

Abstract
The accuracy and precision of fungal molecular identification and classification are challenging, particularly in environmental metabarcoding approaches as these often trade accuracy for efficiency given the large data volumes at hand. In most ecological studies, only a single similarity cutoff value is used for sequence identification. This is not sufficient since the most commonly used DNA markers are known to vary widely in terms of inter‐ and intraspecific variability. We address this problem by presenting a new tool, dnabarcoder, to predict local similarity cutoffs and measure the resolving powers of a biomarker for sequence identification for different clades of fungi. It was shown that the predicted similarity cutoffs varied significantly between the clades of a recently released ITS DNA barcode data set from the CBS culture collection of the Westerdijk Fungal Biodiversity Institute. When classifying a large public fungal ITS data set—the UNITE database—against the barcode data set, the local similarity cutoffs assigned fewer sequences than the traditional cutoffs used in metabarcoding studies. However, the obtained accuracy and precision were significantly improved. Our study showed that it might be better to extract the ITS region from the ITS barcodes to optimize taxonomic assignment accuracy. Furthermore, 15.3, 25.6, and 26.3% of the fungal species of the barcode data set were indistinguishable by full‐length ITS, ITS1, and ITS2, respectively. Except for these indistinguishable species, the resolving powers of full‐length ITS, ITS1, and ITS2 sequences were similar at the species level. Nevertheless, the complete ITS region had a better resolving power at higher taxonomic levels.
Keywords: DNA barcoding, metabarcoding, sequence identification, sequence classification, similarity cutoff, taxonomy
1. INTRODUCTION
Fungi constitute the second largest group of all organisms based on global richness estimates with an estimated 3.8–6 million predicted species, playing fundamental ecological roles as decomposers of organic matter, pathogens, and symbionts (Baldrian et al., 2021; Stajich et al., 2009). Fungal identification is essential for communication purposes and to gain biological insights into the causes, nature, and consequences of environmentally induced changes such as climate changes, spatially and temporarily, for maintaining and improving our health and the natural environment.
The accuracy and precision of fungal identification are challenging as fungi have simple body plans with often morphologically and ecologically obscure or inconspicuous structures (Lücking et al., 2020, 2021). In addition, continuous progress in fungal taxonomy results in a constant stream of reclassifications and new names, which complicates informed decisions on fungal taxonomic delineation. To date, less than a few percent of the estimated number of extant fungal species have been described. Environmental metabarcoding via high‐throughput sequencing has added a new dimension to assessing fungal biodiversity (Taberlet et al., 2012). The metabarcoding approach targets specific genetic markers, barcodes, to provide a taxonomic profile of the environmental community at hand. The nuclear ribosomal internal transcribed spacer (ITS) region was chosen as a universal DNA barcode for fungi (Schoch et al., 2012). Sequences in metabarcoding are typically handled in one of two main ways. In the operational taxonomic unit (OTU; Blaxter et al., 2005) approach, sequences are grouped into approximate species‐level units using sequence similarity, often at 97%–98.5%. A representative sequence of each OTU is then used for taxonomic identification. In the amplicon sequence variant (ASV; Callahan et al., 2017) approach, all unique sequences are retained and are subjected to taxonomic identification. The present study targets the taxonomic identification step and thus pertains to OTU and ASV based approaches alike. Taxonomic identification is typically accomplished by sequence similarity‐based searches against a reference corpus. These searches usually make use of threshold values, so that a sequence that is at least 0.97 (97%) similar to a reference sequence over its full length adopts the species name of the reference sequence. In a study of global soils (Tedersoo et al., 2014), the cutoffs of 0.98, 0.9, 0.85, 0.8, and 0.75 were tentatively used for species, genus, family, order, and class identification, respectively. However, different BLAST algorithms (Altschul et al., 1997) and different barcode data sets can yield different similarity cutoffs.
As more and more fungal DNA barcodes were being generated, it gradually became clear that the use of a single, static threshold value taxonomic identification was problematic. Threshold values that worked well in some parts of the fungal tree of life over‐ or underestimated species boundaries in other parts of the tree (Abarenkov et al., 2016; Vu et al., 2016, 2019). In Vu et al. (2014), we proposed a method to predict similarity cutoffs for sequence identification which was applied to two barcode data sets of yeast and filamentous fungal strains preserved in the CBS collection at the Westerdijk Fungal Biodiversity Institute. Except for species‐level predictions having a confidence measure of ~0.8, at more inclusive (higher) taxonomic levels, the prediction confidence was lower. Vu et al. (2014) thereby lent further weight to the claims of Nilsson et al. (2008) and others that using a single sequence similarity in environmental sequencing and mycology at large may serve to mask and erode significant taxonomic resolution and mycological explanatory power. Different clades will require different similarity cutoffs if resolution and explanatory power are to be maximized.
In this paper, we present dnabarcoder, a tool to help predict a global similarity cutoff for taxonomic identification of sequences in a barcode data set as well as local similarity cutoffs for different clades of the data set. It also contains other components for the analysis, visualization, and classification of barcode data to decide the best similarity cutoffs for fungal sequence identification. For a similarity cutoff in a clade, a confidence measure is computed to evaluate the resolving power of the barcode in that clade. For the evaluation, dnabarcoder was used to predict similarity cutoffs of the filamentous fungal CBS ITS barcode data set of Vu et al. (2019) and to classify the general FASTA release of the UNITE database (Abarenkov et al., 2020; Nilsson et al., 2019). These comparisons were done against the barcode data set with the predicted similarity cutoffs and against the traditional cutoffs used in metabarcoding studies (Tedersoo et al., 2014). We also studied and compared the similarity cutoffs and resolving powers of the complete ITS region, the ITS1 spacer, the 5.8S gene, and the ITS2 spacer. This would contribute significantly to the metabarcoding community as ITS2 is the main biomarker for sequence identification of the environmental samples. As for a real‐life application of dnabarcoder, we reclassified the global soil metabarcoding data set (Tedersoo et al., 2014) against the CBS ITS barcodes with the predicted ITS2 similarity cutoffs to estimate taxon diversity and community structure of the global soil samples based on a curated culture collection.
