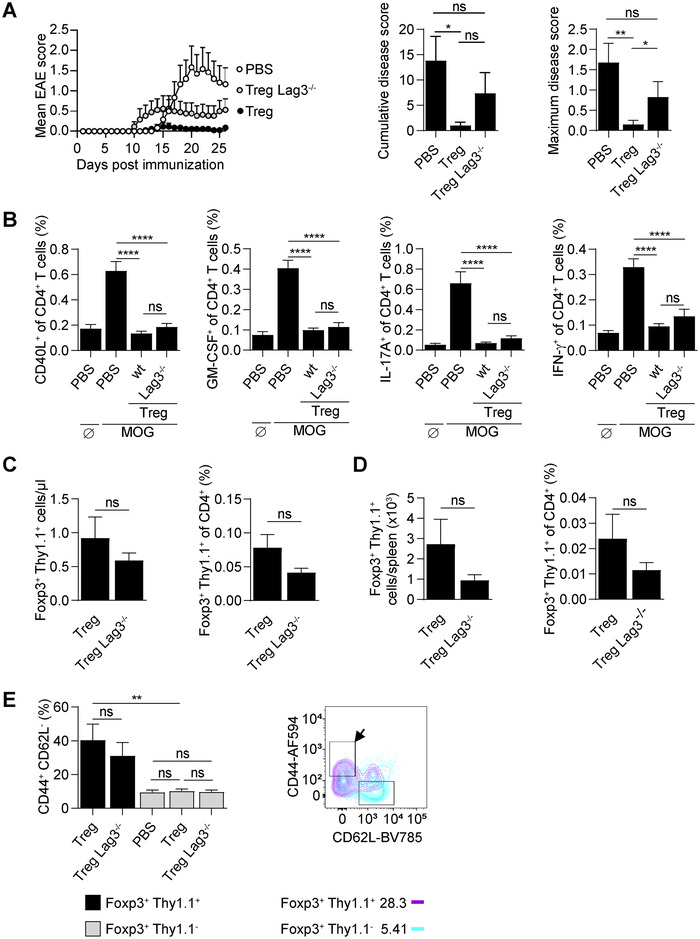
LAG‐3 contributes to the protective function of engineered MOG‐reactive Tregs. Tregs were isolated from WT or Lag3‐deficient mice and transduced with MOG‐reactive TCR of high avidity. A total of 2 × 105 engineered Tregs were adoptively transferred into recipient C57BL/6 mice on day ‐1. One group was left untreated (PBS). On day 0, all mice were immunized to induce EAE. Data are pooled from three experiments, n = 14–17 per group unless stated otherwise. Graphs show mean + SEM. (A) EAE disease courses, cumulative disease scores, and maximum disease scores. (B and C) Nine days p.i., peripheral blood from all mice was collected. Data are pooled from three experiments (n = 17 per group). (B) Peripheral blood cells were stimulated with MOG (35‐55) for 6 h (MOG) or left untreated (Ø; indicated for PBS group). The expression of immune molecules (CD40L, GM‐CSF, IL‐17A, and IFN‐γ) was analyzed by intracellular cytokine staining. (C) Absolute number and percentage of engineered Tregs (Foxp3+Thy1.1+) in peripheral blood. (D) At the end of the experiment (day 35 p.i.), splenocytes were analyzed and the absolute number and percentage of engineered Tregs (Foxp3+Thy1.1+) were determined. (E) Blood cells collected at day 9 p.i. were stained for CD44 and CD62L. Percentage of CD44+CD62L– cells was compared between engineered (Foxp3+Thy1.1+) and endogenous (Foxp3+Thy1.1–) Tregs. Data show mean + SEM. Representative flow cytometry plot shows expression of CD44 and CD62L by engineered (Foxp3+Thy1.1+, magenta) and endogenous (Foxp3+Thy1.1–, cyan) Tregs. The arrow indicates “activated” CD44hiC62Llo cells. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, ns–not significant.
We next analyzed autoreactive CD4+ T‐cell responses in these mice. WT and Lag3‐deficient Tregs both strongly reduced the acute autoreactive CD4+ T‐cell response in treated mice, as shown by the lower abundance of MOG‐reactive CD4+ T cells (identified by the expression of CD40L) and proinflammatory CD4+ T cells (identified by the expression of GM‐CSF, IL‐17A, IFN‐γ) in blood on day 9 p.i. in mice treated with these Tregs compared to untreated control mice (Fig. 2B). In contrast, and even though the disease was still controlled, the treatment had no impact on the long‐term accumulation of autoreactive CD4+ T cells in spleen at the end of the experiment, which were identified either through the upregulation of CD40L or the expression of proinflammatory cytokines upon MOG (35–55) restimulation (Supporting information Fig. S2A). Of note, effector CD4+ T cells do not persist in the CNS of untreated mice at these late time points [24]. Collectively, these data show that adoptive Treg cell therapy suppresses the acute activation of the autoreactive CD4+ T‐cell response, which correlates with the protection from disease. However, they do not interfere with the long‐term accumulation of autoreactive CD4+ T cells in spleen.
We next compared the abundance of MOG‐reactive WT and Lag3‐deficient Tregs in treated mice during the course of the disease. Lag3‐deficient Tregs were present in slightly lower amount than WT Tregs at day 9 (Fig. 2C) and day 35 (Fig. 2D) p.i, yet the difference was not statistically significant.
The TCR‐engineered autoreactive Tregs displayed an activated CD44hiCD62Llo phenotype in treated mice on day 9 p.i. (Fig. 2E) as well as at the end of the experiment (Supporting information Fig. S2B). The Lag3 deficiency did not affect the acquisition of this activated phenotype, and the upregulation of CTLA‐4 in Tregs (Supporting information Fig. S2B).
Collectively, these data demonstrate that LAG‐3 contributes to a modest extent to the protective function of engineered autoreactive Tregs during adoptive cell therapy because its removal partially impairs their protective function.
IL‐10 expression is enhanced in CNS Tregs and crucial for autoreactive Treg‐mediated protection
To define the molecular characteristics of autoreactive Tregs at disease target site in EAE, we performed single RNAseq and bulk transcriptome analyses of Tregs and Tconvs present in CNS at the peak of the disease.
We started by performing single‐cell RNAseq measurements on infiltrated CD45+ from the CNS at EAE peak, and analyzed the CD4+ T‐cell compartment (Fig. 3A). The representation of obtained results using uniform manifold approximation and projection (UMAP) revealed seven clusters of CD4+ T cells within the CNS of mice that developed EAE (Fig. 3A) including a distinct cluster of CD4+Foxp3+ cells (cluster 5) (Fig. 3B). These seven clusters were characterized by different gene expression profiles (Supporting information Fig. S3). In particular, the clusters 1, 3, and 7 showed the highest expression of Ifng, Csf2, and Il17a, which were not found in cluster 5 that instead was the only one to express Foxp3 and had the highest expression level for Il2ra and Ctla4 (Supporting information Fig. S4). The comparison of CD4+Foxp3+ cells and CD4+Foxp3– cells highlighted expected differences, with a higher expression of Treg genes (Tnfrsf4, Izumo1r, Tnfrsf18, Il2ra, Ctla4, Samhd1, as well as Areg and Il10) in Foxp3 + cells and Tconv genes (Ifng, Cxcr6) in Foxp3 – cells (Supporting information Fig. S5A), and identified 220 differentially expressed genes between Foxp3 + and Foxp3 – cells (adj. p‐value <0.05, Supporting information Table S1). Thus, Foxp3 expression faithfully identifies Tregs in the CNS at the peak of EAE. To complement these analyses, we next investigated the transcriptomes of bulk CD4+Foxp3+ Tregs and CD4+Foxp3– Tconvs from the CNS and spleen of Foxp3.eGFP mice at the peak of EAE using eGFP as a surrogate for Foxp3 expression. The comparison of CNS and spleen Tregs from the same mice showed a higher expression of Areg, Ctla2a, Ctla2b, and Cxcr6 in CNS Tregs (Fig. 3C), as well as several differentially expressed transcription factors such as Atf3 (Fig. 3D). Ingenuity analyses of the genes differentially expressed between CNS and spleen Tregs indicated that several of the transcription factors differentially expressed by CNS Tregs might be interconnected including Atf3, Hif1a, Epas1, and Bhlhe40 (Supporting information Fig. S5B). To prioritize the list of genes to investigate functionally, we next identified the 10 genes most upregulated in CNS Tregs compared to spleen Tregs (and vice versa) (Fig. 3E) and the 10 genes most‐upregulated in CNS Tregs compared to CNS Tconvs (and vice versa) (Fig. 3F), which highlighted Areg (amphiregulin) and Il10 as genes markedly upregulated in CNS Tregs: Areg was expressed 87‐fold more in CNS Tregs than in spleen Tregs (Supporting information Table S2) and Il10 was expressed 89‐fold more in CNS Tregs than in CNS Tconvs (Supporting information Table S3). Il10 was also present at a 10‐fold higher level in CNS Tregs than in spleen Tregs in EAE mice (Supporting information Table S2). AREG is a member of the EGF receptor family involved in tissue repair as well as in the development and maintenance of organs [25]. AREG was found to be expressed in Tregs after ischemic stroke, and to contribute to neuronal protection and astrogliosis reduction [26]. IL‐10 has potent suppressive functions on proinflammatory CD4+ T cells as well as myeloid cells, and is involved in the control of EAE progression [27]. The roles of IL‐10 and AREG in the function of antigen receptor‐engineered Tregs have not been defined in CNS autoimmunity.
