

Abstract
Purpose: Reactive microglia are an important hallmark of neuroinflammation. Reactive microglia release various inflammatory mediators, such as cytokines, chemokines, and prostaglandins, which are produced by enzymes like cyclooxygenases (COX). The inducible COX‐2 subtype has been associated with inflammation, whereas the constitutively expressed COX‐1 subtype is generally considered as a housekeeping enzyme. However, recent evidence suggests that COX‐1 can also be upregulated and may play a prominent role in the brain during neuroinflammation. In this review, we summarize the evidence that supports this involvement of COX‐1. Methods: Five databases were used to retrieve relevant studies that addressed COX‐1 in the context of neuroinflammation. The search resulted in 32 articles, describing in vitro, in vivo, post mortem, and in vivo imaging studies that specifically investigated the COX‐1 isoform under such conditions. Results: Reviewed literature generally indicated that the overexpression of COX‐1 was induced by an inflammatory stimulus, which resulted in an increased production of prostaglandin E2. The pharmacological inhibition of COX‐1 was shown to suppress the induction of inflammatory mediators like prostaglandin E2. Positron emission tomography (PET) imaging studies in animal models confirmed the overexpression of COX‐1 during neuroinflammation. The same imaging method, however, could not detect any upregulation of COX‐1 in patients with Alzheimer's disease. Conclusion: Taken together, studies in cultured cells and living rodents suggest that COX‐1 is involved in neuroinflammation. Most postmortem studies on human brains indicate that the concentration of COX‐1‐expressing microglial cells is increased near sites of inflammation. However, evidence for the involvement of COX‐1 in neuroinflammation in the living human brain is still largely lacking.
Keywords: COX‐1, cyclooxygenase, microglia, neuroinflammation, prostaglandins
Significance.
For a long time, only the inducible isoform of cyclooxygenase, COX‐2, was thought to play a role in inflammation, whereas the constitutively expressed isoform, COX‐1, was considered a housekeeping enzyme. Our findings shows there is substantial evidence from preclinical studies and postmortem studies on human brains to support a role of COX‐1 in neuroinflammation. These findings suggest that COX‐1 might be an interesting target for the treatment of brain disorders associated with neuroinflammation.
1. INTRODUCTION
Neuroinflammation is a physiological response that protects the central nervous system from infiltrating pathogens, toxic compounds, and brain damage, but is also associated with various psychiatric and neurodegenerative disorders, such as schizophrenia (Monji et al., 2009; Müller, 2018; Müller et al., 2015; Na et al., 2014; Najjar & Pearlman, 2015), depression (Benedetti et al., 2020; Najjar et al., 2013; Nettis & Pariante, 2020; Troubat et al., 2021), Alzheimer's disease (AD; Calsolaro & Edison, 2016; Heppner et al., 2015; Leng & Edison, 2021; Wyss‐Coray & Rogers, 2012), Parkinson's disease (Bartels & Leenders, 2010; Hirsch & Hunot, 2009; Phani et al., 2012; Tansey et al., 2007; Wang et al., 2021), amyotrophic lateral sclerosis (McCauley & Baloh, 2019; Papadimitriou et al., 2010), and multiple sclerosis (Koudriavtseva & Mainero, 2016; Voet et al., 2019). The biological features of neuroinflammation are diverse and depend on the inflammatory trigger and the stage of the disorder (Grabert et al., 2016; Perry & Teeling, 2013; Saura, 2010). An early characteristic of neuroinflammation is the change of microglia to a reactive phenotype, which is characterized by altered cellular morphology (Marty et al., 1991; Streit, 1996). Inflammatory triggers can stimulate intracellular signaling cascades (e.g., the NF‐κβ pathway) that drive the expression and activation of a variety of inflammation‐associated enzymes (e.g., iNOS, NADPH oxidase, cyclooxygenase, caspases, MMPs) and the subsequent release of cytokines (e.g., IL‐1β, IL‐6, and TNF‐α), prostaglandins, and chemokines (Green et al., 1994; Iñiguez et al., 1999). Cyclooxygenase (COX) is one of the enzymes that produce inflammatory mediators upon the activation of microglia through the biosynthesis of prostaglandins from arachidonic acid (AA; Akundi et al., 2005).
AA is a polyunsaturated fatty acid that is abundantly present in the brain, in particular in the phospholipid membranes of the cells. AA is enzymatically cleaved from phospholipids by phospholipase A2 and phospholipase C. The activation of these phospholipases can occur, when ligands bind to, among others, metabotropic glutamate, interferon‐α, or interferon‐γ receptors (Kim et al., 2015; Martín et al., 2010; Miscia et al., 1997; Ponzoni & Cornaglia‐Ferraris, 1993; Ponzoni et al., 1992).
AA is metabolized via two pathways: the COX pathway and the 5‐lipoxygenase pathway. The COX pathway is the dominant pathway in reactive microglia and macrophages. COX catalyzes the conversion of AA to prostaglandin G2, which is the first and rate‐determining step in the biosynthesis of various prostaglandins (Figure 1). Prostaglandins play a role in a variety of processes related to tissue damage and inflammation, including the recruitment of immune cells, blood vessel dilation, blood clotting, fever, pain, neuroplasticity, and neuronal death (Famitafreshi & Karimian, 2020).
FIGURE 1.
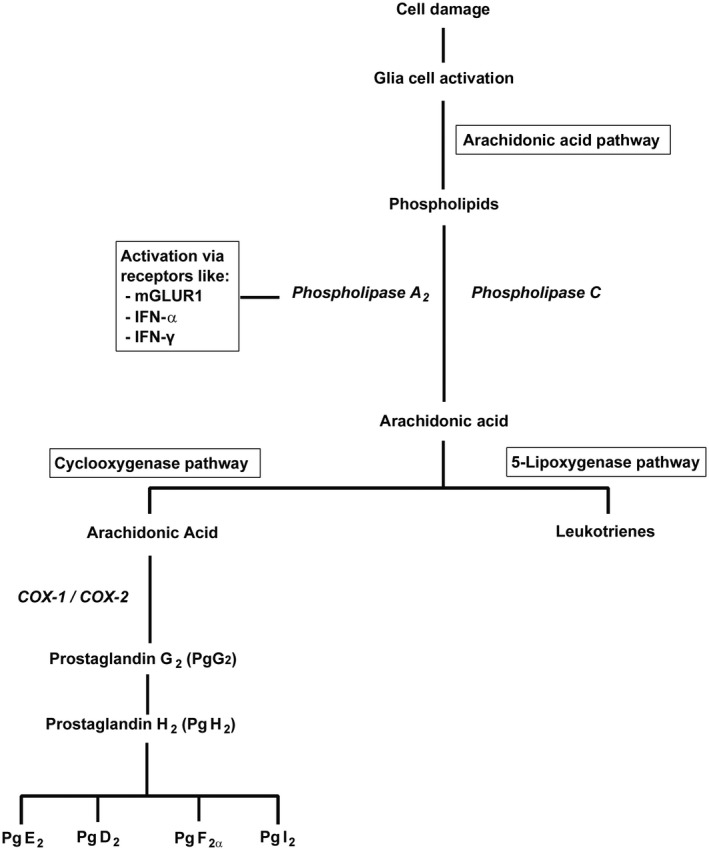
Simplified representation of the pathways that lead to the biosynthesis of prostaglandins and leukotrienes upon activation of glial cells in the course of neuroinflammation
Several members of the COX family have been identified. Initially, COX‐1 and COX‐2 were discovered (Barnett et al., 1994), and more recently also COX‐3 was shown to be present (Chandrasekharan et al., 2002). The main COX isoforms, COX‐1 and COX‐2, share 60% identity in sequence (Rouzer & Marnett, 2009; Tanabe & Tohnai, 2002), but have different regulatory functions and distribution patterns. Both COX‐1 and COX‐2 are located at the inner envelope of nuclear membranes and the luminal surface of the lipid bilayer of the endoplasmic reticulum of human embryonic kidney (HEK293) cells and human lung (CCL210) fibroblasts (Yuan & Smith, 2015), while COX‐2 but not COX‐1 is also present in the Golgi apparatus (Yuan & Smith, 2015), and in the human brain, only COX‐1 can also be found in the cytosol (Kitamura et al., 1999). Because COX‐1 is constitutively expressed in most tissues and is present at constant levels throughout the cell cycle, it has been considered as a housekeeping enzyme. On the other hand, COX‐2 expression is normally restricted to only a few tissues (brain, testis, tracheal epithelia, macula densa of the kidney), but can be rapidly (in 2 to 6 hr) induced during inflammation (Bazan et al., 1994; Smith et al., 1996). For a long time, only COX‐2 was therefore considered to play an important role in inflammation. It should be emphasized that this perspective was largely based on studies that examined the response of tumor cells (glioblastoma, neuroblastoma) or cells from peripheral organs (e.g., fibroblasts, endothelial cells, monocytes, ovarian follicles) to inflammatory stimuli.
In contrast to most peripheral organs, there is a basal expression of COX‐2 in the brain (Hewett et al., 2006; Minghetti, 2004; Slanina & Schweitzer, 2005; Stefanovic et al., 2006; Wang et al., 2005; Yamagata et al., 1993). Some studies reported an upregulation of cerebral COX‐2 under inflammatory conditions (An et al., 2014; Do & Woo, 2018; Lee et al., 2015; Li et al., 2010; Mhillaj et al., 2018; Shrestha et al., 2020). COX‐2 expression in the corpus callosum tissue of postnatal (3‐day old) rats was threefold upregulated, 6 hr after the animals had received two intraperitoneal injections of bacterial lipopolysaccharide (LPS, 1 mg/kg, with an interval of 6 hr [Li et al., 2010]). Intrastriatal lesions made by injection of the excitatory amino acid (RS)‐(tetrazole‐5yl)‐glycine resulted in the expression of COX‐2 by neurons in the vicinity of the lesion and by vascular endothelial cells and monocytes at the center of the lesion (An et al., 2014). Exposure of mice to heat stress caused memory loss and significant signs of neuroinflammation in the hippocampus, including a marked (2.5‐ to threefold) upregulation of COX‐2 after 42 days of heat (Lee et al., 2015). Dextran sodium sulfate‐induced colitis in mice was accompanied by a transient, 3.5‐fold upregulation of COX‐2 in the hypothalamus after 3 days and a modest (20 to 30%), but more persistent upregulation of COX‐2 in the hippocampus (Do & Woo, 2018). A single injection of LPS (10 µg) in the right putamen of rhesus monkeys caused a 32 to 42% increase in the binding potential of the COX‐2 radioligand [11C]MC1 in the entire brain after 1 day, with a subsequent return to baseline values within 8 days (Shrestha et al., 2020). The intracerebroventricular injection of soluble amyloid‐ß in rats resulted in modest (30%) upregulation of COX‐2 in the hippocampus and cognitive impairment after 7 days. Both the COX‐2 upregulation and the memory deficits could be prevented by treating animals with the COX‐2 inhibitor celecoxib (Mhillaj et al., 2018).
Several studies suggest that COX‐1 also plays a role in neuroinflammation (Choi & Bosetti, 2009; Choi et al., 2008; Schwab et al., 2002; Teeling et al., 2010). Changes in burrowing behavior and open field activity of mice after intraperitoneal injection of LPS could be blocked by the selective COX‐1 inhibitor piroxicam but not by the selective COX‐2 inhibitor nimesulide (Teeling et al., 2010). COX‐1‐deficient (−/−) mice showed reduced neuroinflammation in response to ß‐amyloid (Choi & Bosetti, 2009). Genetic deletion or pharmacological blockade of COX‐1 also reduced the inflammatory response and brain injury in mice after intracerebroventricular injection of LPS (Choi et al., 2008). The postmortem analysis of the brain tissue from humans with traumatic brain injury demonstrated a transient upregulation of COX‐1 in endothelial and smooth muscle cells from blood vessels within the lesion and a prolonged accumulation of COX‐1‐expressing microglia and macrophages in areas around the lesion that started within 6 hr and lasted several months (Schwab et al., 2002). There is some evidence that microglia from the brains of patients with Alzheimer's disease do not overexpress COX‐2 and that inhibition of both COX‐1 and COX‐2 is required to effectively reduce the secretion of prostaglandins by such cells (Hoozemans et al., 2001). This raised the presumption that COX‐1 plays a significant role in neuroinflammation (Bosetti & Choi, 2010; Shukuri et al., 2011, 2016). This review aims to survey the evidence for this presumption and to examine if it supports the hypothesis that COX‐1 contributes to the neuropathology of AD.
2. METHODS
2.1. Literature search
We performed computerized literature searches through the PubMed/MEDLINE, Embase, Web of Science, Scopus, and EBSCOhost databases to identify studies addressing COX‐1 in the context of neuroinflammation, published up to November 2020. We used the limits of English language and the search terms consisted of AND/OR combinations of strings such as: “Cyclooxygenase 1”, “Neurodegenerative Diseases”, “Neurogenic Inflammation”, “Microglia”, “Parkinson”, “Alzheimer”, “Schizophrenia” and “Cognitive dysfunction” (see Appendix 1 for the full list of search terms). After the elimination of duplicates and short surveys or conference abstracts, 809 individual articles were retrieved, as represented in Figure 2. In a subsequent round of selection, studies concerning inflammation in tissues of the body outside the brain were removed. Studies concerning neuroinflammation but providing no data on COX‐1 were also excluded. Studies reporting only anti‐inflammatory effects of COX‐2 inhibitors or herbal drugs were removed. Finally, derivative studies, such as review articles, were excluded when they did not contain original experimental data. By this selection procedure, the number of articles was reduced from 809 to 325. The full text of the resulting 325 articles was analyzed. Studies concerning COX‐1 expression in tumors, studies concerning COX‐1 but not reporting changes in its expression or its role in neuroinflammation, and assessments of the impact of nonselective COX inhibitors on the (patho)physiology of the brain were excluded. After this final round of selection, the number was reduced from 325 to 32 studies. Twenty articles concerned the involvement of both COX‐1 and COX‐2 in neuroinflammation, and 12 articles concerned the involvement of COX‐1. These remaining 32 studies are discussed in the following review.
FIGURE 2.