2. MATERIALS AND METHODS
Dnabarcoder consists of five components namely analysis, visualization, prediction, classification, and verification (Figure 1). The components analysis, visualization, and prediction are designed to analyse and predict similarity cutoffs for a reference data set for sequence identification. This reference data set should come in the form of a FASTA file (Pearson & Lipman, 1988) and should contain barcode sequences from as many relevant species as possible. An auxiliary file must contain their full taxonomic classification in a tab‐delimited way (kingdom, phylum, class, and so on). The classification component is used to classify unidentified sequences (DNA barcodes, ASVs, or OTUs) provided in a FASTA file against the reference data set with the predicted similarity cutoffs, while the verification component verifies the classification results. These components are described below. For every function in a component of dnabarcoder, a figure is generated automatically to aid in the interpretation of the results.
FIGURE 1.
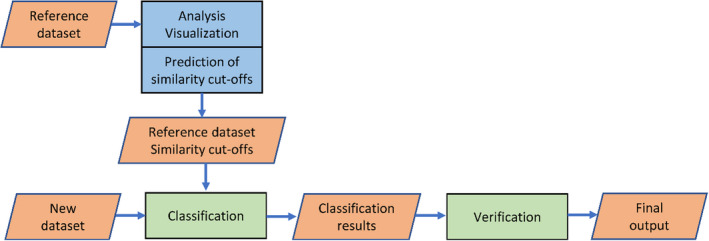
Flow chart of dnabarcoder. The rectangles in blue and green represent the components of dnabarcoder while the parallelograms in orange represent the input and output of those components. The components in blue analyse and predict local similarity cutoffs for a reference data set. The components in green classify a new data set against the reference data set with the predicted similarity cutoffs and verify the results
2.1. Analysis and visualization
The analysis component of dnabarcoder seeks to examine the length, similarity variation, distribution, and taxonomic classification of the sequences of the data set at different taxonomic levels. Sequences are compared using BLAST (Altschul et al., 1997). We used BLAST percent identity as a similarity measure because it is more intuitive for the researchers in the DNA barcoding and metabarcoding community to evaluate how similar two DNA sequences are. In addition, it was shown in Vu et al. (2014) that using Blast percent identity could achieve even a higher confidence measure than using BLAST E‐value when clustering a data set of amidohydrolases protein sequences. A similarity score of two DNA sequences is calculated as the percentage of matches s if the BLAST alignment length l is greater than a given minimum length m. Otherwise, it is recomputed as s*l/m. This is to avoid the problem that a sequence comes out as similar to every other sequence due to its short length. Analysing sequence lengths is important to decide the minimum BLAST alignment length m for computing the similarity score of two DNA sequences. The similarity variation is computed as the minimum and median similarity scores for groups of sequences of the same taxon name at all taxonomic levels (species, genus, family, order, class, and phylum). The similarity variation and distribution of the sequences based on taxa are visualized using Matplotlib (https://matplotlib.org/) while the taxonomic classification is visualized using Krona (Ondov et al., 2011).
The visualization component of dnabarcoder seeks to visualize 2D/3D “embeddings” of the sequences based on DNA sequence comparisons using Matplotlib. The sequences can also be visualized in an interactive web browser using DiVE (Vu et al., 2018) that allows the users to colour the data points based on taxa, zoom in on a group of interest, or filter the data points using the advanced search functionality. Sequences' coordinates in the sequence space are computed based on similarity scores using LargeVis (Tang et al., 2016). Together with the similarity variation, distribution, and taxonomic classification of the sequences, visualization helps the user to examine whether the data set is imbalanced and evaluate the predicted similarity cutoffs and classification results.
2.2. Prediction
The prediction component of dnabarcoder was designed to predict global and local similarity cutoffs for sequence identification based on taxonomic classification ranks. The method used for predicting a global similarity cutoff for a data set was proposed in Vu et al. (2014, 2018) and applied to predict a similarity cutoff for yeasts and filamentous fungi in Vu et al. (2016, 2019). At all taxonomic levels in turn, sequences are clustered with different thresholds. For a threshold, a confidence measure (the F‐measure, Paccanaro et al., 2006) is computed to evaluate the clustering result when comparing it with the clustering based on the taxon names of the sequences. This measure has been widely used in clustering approaches and its formula is described as follows:
Given a set of sequences and taxonomic level, let T = (T 1, …, T m ) be the groups of the sequences based on taxon names, and let G = (G 1, …, G k ) be the groups of the sequences obtained by clustering orthologous sequences. The confidence—F‐measure function F(G,T)—is defined as follows:
where n is the number of the sequences, is the number of sequences in , is the number of sequences in T j , and is the number of sequences in for 1 ≤ i ≤ k and 1 ≤ j ≤ m.
The value of F (G,T) runs between 0 and 1. The higher the value of the confidence (F‐measure), the closer is the grouping of sequences by sequence similarity to the grouping of the sequences based on taxon names. The global similarity cutoff is the threshold that has the highest confidence for sequence identification. For dnabarcoder, the connected components algorithm is used for clustering as it was shown to be accurate in Vu et al. (2014, 2018).
In the ideal situation, groups of sequences with the same taxon name are equally distant from each other (Figure 2a), and therefore, the predicted similarity cutoff has a high confidence measure that is close to 1. In reality, the distribution of fungal barcode sequences is not equal. In some clades (e.g., parts of Fusarium), the groups are closer to each other, while in the other clades (e.g., much of Agaricales), the groups are more distant (Figure 2b). Specifically, in some clades, the sequences are distributed widely (like the red ones in Figure 2b). It was shown in Vu et al. (2016, 2019) that except for the predicted similarity cutoff at the species level having a confidence measure of ~0.8, at higher taxonomic levels, the prediction confidence was lower—at about ~0.6—which was explained by the currently imbalanced fungal taxonomic classification.