Figure 3.
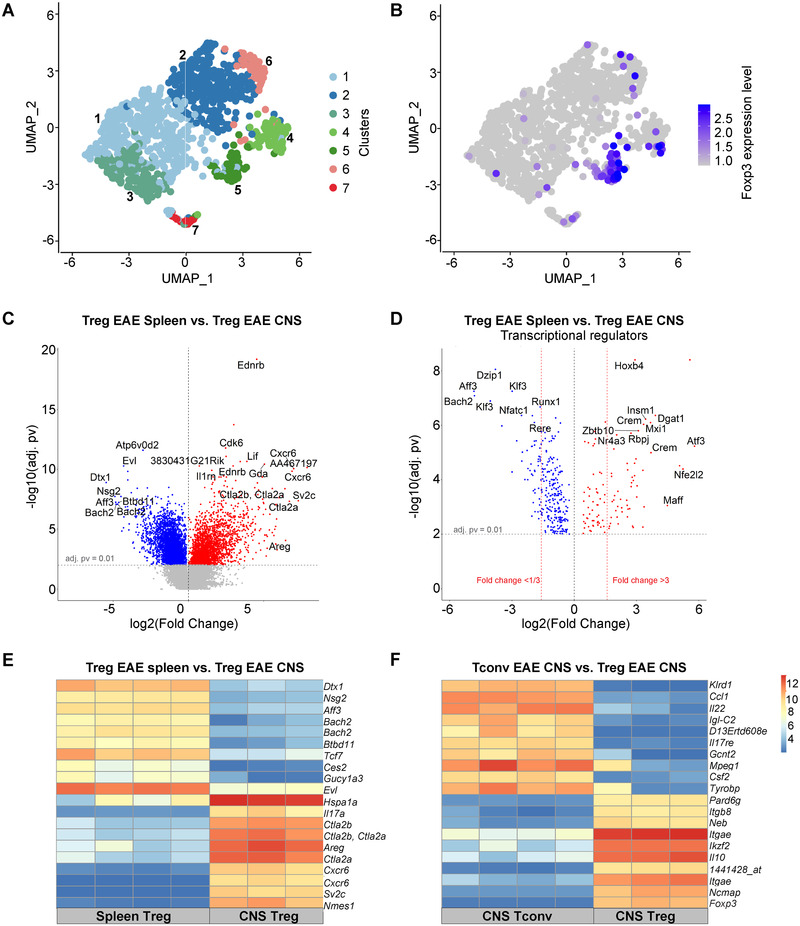
Transcriptome characteristics of CNS Tregs. (A and B) CD45+ cells were sorted from the CNS of C57BL/6 mice at the peak of EAE (day 19) and analyzed by single‐cell RNAseq (10× Genomics). UMAP visualization of CNS‐infiltrating CD4+ T cells colored by cluster assignment (A) and by the expression of Foxp3 (B). (C to F) Tregs and Tconvs were sorted from CNS and spleen of Foxp3.eGFP mice at the peak of EAE according to the expression of CD4 and Foxp3 (Treg: CD4+Foxp3.eGFP+ cells; Tconv: CD4+Foxp3.GFP–). Samples (triplicates or quadruplicates) were hybridized to Affymetrix 430 2.0 whole‐genome mouse arrays. (C) Volcano plot showing differentially expressed genes between spleen Tregs (blue) and CNS Tregs. (D) Volcano plot showing a subset of genes shown in (C) corresponding to differentially expressed transcriptional regulators between spleen Tregs and CNS Tregs (E) mRNA expression levels of 10 genes most upregulated in CNS Tregs and 10 genes most upregulated in spleen Tregs from EAE mice, compared to each other. (F) mRNA expression levels of 10 most upregulated genes in CNS Tregs and 10 most upregulated genes in CNS Tconvs in EAE mice, compared to each other. Expression amounts normalized using the GCRMA are shown (Log2 transformed).
To define the roles of these factors in engineered Tregs, we established tricistronic retroviral vectors encoding for the high‐affinity MOG‐reactive TCR, the Thy1.1 reporter, and miRNAs against Il10 or Areg, respectively (Supporting information Fig. S6A). Six and eight micro RNA (miRNAs) against Il10 and Areg were tested, respectively (Supporting information Fig. S6B). We selected miRNAs reducing IL‐10 and AREG expression by 20‐ and 188‐fold, respectively (Supporting information Fig. S6B). Tregs engineered to express the MOG‐reactive TCR and silenced for IL‐10 production had no protective effect (Fig. 4A), alike Tregs silenced for Ctla4 expression (Fig. 4A), as previously observed [7]. In contrast, Areg silencing did not affect the engineered Treg protective function (Fig. 4A). Engineered Tregs, thus, protect recipient mice from disease through immunological mechanisms.
Figure 4.
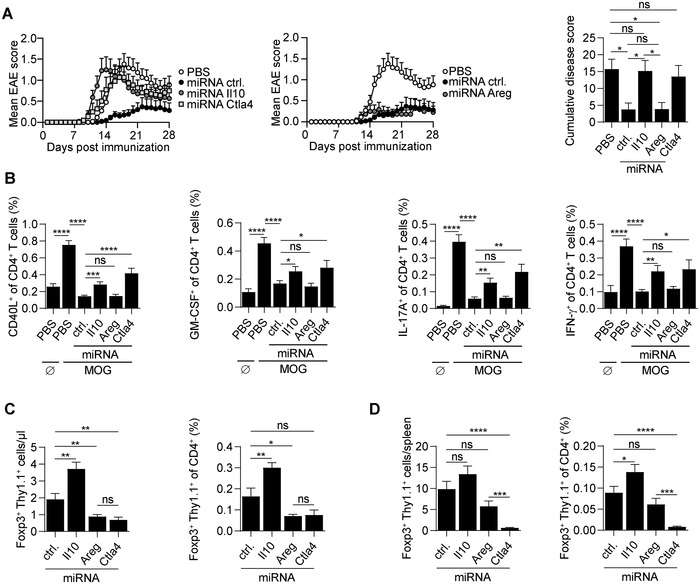
IL‐10 is critically involved in the protective function of autoreactive Tregs. (A) A total of 2 × 105 Tregs engineered to express MOG‐reactive TCR and either control miRNA, miRNA to silence Il10, Areg, or Ctla4 expression were adoptively transferred into C57BL/6 mice on day ‐1. Control groups were treated with PBS. On day 0, mice were immunized with MOG (35‐55) to induce EAE. Data show EAE score and cumulative score. Compilation of four independent experiments (n = 22 per group; mean + SEM). (B) On day 9 p.i., peripheral blood from every mouse was collected. Blood cells were left untreated (Ø) or stimulated with MOG (35‐55) peptide for 6 h (MOG). The expression of immune molecules (CD40L, GM‐CSF, IL‐17A, and IFN‐γ) was analyzed by intracellular cytokine staining. (C) Absolute number and percentage of engineered Tregs (Foxp3+Thy1.1+) in peripheral blood 9 days p.i. (D) Absolute number and percentage of engineered Tregs (Foxp3+Thy1.1+) in spleen at the end of the EAE experiment (day 29 p.i.). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, ns–not significant.