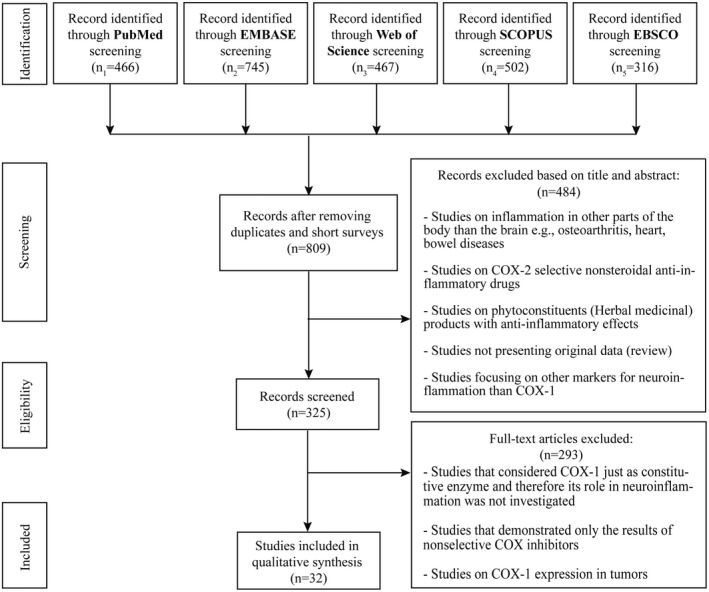
Flow diagram of literature search
3. RESULTS
3.1. In vitro studies
The role of COX‐1 in neuroinflammation was examined in six in vitro studies (Table 1). In the following text, these are discussed in chronological order. In an initial study, human microglia were exposed either to LPS (1 µg/ml), β‐amyloid(1–42) (Aβ1‐42, 10 µM), or AA (1 or 10 µM) (Hoozemans et al., 2002). COX‐1 mRNA or protein expression was not affected by exposure to LPS, Aβ1‐42, AA, or cytokines, whereas COX‐2 expression was induced after exposure of the cells to LPS, but not after exposure to Aβ1‐42, AA, or plaque‐associated cytokines like IL‐1α, IL‐1β, IL‐6, or TNF‐α. The stimulation of cells with LPS or AA resulted in a significant (3.7‐ to 4.4‐fold) increase in PgE2 secretion, whereas exposure to Aβ1‐42 or plaque‐associated cytokines did not have any effect. The COX‐1 selective inhibitor, SC560, and the COX‐2 selective inhibitor, SC‐236 (Figure 3), were used to assess the relative involvement of the COX isoforms in LPS‐ or AA‐induced microglial PgE2 secretion. LPS‐induced PgE2 secretion was strongly (for 98%) inhibited by the COX‐2 selective inhibitor (1 µM SC‐236) and only partially (for 86%) by the COX‐1 selective inhibitor (0.1 µM SC560). On the other hand, AA‐stimulated PgE2 release was only reduced by the COX‐1 selective inhibitor (−64% at 0.1 µM SC560). Thus, inflammatory stimuli may cause an increase in prostaglandin production by the overexpression of COX‐2, or indirectly by stimulating microglia to produce more AA, which is subsequently converted by constitutively expressed COX‐1. COX‐1 inhibitors could (partly) counteract the effects of increased AA production by inhibiting the prostaglandin production catalyzed by constitutively expressed COX‐1 (Hoozemans et al., 2002). In this study, no effect of inflammatory stimuli on COX‐1 expression could be detected, but later, in vitro investigations in murine microglial cell lines reported opposite findings (Calvello et al., 2012, 2017).
TABLE 1.
In vitro studies that investigated the involvement of COX‐1 in neuroinflammation
| Cell type | Stimulus | COX expression | COX inhibitor | Outcome parameter [Number of replicates] | Ref |
|---|---|---|---|---|---|
| Human microglia | LPS | LPS | COX−1 : | IL−1α, IL−1β, IL−6, TNF‐α: = | Hoozemans et al. (2002) |
| β‐Amyloid | COX‐1: = | SC560, | AA‐induced PgE2:↓ | ||
| Arachidonic acid (AA) | COX‐2: ↑ | Valeryl salicylate | LPS‐induced PgE2: ↓ | ||
| β‐Amyloid: | COX‐2: | IL‐1α, IL‐1β, IL‐6, TNF‐α: = | |||
| COX‐1: = | SC‐236 | AA‐induced PgE2: = | |||
| COX‐2: = | LPS‐induced PgE2: ↓ | ||||
| AA: | |||||
| COX‐1: = | [Two or more (up to six) experiments in triplicate] | ||||
| COX‐2: = | |||||
| Chinese hamster ovary cells (CHO‐APPswe) | Transfection COX‐1/2 adenoviral vector | COX‐1: ↑ | Nonselective: Ibuprofen | PgE2 ↓ | Qin et al. (2003) |
| COX‐2: ↑ | β‐Amyloid ↓ | ||||
| [3 experiments in triplicate] | |||||
| Mouse cortical neuron and microglial coculture | Amyloid‐β1−42 HuPrP82‐146 | Not determined | COX‐1: | IL‐6↓ | Bate et al. (2006) |
| SC560 | Neuronal survival↑ | ||||
| FR‐122047 | |||||
| Valeryl salicylate | |||||
| COX‐2: | No effect at low dose | ||||
| LM‐1685 | |||||
| SC‐236 | [3 experiments in triplicate] | ||||
| DuP‐697 | |||||
| Mouse microglia | Medium from astrocytes harvested from α‐synuclein‐overexpressing mice | COX‐1: ↑ | – | [Three independent experiments] | Gu et al. (2010) |
| COX‐2: = | |||||
| Mouse microglial cell line (N13) | LPS | COX‐1: ↑ | COX−1: | Calvello et al. (2012) | |
| COX‐2: ↑ | P6 | COX‐1 ↓ | |||
| P10 | COX‐2: = | ||||
| SC560 | prostaglandin E synthase ↓ | ||||
| Aspirin | PgE2↓ | ||||
| NF‐κβ ↓ | |||||
| NO↓ | |||||
| COX‐2: | COX‐1: = | ||||
| celecoxib | COX‐2 ↓ | ||||
| etoricoxib | NF‐κβ ↓ | ||||
| NO↓ | |||||
| [Three independent experiments] | |||||
| Mouse microglial cell line (BV‐2) | LPS | COX‐1: ↑ | COX‐1: | COX‐1 ↓ | Calvello et al. (2017) |
| P6 | COX‐2: = | ||||
| Mofezolac | PgE2↓ | ||||
| NF‐κB↓ | |||||
| [At least five independent replicates] |
Studies are listed in chronological order.
The underlining in column 3 refers to the stimuli mentioned in column 2. The underlining in column 4 indicated which type of inhibitors (COX1 or COX2 or nonselective), the inhibitors mentioned underneath are.
FIGURE 3.
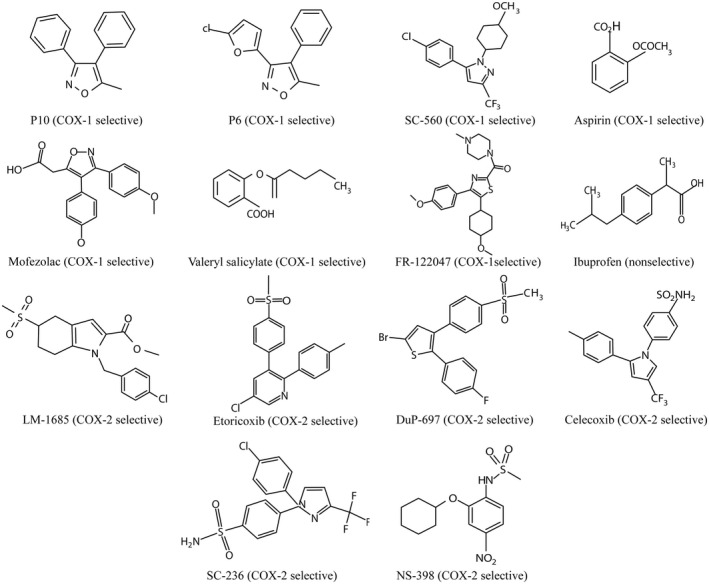
Chemical structures of COX‐1 or COX‐2 selective inhibitors
A second study examined the contribution of the COX enzymes to the production of Aβ peptide in CHO‐APPswe cells transfected with human COX‐1 or COX‐2. The authors showed that transfection with either COX‐1 or COX‐2 resulted in an increased production of PgE2 (4.5‐fold and 6.5‐fold, respectively), which subsequently stimulated the production of Aβ peptides. The nonselective COX inhibitor, ibuprofen, could decrease the production of PgE2 and the amyloid peptides Aβ1‐40 and Aβ1‐42 in both COX‐1 and COX‐2‐overexpressing cells to control levels. This suggested that ibuprofen modulated Aβ peptide generation by inhibiting the COX‐mediated production of PgE2 (Qin et al., 2003).
A third study employed cultures of primary cortical neurons and microglial cells of embryonic mice to model the early stages of neuronal dysfunction and neurodegeneration (Bate et al., 2006). Isolated neurons were cultured for 5 days in neurobasal medium containing B27 components. They were then exposed to amyloid‐β1‐42 (1 to 5 µM) or human prion protein HuPrP82‐146 (1 to 5 µM), and isolated microglial cells were added, in ratios of 1 microglial cell to 2.5, 5, or 10 neurons. Amyloid‐β1‐42 or human prion protein HuPrP82‐146 significantly impaired the survival of neurons in cocultures but not in the absence of microglia. Survival was assessed after 4 days and could be reduced from 100% to about 20%. To define the involvement of the different COX isoforms, the neuron‐microglia cocultures were treated with either a COX‐1 selective (SC560, valeryl salicylate, and FR‐122047) or COX‐2 selective (LM‐1685, SC‐236, and DuP‐697) inhibitor prior to the addition of amyloid‐β1‐42 or HuPrP82‐146. The concentration of the pro‐inflammatory cytokine IL‐6 was reduced (dose dependently, up to 96%) and neuronal survival was increased (in some cases up to control levels) in the presence of a low (nanomolar) concentration of COX‐1 selective inhibitors. COX‐2‐selective inhibitors only demonstrated neuroprotective effects at considerably higher (micromolar) concentrations. IL‐6 was the only cytokine examined in this study. These observations suggest that COX‐1 rather than COX‐2 is involved in neuronal damage induced by amyloid‐β1‐42 or HuPrP82‐146. The authors also demonstrated that neuronal survival was similarly impaired by incubation of the neuron‐microglia coculture with PgD2 as with prion or amyloid peptides. Thus, exposure of neuron‐microglia cocultures to peptides and proteins involved in neurodegenerative diseases activated the COX‐1‐mediated production of PgD2 and subsequently caused neuronal damage (Bate et al., 2006).
A fourth study evaluated COX‐1 expression in transgenic A53T mice that overexpressed an inducible α‐synuclein gene. Cultured microglial cells acquired from postnatal day 1 mice pups were exposed to conditioned medium derived from cultured astrocytes harvested from the A53T mouse brain stem. In microglia exposed to this conditioned medium, COX‐1 expression (mRNA) and IL‐1β levels were significantly upregulated (25‐ and 10‐fold, respectively) while the expression of COX‐2 was unaffected, as compared to levels in microglia cultured in regular control medium (Gu et al., 2010). These results suggest that astrocytes loaded with α‐synuclein release signals that activate microglia and stimulate them to produce COX‐1.
A fifth study (Calvello et al., 2012) stimulated a microglial cell line (N13) with LPS for 24 hr or 48 hr and observed more than threefold increased protein levels of both COX isoforms and 2.5‐ to 2.6‐fold enhanced release of PgE2 in comparison to untreated cells. This finding is in contradiction with the initial study mentioned above (Hoozemans et al., 2002), which reported that LPS did not influence the expression of COX‐1 protein or mRNA. Since cells were stimulated with the same concentration of LPS (1 µg/ml) in both cases, this discrepancy in COX‐1 response could originate from the dissimilar cell type (isolated microglia vs. microglial cell line), species (human vs. mouse), medium (DMEM/Ham's F10 1:1 vs. RPMI 1640), and incubation procedure (24 hr vs. 48 hr) employed in both studies.
The same study showed with immunoblotting that COX‐1 selective inhibitors caused a significant (>70%) reduction of COX‐1 protein in the LPS‐stimulated cells, whereas COX‐2 expression was unaffected. Conversely, COX‐2 inhibitors reduced COX‐2 expression (maximally by 78%), but did not change COX‐1 expression. COX‐1 inhibitors also reduced the expression of cytosolic prostaglandin E synthase (maximally by 47%) and the production of PgE2 (maximally by 64%). Both COX‐1 and COX‐2 selective inhibitors reduced activation of the nuclear transcription factor NF‐κβ and the production of NO (Calvello et al., 2012). The authors concluded that the expression levels of COX‐1 and COX‐2 are independent, that is not affecting each other.
A final study from the same group (Calvello et al., 2017) reported the results of a series of experiments on BV2 microglial cells that were stimulated with LPS. After 48 hr of stimulation, the cells showed a 3.4‐fold increase in COX‐1 protein levels in comparison to unstimulated cells, and a 4.4‐fold increase in the release of PgE2. The COX‐1 selective inhibitors P6 and mofezolac completely counteracted the LPS‐induced increase in COX‐1 expression and release of PgE2 and inhibited NF‐κβ activation. The protein levels of COX‐2 in the same cell lysates were unaffected by the COX‐1 inhibitors, which confirmed that the roles of COX‐1 and COX‐2 were indeed independent.
3.2. Animal studies
Several studies in experimental animals have reported an involvement of COX‐1 in neuroinflammation (Table 2). In an initial study, LPS was intracerebroventricularly injected in COX‐1−/− and wild‐type mice (Choi et al., 2008). Twenty‐four hours later, significantly less neural damage, activation of microglia (−22 to −28%), and expression of PgE2 (−50%), COX‐2 mRNA (−14%), cytokines (IL‐1ß −79% and TNF‐α −76%), and other inflammatory mediators were observed in the knockout animals than in wild‐type controls. The administration of the COX‐1 selective inhibitor SC560 to wild‐type mice prior to LPS injection inhibited the expression of cytokines and prostaglandins in a similar manner as genetic deletion of COX‐1. These results suggested that COX‐1, rather than COX‐2, was the main isoform involved in neuroinflammation. This hypothesis was confirmed by another study (Aid et al., 2008) which used COX‐2‐deficient mice (COX‐2−/−). LPS‐induced neuroinflammation in COX‐2−/− mice led not to a reduction, but an increase in reactive microglia (+40%) and the expression of inflammatory mediators (IL‐1ß + 100%, TNF‐α + 124%) compared to wild‐type mice. This exacerbated inflammatory response in COX‐2 knockout mice indicated that COX‐2 was not a major contributor to the neuroinflammatory response to LPS. A third study investigated the role of COX‐1 in amyloid‐induced neuroinflammation by injecting β‐amyloid (Aβ1‐42) in the lateral ventricle of COX‐1−/− and wild‐type mice. COX‐1−/− knockout mice showed less Aβ1‐42‐induced neuronal damage (−95%), oxidative stress, and glial cell activation (−79%) than wild‐type mice, suggesting that COX‐1 played an important role in amyloid‐induced neuroinflammation (Choi & Bosetti, 2009).