FIGURE 2.
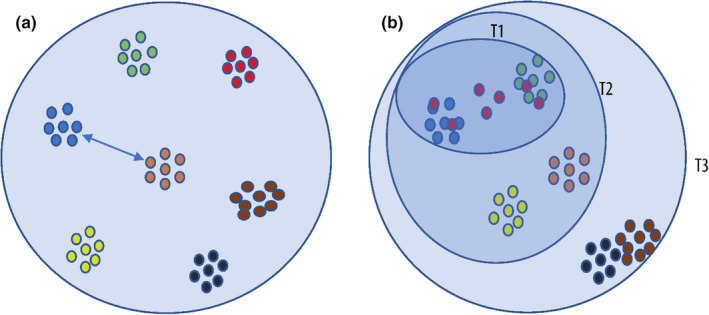
Global similarity cutoffs (a) versus local similarity cutoffs (b). Every small filled (coloured) circle represents a sequence. Sequences with the same taxon name are in the same colour at a taxonomic level l. Figure 2a illustrates the ideal situation that when the groups of sequences of the same taxon name are distant from each other, then this distance (represented by the double arrow) can be predicted with a high confidence measure. Figure 2b Illustrates the distribution of fungal sequences in a more authentic scenario when the distribution of the sequences is not equal. The groups in dark blue and dark brown are very close to each other (bottom right) and would require a high similarity cutoff while the remaining group would require a lower similarity cutoff. T1, T2, and T3 are taxa at the l + 1, l + 2, and l + 3 levels, respectively. The shaded ellipses represent clades of sequences with the same taxon name T1, T2, and T3. The similarity cutoff predicted to separate the groups in clade T1 would have a low confidence measure as the sequences in red are distributed widely. The best similarity cutoff to separate the groups in the clade T1 is the similarity cutoff having the highest confidence measure among the similarity cutoffs predicted for T1, T2, and T3
To overcome this problem, this study proposes the use of local similarity cutoffs for different clades of the data set instead of using only one global similarity cutoff for sequence identification. Suppose that we want to predict a similarity cutoff s to assign a sequence to a taxon name T at the taxonomic level l in Figure 2b. Let T1, T2, and T3 be the higher taxa of T in increasing order, and clades T1, T2, and T3 contain the sequences having the same taxon name T1, T2, and T3, respectively. Let s1, s2, and s3 be the local similarity cutoffs predicted for sequence identification at the taxonomic level l for clades T1, T2, and T3 with a confidence measure of f1, f2, and f3, respectively. Then the best local similarity cutoff s is the similarity cutoff among s1, s2, and s3 that has the highest confidence measure. The reason to consider also clades T2 and T3 into the prediction is to have an optimal solution for clades with some groups that cannot be identified by the current barcodes, or groups that are in need of reclassification (such as the red group in Figure 2b).
Formally, let T be a taxon name at a taxonomic level l, and let T 1, …, T m be the higher taxa of T in increasing taxonomic order. Let s 1, …, s m be the local similarity cutoffs predicted for sequence identification at level l of the clades T 1, …, T m with confidence measures of f 1, …, f m , respectively. The best similarity cutoff for sequence identification at level l of clade T i is s k with i ≤ k ≤ m where f k = max(f i , …, f m ). The best similarity cutoff to assign a sequence to the taxon name T is the best similarity cutoff predicted for sequence identification at level l of clade T 1.
The users of dnabarcoder can select a taxonomic level to predict similarity cutoffs for sequence identification at that level. If higher taxonomic levels are not given, the global cutoff for the whole data set is predicted. Otherwise, the best similarity cutoffs predicted for all the clades of the data set of the given higher taxonomic levels are predicted. To arrive at an optimal prediction, only clades with numbers of sequences and groups greater than n and N, with n = 30 and N = 10 given as defaults, are selected. The output of this prediction is given as a JSON‐formatted file (https://www.json.org/) which can be used as input for the classification of the sequences described in the next section. These similarity cutoffs can also be used for verifying the output of other classification tools (Vu et al., 2020; Wang et al., 2007) to, for example, highlight and remove incorrect classifications.
2.3. Classification and verification
The last component of dnabarcoder was designed to classify a data set against a reference data set. Sequences are compared with the reference sequences to find their best match using BLAST. A sequence is classified to the taxon name of its best match if the obtained similarity score to the best match is greater than or equal to the similarity cutoff predicted for that taxon name. The similarity cutoffs can be predicted by dnabarcoder or provided by the users. If other classification tools (Vu et al., 2020; Wang et al., 2007) are employed, the similarity cutoffs are used to verify the classification results to remove incorrect classifications. The accuracy and precision of classification are computed using Scikit‐learn (Pedregosa et al., 2011).
To further verify the classification results, users can compute multiple sequence alignments and infer phylogenetic trees of the sequences with the reference sequences of the predicted taxon names. A sequence is considered verified if there are at least two reference sequences having the same predicted taxon name, and the branch length of the sequence in the associated phylogenetic tree is shorter than or equal to the maximum branch length of the reference sequences. Clustal‐Omega (Sievers et al., 2011) and IQ‐tree (Nguyen et al., 2015) are used to compute multiple sequence alignments and infer phylogenetic trees, respectively. The phylogenetic analysis is intended as an internal verification step and should not be used to infer phylogenetic trees for publication.