We next analyzed the MOG‐reactive CD4+ T‐cell response in these mice to define the roles of IL‐10 and CTLA‐4 from engineered Tregs. The silencing of Il10 and Ctla4 both impaired the control of the acute autoreactive CD4+ T‐cell response in blood on day 9 p.i., as revealed by the higher abundance of MOG‐reactive CD4+ T cells (CD40L+), including cells producing GM‐CSF, IL‐17A, or IFN‐γ, compared to the group treated with control Tregs (Fig. 4B). The requirement of engineered Treg‐derived IL‐10 and CTLA‐4 for the suppression of both (i) disease course and (ii) early autoreactive CD4+ T‐cell activation, underlines the relationship between these two parameters.
We conclude that engineered Tregs protect recipient mice from disease, and suppress the acute autoreactive CD4+ T‐cell response using nonredundant immunosuppressive mechanisms based on their expression of IL‐10 and CTLA‐4, in addition to LAG‐3.
CTLA‐4 and IL‐10 distinctively control the accumulation of engineered autoreactive Tregs in treated mice
We next evaluated whether Il10, Areg, or Ctla4 silencing affected the engineered Tregs’ accumulation in treated mice. The silencing of Ctla4 reduced the abundance of engineered Tregs in blood at day 9 (Fig. 4C) and day 29 p.i. (Fig. 4D). In contrast, the silencing of Il10 led to an increased abundance of engineered CD4+Foxp3+Thy1.1+ Tregs on both day 9 (Fig. 4C) and day 29 p.i. (Fig. 4D). Engineered Tregs silenced for their intrinsic expression of Ctla4, Areg, or Il10 did not display any defect in their activation, as evaluated by the frequency of CD44+CD62L– cells, and their expression of CTLA‐4 (except for Ctla4 silenced Treg, which displayed the expected reduced CTLA‐4 expression) as well as LAG‐3 (Supporting information Fig. S6C).
The finding that CTLA‐4 controlled the accumulation of engineered autoreactive Tregs was intriguing. To confirm this observation, we used a complementary approach, treating mice that received engineered Tregs with a blocking anti‐CTLA‐4 antibody during the course of the disease. An anti‐PD‐1 treatment was administered as control, using treatment modalities previously reported to exacerbate disease [28, 29]. As expected, mice administered with an anti‐CTLA‐4 or an anti‐PD‐1 using these protocols, without any Treg treatment, displayed a more severe disease than control mice, validating the efficacy of these treatments (Fig. 5A). In contrast, mice treated with Tregs remained fully protected from disease upon the administration of these antibodies, allowing to address the impact of anti‐CTLA‐4 on injected Tregs in a disease context comparable to the group that only received Tregs (Fig. 5A). We, thus, measured the abundance of engineered CD4+Foxp3+Thy1.1+ Tregs, which were all MOG‐reactive, compared to endogenous CD4+Foxp3+Thy1.1– Tregs, which were mostly not reactive against MOG, in anti‐CTLA‐4‐treated and control mice (Fig. 5B). The two anti‐CTLA‐4 injection distinctively affected the abundance of engineered Tregs compared to controls (Fig. 5B), while it had no impact on the abundance of endogenous CD4+Foxp3+Thy1.1– Tregs (Fig. 5C). These treatments did not affect the activation of the engineered Tregs, as evidenced by the proportion of these cells that displayed a PD‐1hiCTLA‐4hi phenotype (Fig. 5D). In combination, these two experimental approaches, namely Ctla4 genetic silencing and antibody‐mediated blockade, demonstrate that CTLA‐4 is involved in the abundance of TCR‐activated Tregs during autoimmune disease.
Figure 5.
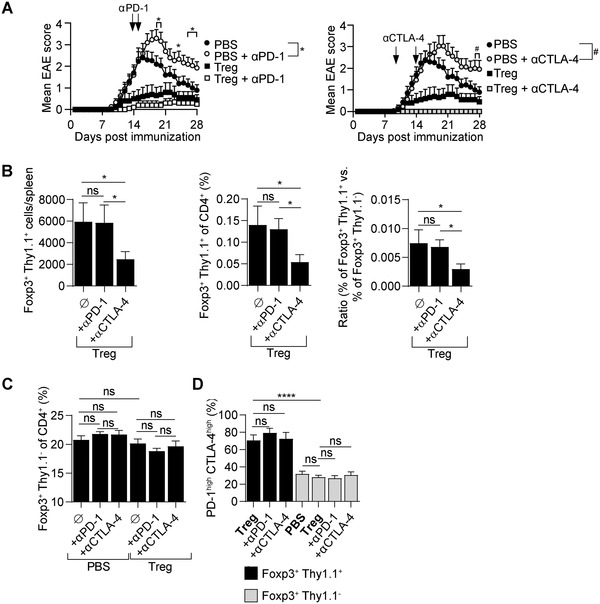
Impact of CTLA‐4 blocker on antigen‐receptor engineered Tregs accumulation during disease. Tregs engineered to express the MOG‐reactive TCR were adoptively transferred into C57BL/6 mice on day ‐1. Control groups were treated with PBS. On day 0, all mice were immunized with MOG (35‐55) to induce EAE. Mice were treated twice with antibodies against CTLA‐4 (days 10 and 14, 0.2 mg/mouse/injection) or PD‐1 (days 14 and 16, 0.2 mg/mouse/injection). At the end of the experiment (day 29 p.i.), splenocytes were analyzed. Data are the compilation of two or three independent experiments. Three experiments were made for groups treated with PBS, PBS + αPD‐1, and PBS + αCTLA‐4 (n = 15 per group). Two experiments were made for groups treated with Treg, Treg+ αPD‐1, and Treg+ αCTLA‐4 (n = 15 per group). Graphs show mean + SEM. (A) EAE disease course. *Designates significant (p < 0.05) difference between groups treated with PBS alone and PBS + antibody against PD‐1. #Designates significant (p < 0.05) difference between groups treated with PBS alone and PBS + antibody against CTLA‐4. (B) Absolute number of engineered Tregs (Foxp3+Thy1.1+) (left), percentage of engineered Tregs (within CD4+) (middle), and ratio of engineered (Thy1.1+) versus endogenous Tregs (Thy1.1–) (right) in spleens at the end of the experiment. Data are the compilation of two independent experiments (n = 9 per group). Groups were compared using one‐tailed t‐test. (C) Percentage of endogenous Tregs (Foxp3+Thy1.1–) in spleens at the end of the experiment. (D) Expression of CTLA‐4 (intracellular staining) and PD‐1 (surface staining) on engineered (Foxp3+Thy1.1+) and endogenous (Foxp3+Thy1.1–) Tregs from spleens at the end of the experiment. ****p < 0.0001, *p < 0.05, ns–not significant.
We conclude that IL‐10 and CTLA‐4 provide nonredundant mechanisms of protection by “monoclonal” engineered autoreactive Tregs during CNS autoimmune disease. These molecules control the priming of the acute autoreactive CD4+ T‐cell response, but differentially influence the accumulation of engineered autoreactive Tregs, underlying their distinct roles in setting the balance between deleterious and protective CD4+ T cells.