TABLE 2.
Animal studies concerning the involvement of COX‐1 in neuroinflammation
| Animal | Stimulus | COX expression | COX inhibitor | Outcome parameter [Number of replicates] | Ref |
|---|---|---|---|---|---|
| COX‐1−/− knockout mice | LPS | COX‐1: ↓ | – | Neuronal damage: ↓ | Choi et al. (2008) |
| Oxidative stress: ↓ | |||||
| Wild‐type mice | LPS | COX‐1: ↑ | COX‐1: | PgE2, PgD2, PgF2α: ↓ | |
| SC560 | Thromboxane B2: ↓ | ||||
| Pro‐inflammatory cytokines: ↓ | |||||
| Pro‐inflammatory chemokines: ↓ | |||||
| [Group size 4 to 6] | |||||
| COX‐1−/− knockout mice | Aβ | COX‐1: ↓ | – | Neuronal damage: ↓ | Choi and Bosetti (2009) |
| Oxidative stress: ↓ | |||||
| Glial cell activation: ↓ | |||||
| [Group size 3 to 4] | |||||
| LPS | COX‐1: ↑ | COX‐1: | PgE2↓ | García‐Bueno et al. (2009) | |
| SC560 | |||||
| COX‐2: | No effect | ||||
| NS398 | |||||
| [Group size 3 to 5] | |||||
| COX‐1−/− knockout mice | LPS | Not determined | – | MMP‐9 ↓ | Aid et al. (2010) |
| MMP‐9 ↑ | |||||
| COX‐2−/− Knockout mice | BBB damage ↑ | ||||
| Wild‐type mice | [Group size 5 to 8] | ||||
| COX‐1−/− knockout mice | LPS | Not determined | – | Leukocyte infiltration ↓ | Choi et al. (2010) |
| Leukocyte infiltration ↑ | |||||
| COX‐2−/− Knockout mice | |||||
| [Group size 3 to 6] | |||||
| Wild‐type mice | |||||
| Wild‐type mice | IL‐1β‐overexpression | COX‐1: ↑ | COX‐1: | PgE2↓ | Matousek et al. (2010) |
| COX‐1−/− knockout mice | COX‐2: = | SC560 | PgE2↓ | ||
| COX‐1: ↓ | [Group size 3 to 9] | ||||
| A53T mice | Mutant α‐synuclein | COX‐1: ↑ | COX‐1: | Life span↑ | Gu et al. (2010) |
| SC560 | |||||
| [Group size 3] | |||||
| Mice | LPS | Not determined | COX‐1: | Open field behavior and burrowing restored after COX‐1 inhibition | Teeling et al. (2010) |
| Piroxicam | Not restored after COX‐2 inhibition | ||||
| Sulindac | |||||
| COX‐2: | |||||
| Nimesulide | [Group size 3 to 5] | ||||
| Niflumic acid | |||||
| Rat | β‐Amyloid | COX‐1: = | COX‐1: | TNF‐α↓ | Dargahi et al. (2011) |
| COX‐2: ↑ | SC560 | PgE2↓ | |||
| Caspase‐3 ↓ | |||||
| Neuronal loss ↓ | |||||
| Astrogliosis ↓ | |||||
| COX‐2 ↓ | |||||
| COX‐2: | TNF‐α↓ | ||||
| NS398 | PgE2↓ | ||||
| Caspase‐3 ↓ | |||||
| Neuronal loss ↓ | |||||
| Astrogliosis ↓ | |||||
| [Group size 3 to 5] | |||||
| COX‐1−/− knockout mice | LPS | Not determined | – | Proliferation,survival, differentiation of hippocampal progenitor cells ↓ in WT, but =in COX‐1−/− mice | Russo et al. (2011) |
| Wild‐type mice | |||||
| [Group size 4 to 6] | |||||
| ME7 mice | Prion protein | COX‐1: ↑ | COX‐1: | IL‐1β: = | Griffin et al. (2013) |
| LPS | SC560 | IL‐6: = | |||
| TNF‐α: = | |||||
| PgE2↓ | |||||
| cognitive deficits↓ | |||||
| COX‐2: | No effect | ||||
| NS398 | |||||
| Prion protein | COX‐1: ↑ | Nonselective | cognitive deficits↓ | ||
| IL‐β | Ibuprofen | [Group size 4 to 7] | |||
| Transgenic AD (3×Tg‐AD) mice | – | Not determined | COX‐1: | TNF‐α↓ | Choi et al. (2013) |
| SC560 | iNOS↓ | ||||
| [Group size 6] | |||||
| Mice | LPS | COX‐1: ↑ | COX‐1: | p‐Iκβα↓ | Calvello et al. (2017) |
| COX‐2: ↑ | Mofezolac | COX‐1 ↓ | |||
| COX‐2: = | |||||
| PgE2 ↓ | |||||
| GFAP ↓ | |||||
| IBA−1 ↓ | |||||
| [At least five independent replicates] | |||||
| Wild‐type mice | Prion protein | COX‐1: ↑ | COX‐1: | PgE2↓ | Nazmi et al. (2019) |
| IFNAR1−/− knockout mice | COX‐1: = | SC560 | PgE2↓ | ||
| Neuronal loss ↓ | |||||
| Disease progression ↓ | |||||
| [Group size 3 to 7] | |||||
| COX‐1 knockout mice | Social defeat | Not determined | COX‐1: | PgE2↓ | Nie et al. (2019) |
| TLR double knockout mice | SC560 | COX‐1 ↓ | |||
| COX‐2: | PgE2↓ | ||||
| SC236 | COX‐2: = | ||||
| [Group size 7] |
Studies are listed in chronological order.
The underlining in column 4 indicated which type of inhibitors (COX1 or COX2 or nonselective), the inhibitors mentioned underneath are.
Later studies from the same group examined the role of the COX isozymes in blood–brain barrier (BBB) disruption during neuroinflammation. Matrix metalloproteinase‐9 activity (MMP‐9) after intracerebroventricular injection of LPS was decreased in COX‐1−/− mice (−24% in cortex and −29% in hippocampus), but significantly increased in COX‐2−/− mice (+38% in cortex and +33% in hippocampus) compared to wild‐type controls (Aid et al., 2010). Similar changes were observed in the activity of MMP‐3, and in this case, both the decreases in COX‐1−/− and increases in COX‐2−/− animals were statistically significant. As a consequence of this modulation of MMP activity, LPS‐induced disruption of the BBB was reduced in COX‐1−/− mice (−26% in whole brain and −31% in hippocampus), but significantly increased in COX‐2−/− mice (+114% in whole brain and +180% in hippocampus; Aid et al., 2010). Thus, COX‐2 activity seemed necessary to maintain BBB integrity after an inflammatory stimulus, whereas COX‐1 exacerbated the LPS‐induced BBB damage. In a follow‐up study, ionized calcium‐binding adapter molecule‐1 (Iba‐1, a microglia marker), myeloperoxidase, 7/4 (neutrophil markers), and various cytokines were quantified in wild‐type and knockout mice after intracerebroventricular injection of LPS. The number of neutrophils in the brain after LPS administration was 10‐fold reduced in COX‐1−/− but 50% increased in COX‐2−/− mice, compared to wild‐type controls. The data of this study suggested that inhibition of COX‐1 reduced but inhibition of COX‐2 increased the neuroinflammatory response and leukocyte recruitment to the brain (Choi et al., 2010).
In a study from another group, a systemic (i.v.) injection of LPS was applied to induce neuroinflammation in rats, and COX‐1 immunoreactivity in endothelial, perivascular, and parenchymal microglial cells was subsequently determined. The expression of the COX‐1 protein in endothelial cells of the brain was fivefold enhanced, 1 to 3 hr after the LPS injection. A 3.5‐fold increase in cerebral PgE2 secretion was also observed during this early phase. These effects were attenuated by intracerebral treatment with the COX‐1 selective inhibitor SC560, whereas COX‐2 inhibitors did not reduce the secretion of PgE2. These findings suggested a pro‐inflammatory role of COX‐1 in neuroinflammation (García‐Bueno et al., 2009).
A study in a transgenic mouse model showed that the overexpression of IL‐1β was accompanied by a significant increase in COX‐1 mRNA (2.4‐fold after 1 week, 2.9‐fold after 2 months), but not by an increase in COX‐2 mRNA or protein levels. Studies in COX‐1 knockout mice revealed that deletion of COX‐1 abolishes the IL‐1β‐mediated (>2‐fold) increase in PgE2 release and neuroinflammation. Pharmacological treatment with the COX‐1 selective inhibitor SC560 caused a similar reduction in the IL‐1β‐induced release of PgE2 as genetic deletion of COX‐1. The immunohistochemical staining of brain tissue of transgenic mice with IL‐1β activation demonstrated about 40% lower levels of glial fibrillary acidic protein (GFAP) mRNA in COX‐1 knockout mice compared to wild‐type mice. These data suggested that COX‐1 was involved in astrogliosis (Matousek et al., 2010).
A therapy‐oriented study investigated the role of COX‐1 in a transgenic animal model of Parkinson's disease that selectively expressed mutant α‐synuclein in astrocytes (Gu et al., 2010). Symptomatic mice showed a significant increase in reactive microglia (highest value in brain stem, 4.8‐fold) along with an upregulation of COX‐1 mRNA (2.4‐fold). When these transgenic A53T mice were treated with a single daily dose of the selective COX‐1 inhibitor SC560 for 8 days, a significant extension of their life span was observed. These results suggest that COX‐1 plays a pivotal role in α‐synuclein‐induced neurodegeneration.
In another study, a solution of amyloid‐ß (Aß) was bilaterally injected into the prefrontal cortex of Wistar rats. Thirteen days after the Aβ injection, an inflammatory response (6.3‐fold increase in TNF‐α, >10‐fold increase in PgE2) and a 2.4‐fold increase in COX‐2 protein were observed, while COX‐1 protein levels remained unaffected. A fixed intracerebroventricular cannula was surgically inserted in the animals, to enable repetitive administration of a COX‐1 or COX‐2 selective inhibitor. The chronic administration of the COX‐1 selective inhibitor, SC560, the COX‐2 selective inhibitor, NS398, or a combination of both equally attenuated the neuroinflammatory response to Aβ, as demonstrated by a reduced overexpression of TNF‐α and PgE2 (now both <2‐fold), attenuated caspase‐3 activation, reduced neural loss and diminished astrogliosis. Treatment with a selective COX‐1 inhibitor completely blocked the Aβ‐induced overexpression of COX‐2. The authors concluded that both COX isoforms are involved in neuroinflammation, but COX‐1 is essential for the induction of COX‐2 (Dargahi et al., 2011).
An interesting study demonstrated that the proliferation, survival, and differentiation of hippocampal progenitor cells was significantly reduced (−34 to −35%) in wild‐type but not significantly altered in COX‐1−/− mice after intracerebroventricular administration of LPS (Russo et al., 2011). Thus, COX‐1 appeared to be involved in the inhibition of hippocampal neurogenesis induced by LPS.
The link between COX‐1, neuroinflammation, and cognitive deficits was explored in the ME7 progressive neurodegeneration mouse model for prion disease. An intraperitoneal injection of LPS was used as an additional systemic inflammatory stimulus. This animal model is characterized by cognitive dysfunction, glial cell activation, COX‐1 overexpression in microglia and perivascular macrophages, and a concomitant increase in the expression of PgE2. The administration (i.p.) of the COX‐1 selective inhibitor SC560 60 min prior to LPS injection did not reduce the LPS‐induced increase in pro‐inflammatory markers, such as IL‐1β, IL‐6, TNF‐α, but attenuated the response of hippocampal and thalamic PgE2 levels (by at least 85%), and reduced LPS‐induced cognitive deficits. In contrast to the COX‐1 inhibitor, the COX‐2‐specific inhibitor NS398 did not have any modulating effect on PgE2 release or cognition, suggesting that the PgE2 overexpression in thalamus and hippocampus was COX‐1 dependent. Another group of ME7 animals was used to examine the effect of intraperitoneal administration of IL‐1β on the induction of PgE2 synthesis. In this group of animals, systematic administration of IL‐1β was sufficient to replicate the effect of LPS on working memory deficits. Pretreatment with the nonselective COX inhibitor, ibuprofen, counteracted the effect of IL‐1β. Data from this study suggested that COX‐1‐dependent prostaglandin expression in the context of neuroinflammation may lead to deficits in working memory (Griffin et al., 2013).
Another study examined the role of COX‐1 in microglial activation and the correlation of amyloid and tau pathology with neuroinflammation in the 3×Tg‐AD transgenic mouse model of AD. These mice express the mutant human genes PS1M146V, APPswe, and TauP301L (Choi et al., 2013). The animals received a single daily dose of the COX‐1 inhibitor SC560 for 8 days, at the age of 20 months, when they showed memory deficits and deposition of amyloid and phosphorylated tau. SC560 administration altered the microglial activation phenotype and reduced the levels of TNF‐α (−43%) and iNOS (−52%), but did not change the levels of full‐length amyloid precursor protein. Behavioral tests showed that SC560 administration improved spatial learning and memory retention in 3×Tg‐AD mice. These results indicated that COX‐1 played a significant role in neuroinflammation in this mouse model of AD.
In a subsequent study, LPS was injected in the lateral cerebral ventricle of adult male mice to evoke an inflammatory response (Calvello et al., 2017). Animals were treated with a daily 6 mg/kg dose (i.p.) of the selective COX‐1 inhibitor, mofezolac, for 10 days, starting 7 days before LPS injection. Three days after the administration of LPS, the regional protein levels of both COX‐1 and COX‐2 were increased in control mice (8‐ to 21 fold, and 2.5‐ to 46‐fold, respectively). The administration of mofezolac selectively prevented the upregulation of COX‐1, but not of COX‐2, in all examined brain regions (caudate‐putamen, hippocampus, frontal lobe, and substantia nigra). The LPS‐induced increase in the release of PgE2 was completely inhibited by mofezolac, suggesting that COX‐1, rather than COX‐2, was responsible for the increased production of PgE2. Treatment with mofezolac reduced the LPS‐induced increase in the expression of two markers of inflammation, GFAP (activated astrocytes) and IBA‐1 (expressed by microglia). The COX‐1 inhibitor also diminished the LPS‐induced increase in p‐Iκβα expression, indicating that activation of the NF‐κβ pathway was inhibited by mofezolac.