2.4. Materials
2.4.1. The CBSITS barcode data set
We used the CBSITS data set (Vu et al., 2019) representing 40% of the cultured filamentous fungi of the CBS collection preserved at the WI as a reference data set for the evaluation of dnabarcoder. The whole ITS sequences were generated in a DNA barcoding project (Vu et al., 2012) using the forward and backward ITS5 and ITS4 primers, containing partial 18S, complete ITS1‐5.8S‐ITS2, and partial LSU sequences (Stielow et al., 2015). They were manually checked and curated by the experts at the WI and deposited to GenBank under the BioProject number PRJNA422523 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA422523). The taxonomic classifications of the CBSITS sequences were downloaded from the WI‐CBS collection and MycoBank (Robert et al., 2013) in October 2021 and are given in the Supporting Information file MBclassification.xlsx. To study the similarity cutoffs and resolving powers of different ITS regions, complete ITS, ITS1, ITS2, and 5.8S sequences were extracted from the CBSITS data set using ITSx version 1.1.1 (http://microbiology.se/software/itsx). The obtained data sets were labelled as CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S, respectively.
2.4.2. The UNITE data set
The UNITE data set consisting of sequences of the UNITE general FASTA release (Abarenkov et al., 2020) was classified against the CBSITS data set for the evaluation of dnabarcoder. To obtain a fair evaluation, sequences of the CBSITS data set that were also present in the UNITE data set were removed from the latter, and all sequences were updated with the current names from MycoBank to reduce the impact of synonyms.
2.4.3. The global soil data set
As for an application of dnabarcoder, we reclassified the global soil data set of Tedersoo et al. (2014) consisting of 50,589 of nonsingleton OTUs against the CBSITS2 data set with the predicted ITS2 similarity cutoffs to study the taxon diversity and community of the global soil samples based on a culture collection. The global soil samples were collected from 365 sites of 11 biomes Arctic tundra (AT), Grassland and shrubland (GS), Dry tropical forests (DTF), Mediterranean (MED), Boreal forests (BF), Tropical montane forests (TMF), Savannas (SAV), Southern temperate forests (STF), Temperate coniferous forests (TCF), Temperate deciduous forests (TDF), and Moist tropical forests (MTF) across the world. The sequences were obtained as OTU representatives with a threshold of 0.98, and classified to the genus, family, order, and class levels based on the reference sequences of the UNITE + INSDC data set available at the time of the study with given thresholds of 0.9, 0.85, 0.8, and 0.75, respectively (Tedersoo et al., 2014). To optimize the accuracy of taxonomic identification, we extracted only the ITS2 regions from the global soil data set using ITSx. Out of the initial 50,589 sequences, 42,626 remained.
Table 1 shows the sequence and group numbers of all data sets at each taxonomic level (species, genus, family, order, and class).
TABLE 1.
Sequence and group numbers at each taxonomic level (species, genus, family, order, and class) of the CBS ITS, CBSITScomplete, CBSITS1, CBSITS2, CBS5.8S, UNITE, and global soil data sets
| Data set | Seq. no | Species level | Genus level | Family level | Order level | Class level | Phylum level | |
|---|---|---|---|---|---|---|---|---|
| CBSITS | 11,715 | Seq. no. | 11,714 | 11,694 | 11,037 | 11,278 | 11,496 | 11,676 |
| Group no. | 5846 | 1658 | 412 | 134 | 36 | 10 | ||
| CBSITS complete | 7965 | Seq. no. | 7965 | 7908 | 7432 | 7656 | 7785 | 7895 |
| Group no. | 4294 | 1334 | 353 | 115 | 31 | 8 | ||
| CBSITS1 | 11,680 | Seq. no. | 11,680 | 11,615 | 10,899 | 11,195 | 11,405 | 11,596 |
| Group no. | 6069 | 1644 | 399 | 130 | 36 | 10 | ||
| CBSITS2 | 11,674 | Seq. no. | 11,674 | 11,609 | 10,897 | 11,190 | 11,400 | 11,590 |
| Group no. | 6064 | 1639 | 398 | 130 | 36 | 10 | ||
| CBS5.8S | 11,643 | Seq. no. | 11,643 | 11,578 | 10,866 | 11,159 | 11,369 | 11,559 |
| Group no. | 6053 | 1638 | 398 | 130 | 36 | 10 | ||
| UNITE | 47,214 | Seq. no | 26,500 | 36,505 | 40,746 | 43,755 | 44,592 | 45,358 |
| Group no. | 19,067 | 3743 | 829 | 292 | 82 | 20 | ||
| Global soil | 42,626 | Seq. no | 1864 | 14,258 | 21,687 | 33,006 | 39,375 | 39,376 |
| Group no. | 1397 | 984 | 293 | 150 | 72 | 19 |
2.5. Implementation
Dnabarcoder was implemented in Python (version 3.7) using Blastn (version 2.6.0). Matplotlib was used to interpret the results of all functions in dnabarcoder. For taxonomic classification, Krona was downloaded from https://github.com/marbl/Krona/wiki. For the visualization of sequences based on similarity scores, LargeVis (https://github.com/rugantio/LargeVis‐python3) and DiVE (https://nlesc.github.io/DiVE) were installed. We used the scikit‐learn library (https://scikit‐learn.org/) to compute the accuracy and precision of the classification. For verification, Clustal‐Omega v. 1.2.4 and IQ‐tree v. 1.6.1 were employed. The source code and manual of dnabarcoder are available at https://github.com/vuthuyduong/dnabarcoder. Dnabarcoder is licensed under the Apache Licence version 2.0.
3. RESULTS
We evaluated dnabarcoder through (1) analysing and predicting global and local similarity cutoffs for the reference CBSITS data set (Vu et al., 2019); (2) classifying the UNITE data set against the CBSITS data set with the predicted cutoffs and the traditional cutoffs used in metabarcoding studies; (3) comparing the obtained accuracies and precisions for the evaluation; (4) computing and comparing similarity cutoffs and resolving powers of full‐length ITS, ITS1, and ITS2; and (5) as an application of dnabarcoder, we reclassified the global soil data set against the CBSITS2 data set using the predicted ITS2 similarity cutoffs to estimate taxon diversity and community structure of the global soil samples.