Discussion
The development of cell therapies with antigen receptor‐engineered Tregs is one of the most promising paths to reach antigen‐specific immune treatments restoring organ‐specific immune tolerance in a safe and efficient way. Deciphering the mode of action of antigen receptor‐engineered Tregs is crucial to optimize their therapeutic application in distinct clinical entities. Here, we have addressed how TCR‐engineered autoreactive Tregs provide protection against CNS autoimmunity in cell therapy.
As starting point, we analyzed the characteristics of disease‐relevant Tregs during EAE, measuring the expression of TCR‐controlled cell‐surface receptors on engineered Tregs in LN, and defining the transcriptome of CNS Tregs at the peak of the disease. In LN, autoreactive‐engineered Tregs upregulated the expression of CD25, CD152 (CTLA‐4), CD279 (PD‐1), and LAG‐3. Then, we showed that LAG‐3 and CTLA‐4 contributed to their protective function. In CNS, Tregs displayed an increased expression of several genes compared to their spleen counterparts, including Areg, Il10, the chemokine receptor Cxcr6, which also marks encephalitogenic CD4+ T cells [30], the cysteine protease inhibitors Ctla2a and Ctla2b, which can control cathepsin L in the CNS [31], the IL‐1 receptor antagonist (Il1rn), and the receptor for endothelin (Ednrb). These molecules might all contribute to the protective functions of Tregs in this disease. In demyelinating lesions, endothelin is produced by reactive astrocytes and modulate oligodendrocyte progenitor cell differentiation [32]. At steady state, it is mainly produced by vascular endothelial cells, which modulates the myelinating capacity of mature oligodendrocytes [33]. CNS Tregs also displayed a higher expression of transcription factors potentially involved in their protective function, including Atf3, which contributed to Il10 expression in CD4+ T cells [34], and Hif1a as well as Epas1 (also known as Hif2a), which were involved in cell adaptation to hypoxia [35].
To initiate the systematic assessment of these molecules, we tested here the roles played by several factors using multicistronic retroviral vectors enabling the expression of an autoreactive TCR, miRNA for selected genes, and a reporter gene permitting to track selectively the fate of the engineered Tregs [36]. Engineered MOG‐reactive Tregs relied on IL‐10, CTLA‐4, and to a minor extent LAG‐3 to control disease. The involvement of these three molecules in the protective function of engineered Tregs underlined the remarkable capacity of such cell therapy to appropriately provide in time and space nonredundant mechanisms of immune regulation. In addition, we observed that Ctla4‐silenced autoreactive Tregs displayed an impaired accumulation in recipient mice. The effect of Ctla4 silencing on disease‐relevant Tregs was corroborated by results obtained with a blocking anti‐CTLA‐4 antibody. These findings suggest that CTLA‐4 intrinsically provides activated Tregs with key survival or proliferative signals, which are not necessary for homeostatic Treg maintenance. According to this notion, CTLA‐4 would contribute to the regulatory function of activated Tregs in two ways: (1) the engagement of CD80/CD86 on APCs and (2) the provision of survival or proliferation signals to activated Tregs. Intriguingly, Tregs expressing a transgenic myelin basic protein‐reactive TCR suppressed CNS autoimmunity in a Ctla4‐independent manner [37, 38]. These opposite findings might relate to the different methodologies used in the two studies. Here, we used polyclonal Tregs that developed in a normal environment, and were subsequently engineered in vitro to silence Ctla4 expression. In contrast, Tregs that developed in a fully Ctla4‐deficient genetic background might carry distinct properties. These observations underline the need to systematically analyze the mechanisms underpinning the protective function of antigen receptor‐engineered Tregs in immune‐mediated inflammatory diseases. Remarkably, the transient blockade of CTLA‐4 using antibodies did not intercept the beneficial effect of the administered Tregs, suggesting that such Tregs might be relevant to prevent unwanted immunopathology in cancer patients treated by checkpoint blockers.
It was unexpected that adoptive Treg therapy had no impact on the long‐term accumulation of autoreactive Tconvs in spleen including their ability to produce proinflammatory cytokines. Although the pathogenic function of these CD4+ T cells was not tested, and the mice remained protected, it cannot be excluded that their accumulation might eventually facilitate the occurrence of disease relapse upon exposition to certain environmental factors such as infectious agents. Accordingly, adoptive Treg therapy might benefit from combination with therapeutics that remove autoreactive Tconvs. Supporting this “double hit” strategy, the co‐application of Tregs and immunosuppressive drugs led to remarkable results in a small trial on liver transplantation involving 10 patients [39]. The immunosuppressants used in this case were cyclophosphamide, and inhibitors of calcineurin or mTOR. The immunosuppressive treatment could be discontinued as early as 18 months post‐transplantation in the majority of patients, which then remained drug free for more than 12 months, suggesting the establishment of operational tolerance [39]. A comparable protocol prevented renal allograft rejection in rhesus monkeys, and the single omission of cyclophosphamide led to acute graft rejection [40]. Cyclophosphamide can deplete alloantigen‐reactive Tconvs [41]. This study also supports the notion that adoptive Treg therapy might benefit from supportive combination with “Treg friendly” immunosuppressive drugs. Combination therapy using carefully chosen immunosuppressive drugs could, thus, greatly advance the success of adoptive Treg therapy in the clinic.
Future studies will be required to examine the role of genes differentially expressed in CNS Tregs identified here through scRNAseq and RNAseq analyses in the protective function of these cells during CNS autoimmunity. This includes Ctla2a, Ctla2b, Il1rn, and Ednrb, as well as transcription factors potentially implicated in the adaptation of Tregs to the inflamed CNS environment and Il10 transcription. Intiguingly, the RNAseq comparison of spleen and CNS Tregs highlighted Il17a as a gene overexpressed in CNS Tregs. The scRNAseq analysis of CNS Tregs, however, failed to detect Il17a expression in Tregs, even though it could be seen in some Foxp3 – CD4+ T‐cell clusters in CNS. It is possible that the differential abundance of Il17a between bulk spleen and CNS Tregs reflected a contamination of CNS Tregs by other Il17a ‐expressing cells from CNS, or that CNS Tregs transcribe Il17a at low level. Our data do not allow to discriminate between these possibilities. However, our previous study demonstrated that CNS Tregs do not express IL‐17 protein, and are refractory to conversion toward an IL‐17‐producing phenotype [42].
In conclusion, this study shows that engineered autoreactive Tregs protect from CNS autoimmunity via multiple mechanisms, including IL‐10, LAG‐3, and CTLA‐4, which act in nonredundant manners. Adoptive cell therapy is, thus, unique in its capacity to deliver complementary mechanisms at the right time and in the right place to suppress undesirable immune responses. Remarkably, engineered Tregs could block disease onset, underlining their high potential. However, they did not influence the long‐term accumulation of autoreactive Tconvs in treated mice, which, even though the latter remained protected, might be a limitation to their impact in enduring chronic diseases.
Methods
Mice
C57BL/6 mice were purchased from Janvier Labs and Taconic Biosciences A/S. Foxp3‐eGFP mice [43] were maintained at the University of Edinburgh. Lag3 –/– mice [44, 45] were bred at INEM. Eight to twelve weeks old female mice were used in all experiments. All mice were bred under specific pathogen‐free conditions. All experiments were reviewed and approved by appropriate institutional review committees and conducted according to French and UK legislations, in compliance with European Community Council Directive 68/609/EEC Guidelines.
Cell lines and culturing conditions
HEK293T were cultured in DMEM medium (Invitrogen, Waltham, MA, USA) supplemented with 10% FCS (PAN Biotech, #P30‐2102), 100 IU/mL penicillin/streptomycin (Gibco, #15140‐122), and 50 μM 2‐ME (Gibco, #31350‐010). NIH/3T3 cells were cultured in RPMI medium 1640‐GlutaMAX (Invitrogen, #61870010), supplemented with 10% FCS, 100 IU/mL penicillin/streptomycin, and 50 μM 2‐ME.