Another study aimed to investigate whether interferon is involved in the prion protein‐induced upregulation of COX‐1. Scrapie (ME7 strain)‐infected brain homogenate was inoculated in wild‐type and heterodimeric interferon α/β receptor 1‐deficient (IFNAR1−/−) mice. COX‐1 mRNA was significantly (3.4‐fold) elevated in ME7‐treated wild‐type mice, but not in ME7‐treated IFNAR1−/− mice. Treatment of wild‐type mice with ME7 resulted in a 2.1‐fold increase in the production of PgE2, which was completely blocked after treatment with the COX‐1 inhibitor SC560. Deletion of the IFNAR1 gene resulted in a similar inhibition of the ME7‐induced overproduction of PgE2. These results indicated that the interferon signaling pathway is involved in the prion protein‐induced overexpression of COX‐1 and could thus indirectly affect the production of PgE2 (Nazmi et al., 2019).
A final study examined the role of Toll‐like receptors (TLR) in neuroinflammation and microglial activation in COX‐1 (Ptgs1) knockout mice and TLR 2/4 (Tlr2/4) double knockout mice exposed to repeated social defeat stress (SDS) for 10 days (Nie et al., 2019). SDS induced PgE2 release (increase +53% above control levels in the whole brain) of wild‐type mice, but not COX‐1 or TLR 2/4 knockout mice. The administration of either the COX‐1 selective inhibitor, SC560, or the COX‐2 selective inhibitor, SC‐236, could suppress the SDS‐induced PgE2 release. PgE2 release in cortical regions was not affected by SDS in any of the mouse strains. The study findings indicated an involvement of TLR 2/4 in the SDS‐induced expression of COX‐1, since COX‐1 mRNA in subcortical regions was significantly (~35%) lower in TLR 2/4 knockout mice than in wild‐type mice after SDS, while COX‐2 mRNA levels were not significantly different. These data suggested a significant role for TLR in the SDS‐induced COX‐1‐mediated release of PgE2.
3.3. Postmortem studies in humans
A very early study examined the levels of COX‐1 mRNA in subcortical white matter of human immunodeficiency virus (HIV)‐positive individuals with and without dementia (Table 3) (Griffin et al., 1994). Subjects with dementia showed an almost twofold higher expression of the COX‐1 gene than nondemented subjects, but the difference did not reach statistical significance because of a high interindividual variability in the patient groups. Yet, this observation suggested a potential involvement of COX‐1 in neuroinflammatory damage to the human brain.
TABLE 3.
Postmortem studies on human brain samples concerning the involvement of COX‐1 in neuroinflammation
| Diagnosis | Sample size | Sampled regions | Observed outcome | Ref. |
|---|---|---|---|---|
| HIV infection | n (with dementia) = 11 | Subcortical white matter | COX‐1 ↑ in HIV‐positive individuals with dementia compared to nondemented patients | Griffin et al. (1994) |
| n (without dementia) = 7 | ||||
| AD | n (AD) = 5 | Entorhinal cortex, hippocampus, midtemporal gyrus, substantia nigra, and thalamus | COX‐1= | Yasojima et al. (1999) |
| n (control) = 5 | COX‐2↑: 2–3 fold | |||
| AD | n (AD) = 7 | Temporal cortex | COX‐1↑: in cytosolic and particulate fraction (40%–50%) | Kitamura et al. (1999) |
| n (control) = 6 | COX‐2↑: in particulate fraction only (80%) | |||
| AD | n (AD) = 10 | Hippocampus, cortex | COX‐1 expression: = | Yermakova et al. (1999) |
| n (control) = 10 | Number of COX‐1 positive microglia: ↑ | |||
| AD | n (AD) = 6 | Frontal cortex | Number of COX‐1‐positive microglia↑ Number of COX‐2‐positive neurons↑ | Hoozemans et al. (2001) |
| n (control) = 6 | ||||
| Traumatic brain injury | n = 31 | Frontal, frontoparietal, frontotemporal, frontobasal, occipital, temporobasal, parietotemporal, parieto‐occipital | COX‐1 expression in vessel endothelial and smooth muscle cells↑ Number of COX‐1‐positive microglia↑ | Schwab et al. (2002) |
| Sporadic Creutzfeldt‐Jakob disease | n (CJ) = 8 | Cortex | Number of COX‐1‐expressing microglia: ↑ | Deininger et al. (2003) |
| n (control) = 4 | Number of COX‐2‐expressing neurons and astrocytes: ↑ | |||
| Bipolar disorder | n (BD) = 15 | Several regions | COX‐1:= | Maida et al. (2006) |
| Major depression | n (DD) = 15 | COX‐2:= | ||
| Schizophrenia | n (SZ) = 15 | |||
| Bipolar disorder | n (BD) = 10 | Frontal cortex | COX‐1↓ | Kim et al. (2011) |
| n (control) = 10 | COX‐2↑ | |||
| AA↑ |
Studies are listed in chronological order.
A later study evaluated the expression of COX‐1 and COX‐2 in postmortem brains of five patients with AD and five healthy controls (Yasojima et al., 1999). This study reported that both COX‐1 and COX‐2 were constitutively expressed in the brains of patients with AD and control subjects. COX‐1 mRNA was slightly (<2‐fold) higher in the brain of patients with AD, but this difference was not statistically significant in any brain region. In contrast, COX‐2 mRNA levels were significantly (twofold to threefold) upregulated in several regions of the brain of patients with AD, in particular in entorhinal cortex, hippocampus, midtemporal gyrus, substantia nigra, and thalamus (Yasojima et al., 1999).
Another study on postmortem brain tissue of seven patients clinically diagnosed with AD and six controls that was published in the same year reported that COX‐1 protein levels in both the cytosolic and particulate fraction of the temporal cortex of patients with AD were significantly (40%–50%) increased compared to controls (Kitamura et al., 1999). The expression of COX‐2 was also significantly increased in the brain of patients with AD (by approximately 80%), but only in the particulate fraction of the temporal cortex.
A third study examined hippocampus and cortical tissue of 10 patients clinically diagnosed with AD and 10 control subjects (Yermakova et al., 1999). The authors observed COX‐1 immunoreactivity in hippocampal and cortical neurons and also in microglial cells, but not in astrocytes. The amount of COX‐1 protein in microglia was not dependent on their activation state. However, since the density of microglia is increased in the cortex of patients with AD, an absolute increase in the concentration of COX‐1 immunoreactive cells was observed in AD (+38 and +31% in cortical layers II and III, respectively).
In a fourth study, the temporal and frontal cortex of patients with AD and nondemented controls was investigated postmortem using immunohistochemistry (Hoozemans et al., 2001). The expression of COX‐1 in microglial cells of white and gray matter of patients with AD and healthy controls was determined. The number of COX‐1‐positive microglial cells in frontal and temporal cortex was higher in patients with AD than in controls, and the COX‐1‐positive microglia in patients with AD were mainly located in the proximity of Aβ plaques. Neural expression of COX‐2 was detected, with a higher density of COX‐2‐positive neurons in the brains of patients with AD, as compared to the control group. The number of COX‐2‐positive cells in the cortex of patients with AD was almost threefold higher than in nondemented controls (9.5 ± 1.3 vs. 3.6 ± 1.5%, respectively), and was suggested to be an early indication of AD pathology occurring before the formation of plaques or tangles. The distinct expression patterns of COX‐1 and COX‐2 that were observed in this study may indicate that the two COX isoforms are involved in different cellular processes in the AD brain.
A fifth study investigated the localization of COX‐1 and COX‐2 in cortical tissue of four control subjects and eight patients diagnosed with sporadic Creutzfeldt‐Jakob disease (Deininger et al., 2003). In healthy brains, COX‐1 protein was observed in microglial cells and occasionally in neurons, whereas COX‐2 protein was found in neurons and occasionally in endothelial cells. In patients with Creutzfeldt‐Jakob disease, significantly more COX‐1‐expressing microglial cells adjacent to hypertrophic neurons were observed (mean labeling scores 2.6 ± 0.18 vs. 1.5 ± 0.5 in healthy controls). The number of COX‐2‐expressing cells was also significantly higher in patients than in controls (mean labeling scores 2.5 ± 0.19 vs. 0.5 ± 0.29, respectively), in particular in neurons and astrocytes of brain regions with severe tissue damage. Based on their results, the authors concluded that COX‐1‐expressing microglia and COX‐2‐expressing neurons are involved in the pathology of sporadic Creutzfeldt‐Jakob disease.
The expression of COX‐1 has not only been investigated in postmortem brain samples of patients with neurodegenerative diseases, but also in patients with psychiatric disorders. One study examined the AA cascade which leads to the generation of prostaglandins (Maida et al., 2006). The authors quantified the regional expression of COX‐1, COX‐2, and cytosolic prostaglandin E synthase in postmortem brain tissues of normal subjects and patients with bipolar disorder (BD), major depressive disorder (MDD), and schizophrenia. Immunohistochemical and western blot analysis did not reveal any significant differences in COX‐1 or COX‐2 protein levels for any brain region between the patient groups. However, a significant decrease (−30 to −35%) in the expression levels of cPGES protein was found in the frontal cortex of patients with BD, MDD and schizophrenia relative to healthy controls. In patients with BD, a 38% decrease was also noted in the temporal cortex. Medicated patients did not show any differences in the expression level of COX‐2 and cPGES compared to controls, except for a 27% decrease in cPGES in the frontal cortex of patients with BP that approached significance (p = 0.054). Nonmedicated patients showed a marginally significant increase in COX‐2 expression (p = 0.047) and a significantly (−62%) reduced cPGES expression in frontal cortex (p ≤ 0.001), compared to controls.
A final study measured several enzymes of the AA cascade in postmortem frontal cortex from 10 patients with BD and 10 age‐matched controls (Kim et al., 2011). Cytosolic phospholipase A2, secretory phospholipase A2, COX‐2, and membrane prostaglandin E synthase protein and mRNA levels were significantly elevated in the frontal cortex of patients with BD(protein +87, +92, +82, and +71%, mRNA threefold, sixfold, 3.4‐fold, and 2.6‐fold, respectively), while protein and mRNA levels of COX‐1 and cytosolic prostaglandin E synthase were significantly reduced (protein −40% and −54%, mRNA −40% and −24%, respectively), compared to controls. These results suggested that the AA cascade was upregulated in patients with BD, which could be related to underlying excitotoxicity and neuroinflammation. This process seemed to mainly involve the COX‐2 pathway, whereas the COX‐1 pathway was inactivated as a compensatory response.
3.4. PET imaging of COX‐1 expression
Advanced neuroimaging techniques allow the assessment of physiological and pathological alterations in specific biologic targets in living subjects. This includes the quantification of inflammatory markers (Lancelot & Zimmer, 2010). Positron emission tomography (PET) is a noninvasive molecular imaging modality with high sensitivity for detecting small changes in the biological target of interest (Cagnin et al., 2007; Kannan et al., 2009; Meyer et al., 2020). If positron‐emitting radioligands with sufficient target affinity, target specificity, and passage across the BBB would become widely available, PET could be used to investigate and elucidate whether COX‐1 is involved in neuroinflammatory processes. Several candidate PET tracers for imaging of COX expression have been developed. Most candidate PET tracers were developed for COX‐2 imaging since COX‐2 was considered as the inducible isozyme involved in neuroinflammation. The success of these tracers has been limited (de Vries et al., 2003, 2008; Gao et al., 2009; Laube et al., 2013; Majo et al., 2005; Shrestha et al., 2020; Uddin et al., 2011; Wuest et al., 2008). Only in the last decade, scientists have started to develop PET tracers for COX‐1 (Figure 4; Table 4).
FIGURE 4.
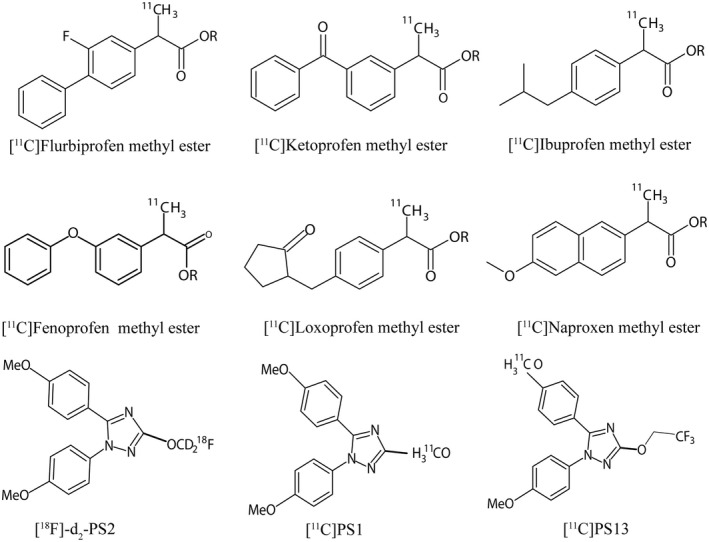
Structure of PET radiotracers developed targeting COX‐1 expression
TABLE 4.
PET studies with radioligands targeting COX‐1
| Species | COX−1 induction | Sample size (per group) | Tracer | Ref. |
|---|---|---|---|---|
| Rats | Intracerebral LPS injection | n = 2 to 3 | [11C]Ketoprofen methyl ester, | Takashima‐Hirano et al. (2010) |
| [11C]Flurbiprofen methyl ester, | [11C]Ibuprofen methyl ester, | |||
| n = 4 to 5 in each group of the blocking study | [11C]Naproxen methyl ester, | |||
| [11C]Fenoprofen methyl ester, | ||||
| [11C]Loxoprofen methyl ester, | ||||
| Mice, knockout | COX‐1 or COX‐2 deficient | n = 6 | [11C]KTP‐Me | Shukuri et al. (2011) |
| n = 4 | ||||
| Rats | Intrastriatal injection of LPS or quinolinic acid | n = 3 | ||
| Humans | Healthy subjects | n = 6 | [11C]KTP‐Me | Ohnishi et al. (2014) |
| Humans | Healthy controls | n = 7 | [11C]KTP‐Me | Ohnishi et al. (2016) |
| Patients with AD, MCI | n = 3 | |||
| n = 5 | ||||
| Rats | Intracerebral LPS injection | n = 3, | (R)‐[11C]KTP‐Me, | Shukuri et al. (2016) |
| n = 4, |
(S)‐[11C]KTP‐Me, (RS)‐[11C]KTP‐Me, |
|||
| 8 months, n = 3; | ||||
| Mice, APP‐Tg | Transgenic animal | 13 months, n = 4; | ||
| 16 months, n = 5; | ||||
| 24 months, n = 4 | ||||
| Rhesus monkey | Healthy animals | n = 1 | [2H2,18F]PS2, | Shrestha et al. (2018; Singh et al. (2018) |
| n = 1 | [11C]PS1, | |||
| n = 3 | [11C]PS13, | |||
| Human | Healthy subjects | n = 10 | [11C]PS13 | Kim et al. (2020) |
| Mice, ICRscid | Xenografts | n = 6 | [11C]PS13 | Boyle et al. (2021) |
| n = 2 (pretreatment) | ||||
| Rhesus monkey | Healthy animals | n = 2 (pretreatment) | [11F]PS13 | Taddei et al. (2021) |
| n = 1 (whole‐body scan) |
Studies are listed in chronological order.