3.1. Predicting similarity cutoffs for the filamentous fungal CBS ITS barcodes
3.1.1. Analysis and visualization of the CBS ITS data set
The taxonomic classification of the CBSITS data set figure is given in the supplementary file CBSITS.krona.html. The distribution of the sequences at all taxonomic levels are given in Figure S1, showing that the taxonomic classification of the CBSITS data set was imbalanced as the five largest groups at the genus, family, order, and class levels contained more than 17, 30, 49, and 89% of the sequences, respectively.
The CBSITS sequence lengths varied from 300–5600 bases. However, a majority of the ITS barcodes had lengths in the 400–800 base interval (Figure S2). In particular, 99.5% of the barcodes had a sequence length greater than 400 bases. Thus, when comparing the sequences of the data set with BLAST, the minimum alignment length was set to 400.
Figure 3 shows that the minimum and median similarity scores within the groups of the CBSITS data set at all taxonomic levels varied significantly, ranging from 0.4–1 at the species level and from 0–1 at the higher taxonomic levels. However, the median values of these scores were high. At the species and genus levels, they were 1. At the family, order, and class levels, they were (0.8936 and 0.9496), (0.8332 and 0.9212), and (0.3727 and 0.8343), respectively.
FIGURE 3.
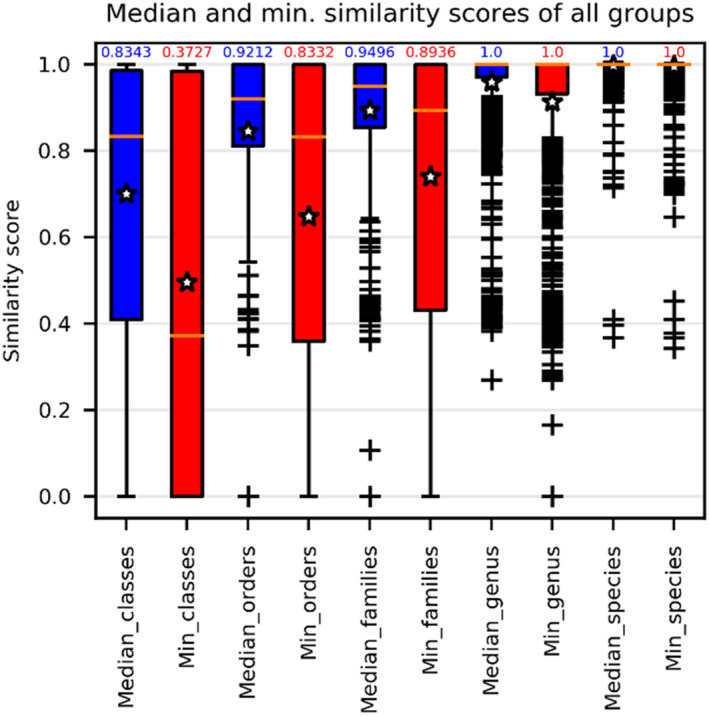
The median (in blue) and minimum (in red) similarity scores computed for all groups of the reference filamentous fungal CBS ITS barcode data set at different taxonomic levels. The numbers above the bars are the median values of the associated similarity scores. The stars indicate the average values, whereas the horizontal lines indicate the median values of these similarity scores
The visualization of the sequences based on similarity scores is given in Figure 4 in which sequences of the same taxonomic class have the same colour. Although the taxonomic classes were distinct from each other in the figure, some classes were closer to each other than the rest. These results suggest that it is unrealistic to expect any single cutoff threshold value to work equally well across the full fungal kingdom
FIGURE 4.
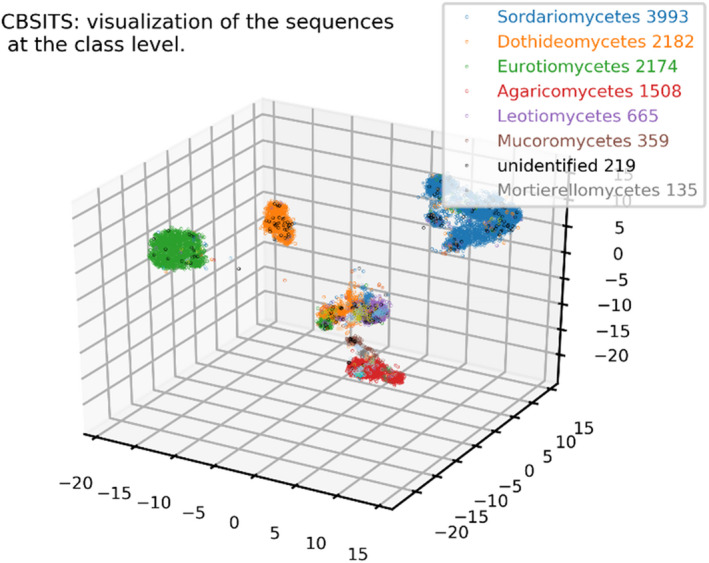
The visualization of the sequences of the reference filamentous fungal CBS ITS barcode data set based on DNA sequence comparisons. Sequence comparisons were done using BLAST (Altschul et al., 1997). Based on the obtained similarity matrix, sequence coordinates were computed using LargeVis (Tang et al., 2016) and visualized using the mplot3d toolkit of matplotlib. The numbers on the axes are the tick values of the axes. Sequences were coloured based on class name. The unidentified sequences were coloured in black. The number of classes to display was set to 8. Note that sequences can also be visualized with DiVE, an interactive web‐based visualization component of fMLC (Vu et al., 2018)
3.1.2. Prediction of global and local similarity cutoffs for the CBSITS data set
Figure 5 shows the global similarity cutoffs for sequence identification of the CBSITS barcode data set at all taxonomic levels. The sequences were clustered at thresholds ranging from 0.9 to 1 at the species level and from 0.7 to 1 at higher taxonomic levels, with a step size of 0.001. For each threshold, a confidence measure (F‐measure, Paccanaro et al., 2006) was computed to evaluate the clustering result. Note that for the prediction at the species level, we removed 1373 sequences of (623, 10.65%) indistinguishable species (distinct species with identical ITS sequences; Abarenkov et al., 2016) that came out in the same group when clustering the barcode data set with a 100% similarity score, to reduce the impact caused by these distinct species with identical ITS sequences on the prediction. The global similarity cutoffs predicted at the species, genus, family, order, and class level were 0.994, 0.955, 0.936, 0.922, and 0.922, respectively. Except for the species level that had a confidence measure of 0.83, at the higher taxonomic levels, the obtained confidences were low (<0.66) which was also observed in Vu et al. (2019).