EAE induction
MOG (35–55) (MEVGWYRSPFSRVVHLYRNGK) peptide was from Charité Universitätsmedizin (Berlin, Germany) or Proteogenix (Schiltigheim, France). C57BL/6 mice were immunized subcutaneously with 100 μg of MOG(35‐55) in CFA (Sigma, #F5881‐10ML) containing 400 μg of Mycobacterium tuberculosis H37RA (BD Difco, #231141), and administered with pertussis toxin (Sigma, #P‐2980) i.p. on the day of the immunization and 2 days later, as described in Ref. [27]. Clinical EAE signs were assessed daily using a scoring range from 0 to 6 (0–healthy; 1–flaccid tail; 2–impaired righting reflex and/or abnormal gait; 3–partial hind leg paralysis; 4–total hind leg paralysis; 5–hind leg paralysis with partial front leg paralysis; 6–moribund or dead).
Isolation of CNS mononuclear cells from CNS
Mice were euthanized by exposure to CO2 and perfused. Brain and spinal cord were removed and digested with Collagenase IV (Gibco, #17104‐019) and DNAse (Sigma, #DN25), followed by mechanical disaggregation to obtain a single‐cell suspension. Mononuclear cells were isolated on a 30% Percoll gradient (GE Healthcare, #17‐0891‐02) by centrifugation for 20 min at 2000 rpm (without break).
Treatment with anti‐CTLA‐4 and anti‐PD‐1 antibodies
Antibodies against CTLA‐4 (BioXcell, clone 9D9, #BE0164) were administered on days 10 and 14, or PD‐1 (BioXcell, clone RMP1‐14, #BE0146) on days 14 and 16, (both at 0.2 mg/mouse/injection).
Tregs isolation
Tregs were isolated from spleens and LNs using EasySep Mouse CD4+CD25+ Regulatory T Cell Isolation Kit II (Stemcell Technologies, #18783RF) for the experiments shown in Fig. 1, and the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec, #130‐091‐041) for all other data using automated cell separators Robosep or AutoMACS, respectively, following the manufacturers’ instructions. Treg purity after isolation was around 90 and 94% with the Stemcell Technologies and Miltenyi Biotec approaches, respectively. The proportion of Foxp3+Thy1.1+ T cells at the end of the transduction culture was 40 and 80% with the Stemcell Technologies and Miltenyi Biotec approaches, respectively. The final cultures obtained with these two methodologies contained around 60 and 14% Thy1.1+Foxp3– T cells with the Stemcell Technologies and Miltenyi Biotec approaches, respectively.
Plasmids
MP71 bicistronic retroviral vectors [46] coding for the high‐avidity MOG reactive TCR T6‐106 and the Thy1.1 (CD90.1) surface marker (MP71‐T6‐106‐Thy1.1), and tricistronic MP71 vectors [36] additionally coding for miRNAs were described previously [7]. TCR‐β, TCR‐α, and Thy1.1 were separated by p2A and t2A peptides. MP71 vectors expressing miRNA against Il10 or Areg were tested for their capacity to silence the expression of mouse IL‐10 or AREG using pMIG‐R1 vectors containing the cDNA for these genes and their respective 3’UTR sequences cloned via XhoI and NotI restriction sites to obtain pMig‐IL‐10‐IRES‐eGFP and pMig‐AREG‐IRES‐eGFP.
Sequences of miRNA for silencing of IL‐10, AREG, CTLA‐4, and control miRNA
Antisense sequences were designed by BLOCK‐iT RNAi Designer (Invitrogen), Custom siRNA Design Service (Sigma), or Whitehead siRNA designing tool (Whitehead Institute for Biomedical Research). Sequences were inserted into one or two miRNA environments before the T6‐106 TCR start codon. The most effective miRNA antisense sequences for IL‐10, AREG, and control miRNA were as follows, TTA AGG AGT CGG TTA GCA GTA, TGA GCT CCA AAG CAG CTG CAT and AAA TTA TTA GCG CTA TCG CGC, respectively. To silence the expression of CTLA‐4, siRNAs against two different target sequences were inserted: AAC ACT ACA ACC TTT GGA ACC and TTC ATA AAC GGC CTT TCA GTT.
Retroviral transduction and adoptive transfer
Viral particles were produced using HEK293T cells transfected with MP71 or pMIG vectors together with packaging plasmids (pCMV‐Gag‐Pol and pENV‐LTR) using CaPO4. One day after transfection, the medium was changed for a fresh one (RPMI1640–GlutaMAX, 10% FCS, 1 mM sodium pyruvate, 1 mM MEM nonessential amino acids, 1 mM HEPES, 50 μM 2‐ME), and 24 h later viral supernatants were harvested to be used for transduction.
Enriched CD25+CD4+ cells were transferred into RPMI1640‐GlutaMAX (Invitrogen, #61870010) medium supplemented with 10% FCS (PAN Biotech, #P30‐2102), 1 mM sodium pyruvate (Invitrogen, #11360070), 1 mM MEM nonessential amino acids (Invitrogen, #11140035), 1 mM HEPES, (Dominique Dutcher, #L0180‐100), 100 IU/mL penicillin/streptomycin (Gibco, #15140‐122), 50 μM 2‐ME (Gibco, #31350‐010), and 750 U/mL recombinant human IL‐2 (Miltenyi Biotec, #130‐097‐745) and seeded onto nontissue culture plates (Greiner Bio‐One, #662102) coated with 1 μg/mL of anti‐mCD3 (BD Pharmingen, clone 145‐2C11, #553057) and 1 μg/mL of anti‐mCD28 (BD Pharmingen, clone 37.51, #553294).
Cells were transduced on days 2 and 3 postisolation by spinoculation (800 g, 32°C, 90 min) and spinoculation (3000 g, 4°C, 120 min) on Retronectin (TaKaRa, #T100B) coated plates, respectively. At each transduction step, protamine sulfate (Sigma, #P4505) at 5 μg/mL was added. Engineered Treg cells were harvested 6 days after isolation and used for adoptive transfer. Cells were washed in DPBS and transferred i.v. into mice one day before EAE induction. For the purpose of LN analysis, mice received 2 × 106 engineered Treg cells that were labeled with proliferation dye eBioscience Cell Proliferation Dye eFluor 450 (Invitrogen, #450 65‐0842‐90). For the prophylactic treatment, mice received 0.5 × 106 cells.
To test the silencing efficiency of miRNAs against IL‐10 and AREG, the NIH/3T3 cells were simultaneously transduced with two kinds of viral particles, one for expression of IL‐10 (pMig‐IL‐10‐IRES‐eGFP) or AREG (pMig‐AREG‐IRES‐eGFP) and the second one for expression of high‐avidity MOG reactive TCR (T6‐106) + miRNA. One day before transduction, NIH/3T3 cells were seeded onto cell culture‐treated 12‐well plates (Greiner‐Bio‐One, #665180) at 6 × 104 cells per well. Cells were transduced by spinoculation (800 g, 32°C, 90 min) at the presence of 5 μg/mL of protamine sulfate (Sigma, #P4505).
Single cell RNAseq analyses
EAE was induced in C57BL/6 mice. At the peak of disease, mice were sacrificed. CNS (brains and spinal cords) were isolated from two batches (four mice each). After tissue digestion, viable CD45+ cells were sorted by FACS for sequencing. The scRNA‐seq libraries were generated using Chromium Single Cell Next GEM 3′ Library & Gel Bead Kit version 3.1 with Feature Barcode Technology (10× Genomics) according to the manufacturer's protocol. Gene expression (mRNA) and ADT libraries were generated. Briefly, cells were counted, diluted at 1000 cells/μL in PBS + 0.04% BSA and 20 000 cells were loaded in the 10× chromium controller to generate single‐cell gel beads in emulsion. After RT, gel beads in emulsion were disrupted. Barcoded complementary DNA was isolated and amplified by PCR. Following fragmentation, end repair and A‐tailing, sample indexes were added during index PCR. The purified libraries were sequenced on a Novaseq (Illumina) with 28 cycles of read 1, 8 cycles of i7 index and 91 cycles of read 2, targeting a median depth of 45 000 reads per cell for gene expression and 5000 reads per cell for ADT libraries.