Japanese radiochemists radiolabeled six nonsteroidal anti‐inflammatory drugs (NSAIDs: ibuprofen, naproxen, flurbiprofen, fenoprofen, ketoprofen, and loxoprofen) and their methyl ester prodrugs with the positron‐emitting isotope carbon‐11 and evaluated them as potential PET tracers for imaging COX‐1 expression in a rat model of neuroinflammation induced by unilateral injection of LPS into the striatum (Takashima‐Hirano et al., 2010). All labeled methyl ester prodrug compounds except [11C]ibuprofen methyl ester showed high uptake in the vicinity of the LPS injection site. The highest accumulation in the inflamed striatum was observed for [11C]ketoprofen methyl ester. The authors performed postmortem immunohistochemistry to confirm upregulation of COX‐1 at the LPS injection site. The specificity of [11C]ketoprofen methyl ester for COX‐1 was demonstrated in blocking studies, in which the animals were pretreated with an excess of unlabeled ketoprofen methyl ester. As expected, this pretreatment resulted in a significant (−42%) decrease in tracer accumulation at the site of LPS injection, since the unlabeled and labeled molecule competed for the same target. In contrast to its methyl ester prodrug, [11C]ketoprofen did not show any accumulation at the LPS injection site in this animal model. The authors explained the lack of detectable accumulation of [11C]ketoprofen at the injection site by a low penetration of ketoprofen across the BBB (Table 4).
The same group further evaluated the active (S)‐enantiomer of [11C]ketoprofen methyl ester ((S)‐[11C]KTP‐Me) in COX‐1‐ or COX‐2‐deficient mice to define COX subtype selectivity. (S)‐[11C]KTP‐Me showed highest uptake in the hippocampus, cerebral cortex, and cerebellum of wild‐type mice. Significantly (up to 50%), lower uptake of the tracer was observed in the brain of COX‐1‐/‐ knockout mice, whereas tracer uptake in COX‐2−/− knockout mice was similar to that in wild‐type controls, demonstrating the selectivity of (S)‐[11C]KTP‐Me for COX‐1 (Shukuri et al., 2011). The tracer was subsequently evaluated in a neuroinflammation model, generated by unilateral injection of either LPS or quinolinic acid into the striatum of rats. The PET scans showed high uptake of (S)‐[11C]KTP‐Me around the injection site. Tracer accumulation was visible after 6 hr, peaked after 1 day and had returned to baseline 1 week after stereotactic injection of the toxin (Shukuri et al., 2011). Postmortem analysis by immunohistochemistry confirmed that COX‐1 expression was increased by the same time course as tracer uptake in the PET scan. The pharmacologically active enantiomer (S)‐[11C]KTP‐Me demonstrated higher uptake at the site of neuroinflammation than racemic [11C]KTP‐Me or the less active enantiomer (R)‐[11C]KTP‐Me (+49% and +115%, respectively [Shukuri et al., 2016]).
PET imaging with (S)‐[11C]KTP‐Me has also been used to explore the involvement of COX‐1 in disease progression in amyloid precursor protein transgenic (APP‐Tg) mice, an animal model of AD. A significant increase in the brain uptake of the tracer was noted as the disease progressed (twofold in hippocampus, between ages 8 and 24 months). This increase was in agreement with histopathology, which confirmed the presence of a time‐dependent increase in Aβ plaques and reactive microglia. Ex vivo analysis of brain tissue confirmed that increased accumulation of (S)‐[11C]KTP‐Me was indeed associated with increased COX‐1 expression by reactive microglia surrounding Aβ plaques in the hippocampus and frontal cortex.
A first‐in‐human PET study was performed with racemic [11C]KTP‐Me in six healthy male volunteers (Ohnishi et al., 2014). The study indicated that [11C]KTP‐Me could be safely applied to human subjects, but was rapidly metabolized to [11C]ketoprofen (within 2–3 min). The same group (Ohnishi et al., 2016) applied racemic [11C]KTP‐Me PET to study COX‐1 expression in patients with AD or mild cognitive impairment (MCI), but did not find any significant differences in tracer uptake between healthy volunteers and amyloid‐positive subjects with MCI or AD. The lack of specific tracer uptake in this clinical study could be due to the absence of overexpression of COX‐1 in cognitively impaired subjects or to insufficient sensitivity of the PET tracer to detect a rather minor overexpression. Rapid conversion of the methyl ester prodrug to [11C]ketoprofen and limited ability of [11C]‐ketoprofen to cross the BBB could also explain the absence of specific tracer uptake.
Researchers from the National Institutes of Health developed novel PET tracers for imaging of COX‐1, namely [11C]PS1, [11C]PS13, and [18F]PS2 (Kim et al., 2018; Shrestha et al., 2018; Singh et al., 2018). [11C]PS13 showed rather high uptake and considerable specific binding to COX‐1 in the healthy monkey brain. Pretreatment of the animals with unlabeled PS13 at a dose of 1 mg/kg showed that 55% of the brain uptake of the tracer represented specific binding. The brain uptake of [11C]PS13 was later compared to that of [11C]MC1, a successful radioligand for imaging and quantification of COX‐2 (Shrestha et al., 2020). In contrast to [11C]PS13, the COX‐2 tracer [11C]MC1 did not show any specific binding in the healthy monkey brain, although specific binding was observed in ovary tissue.
A first‐in‐human PET study with [11C]PS13 has been performed in 10 healthy volunteers. The hippocampus and occipital cortex showed the highest tracer uptake, followed by the pericentral cortex. A significant correlation was observed between COX‐1 mRNA levels provided by the Allen Human Brain Atlas and the regional distribution volumes of the tracer, suggesting that the regional uptake of [11C]PS13 indeed reflected regional COX‐1 density in the human brain (Kim et al., 2020).
Additional proof for the target specificity of [11C]PS13 was acquired in a PET study in mice bearing ovarian cancer xenografts (Boyle et al., 2021). COX‐1 overexpression in ovarian cancer is associated with tumor progression (Malerba et al., 2020). ICRscid mice with xenografts were scanned at baseline and after pretreatment with 2 mg of ketoprofen in 20 µl dimethylsulfoxide (i.p). The tracer could visualize all tumors, and tumor uptake was 2.7‐fold decreased after pretreatment with ketoprofen, which indicated significant engagement of [11C]PS13 with its COX‐1 target (Boyle et al., 2021).
Because of the short half‐life of carbon‐11 (20.4 min), 11C‐labeled tracers cannot be distributed to remote imaging centers. After the promising results acquired with [11C]PS13, the same group thus attempted to radiolabel PS13 with 18F (half‐life 109.8 min) (Taddei et al., 2021). The resulting [18F]PS13 was used to make two brain scans (at baseline and after pretreatment with unlabeled PS13) in two rhesus monkeys and a single whole‐body scan in one monkey. A high baseline uptake of the tracer was observed in the prefrontal and parietal regions of the cortex, and tracer uptake was significantly decreased after target blocking with PS13 (0.3 mg/kg, i.v.). However, the molar activity of the acquired [18F]PS13 was low (7.9 GBq/µmol) resulting in a high carrier dose at baseline and a small specific binding fraction of [18F]PS13 in the monkey brain (about 33% vs. 55% for [11C]PS13). The radiochemical procedures for production of [18F]PS13 should thus be improved to acquire [18F]PS13 with a higher molar activity. Studies with [18F]PS13 in animal models or patients with neuroinflammation have not yet been reported.
3.5. Summary of literature findings
All in vitro studies with cells derived from animals (Bate et al., 2006; Calvello et al., 2012, 2017; Gu et al., 2010; Qin et al., 2003) showed an increase in COX‐1 expression or suggested an important role of COX‐1 after exposure to an inflammatory stimulus. This overexpression of COX‐1 was accompanied by an increase in the production of PgE2. Selective COX‐1 inhibitors could prevent COX‐1 overexpression, reduce PgE2 release and moderate its downstream effects like amyloid deposition and neuronal damage. In contrast, a single in vitro study with human microglial cells (Hoozemans et al., 2002) indicated that COX‐1 expression was not affected by an inflammatory stimulus. Yet, selective COX‐1 inhibitors could reduce the production of PgE2 also in human microglia (Hoozemans et al., 2002).
Studies in experimental animals showed that inflammatory stimuli like LPS, Aβ, IL‐1β, and social stress induced the overexpression of COX‐1 (Calvello et al., 2017; García‐Bueno et al., 2009; Griffin et al., 2013; Gu et al., 2010; Matousek et al., 2010; Nazmi et al., 2019; Nie et al., 2019), probably via activation of TLR (Nie et al., 2019). Most studies also showed that pharmacological inhibition of COX‐1 had an anti‐inflammatory effect and resulted in suppression of the induction of inflammatory mediators like PgE2 and TNF‐α, inhibition of the NF‐κβ pathway, neuroprotection and prevention of cognitive deficits and behavioral changes (Calvello et al., 2017; Choi et al., 2008, 2013; Dargahi et al., 2011; García‐Bueno et al., 2009; Griffin et al., 2013; Gu et al., 2010; Matousek et al., 2010; Nazmi et al., 2019; Nie et al., 2019; Teeling et al., 2010). Interestingly, COX‐1 appeared to be required for the inflammation‐induced upregulation of COX‐2, thus both COX‐1 and COX‐2 seem to play a role in the inflammatory response of the brain (Choi et al., 2008; Dargahi et al., 2011).
Postmortem analysis of tissue samples demonstrated COX‐1 (Deininger et al., 2003; Kitamura et al., 1999; Schwab et al., 2002; Yermakova et al., 1999) and COX‐2 (Deininger et al., 2003; Hoozemans et al., 2001; Kitamura et al., 1999; Yasojima et al., 1999) expression in both the healthy and diseased human brain. These analyses suggested that COX‐1 was predominantly expressed by microglia and occasionally by neurons, while COX‐2 expression was mainly observed in neurons and astrocytes. Although higher COX‐1 levels were observed in brain tissue from patients with neurodegenerative diseases than from healthy controls, evidence suggested that this was mainly due to an increased number of COX‐1 expressing microglial cells near sites with brain injury, rather than upregulation of COX‐1 expression within the microglia (Deininger et al., 2003; Schwab et al., 2002; Yermakova et al., 1999). Postmortem studies did not provide any evidence for COX‐1 involvement in the neuroinflammation of psychiatric patients (Kim et al., 2011; Maida et al., 2006).
Preclinical PET imaging has provided evidence for an upregulation of COX‐1 expression in animal models of neuroinflammation (Shukuri et al., 2011, 2016; Takashima‐Hirano et al., 2010). However, clinical PET studies could not detect any upregulation of COX‐1 in patients with AD or MCI (Ohnishi et al., 2016). Whether this discrepancy is caused by methodological issues (insufficient sensitivity of the PET technique) or by the absence of COX‐1 overexpression in the diseased human brain remains to be investigated.
4. DISCUSSION
A single study in cultured adult human microglia (Hoozemans et al., 2002) suggested that increased release of PgE2 by these cells was not due to the overexpression of COX‐1, but to increased levels of the COX substrate AA. These results were in contrast to studies in cells from experimental animals which reported an overexpression of COX‐1 after exposure to inflammatory stimuli (Bate et al., 2006; Calvello et al., 2012, 2017; Gu et al., 2010; Qin et al., 2003).
Various explanations have been offered to explain this discrepancy between studies. One publication (Choi et al., 2013) suggested that the relative contributions of COX‐1 and COX‐2 to the cellular response could be time dependent since alterations in microglia and neurons change as a function of time. The hypothesis is in line with some animal studies which showed an increase in cerebral PgE2 secretion only in the early phase after administration of LPS (García‐Bueno et al., 2009) and with the outcome of PET scans which could visualize COX‐1 expression in the rat brain only in the first 24 hr after LPS injection (Shukuri et al., 2011).
Another explanation for the contradictory results could be the existence of species differences between rodents and humans. Results from imaging studies have indicated that such differences may indeed exist. PET imaging with the tracer [11C]KTP‐Me could clearly detect LPS‐induced COX‐1 overexpression in rodents (Shukuri et al., 2011, 2016; Takashima‐Hirano et al., 2010), but was unable to detect COX‐1 overexpression in humans, in particular in patients with AD or MCI (Ohnishi et al., 2016). On the other hand, postmortem studies on human brain samples showed an increase in the density of COX‐1‐expressing microglia in patients with neurodegenerative diseases like AD, in particular in the proximity of brain injury or protein deposits (Deininger et al., 2003; Hoozemans et al., 2001; Kitamura et al., 1999; Yermakova et al., 1999). A similar increase was not observed in samples from patients with psychiatric disorders, probably because psychiatric disorders are associated with less neuronal damage than neurodegenerative disease (Kim et al., 2011; Maida et al., 2006). The failure of PET imaging to detect COX‐1 overexpression in AD may have been caused by a dispersed pattern of localized increases in microglial density around amyloid plaques. Such increases may be too small—in a spatial sense—to be detected with PET, and/or the affinity of the existing PET tracers for COX‐1 may be too low to detect them. Postmortem studies did not provide evidence for any upregulation of COX‐1 expression in individual microglial cells (Deininger et al., 2003; Hoozemans et al., 2001; Kitamura et al., 1999; Yermakova et al., 1999), which is in agreement with the results of the single in vitro study on human microglia (Hoozemans et al., 2002).