FIGURE 5.
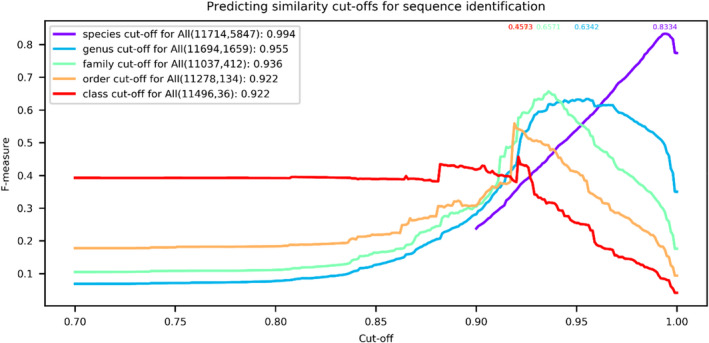
The prediction of global similarity cutoffs for sequence identification at different taxonomic levels for the reference filamentous fungal CBS ITS barcode data set. The number of sequences and groups at the associated taxonomic level are given in parentheses. The global similarity cutoffs predicted for sequence identification at the associated taxonomic level are given to the right of the parentheses. The numbers above the curves are the highest confidence measures obtained for the global similarity cutoffs
The local and best similarity cutoffs predicted for all clades of the CBSITS data set at all different taxonomic levels are given in Table S1. They are also given in the Supporting Information file CBSITS.cutoffs.json. Note that only the clades with more than 30 sequences and 10 groups were selected for the prediction. Figure 6 and Table S1 show that the local similarity cutoffs varied significantly, viz. between 0.927–0.999 for the species level, 0.83–0.99 for the genus level, and 0.83–0.936 for the family level. The corresponding median values were 0.993, 0.935, and 0.895, respectively. For species identification, the confidence measures obtained for the predicted similarity cutoffs were high, between 0.704–1 with a median value of 0.8956. Specifically, genera such as Scytinostroma, Arthroderma, Sarocladium, Exophiala, Ramularia, Diaporthe, Epichloe, Vararia, Microascus, Hypomyces, Acremonium, Sporothrix, Peniophora, Rhizoctonia, Ophiostoma, Mycena, Pseudocercospora, and Mucor had small proportions of indistinguishable species (<6%) and high confidence measures (>0.9), indicating that species in these genera were sequence‐wise distinct from each other. For genus identification, the confidence values of the predicted similarity cutoffs varied between 0.5576–0.9563 with a median value of 0.7489. The family Agaricaceae had a very high confidence measure of 0.9563 for predicting a similarity cutoff of 0.893 for genus identification. For family identification, the confidence values of the predicted similarity cutoffs varied between 0.611–0.8256, with a median value of 0.7345.
FIGURE 6.
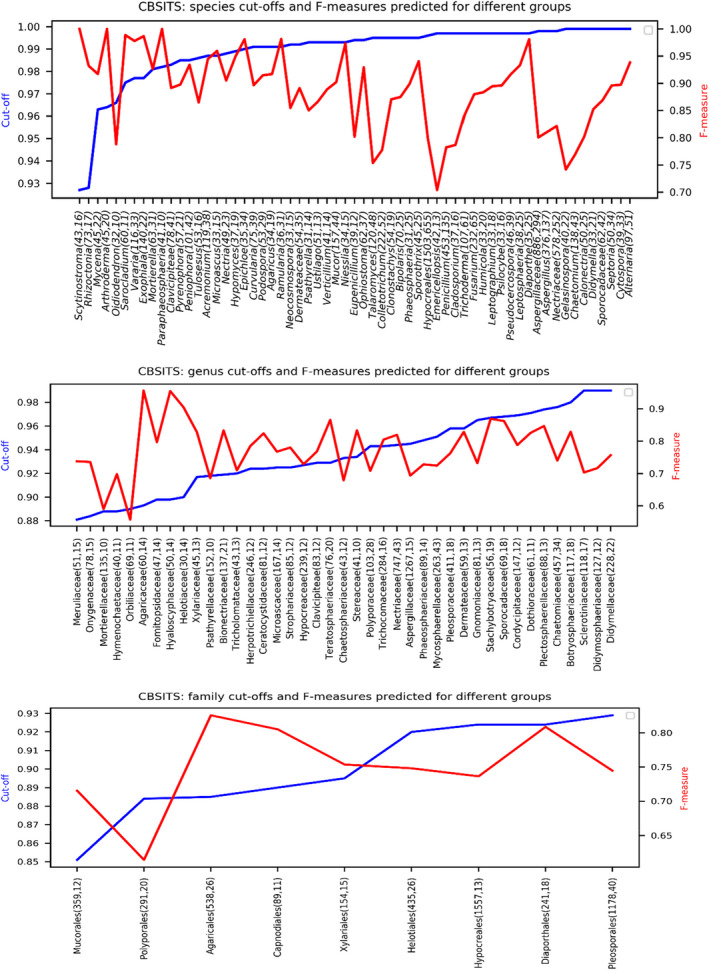
The prediction of local similarity cutoffs for species, genus, and family identification in the genera, families, and orders of the reference filamentous fungal CBSITS data set, respectively. The numbers in parentheses are the numbers of sequences and groups of the associated clades. Only clades with more than 30 sequences and 10 groups were included in the prediction
For order‐level identification, the similarity cutoffs predicted for Sordariomycetes, Agaricomycetes, Dothideomycetes, Ascomycota, and Basidiomycota were 0.922, 0.869, 0.927, 0.922, and 0.869. The confidence values were 0.61, 0.5861, 0.673, 0.5957, and 0.61, respectively. For class identification, the similarity cutoffs predicted for Ascomycota was 0.922 with a low confidence measure of 0.497, while for Basidiomycota, the similarity cutoff was 0.675 with a high confidence measure of 0.9614.