Sequencing reads were demultiplexed and aligned to the mouse reference genome (mm10, release 98, built from Ensembl sources), using the CellRanger Pipeline version 6.0.0. Filtered unique molecular identifier (UMI) count matrices were used for downstream analysis using the Seurat workflow [47].
Low‐quality cells were excluded if they fulfilled one or more of the following criteria: (i) number of genes expressed <200, (ii) number of genes expressed >2500, (iii) ≥5% of the total UMIs mapped to mitochondrial RNA. Data from the two mice batches were integrated using the Seurat version 3 SCTransform integration [48] and dimensionality reduction and clustering were performed using default Seurat parameters. After Principal Component Analysis, cells were clustered using the FindClusters function with a resolution of 0.5. UMAP was performed using the RunUMAP function on the selected PCs and the UMAP coordinates were used for cell visualization. Clusters annotation was done manually based on the expression of a large number of hallmark genes, for example, Cd3 and Cd4 for T cells, S100a6 for granulocytes and Hexb, Cst3 and Sall1 for microglia. CD4+ T cell clusters were isolated and submitted to another round of clustering to identify subpopulations based on differential gene expression. Cluster of Tregs was manually annotated based on Foxp3 expression. Heatmap was generated with the Seurat function DoHeatmap using the top 10 markers expressed in each cell cluster. Violin plots were generated with the Seurat function VlnPlot using selected markers per cluster. Differential gene expression analysis was performed using the Seurat FindMarkers function and the MAST package (v1.8.2) [49]. Genes were considered differentially expressed when adjusted p‐value was <0.05 and a log2‐fold change was more than 0.5.
Gene array analysis
EAE was induced in Foxp3‐eGFP mice. At the peak of the disease (day 16–18), mice were sacrificed. CNS and spleens were collected and lymphocytes isolated, as previously described [42]. Treg and Tconv cells were sorted by flow cytometry according to the expression of CD4 and Foxp3‐GFP (Treg: CD11b–CD4+Foxp3.GFP+ cells; Tconv: CD11b–CD4+Foxp3.GFP–). The same populations were isolated from the spleen of naïve mice. GFP+ cells were double sorted to attain the best purity. Purities were 99.5% or higher. Six different populations were compared (Treg from naïve spleen, Tconv from naïve spleen, Treg from EAE spleen, Tconv from EAE spleen, Treg from EAE CNS, Tconv from EAE CNS). Samples were processed in total and hybridized to Affymetrix 430 2.0 whole‐genome mouse arrays using standard Affymetrix protocol after quality control (Agilent 2100 Bio‐analyzer and quantification with NanoDrop ND‐1000 spectrophotometer) [50].
Antibodies and flow cytometry
Flow cytometry analyses were conducted according to the guidelines for the use of flow cytometry and cell sorting in immunological studies [51]. Fc receptors were blocked with anti‐mouse CD16/CD32 (2.4G2, InVivoMAb anti‐mouse CD16/CD32, 1:800). Viability staining was done with Fixable viability stain FVS 575V (BD Bioscience, San Jose, CA, USA, #565694: 1:2000). To track their proliferation, cells were labeled with eBioscience Cell Proliferation Dye eFluor 450 (Invitrogen, #65‐0842‐85). Surface staining was performed with mAbs against CD4 (RM4‐5. 1:300), CD25 (PC61.5, 1:200), CD3 (17A2 1:200), Thy1.1 (OX‐7, 1:200), CD62L (MEL‐14, 1:200), CD44 (IM7: 1:200), LAG‐3 (C9B7W: 1:150), CD121b (4E2: 1:200), PD‐1 (RMP1‐30, 1:200), CD83 (Michel‐19, 1:200), CD200 (OX‐90, 1:200). For intracellular staining, cells were fixed and permeabilized with eBioscience Foxp3/Transcription Factor Staining Buffer Set (Invitrogen, #00‐5523‐00) and stained with mAbs against Foxp3 (FJK‐16s, 1:125), CTLA‐4 (UC10‐4B9, 1:200), CD40L (MR1, 1:100), IFN‐γ (XMG1.2, 1:100), GM‐CSF (MP1‐22E9, 1:100), IL‐17A (TC11‐18H10.1, 1:200). Cell counting in blood, spleen, and brain samples was performed with Precision Count Beads (BioLegend, San Diego, CA, USA, #424902). Antibodies and staining reagents were from BioLegend, Invitrogen–Thermo Fischer Scientific or BD Biosciences. Data were acquired on BD LSRFortessa SORP and analyzed with FlowJo software (FlowJo LLC, Ashland, OR, USA). The gating strategy is explained in Supporting information Fig. S7.
Cytokine detection in supernatants
AREG concentration in the supernatants from NIH/3T3 cells and transduced Treg cells was determined by ELISA using DuoSet ELISA Mouse Amphiregulin (R&D Systems, #DY989) according to the manufacturer's instructions. IL‐10 concentration in supernatants from NIH/3T3 cells was determined using Bio‐Plex system (BioRad). Assay beads were provided in the IL‐10 kit (#171G5009M). IL‐10 standard was provided in Bio‐Plex‐Pro Mouse Cytokine Standards group I, 23‐plex (#171I50001).
Intracellular cytokine detection
Expression of immune mediators (CD40L, GM‐CSF, IL‐17A, IFN‐γ) by MOG‐reactive CD4+ T cells was detected in blood and spleens. Nine days p.i. blood was collected into heparin‐coated tubes (Greiner Bio‐One, #454089) and subjected to RBC lysis with Red Blood Cell Lysing Buffer Hybri‐Max (Sigma, #R7757). Spleens were collected at the end of the experiment and a single‐cell suspension was prepared by smashing the organs through 30 μm cell strainers (BD Falcon, #352340), followed by RBC lysis. Cells were then seeded onto 96‐well cell culture plates (Greiner Bio‐One, #655182) at 6 × 105 cells/well for cells from blood and 8 × 105 cells/well for spleens. Subsequently, cells were stimulated with 30 μg/mL of MOG (35‐55) peptide in the presence of GolgiStop (BD Bioscience). After 6 h, surface and intracellular staining were performed using eBioscience Foxp3/Transcription Factor Staining Buffer Set (Invitrogen, #00‐5523‐00).
Statistical analyses
Data presentation and statistical analyses were performed using GraphPad Prism (GraphPad Software, USA). Graphs show mean + SEM (unless stated otherwise). Data were represented as mean ± SEM. Groups were compared using two‐tailed t‐test (or one‐tail t‐test where stated). Cumulative disease scores and data from peptide stimulations were analyzed with one‐way ANOVA and subsequent Turkey's multiple comparisons test to compare differences between groups. ns > 0.05, *p < 0.05, **p < 0.01; ***p < 0.001, ****p < 0.0001
Conflict of interest
The authors declare no conflict of interest.
Author contributions
J.P. designed the study, performed experiments, analyzed and interpreted data, and wrote the manuscript. R.O.C., S.M.A., B.M., and M.E‐B. performed experiments. M.M, M.L., L.J., P.B., and M.E‐B. analyzed and interpreted transcriptomic data. M.B. and W.U. provided key resources for the study. R.L. and S.F. designed the study and experiments, analyzed and interpreted data, and wrote the manuscript.
Materials and Correspondence
Correspondence and requests for materials should be addressed to S.F.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202249845.
Abbreviations
- AREG
amphiregulin
- FOXP3
forkhead box P3
- miRNA
micro RNA
- MOG
myelin oligodendrocyte glycoprotein
- p.i.
post‐immunization
- Tconv
T‐conventional cells
- UMAP
uniform manifold approximation and projection
Supporting information
Supplementary Figure 1 (related to Fig. 1): Surface expression of CD83, CD121b, and CD200 does not differ significantly between engineered and endogenous Tregs.
Supplementary Figure 2, related to Fig. 2: LAG‐3 contributes to the protective function of engineered MOG‐reactive Tregs.