Yet, several animal studies have suggested a considerable role for COX‐1 in the neuroinflammatory cascade. The injection of (endo)toxins like LPS, Aβ, or prion protein generally resulted in the activation of glial cells, increased expression levels of COX‐1, and release of PgE2. Not only microglia, but also astrocytes appeared to be involved in the upregulation of COX‐1 activity during the neuroinflammatory process in animal models of neurodegenerative disease (Calvello et al., 2017; Gu et al., 2010; Matousek et al., 2010). COX‐1‐dependent prostaglandin overexpression in the inflamed brain led to cognitive dysfunction and memory problems in rodents. The genetic deletion of COX‐1 or administration of a COX‐1 selective inhibitor prevented the overproduction of prostaglandins, improved memory retention, and prevented cognitive impairment in animal models of neuroinflammation (Calvello et al., 2017; Choi et al., 2008, 2013; Choi & Bosetti, 2009; Dargahi et al., 2011; García‐Bueno et al., 2009; Griffin et al., 2013; Gu et al., 2010; Matousek et al., 2010; Nie et al., 2019). In one animal model of neurodegeneration, the life span of the animals could be extended by the inhibition of COX‐1 with SC560 (Gu et al., 2010).
Such results from animal studies suggest that interventions with selective COX‐1 inhibitors could be tested in humans. However, there may be safety risks associated with such trials, since prostaglandins are involved in vascular dilation and blood clotting. A clinical trial in patients with AD who received the COX‐1 selective inhibitor, aspirin, as part of a multicomponent treatment protocol observed an increased risk of intracerebral hemorrhages compared to the control group (Thoonsen et al., 2010). The authors reasoned that this was due to cerebral amyloid angiopathy in AD. This explanation of their data was supported by a high prevalence of AD‐associated microbleeds observed in neuroimaging (Pettersen et al., 2008). The first fully randomized clinical trial which evaluated the impact of aspirin on dementia in AD did not demonstrate any significant improvement in cognition (Bentham et al., 2008), which could be due to the advanced stage of neurodegeneration in the patient group.
Animal studies have indicated that the induction of COX‐2 expression by inflammatory stimuli is dependent on the presence of COX‐1. In COX‐1−/− knockout mice, the expression of COX‐2 was significantly reduced (Aid et al., 2008; Choi & Bosetti, 2009; Choi et al., 2008), and pharmacological inhibition of COX‐1 could prevent the upregulation of COX‐2 (Dargahi et al., 2011). However, in vitro studies suggested that COX‐1 and COX‐2 were independently expressed by (immortalized) microglia (Calvello et al., 2012). Possibly, changes in COX‐1 and COX‐2 activity occur independently during the early phase of neuroinflammation. The overexpression of COX‐1 in microglia may have a pro‐inflammatory effect in this early phase (Calvello et al., 2012). The pro‐inflammatory function may be taken over by COX‐2 in later phases, when the expression of COX‐1 and COX‐2 become interdependent (Aid et al., 2008; Dargahi et al., 2011; Jeohn et al., 1998; Suzumura et al., 2006).
Evidently, further research needs to be carried out to unravel the time‐dependent roles of COX‐1 and COX‐2 in neuroinflammation associated with neurological diseases. The development of selective PET tracers with higher affinity to COX‐1 than the existing radiopharmaceuticals could result in greater ability of the PET technique to detect small or highly localized increases of COX‐1 in the diseased human brain, and lead to greater understanding of the role of COX‐1 in human pathophysiology.
5. CONCLUSION
Most literature reviewed in the current study indicates that COX‐1 is overexpressed by microglia, endothelial and perivascular cells during neuroinflammation in animals. The overexpression of COX‐1 appears to be an early event in the inflammatory process. This upregulation of COX‐1 is believed to result in an increased production of prostaglandins, such as PgE2 and PgD2, which have a detrimental effect on the formation of amyloid‐β plaques, neuronal loss, and cognitive functioning. Some studies suggest that the upregulation of COX‐1 is required for the overexpression of COX‐2. Although the exact mechanism of COX‐1 upregulation in reactive microglia remains unclear, there is some evidence that TLR and the interferon signaling pathway are involved. Based on such preclinical findings, COX‐1 may be considered as a therapeutic target for the treatment of neuroinflammation associated with several disorders of the human brain. However, the preclinical findings need first to be confirmed in patients. Evidence for an active role of COX‐1 in human neuroinflammation is still scarce. Postmortem studies have shown an increased density of COX‐1‐expressing microglia in the proximity of brain injury in tissue samples of patients with neurodegenerative diseases, but imaging studies could not yet confirm this observation in living subjects, and clinical trials with COX‐1 inhibitors in patients have been unsuccessful.
CONFLICT OF INTEREST
None of the authors have competing interests.
AUTHOR CONTRIBUTIONS
NG and AvW performed the investigation of the literature, data curation and writing of the first draft, editing and reviewing of the manuscript. EdV and JD were involved in the conceptualization of the study, supervision and reviewing of the manuscript. RD contributed to the editing and reviewing of the manuscript.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jnr.24934.
Supporting information
Transparent Peer Review Report
Supplementary Material
Ghazanfari, N. , van Waarde, A. , Dierckx, R. A. J. O. , Doorduin, J. , & de Vries, E. F. J. (2021). Is cyclooxygenase‐1 involved in neuroinflammation? Journal of Neuroscience Research, 99, 2976–2998. 10.1002/jnr.24934
Edited by Patricia Schuck and Cristina Ghiani. Reviewed by Ayse Er and Elisa Martelletti.
REFERENCES
- Aid, S. , Langenbach, R. , & Bosetti, F. (2008). Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase‐2. Journal of Neuroinflammation, 5, 17. 10.1186/1742-2094-5-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid, S. , Silva, A. C. , Candelario‐Jalil, E. , Choi, S. H. , Rosenberg, G. A. , & Bosetti, F. (2010). Cyclooxygenase‐1 and ‐2 differentially modulate lipopolysaccharide‐induced blood‐brain barrier disruption through matrix metalloproteinase activity. Journal of Cerebral Blood Flow and Metabolism, 30(2), 370–380. 10.1038/jcbfm.2009.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akundi, R. S. , Candelario‐Jalil, E. , Hess, S. , Hüll, M. , Lieb, K. , Gebicke‐Haerter, P. J. , & Fiebich, B. L. (2005). Signal transduction pathways regulating cyclooxygenase‐2 in lipopolysaccharide‐activated primary rat microglia. Glia, 51(3), 199–208. 10.1002/glia.20198 [DOI] [PubMed] [Google Scholar]
- An, Y. , Belevych, N. , Wang, Y. , Zhang, H. , Herschman, H. , Chen, Q. , & Quan, N. (2014). Neuronal and nonneuronal COX‐2 expression confers neurotoxic and neuroprotective phenotypes in response to excitotoxin challenge. Journal of Neuroscience Research, 92(4), 486–495. 10.1002/jnr.23317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett, J. , Chow, J. , Ives, D. , Chiou, M. , Mackenzie, R. , Osen, E. , Nguyen, B. , Tsing, S. , Bach, C. , Freire, J. , Chan, H. , Sigal, E. , & Ramesha, C. (1994). Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochimica et Biophysica Acta, 1209(1), 130–139. 10.1016/0167-4838(94)90148-1 [DOI] [PubMed] [Google Scholar]
- Bartels, A. L. , & Leenders, K. L. (2010). Cyclooxygenase and neuroinflammation in Parkinson's disease neurodegeneration. Current Neuropharmacology, 8(1), 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate, C. , Kempster, S. , & Williams, A. (2006). Prostaglandin D2 mediates neuronal damage by amyloid‐beta or prions which activates microglial cells. Neuropharmacology, 50(2), 229–237. [DOI] [PubMed] [Google Scholar]
- Bazan, N. G. , Fletcher, B. S. , Herschman, H. R. , & Mukherjee, P. K. (1994). Platelet‐activating factor and retinoic acid synergistically activate the inducible prostaglandin synthase gene. Proceedings of the National Academy of Sciences of the United States of America, 91(12), 5252–5256. 10.1073/pnas.91.12.5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti, F. , Aggio, V. , Pratesi, M. L. , Greco, G. , & Furlan, R. (2020). Neuroinflammation in bipolar depression. Frontiers in Psychiatry, 11, 71. 10.3389/fpsyt.2020.00071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentham, P. , Gray, R. , Sellwood, E. , Hills, R. , Crome, P. , & Raftery, J. (2008). Aspirin in Alzheimer's disease (AD2000): A randomised open‐label trial. The Lancet Neurology, 7(1), 41–49. [DOI] [PubMed] [Google Scholar]
- Bosetti, F. , & Choi, S. H. (2010). Rethinking the role of cyclooxygenase‐1 in neuroinflammation: More than homeostasis. Cell Cycle, 9(15), 2919–2920. 10.4161/cc.9.15.12715 [DOI] [PubMed] [Google Scholar]
- Boyle, A. J. , Tong, J. , Zoghbi, S. , Pike, V. W. , Innis, R. , & Vasdev, N. (2021). Repurposing [(11)C]PS13 for PET imaging of cyclooxygenase‐1 (COX‐1) in ovarian cancer xenograft mouse models. Journal of Nuclear Medicine, 62(5), 665–668. 10.2967/jnumed.120.249367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnin, A. , Kassiou, M. , Meikle, S. R. , & Banati, R. B. (2007). Positron emission tomography imaging of neuroinflammation. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics, 4(3), 443–452. 10.1016/j.nurt.2007.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calsolaro, V. , & Edison, P. (2016). Neuroinflammation in Alzheimer's disease: Current evidence and future directions. Alzheimer's & Dementia: The Journal of the Alzheimer's Association, 12(6), 719–732. 10.1016/j.jalz.2016.02.010 [DOI] [PubMed] [Google Scholar]
- Calvello, R. , Lofrumento, D. D. , Perrone, M. G. , Cianciulli, A. , Salvatore, R. , Vitale, P. , De Nuccio, F. , Giannotti, L. , Nicolardi, G. , Panaro, M. A. , & Scilimati, A. (2017). Highly selective cyclooxygenase‐1 inhibitors P6 and Mofezolac counteract inflammatory state both in vitro and in vivo models of neuroinflammation. Frontiers in Neurology, 8, 251. 10.3389/fneur.2017.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvello, R. , Panaro, M. A. , Carbone, M. L. , Cianciulli, A. , Perrone, M. G. , Vitale, P. , Malerba, P. , & Scilimati, A. (2012). Novel selective COX‐1 inhibitors suppress neuroinflammatory mediators in LPS‐stimulated N13 microglial cells. Pharmacological Research, 65(1), 137–148. 10.1016/j.phrs.2011.09.009 [DOI] [PubMed] [Google Scholar]
- Chandrasekharan, N. V. , Dai, H. , Roos, K. L. , Evanson, N. K. , Tomsik, J. , Elton, T. S. , & Simmons, D. L. (2002). COX‐3, a cyclooxygenase‐1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proceedings of the National Academy of Sciences of the United States of America, 99(21), 13926–13931. 10.1073/pnas.162468699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. , Aid, S. , Caracciolo, L. , Minami, S. S. , Niikura, T. , Matsuoka, Y. , Turner, R. S. , Mattson, M. P. , & Bosetti, F. (2013). Cyclooxygenase‐1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer's disease. Journal of Neurochemistry, 124(1), 59–68. 10.1111/jnc.12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. , Aid, S. , Choi, U. , & Bosetti, F. (2010). Cyclooxygenases‐1 and ‐2 differentially modulate leukocyte recruitment into the inflamed brain. The Pharmacogenomics Journal, 10(5), 448–457. 10.1038/tpj.2009.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. , & Bosetti, F. (2009). Cyclooxygenase‐1 null mice show reduced neuroinflammation in response to beta‐amyloid. Aging, 1(2), 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. , Langenbach, R. , & Bosetti, F. (2008). Genetic deletion or pharmacological inhibition of cyclooxygenase‐1 attenuate lipopolysaccharide‐induced inflammatory response and brain injury. The FASEB Journal, 22(5), 1491–1501. 10.1096/fj.07-9411com [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargahi, L. , Nasiraei‐Moghadam, S. , Abdi, A. , Khalaj, L. , Moradi, F. , & Ahmadiani, A. (2011). Cyclooxygenase (COX)‐1 activity precedes the COX‐2 induction in Aß‐induced neuroinflammation. Journal of Molecular Neuroscience, 45(1), 10–21. [DOI] [PubMed] [Google Scholar]
- de Vries, E. F. , Doorduin, J. , Dierckx, R. A. , & van Waarde, A. (2008). Evaluation of [(11)C]rofecoxib as PET tracer for cyclooxygenase 2 overexpression in rat models of inflammation. Nuclear Medicine and Biology, 35(1), 35–42. 10.1016/j.nucmedbio.2007.07.015 [DOI] [PubMed] [Google Scholar]
- de Vries, E. F. , van Waarde, A. , Buursma, A. R. , & Vaalburg, W. (2003). Synthesis and in vivo evaluation of 18F‐desbromo‐DuP‐697 as a PET tracer for cyclooxygenase‐2 expression. Journal of Nuclear Medicine, 44(10), 1700–1706. [PubMed] [Google Scholar]