Most of the taxa (239/280, 85.36%) had a confidence measure greater than the global confidence measure predicted for the whole data set except for some genera such as Aspergillus, Calonectria, Colletotrichum, Chaetomium, Penicillium, and Talaromyces, and their higher classifications at the species level. This could be explained by the fact that these genera contained multiple subclades and species complexes that are indistinguishable by ITS (Abarenkov et al., 2016).
A total of 209/280 (74.64%) taxa had a similarity cutoff equal to the best similarity cutoff (with the maximum confidence measure). When considering only the taxa at the taxonomic level l + 1 for predicting similarity cutoffs for sequence identification at the taxonomic level l, only 28/110 (25.45%) taxa had a similarity cutoff different from the best similarity cutoff (see Table S2). Among them, except for the two families Mortierellaceae and Orbiliaceae that had a low confidence measure of 0.5905 and 0.5576 for predicting sequence identification at the genus level, the other taxa had a similarity cutoff less than 0.05 different from the best similarity cutoff. Predicting the best similarity cutoffs for the clades of very large data sets would be computationally very expensive, and our results suggest that using the similarity cutoffs predicted for the clades of the taxa at the immediately higher taxonomic level would be a suitable alternative.
3.2. Accuracy and precision of the classification of the UNITE data set against the CBSITS data set
We classified the UNITE data set against the reference CBSITS data set using the global similarity cutoff predicted for the whole barcode data set, the best local similarity cutoffs predicted for different clades of the data set, and the traditional similarity cutoffs used in metabarcoding studies. The traditional similarity cutoffs for sequences identification at the taxonomic genus, family, order, and class level were taken from Tedersoo et al. (2014) which were 0.9, 0.85, 0.8, and 0.75, respectively. At the species level, we employed the similarity cutoffs of 0.97, 0.975, 0.98, 0.985, 0.99, and 0.995 proposed by UNITE for the species hypotheses (Koljalg et al., 2013). The obtained accuracy and precision values were compared for the evaluation of dnabarcoder.
Table 2 shows the number of sequences (A_seqno), accuracy (A_accuracy), and precision (A_precision) obtained by classifying the UNITE data set against the CBSITS data set. When considering only UNITE sequences whose taxon name was represented by at least one sequence in the CBSITS data set, these values were designated B_seqno, B_accuracy, and B_precision, respectively. Table 2 shows that the global similarity cutoffs had the highest accuracies and precisions obtained at most taxonomic levels but assigned the least numbers of sequences. The traditional cutoffs assigned much more sequences than the predicted cutoffs in most cases. However, the obtained accuracies and precisions were 7.08%–8.3% and 16.45%–20.39% lower than the ones obtained by the local cutoffs at the genus and higher taxonomic levels. At the species level, the accuracies and precisions obtained by the similarity cutoffs ranging from 0.97 to 0.99 were 3.35%–12.94% and 4.02%–18.35% lower than the ones obtained by the local cutoff. The accuracy and precision obtained by the similarity cutoff of 0.995 were 0.32 and 1.54% higher than the accuracy and precision obtained by the local similarity cutoff. However, 696 fewer sequences were assigned this way. Compared with the global cutoffs, the local similarity cutoffs assigned 1–24% more sequences with slightly lower accuracies and precisions (up to 2.81 and 5.6%).
TABLE 2.
Accuracy and precision of classifying the UNITE general release data set against the reference filamentous fungal CBSITS barcode data set
| Level | Cutoff | A_seqno | A_accuracy | A_precision | B_seqno | B_accuracy | B_precision |
|---|---|---|---|---|---|---|---|
| Species | 0.97 | 8529 | 0.5224 | 0.401 | 4738 | 0.7526 | 0.7953 |
| 0.975 | 7726 | 0.5441 | 0.4334 | 4583 | 0.7556 | 0.8026 | |
| 0.98 | 7005 | 0.5633 | 0.4627 | 4401 | 0.7569 | 0.8073 | |
| 0.985 | 6385 | 0.594 | 0.5 | 4217 | 0.7631 | 0.8182 | |
| 0.99 | 5795 | 0.6183 | 0.5443 | 4019 | 0.7621 | 0.8187 | |
| 0.995 | 4864 | 0.655 | 0.5999 | 3592 | 0.7706 | 0.8274 | |
| 0.994 | 5089 | 0.6488 | 0.59 | 3720 | 0.7688 | 0.8279 | |
| best | 5560 | 0.6518 | 0.5845 | 3892 | 0.7749 | 0.8285 | |
| Genus | 0.9 | 18,576 | 0.745 | 0.4377 | 13,453 | 0.8231 | 0.7072 |
| 0.955 | 10,785 | 0.8561 | 0.6578 | 8924 | 0.8842 | 0.7952 | |
| best | 13,408 | 0.828 | 0.6022 | 10,833 | 0.8661 | 0.7726 | |
| Family | 0.85 | 24,693 | 0.8219 | 0.5513 | 20,739 | 0.8658 | 0.7775 |
| 0.936 | 12,778 | 0.9081 | 0.7658 | 11,513 | 0.9164 | 0.8429 | |
| best | 15,205 | 0.8927 | 0.7289 | 13,518 | 0.9061 | 0.8289 | |
| Order | 0.8 | 31,044 | 0.8732 | 0.5565 | 27,850 | 0.9202 | 0.8037 |
| 0.922 | 14,815 | 0.9491 | 0.776 | 14,043 | 0.9505 | 0.8405 | |
| best | 18,809 | 0.9494 | 0.7604 | 17,971 | 0.9506 | 0.8296 | |
| Class | 0.75 | 33,252 | 0.9038 | 0.5941 | 31,853 | 0.9057 | 0.7798 |
| 0.922 | 15,135 | 0.9689 | 0.8344 | 14,545 | 0.9691 | 0.8876 | |
| best | 26,345 | 0.9774 | 0.7918 | 25,634 | 0.9776 | 0.8641 |
Notes: Similarity cutoffs for traditional (regular font), global (italics), and best local (bold) are shown for each taxonomic level. Columns prefixed with a refer to the number, accuracy, and precision of the classified sequences. Columns prefixed with B refer to the number, accuracy, and precision of the classified sequences whose taxon name was represented by at least one sequence in the CBSITS data set.