Supplementary Figure 3, related to Fig. 3: Transcriptome characteristics of CNS Tregs
Supplementary Figure 4, related to Fig. 3: Transcriptome characteristics of CNS Tregs
Supplementary Figure 5, related to Fig. 3: Transcriptome characteristics of CNS Tregs
Supplementary Figure 6, related to Fig. 4: IL‐10 is critically involved in the protective function of autoreactive Tregs
Supplementary Figure 7: Gating strategy
Supplement Material
Supplement Material
Supplement Material
Acknowledgments
This research was supported by the French Research Agency (ANR TCRinMS), an AXA Chair in Translational Immunology, and Fondation pour l'aide à la recherche sue la sclérose en plaques (ARSEP). We thank J. Kirsch, H. Schliemann, and the Regine von Ramin Laboratory (DRFZ) for technical help.
Data availability statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files or are available from the corresponding author upon reasonable request. ScRNAseq transcriptome data are available on ArrayExpress database under the accession number E‐MTAB‐11294. Transcriptome data are available on GEO database under the accession number GSE164460. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164460
References
- 1. Brunkow, M. E. , Jeffery, E. W. , Hjerrild, K. A. , Paeper, B. , Clark, L. B. , Yasayko, S. A. , Wilkinson, J. E. et al., Disruption of a new forkhead/winged‐helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001. 27: 68–73. [DOI] [PubMed] [Google Scholar]
- 2. Raffin, C. , Vo, L. T. and Bluestone, J. A. , Treg cell‐based therapies: challenges and perspectives. Nat. Rev. Immunol. 2020. 20: 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bluestone, J. A. , Buckner, J. H. , Fitch, M. , Gitelman, S. E. , Gupta, S. , Hellerstein, M. K. , Herold, K. C. et al., Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015. 7: 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marek‐Trzonkowska, N. , Mysliwiec, M. , Dobyszuk, A. , Grabowska, M. , Derkowska, I. , Juscinska, J. , Owczuk, R. et al., Therapy of type 1 diabetes with CD4(+)CD25(high)CD127‐regulatory T cells prolongs survival of pancreatic islets—results of one year follow‐up. Clin. Immunol. 2014. 153: 23–30. [DOI] [PubMed] [Google Scholar]
- 5. Romano, M. , Tung, S. L. , Smyth, L. A. and Lombardi, G. , Treg therapy in transplantation: a general overview. Transpl. Int. 2017. 30: 745–753. [DOI] [PubMed] [Google Scholar]
- 6. Romano, M. , Fanelli, G. , Albany, C. J. , Giganti, G. and Lombardi, G. , Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front. Immunol. 2019. 10: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kieback, E. , Hilgenberg, E. , Stervbo, U. , Lampropoulou, V. , Shen, P. , Bunse, M. , Jaimes, Y. et al., Thymus‐derived regulatory T cells are positively selected on natural self‐antigen through cognate interactions of high functional avidity. Immunity 2016. 44: 1114–1126. [DOI] [PubMed] [Google Scholar]
- 8. Nowak, A. , Lock, D. , Bacher, P. , Hohnstein, T. , Vogt, K. , Gottfreund, J. , Giehr, P. et al., CD137+CD154‐ expression as a regulatory T cell (Treg)‐specific activation signature for identification and sorting of stable human Tregs from in vitro expansion cultures. Front. Immunol. 2018. 9: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pohar, J. , Simon, Q. and Fillatreau, S. , Antigen‐specificity in the thymic development and peripheral activity of CD4(+)FOXP3(+) T regulatory cells. Front. Immunol. 2018. 9: 1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsang, J. Y. , Ratnasothy, K. , Li, D. , Chen, Y. , Bucy, R. P. , Lau, K. F. , Smyth, L. et al., The potency of allospecific Tregs cells appears to correlate with T cell receptor functional avidity. Am. J. Transplant. 2011. 11: 1610–1620. [DOI] [PubMed] [Google Scholar]
- 11. Malviya, M. , Saoudi, A. , Bauer, J. , Fillatreau, S. and Liblau, R. , Treatment of experimental autoimmune encephalomyelitis with engineered bi‐specific Foxp3+ regulatory CD4+ T cells. J. Autoimmun. 2020. 108: 102401. [DOI] [PubMed] [Google Scholar]
- 12. Li, M. O. and Rudensky, A. Y. , T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016. 16: 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sidwell, T. , Liao, Y. , Garnham, A. L. , Vasanthakumar, A. , Gloury, R. , Blume, J. , Teh, P. P. et al., Attenuation of TCR‐induced transcription by Bach2 controls regulatory T cell differentiation and homeostasis. Nat. Commun. 2020. 11: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt, A. , Oberle, N. and Krammer, P. H. , Molecular mechanisms of treg‐mediated T cell suppression. Front. Immunol. 2012. 3: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rubtsov, Y. P. , Rasmussen, J. P. , Chi, E. Y. , Fontenot, J. , Castelli, L. , Ye, X. , Treuting, P. et al., Regulatory T cell‐derived interleukin‐10 limits inflammation at environmental interfaces. Immunity 2008. 28: 546–558. [DOI] [PubMed] [Google Scholar]
- 16. Wang, W. W. , Wang, Y. , Li, K. , Tadagavadi, R. , Friedrichs, W. E. , Budatha, M. and Reeves, W. B. , IL‐10 from dendritic cells but not from T regulatory cells protects against cisplatin‐induced nephrotoxicity. PLoS One 2020. 15: e0238816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee, H. , Nho, D. , Chung, H. S. , Lee, H. , Shin, M. K. , Kim, S. H. and Bae, H. , CD4+CD25+ regulatory T cells attenuate cisplatin‐induced nephrotoxicity in mice. Kidney Int. 2010. 78: 1100–1109. [DOI] [PubMed] [Google Scholar]
- 18. Panduro, M. , Benoist, C. and Mathis, D. , Tissue Tregs. Annu. Rev. Immunol. 2016. 34: 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li, C. , DiSpirito, J. R. , Zemmour, D. , Spallanzani, R. G. , Kuswanto, W. , Benoist, C. and Mathis, D. , TCR transgenic mice reveal stepwise, Multi‐site acquisition of the distinctive Fat‐treg phenotype. Cell 2018. 174: 285–299 e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hui, S. P. , Sheng, D. Z. , Sugimoto, K. , Gonzalez‐Rajal, A. , Nakagawa, S. , Hesselson, D. and Kikuchi, K. , Zebrafish regulatory T cells mediate organ‐specific regenerative programs. Dev. Cell 2017. 43: 659–672. [DOI] [PubMed] [Google Scholar]
- 21. Levine, A. G. , Arvey, A. , Jin, W. and Rudensky, A. Y. , Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol. 2014. 15: 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang, C. T. , Workman, C. J. , Flies, D. , Pan, X. , Marson, A. L. , Zhou, G. , Hipkiss, E. L. et al., Role of LAG‐3 in regulatory T cells. Immunity 2004. 21: 503–513. [DOI] [PubMed] [Google Scholar]
- 23. Zhang, Q. , Chikina, M. , Szymczak‐Workman, A. L. , Horne, W. , Kolls, J. K. , Vignali, K. M. , Normolle, D. et al., LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2017. 2: eaah4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoehlig, K. , Shen, P. , Lampropoulou, V. , Roch, T. , Malissen, B. , O'Connor, R. , Ries, S. et al., Activation of CD4(+) Foxp3(+) regulatory T cells proceeds normally in the absence of B cells during EAE. Eur. J. Immunol. 2012. 42: 1164–1173. [DOI] [PubMed] [Google Scholar]
- 25. Berasain, C. and Avila, M. A. , Amphiregulin. Semin. Cell Dev. Biol. 2014. 28: 31–41. [DOI] [PubMed] [Google Scholar]
- 26. Ito, M. , Komai, K. , Mise‐Omata, S. , Iizuka‐Koga, M. , Noguchi, Y. , Kondo, T. , Sakai, R. et al., Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019. 565: 246–250. [DOI] [PubMed] [Google Scholar]
- 27. Fillatreau, S. , Sweenie, C. H. , McGeachy, M. J. , Gray, D. and Anderton, S. M. , B cells regulate autoimmunity by provision of IL‐10. Nat. Immunol. 2002. 3: 944–950. [DOI] [PubMed] [Google Scholar]