- Deininger, M. H. , Bekure‐Nemariam, K. , Trautmann, K. , Morgalla, M. , Meyermann, R. , & Schluesener, H. J. (2003). Cyclooxygenase‐1 and ‐2 in brains of patients who died with sporadic Creutzfeldt‐Jakob disease. Journal of Molecular Neuroscience, 20(1), 25–30. 10.1385/JMN:20:1:25 [DOI] [PubMed] [Google Scholar]
- Do, J. , & Woo, J. (2018). From gut to brain: Alteration in inflammation markers in the brain of dextran sodium sulfate‐induced colitis model mice. Clinical Psychopharmacology and Neuroscience, 16(4), 422–433. 10.9758/cpn.2018.16.4.422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famitafreshi, H. , & Karimian, M. (2020). Prostaglandins as the agents that modulate the course of brain disorders. Degenerative Neurological and Neuromuscular Disease, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, M. , Wang, M. , Miller, K. D. , Hutchins, G. D. , & Zheng, Q. H. (2009). Synthesis of carbon‐11 labeled celecoxib derivatives as new candidate PET radioligands for imaging of inflammation. Applied Radiation and Isotopes, 67(11), 2019–2024. 10.1016/j.apradiso.2009.07.022 [DOI] [PubMed] [Google Scholar]
- García‐Bueno, B. , Serrats, J. , & Sawchenko, P. E. (2009). Cerebrovascular cyclooxygenase‐1 expression, regulation, and role in hypothalamic‐pituitary‐adrenal axis activation by inflammatory stimuli. Journal of Neuroscience, 29(41), 12970–12981. 10.1523/JNEUROSCI.2373-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabert, K. , Michoel, T. , Karavolos, M. H. , Clohisey, S. , Baillie, J. K. , Stevens, M. P. , Freeman, T. C. , Summers, K. M. , & McColl, B. W. (2016). Microglial brain region‐dependent diversity and selective regional sensitivities to aging. Nature Neuroscience, 19(3), 504–516. 10.1038/nn.4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, S. J. , Scheller, L. F. , Marletta, M. A. , Seguin, M. C. , Klotz, F. W. , Slayter, M. , Nelson, B. J. , & Nacy, C. A. (1994). Nitric oxide: Cytokine‐regulation of nitric oxide in host resistance to intracellular pathogens. Immunology Letters, 43(1–2), 87–94. 10.1016/0165-2478(94)00158-8 [DOI] [PubMed] [Google Scholar]
- Griffin, D. E. , Wesselingh, S. L. , & McArthur, J. C. (1994). Elevated central nervous system prostaglandins in human immunodeficiency virus‐associated dementia. Annals of Neurology, 35(5), 592–597. 10.1002/ana.410350513 [DOI] [PubMed] [Google Scholar]
- Griffin, É. W. , Skelly, D. T. , Murray, C. L. , & Cunningham, C. (2013). Cyclooxygenase‐1‐dependent prostaglandins mediate susceptibility to systemic inflammation‐induced acute cognitive dysfunction. Journal of Neuroscience, 33(38), 15248–15258. 10.1523/JNEUROSCI.6361-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, X. L. , Long, C. X. , Sun, L. , Xie, C. , Lin, X. , & Cai, H. (2010). Astrocytic expression of Parkinson's disease‐related A53T alpha‐synuclein causes neurodegeneration in mice. Molecular Brain, 3, 12. 10.1186/1756-6606-3-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner, F. L. , Ransohoff, R. M. , & Becher, B. (2015). Immune attack: The role of inflammation in Alzheimer disease. Nature Reviews Neuroscience, 16(6), 358–372. 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- Hewett, S. J. , Bell, S. C. , & Hewett, J. A. (2006). Contributions of cyclooxygenase‐2 to neuroplasticity and neuropathology of the central nervous system. Pharmacology & Therapeutics, 112(2), 335–357. 10.1016/j.pharmthera.2005.04.011 [DOI] [PubMed] [Google Scholar]
- Hirsch, E. C. , & Hunot, S. (2009). Neuroinflammation in Parkinson's disease: A target for neuroprotection? The Lancet Neurology, 8(4), 382–397. [DOI] [PubMed] [Google Scholar]
- Hoozemans, J. J. , Rozemuller, A. J. , Janssen, I. , De Groot, C. J. , Veerhuis, R. , & Eikelenboom, P. (2001). Cyclooxygenase expression in microglia and neurons in Alzheimer's disease and control brain. Acta Neuropathologica, 101(1), 2–8. 10.1007/s004010000251 [DOI] [PubMed] [Google Scholar]
- Hoozemans, J. J. , Veerhuis, R. , Janssen, I. , van Elk, E. J. , Rozemuller, A. J. , & Eikelenboom, P. (2002). The role of cyclo‐oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: Implications for Alzheimer's disease. Brain Research, 951(2), 218–226. 10.1016/S0006-8993(02)03164-5 [DOI] [PubMed] [Google Scholar]
- Iñiguez, M. A. , Punzón, C. , & Fresno, M. (1999). Induction of cyclooxygenase‐2 on activated T lymphocytes: Regulation of T cell activation by cyclooxygenase‐2 inhibitors. Journal of Immunology, 163(1), 111–119. [PubMed] [Google Scholar]
- Jeohn, G. H. , Kong, L. Y. , Wilson, B. , Hudson, P. , & Hong, J. S. (1998). Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. Journal of Neuroimmunology, 85(1), 1–10. 10.1016/S0165-5728(97)00204-X [DOI] [PubMed] [Google Scholar]
- Kannan, S. , Balakrishnan, B. , Muzik, O. , Romero, R. , & Chugani, D. (2009). Positron emission tomography imaging of neuroinflammation. Journal of Child Neurology, 24(9), 1190–1199. 10.1177/0883073809338063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. H. , Lee, K. H. , Lee, D. , Han, Y. E. , Lee, S. H. , Sohn, J. W. , & Ho, W. K. (2015). Costimulation of AMPA and metabotropic glutamate receptors underlies phospholipase C activation by glutamate in hippocampus. Journal of Neuroscience, 35(16), 6401–6412. 10.1523/JNEUROSCI.4208-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. W. , Rapoport, S. I. , & Rao, J. S. (2011). Altered arachidonic acid cascade enzymes in postmortem brain from bipolar disorder patients. Molecular Psychiatry, 16(4), 419–428. 10.1038/mp.2009.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. J. , Lee, J. H. , Juarez, A. F. , Hong, J. , Miller, W. , Telu, S. , Singh, P. , Cortes, M. Y. , Henry, K. , Tye, G. L. , Frankland, M. P. , Montero Santamaria, J. A. , Liow, J. S. , Zoghbi, S. S. , Fujita, M. , Pike, V. W. , & Innis, R. B. (2020). First‐in‐human evaluation of [(11)C]PS13, a novel PET radioligand, to quantify cyclooxygenase‐1 in the brain. European Journal of Nuclear Medicine and Molecular Imaging, 47(13), 3143–3151. 10.1007/s00259-020-04855-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. J. , Shrestha, S. S. , Cortes, M. , Singh, P. , Morse, C. , Liow, J. S. , Gladding, R. L. , Brouwer, C. , Henry, K. , Gallagher, E. , Tye, G. L. , Zoghbi, S. S. , Fujita, M. , Pike, V. W. , & Innis, R. B. (2018). Evaluation of two potent and selective PET radioligands to image COX‐1 and COX‐2 in rhesus monkeys. Journal of Nuclear Medicine, 59(12), 1907–1912. 10.2967/jnumed.118.211144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura, Y. , Shimohama, S. , Koike, H. , Kakimura, J. , Matsuoka, Y. , Nomura, Y. , Gebicke‐Haerter, P. J. , & Taniguchi, T. (1999). Increased expression of cyclooxygenases and peroxisome proliferator‐activated receptor‐gamma in Alzheimer's disease brains. Biochemical and Biophysical Research Communications, 254(3), 582–586. [DOI] [PubMed] [Google Scholar]
- Koudriavtseva, T. , & Mainero, C. (2016). Neuroinflammation, neurodegeneration and regeneration in multiple sclerosis: Intercorrelated manifestations of the immune response. Neural Regeneration Research, 11(11), 1727–1730. 10.4103/1673-5374.194804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancelot, S. , & Zimmer, L. (2010). Small‐animal positron emission tomography as a tool for neuropharmacology. Trends in Pharmacological Sciences, 31(9), 411–417. 10.1016/j.tips.2010.06.002 [DOI] [PubMed] [Google Scholar]
- Laube, M. , Kniess, T. , & Pietzsch, J. (2013). Radiolabeled COX‐2 inhibitors for non‐invasive visualization of COX‐2 expression and activity—A critical update. Molecules, 18(6), 6311–6355. 10.3390/molecules18066311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W. , Moon, M. , Kim, H. G. , Lee, T. H. , & Oh, M. S. (2015). Heat stress‐induced memory impairment is associated with neuroinflammation in mice. Journal of Neuroinflammation, 12, 102. 10.1186/s12974-015-0324-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng, F. , & Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nature Reviews Neurology, 17(3), 157–172. 10.1038/s41582-020-00435-y [DOI] [PubMed] [Google Scholar]
- Li, P. , Kaur, C. , Lu, J. , Sivakumar, V. , Dheen, S. T. , & Ling, E. A. (2010). Expression of cyclooxygenase‐2 and microsomal prostaglandin‐E synthase in amoeboid microglial cells in the developing brain and effects of cyclooxygenase‐2 neutralization on BV‐2 microglial cells. Journal of Neuroscience Research, 88(7), 1577–1594. [DOI] [PubMed] [Google Scholar]
- Maida, M. E. , Hurley, S. D. , Daeschner, J. A. , Moore, A. H. , & O'Banion, M. K. (2006). Cytosolic prostaglandin E2 synthase (cPGES) expression is decreased in discrete cortical regions in psychiatric disease. Brain Research, 1103(1), 164–172. 10.1016/j.brainres.2006.05.048 [DOI] [PubMed] [Google Scholar]
- Majo, V. J. , Prabhakaran, J. , Simpson, N. R. , Van Heertum, R. L. , Mann, J. J. , & Kumar, J. S. (2005). A general method for the synthesis of aryl [11C]methylsulfones: Potential PET probes for imaging cyclooxygenase‐2 expression. Bioorganic & Medicinal Chemistry Letters, 15(19), 4268–4271. 10.1016/j.bmcl.2005.06.080 [DOI] [PubMed] [Google Scholar]
- Malerba, P. , Crews, B. C. , Ghebreselasie, K. , Daniel, C. K. , Jashim, E. , Aleem, A. M. , Salam, R. A. , Marnett, L. J. , & Uddin, M. J. (2020). Targeted detection of cyclooxygenase‐1 in ovarian cancer. ACS Medicinal Chemistry Letters, 11(10), 1837–1842. 10.1021/acsmedchemlett.9b00280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín, R. , Durroux, T. , Ciruela, F. , Torres, M. , Pin, J. P. , & Sánchez‐Prieto, J. (2010). The metabotropic glutamate receptor mGlu7 activates phospholipase C, translocates munc‐13‐1 protein, and potentiates glutamate release at cerebrocortical nerve terminals. Journal of Biological Chemistry, 285(23), 17907–17917. 10.1074/jbc.M109.080838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty, S. , Dusart, I. , & Peschanski, M. (1991). Glial changes following an excitotoxic lesion in the CNS–I. Microglia/macrophages. Neuroscience, 45(3), 529–539. 10.1016/0306-4522(91)90268-S [DOI] [PubMed] [Google Scholar]
- Matousek, S. B. , Hein, A. M. , Shaftel, S. S. , Olschowka, J. A. , Kyrkanides, S. , & O'Banion, M. K. (2010). Cyclooxygenase‐1 mediates prostaglandin E(2) elevation and contextual memory impairment in a model of sustained hippocampal interleukin‐1beta expression. Journal of Neurochemistry, 114(1), 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley, M. E. , & Baloh, R. H. (2019). Inflammation in ALS/FTD pathogenesis. Acta Neuropathologica, 137(5), 715–730. 10.1007/s00401-018-1933-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, J. H. , Cervenka, S. , Kim, M. J. , Kreisl, W. C. , Henter, I. D. , & Innis, R. B. (2020). Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry, 7(12), 1064–1074. 10.1016/S2215-0366(20)30255-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhillaj, E. , Morgese, M. G. , Tucci, P. , Furiano, A. , Luongo, L. , Bove, M. , Maione, S. , Cuomo, V. , Schiavone, S. , & Trabace, L. (2018). Celecoxib prevents cognitive impairment and neuroinflammation in soluble amyloid ß‐treated rats. Neuroscience, 372, 58–73. [DOI] [PubMed] [Google Scholar]
- Minghetti, L. (2004). Cyclooxygenase‐2 (COX‐2) in inflammatory and degenerative brain diseases. Journal of Neuropathology and Experimental Neurology, 63(9), 901–910. 10.1093/jnen/63.9.901 [DOI] [PubMed] [Google Scholar]
- Miscia, S. , Di, B. A. , Rana, R. A. , & Cataldi, A. (1997). Phospholipase C gamma 1 overexpression and activation induced by interferon beta in human T lymphocytes: An ISGF3‐independent response. Cytokine, 9(9), 660–665. [DOI] [PubMed] [Google Scholar]
- Monji, A. , Kato, T. , & Kanba, S. (2009). Cytokines and schizophrenia: Microglia hypothesis of schizophrenia. Psychiatry and Clinical Neurosciences, 63(3), 257–265. 10.1111/j.1440-1819.2009.01945.x [DOI] [PubMed] [Google Scholar]
- Müller, N. (2018). Inflammation in schizophrenia: Pathogenetic aspects and therapeutic considerations. Schizophrenia Bulletin, 44(5), 973–982. 10.1093/schbul/sby024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, N. , Weidinger, E. , Leitner, B. , & Schwarz, M. J. (2015). The role of inflammation in schizophrenia. Frontiers in Neuroscience, 9, 372. 10.3389/fnins.2015.00372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na, K. S. , Jung, H. Y. , & Kim, Y. K. (2014). The role of pro‐inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Progress in Neuro‐Psychopharmacology and Biological Psychiatry, 48, 277–286. [DOI] [PubMed] [Google Scholar]
- Najjar, S. , & Pearlman, D. M. (2015). Neuroinflammation and white matter pathology in schizophrenia: Systematic review. Schizophrenia Research, 161(1), 102–112. 10.1016/j.schres.2014.04.041 [DOI] [PubMed] [Google Scholar]
- Najjar, S. , Pearlman, D. M. , Alper, K. , Najjar, A. , & Devinsky, O. (2013). Neuroinflammation and psychiatric illness. Journal of Neuroinflammation, 10, 43. 10.1186/1742-2094-10-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazmi, A. , Field, R. H. , Griffin, E. W. , Haugh, O. , Hennessy, E. , Cox, D. , Reis, R. , Tortorelli, L. , Murray, C. L. , Lopez‐Rodriguez, A. B. , Jin, L. , Lavelle, E. C. , Dunne, A. , & Cunningham, C. (2019). Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia, 67(7), 1254–1276. 10.1002/glia.23592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettis, M. A. , & Pariante, C. M. (2020). Is there neuroinflammation in depression? Understanding the link between the brain and the peripheral immune system in depression. International Review of Neurobiology, 152, 23–40. [DOI] [PubMed] [Google Scholar]
- Nie, X. , Kitaoka, S. , Shinohara, M. , Kakizuka, A. , Narumiya, S. , & Furuyashiki, T. (2019). Roles of Toll‐like receptor 2/4, monoacylglycerol lipase, and cyclooxygenase in social defeat stress‐induced prostaglandin E(2) synthesis in the brain and their behavioral relevance. Scientific Reports, 9(1), 17548. 10.1038/s41598-019-54082-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi, A. , Senda, M. , Yamane, T. , Mikami, T. , Nishida, H. , Nishio, T. , Akamatsu, G. , Ikari, Y. , Kimoto, S. , Aita, K. , Sasaki, M. , Shinkawa, H. , Yamamoto, Y. , Shukuri, M. , Mawatari, A. , Doi, H. , Watanabe, Y. , & Onoe, H. (2016). Exploratory human PET study of the effectiveness of (11)C‐ketoprofen methyl ester, a potential biomarker of neuroinflammatory processes in Alzheimer's disease. Nuclear Medicine and Biology, 43(7), 438–444. 10.1016/j.nucmedbio.2016.04.005 [DOI] [PubMed] [Google Scholar]
- Ohnishi, A. , Senda, M. , Yamane, T. , Sasaki, M. , Mikami, T. , Nishio, T. , Ikari, Y. , Nishida, H. , Shukuri, M. , Takashima, T. , Mawatari, A. , Doi, H. , Watanabe, Y. , & Onoe, H. (2014). Human whole‐body biodistribution and dosimetry of a new PET tracer, [(11)C]ketoprofen methyl ester, for imagings of neuroinflammation. Nuclear Medicine and Biology, 41(7), 594–599. 10.1016/j.nucmedbio.2014.04.008 [DOI] [PubMed] [Google Scholar]
- Papadimitriou, D. , Le Verche, V. , Jacquier, A. , Ikiz, B. , Przedborski, S. , & Re, D. B. (2010). Inflammation in ALS and SMA: Sorting out the good from the evil. Neurobiology of Diseases, 37(3), 493–502. 10.1016/j.nbd.2009.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, V. H. , & Teeling, J. (2013). Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Seminars in Immunopathology, 35(5), 601–612. 10.1007/s00281-013-0382-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen, J. A. , Sathiyamoorthy, G. , Gao, F. Q. , Szilagyi, G. , Nadkarni, N. K. , St George‐Hyslop, P. , Rogaeva, E. , & Black, S. E. (2008). Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Archives of Neurology, 65(6), 790–795. 10.1001/archneur.65.6.790 [DOI] [PubMed] [Google Scholar]
- Phani, S. , Loike, J. D. , & Przedborski, S. (2012). Neurodegeneration and inflammation in Parkinson's disease. Parkinsonism & Related Disorders, 18(Suppl. 1), S207–S209. 10.1016/S1353-8020(11)70064-5 [DOI] [PubMed] [Google Scholar]
- Ponzoni, M. , & Cornaglia‐Ferraris, P. (1993). Interferon‐gamma‐stimulated and GTP‐binding‐proteins‐mediated phospholipase A2 activation in human neuroblasts. Biochemical Journal, 294(Pt 3), 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzoni, M. , Montaldo, P. G. , & Cornaglia‐Ferraris, P. (1992). Stimulation of receptor‐coupled phospholipase A2 by interferon‐gamma. FEBS Letters, 310(1), 17–21. [DOI] [PubMed] [Google Scholar]
- Qin, W. , Ho, L. , Pompl, P. N. , Peng, Y. , Zhao, Z. , Xiang, Z. , Robakis, N. K. , Shioi, J. , Suh, J. , & Pasinetti, G. M. (2003). Cyclooxygenase (COX)‐2 and COX‐1 potentiate beta‐amyloid peptide generation through mechanisms that involve gamma‐secretase activity. Journal of Biological Chemistry, 278(51), 50970–50977. [DOI] [PubMed] [Google Scholar]
- Rouzer, C. A. , & Marnett, L. J. (2009). Cyclooxygenases: Structural and functional insights. Journal of Lipid Research, 50(Suppl.), S29–S34. 10.1194/jlr.R800042-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, I. , Amornphimoltham, P. , Weigert, R. , Barlati, S. , & Bosetti, F. (2011). Cyclooxygenase‐1 is involved in the inhibition of hippocampal neurogenesis after lipopolysaccharide‐induced neuroinflammation. Cell Cycle, 10(15), 2568–2573. 10.4161/cc.10.15.15946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura, C. A. (2010). Presenilin/gamma‐secretase and inflammation. Frontiers in Aging Neuroscience, 2, 16. 10.3389/fnagi.2010.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, J. M. , Beschorner, R. , Meyermann, R. , Gözalan, F. , & Schluesener, H. J. (2002). Persistent accumulation of cyclooxygenase‐1‐expressing microglial cells and macrophages and transient upregulation by endothelium in human brain injury. Journal of Neurosurgery, 96(5), 892–899. 10.3171/jns.2002.96.5.0892 [DOI] [PubMed] [Google Scholar]
- Shrestha, S. , Kim, M. J. , Eldridge, M. , Lehmann, M. L. , Frankland, M. , Liow, J. S. , Yu, Z. X. , Cortes‐Salva, M. , Telu, S. , Henter, I. D. , Gallagher, E. , Lee, J. H. , Fredericks, J. M. , Poffenberger, C. , Tye, G. , Ruiz‐Perdomo, Y. , Anaya, F. J. , Montero Santamaria, J. A. , Gladding, R. L. , … Innis, R. B. (2020). PET measurement of cyclooxygenase‐2 using a novel radioligand: Upregulation in primate neuroinflammation and first‐in‐human study. Journal of Neuroinflammation, 17(1), 140. 10.1186/s12974-020-01804-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha, S. , Singh, P. , Cortes‐Salva, M. Y. , Jenko, K. J. , Ikawa, M. , Kim, M. J. , Kobayashi, M. , Morse, C. L. , Gladding, R. L. , Liow, J. S. , Zoghbi, S. S. , Fujita, M. , Innis, R. B. , & Pike, V. W. (2018). 3‐Substituted 1,5‐diaryl‐1 H‐1,2,4‐triazoles as prospective PET radioligands for imaging brain COX‐1 in monkey. Part 2: Selection and evaluation of [(11)C]PS13 for quantitative imaging. ACS Chemical Neuroscience, 9(11), 2620–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukuri, M. , Mawatari, A. , Ohno, M. , Suzuki, M. , Doi, H. , Watanabe, Y. , & Onoe, H. (2016). Detection of cyclooxygenase‐1 in activated microglia during amyloid plaque progression: PET studies in Alzheimer's disease model mice. Journal of Nuclear Medicine, 57(2), 291–296. 10.2967/jnumed.115.166116 [DOI] [PubMed] [Google Scholar]
- Shukuri, M. , Takashima‐Hirano, M. , Tokuda, K. , Takashima, T. , Matsumura, K. , Inoue, O. , Doi, H. , Suzuki, M. , Watanabe, Y. , & Onoe, H. (2011). In vivo expression of cyclooxygenase‐1 in activated microglia and macrophages during neuroinflammation visualized by PET with 11C‐ketoprofen methyl ester. Journal of Nuclear Medicine, 52(7), 1094–1101. [DOI] [PubMed] [Google Scholar]
- Singh, P. , Shrestha, S. , Cortes‐Salva, M. Y. , Jenko, K. J. , Zoghbi, S. S. , Morse, C. L. , Innis, R. B. , & Pike, V. W. (2018). 3‐Substituted 1,5‐diaryl‐1 H‐1,2,4‐triazoles as prospective PET radioligands for imaging brain COX‐1 in monkey. Part 1: Synthesis and pharmacology. ACS Chemical Neuroscience, 9(11), 2610–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slanina, K. A. , & Schweitzer, P. (2005). Inhibition of cyclooxygenase‐2 elicits a CB1‐mediated decrease of excitatory transmission in rat CA1 hippocampus. Neuropharmacology, 49(5), 653–659. 10.1016/j.neuropharm.2005.04.019 [DOI] [PubMed] [Google Scholar]
- Smith, W. L. , Garavito, R. M. , & DeWitt, D. L. (1996). Prostaglandin endoperoxide H synthases (cyclooxygenases)‐1 and ‐2. Journal of Biological Chemistry, 271(52), 33157–33160. 10.1074/jbc.271.52.33157 [DOI] [PubMed] [Google Scholar]
- Stefanovic, B. , Bosetti, F. , & Silva, A. C. (2006). Modulatory role of cyclooxygenase‐2 in cerebrovascular coupling. NeuroImage, 32(1), 23–32. 10.1016/j.neuroimage.2006.03.014 [DOI] [PubMed] [Google Scholar]
- Streit, W. J. (1996). The role of microglia in brain injury. Neurotoxicology, 17(3–4), 671–678. [PubMed] [Google Scholar]
- Suzumura, A. , Takeuchi, H. , Zhang, G. , Kuno, R. , & Mizuno, T . (2006). Roles of glia‐derived cytokines on neuronal degeneration and regeneration. Annals of the New York Academy of Sciences, 1088, 219–229. [DOI] [PubMed] [Google Scholar]
- Taddei, C. , Morse, C. L. , Kim, M. J. , Liow, J. S. , Montero, S. J. , Zhang, A. , Manly, L. S. , Zanotti‐Fregonara, P. , Gladding, R. L. , Zoghbi, S. S. , Innis, R. B. , & Pike, V. W. (2021). Synthesis of [(18)F]PS13 and evaluation as a PET radioligand for cyclooxygenase‐1 in monkey. ACS Chemical Neuroscience, 12(3), 517–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima‐Hirano, M. , Shukuri, M. , Takashima, T. , Goto, M. , Wada, Y. , Watanabe, Y. , Onoe, H. , Doi, H. , & Suzuki, M. (2010). General method for the (11)C‐labeling of 2‐arylpropionic acids and their esters: Construction of a PET tracer library for a study of biological events involved in COXs expression. Chemistry, 16(14), 4250–4258. [DOI] [PubMed] [Google Scholar]
- Tanabe, T. , & Tohnai, N. (2002). Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins & Other Lipid Mediators, 68–69, 95–114. 10.1016/S0090-6980(02)00024-2 [DOI] [PubMed] [Google Scholar]
- Tansey, M. G. , McCoy, M. K. , & Frank‐Cannon, T. C. (2007). Neuroinflammatory mechanisms in Parkinson's disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Experimental Neurology, 208(1), 1–25. 10.1016/j.expneurol.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling, J. L. , Cunningham, C. , Newman, T. A. , & Perry, V. H. (2010). The effect of non‐steroidal anti‐inflammatory agents on behavioural changes and cytokine production following systemic inflammation: Implications for a role of COX‐1. Brain, Behavior, and Immunity, 24(3), 409–419. 10.1016/j.bbi.2009.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoonsen, H. , Richard, E. , Bentham, P. , Gray, R. , van Geloven, N. , De Haan, R. J. , Van Gool, W. A. , & Nederkoorn, P. J. (2010). Aspirin in Alzheimer's disease: Increased risk of intracerebral hemorrhage: Cause for concern? Stroke, 41(11), 2690–2692. 10.1161/STROKEAHA.109.576975 [DOI] [PubMed] [Google Scholar]
- Troubat, R. , Barone, P. , Leman, S. , Desmidt, T. , Cressant, A. , Atanasova, B. , Brizard, B. , El, H. W. , Surget, A. , Belzung, C. , & Camus, V. (2021). Neuroinflammation and depression: A review. European Journal of Neuroscience, 53(1), 151–171. 10.1111/ejn.14720 [DOI] [PubMed] [Google Scholar]
- Uddin, M. J. , Crews, B. C. , Ghebreselasie, K. , Huda, I. , Kingsley, P. J. , Ansari, M. S. , Tantawy, M. N. , Reese, J. , & Marnett, L. J. (2011). Fluorinated COX‐2 inhibitors as agents in PET imaging of inflammation and cancer. Cancer Prevention Research, 4(10), 1536–1545. 10.1158/1940-6207.CAPR-11-0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet, S. , Prinz, M. , & van Loo, G. (2019). Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends in Molecular Medicine, 25(2), 112–123. 10.1016/j.molmed.2018.11.005 [DOI] [PubMed] [Google Scholar]
- Wang, H. , Hitron, I. M. , Iadecola, C. , & Pickel, V. M. (2005). Synaptic and vascular associations of neurons containing cyclooxygenase‐2 and nitric oxide synthase in rat somatosensory cortex. Cerebral Cortex, 15(8), 1250–1260. 10.1093/cercor/bhi008 [DOI] [PubMed] [Google Scholar]
- Wang, T. , Shi, C. , Luo, H. , Zheng, H. , Fan, L. , Tang, M. , Su, Y. , Yang, J. , Mao, C. , & Xu, Y. (2021). Neuroinflammation in Parkinson's disease: Triggers, mechanisms, and immunotherapies. Neuroscientist. 10.1177/1073858421991066 [DOI] [PubMed] [Google Scholar]
- Wuest, F. , Kniess, T. , Bergmann, R. , & Pietzsch, J. (2008). Synthesis and evaluation in vitro and in vivo of a 11C‐labeled cyclooxygenase‐2 (COX‐2) inhibitor. Bioorganic & Medicinal Chemistry, 16(16), 7662–7670. 10.1016/j.bmc.2008.07.016 [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray, T. , & Rogers, J. (2012). Inflammation in Alzheimer disease‐a brief review of the basic science and clinical literature. Cold Spring Harbor Perspectives in Medicine, 2(1), a006346. 10.1101/cshperspect.a006346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata, K. , Andreasson, K. I. , Kaufmann, W. E. , Barnes, C. A. , & Worley, P. F. (1993). Expression of a mitogen‐inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron, 11(2), 371–386. 10.1016/0896-6273(93)90192-T [DOI] [PubMed] [Google Scholar]
- Yasojima, K. , Schwab, C. , McGeer, E. G. , & McGeer, P. L. (1999). Distribution of cyclooxygenase‐1 and cyclooxygenase‐2 mRNAs and proteins in human brain and peripheral organs. Brain Research, 830(2), 226–236. 10.1016/S0006-8993(99)01389-X [DOI] [PubMed] [Google Scholar]
- Yermakova, A. V. , Rollins, J. , Callahan, L. M. , Rogers, J. , & O'Banion, M. K. (1999). Cyclooxygenase‐1 in human Alzheimer and control brain: Quantitative analysis of expression by microglia and CA3 hippocampal neurons. Journal of Neuropathology and Experimental Neurology, 58(11), 1135–1146. 10.1097/00005072-199911000-00003 [DOI] [PubMed] [Google Scholar]
- Yuan, C. , & Smith, W. L. (2015). A cyclooxygenase‐2‐dependent prostaglandin E2 biosynthetic system in the Golgi apparatus. Journal of Biological Chemistry, 290(9), 5606–5620. 10.1074/jbc.M114.632463 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparent Peer Review Report
Supplementary Material