- 28. Salama, A. D. , Chitnis, T. , Imitola, J. , Ansari, M. J. , Akiba, H. , Tushima, F. , Azuma, M. et al., Critical role of the programmed death‐1 (PD‐1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2003. 198: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hurwitz, A. A. , Sullivan, T. J. , Sobel, R. A. and Allison, J. P. , Cytotoxic T lymphocyte antigen‐4 (CTLA‐4) limits the expansion of encephalitogenic T cells in experimental autoimmune encephalomyelitis (EAE)‐resistant BALB/c mice. Proc. Natl. Acad. Sci. USA. 2002. 99: 3013–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hou, L. , Rao, D. A. , Yuki, K. , Cooley, J. , Henderson, L. A. , Jonsson, A. H. , Kaiserman, D. et al., SerpinB1 controls encephalitogenic T helper cells in neuroinflammation. Proc. Natl. Acad. Sci. USA. 2019. 116: 20635–20643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luziga, C. , Nga, B. T. , Mbassa, G. and Yamamoto, Y. , Cathepsin L coexists with cytotoxic T‐lymphocyte Antigen‐2 alpha in distinct regions of the mouse brain. Acta Histochem. 2016. 118: 704–710. [DOI] [PubMed] [Google Scholar]
- 32. Hammond, T. R. , Gadea, A. , Dupree, J. , Kerninon, C. , Nait‐Oumesmar, B. , Aguirre, A. and Gallo, V. , Astrocyte‐derived endothelin‐1 inhibits remyelination through notch activation. Neuron 2014. 81: 588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swire, M. , Kotelevtsev, Y. , Webb, D. J. , Lyons, D. A. and Ffrench‐Constant, C. , Endothelin signalling mediates experience‐dependent myelination in the CNS. Elife 2019. 8: e49493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang, H. , Madi, A. , Yosef, N. , Chihara, N. , Awasthi, A. , Pot, C. , Lambden, C. et al., An IL‐27‐driven transcriptional network identifies regulators of IL‐10 expression across T helper cell subsets. Cell Rep. 2020. 33: 108433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clambey, E. T. , McNamee, E. N. , Westrich, J. A. , Glover, L. E. , Campbell, E. L. , Jedlicka, P. , de Zoeten, E. F. et al., Hypoxia‐inducible factor‐1 alpha‐dependent induction of FoxP3 drives regulatory T‐cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA. 2012. 109: E2784–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bunse, M. , Bendle, G. M. , Linnemann, C. , Bies, L. , Schulz, S. , Schumacher, T. N. and Uckert, W. , RNAi‐mediated TCR knockdown prevents autoimmunity in mice caused by mixed TCR dimers following TCR gene transfer. Mol. Ther. 2014. 22: 1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Verhagen, J. , Gabrysova, L. , Minaee, S. , Sabatos, C. A. , Anderson, G. , Sharpe, A. H. and Wraith, D. C. , Enhanced selection of FoxP3+ T‐regulatory cells protects CTLA‐4‐deficient mice from CNS autoimmune disease. Proc. Natl. Acad. Sci. USA. 2009. 106: 3306–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paterson, A. M. , Lovitch, S. B. , Sage, P. T. , Juneja, V. R. , Lee, Y. , Trombley, J. D. , Arancibia‐Carcamo, C. V. et al., Deletion of CTLA‐4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med. 2015. 212: 1603–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Todo, S. , Yamashita, K. , Goto, R. , Zaitsu, M. , Nagatsu, A. , Oura, T. , Watanabe, M. et al., A pilot study of operational tolerance with a regulatory T‐cell‐based cell therapy in living donor liver transplantation. Hepatology 2016. 64: 632–643. [DOI] [PubMed] [Google Scholar]
- 40. Bashuda, H. , Kimikawa, M. , Seino, K. , Kato, Y. , Ono, F. , Shimizu, A. , Yagita, H. et al., Renal allograft rejection is prevented by adoptive transfer of anergic T cells in nonhuman primates. J. Clin. Invest. 2005. 115: 1896–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luznik, L. , O'Donnell, P. V. and Fuchs, E. J. , Post‐transplantation cyclophosphamide for tolerance induction in HLA‐haploidentical bone marrow transplantation. Semin. Oncol. 2012. 39: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Connor, R. A. , Floess, S. , Huehn, J. , Jones, S. A. and Anderton, S. M. , Foxp3(+) Treg cells in the inflamed CNS are insensitive to IL‐6‐driven IL‐17 production. Eur. J. Immunol. 2012. 42: 1174–1179. [DOI] [PubMed] [Google Scholar]
- 43. Fontenot, J. D. , Rasmussen, J. P. , Williams, L. M. , Dooley, J. L. , Farr, A. G. and Rudensky, A. Y. , Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005. 22: 329–341. [DOI] [PubMed] [Google Scholar]
- 44. Miyazaki, T. , Dierich, A. , Benoist, C. and Mathis, D. , Independent modes of natural killing distinguished in mice lacking Lag3. Science 1996. 272: 405–408. [DOI] [PubMed] [Google Scholar]
- 45. Workman, C. J. , Cauley, L. S. , Kim, I. J. , Blackman, M. A. , Woodland, D. L. and Vignali, D. A. , Lymphocyte activation gene‐3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J. Immunol. 2004. 172: 5450–5455. [DOI] [PubMed] [Google Scholar]
- 46. Engels, B. , Cam, H. , Schuler, T. , Indraccolo, S. , Gladow, M. , Baum, C. , Blankenstein, T. et al., Retroviral vectors for high‐level transgene expression in T lymphocytes. Hum. Gene. Ther. 2003. 14: 1155–1168. [DOI] [PubMed] [Google Scholar]
- 47. Satija, R. , Farrell, J. A. , Gennert, D. , Schier, A. F. and Regev, A. , Spatial reconstruction of single‐cell gene expression data. Nat. Biotechnol. 2015. 33: 495–U206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stuart, T. , Butler, A. , Hoffman, P. , Hafemeister, C. , Papalexi, E. , Mauck, W. M. , Hao, Y. H. et al., Comprehensive integration of Single‐cell data. Cell 2019. 177: 1888–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Finak, G. , McDavid, A. , Yajima, M. , Deng, J. Y. , Gersuk, V. , Shalek, A. K. , Slichter, C. K. et al., MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single‐cell RNA sequencing data. Genome Biol. 2015. 16. 10.1186/s13059-015-0844-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lino, A. C. , Dang, V. D. , Lampropoulou, V. , Welle, A. , Joedicke, J. , Pohar, J. , Simon, Q. et al., LAG‐3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity 2018. 49: 120–133 e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cossarizza, A. , Chang, H. D. , Radbruch, A. , Abrignani, S. , Addo, R. , Akdis, M. , Andrä, I. et al., Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur. J. Immunol. 2021. 51: 2708–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 (related to Fig. 1): Surface expression of CD83, CD121b, and CD200 does not differ significantly between engineered and endogenous Tregs.
Supplementary Figure 2, related to Fig. 2: LAG‐3 contributes to the protective function of engineered MOG‐reactive Tregs.
Supplementary Figure 3, related to Fig. 3: Transcriptome characteristics of CNS Tregs
Supplementary Figure 4, related to Fig. 3: Transcriptome characteristics of CNS Tregs
Supplementary Figure 5, related to Fig. 3: Transcriptome characteristics of CNS Tregs
Supplementary Figure 6, related to Fig. 4: IL‐10 is critically involved in the protective function of autoreactive Tregs
Supplementary Figure 7: Gating strategy
Supplement Material
Supplement Material
Supplement Material
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files or are available from the corresponding author upon reasonable request. ScRNAseq transcriptome data are available on ArrayExpress database under the accession number E‐MTAB‐11294. Transcriptome data are available on GEO database under the accession number GSE164460. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164460