Overall, the obtained precisions were low. They increased significantly when considering only sequences that were represented by sequences with the same taxon name in the barcode data set. This shows that in addition to the complication that some sequences were annotated incorrectly (Bensch et al., 2020; Hofstetter et al., 2019), many species names were not updated but rather represented legacy concepts, and it was also apparent that some species complexes were indistinguishable by the ITS region. The identification of other sequences suffered from the lack of available reference sequences. This highlights the necessity for publicly available, authenticated reference sequences such as those provided by the NCBI RefSeq Targeted Loci Project (Schoch et al., 2014).
The classification of the UNITE data set up to the class level using the predicted best cutoffs is given in the Supporting Information file UNITE.CBSITS_BLAST.krona.html file. The numbers of classified and unclassified sequences at each taxonomic level are given in Table 3.
TABLE 3.
Number of classified and unclassified sequences of the UNITE data set against the CBS ITS data set with the best local cutoffs at each taxonomic level
| Level | Number of classified sequences | Number of unclassified sequences |
|---|---|---|
| Species | 5560 (11.78%) | 41,654 (88.22%) |
| Genus | 13,408 (28.4%) | 33,806 (71.6%) |
| Family | 15,205 (32.2%) | 32,009 (67.8%) |
| Order | 18,809 (39.84%) | 28,405 (60.16%) |
| Class | 26,345 (55.8%) | 20,869 (44.2%) |
To further verify the classification result of the sequences at the species level, multiple sequence alignments and phylogenetic trees were computed for the classified sequences with the reference sequences of the same taxon names. There were 2408 (43.32%) sequences without multiple sequence alignments because the number of relevant reference sequences was less than 3. A total of 2541 (45.71%) sequences were verified as their branch length in the associated phylogenetic tree was less than the maximum branch length of the reference sequences. The number of sequences with a branch length greater than the maximum branch length of the reference sequences was 610 (10.97%). The verifications based on multiple sequence alignments and phylogenetic trees of the classification results at the genus and higher taxonomic levels were not made due to time restraints.
3.3. The similarity cutoffs and resolving powers of complete ITS, ITS1, and ITS2
3.3.1. Analysis and visualization of the CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S data sets
The sequence lengths of the CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S data sets varied from 353–939, 5–779, 5–689, and 119–159 bases in which 99.5, 99.957, 99.82, and 99.92% of the sequences had a length of more than 400, 50, 50, and 150 bases, respectively (Figure S3). When comparing the sequences, the minimum BLAST alignment lengths of the CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S data sets were set to 400, 50, 50, and 150 bases, respectively.
Similar to CBSITS, the minimum and median similarity scores within the groups of CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S at all taxonomic levels varied significantly (Figure S4), between 0.34– and 0.39–1 for CBSITScomplete at the species level and between 0–1 for the other data sets and at the higher taxonomic levels, suggesting a wide range for a (complete ITS, ITS1, 5.8S, and ITS2) similarity cutoff for fungal identification at all taxonomic levels. The distributions of the sequences of the data sets based on BLAST similarity scores are given in Figure S5. Again, it was shown that it is unrealistic to expect any single cutoff threshold value for the complete ITS region, the ITS1 spacer, the 5.8S gene, and the ITS2 spacer to work equally well across the full fungal kingdom.
3.3.2. Global and local similarity cutoffs of the CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S data sets
Figure 7 shows the prediction of the global similarity cutoffs at each taxonomic level of the CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S. The predicted global similarity cutoffs and their associated confidence measures are given in Table 4. Note that for the prediction at the species level, 1058 (13.28%), 2833 (24.25%), 3030 (25.9%), and 4969 (42.7%) sequences of 573 (13.34%), 1554 (25.6%), 1599 (26.36%), and 2526 (41.73%) indistinguishable species were removed from the CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S data sets, respectively.
FIGURE 7.
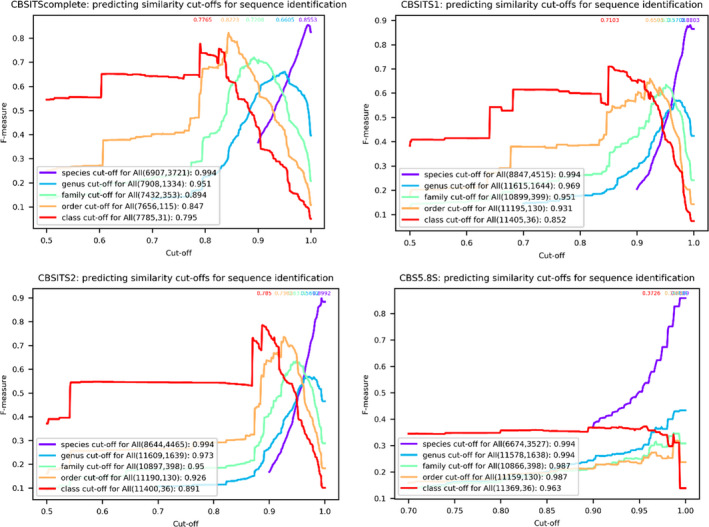
The prediction of global similarity cutoffs for sequence identification at different taxonomic levels for the reference CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S data sets. The number of sequences and groups at the associated taxonomic level are given in parentheses. The global similarity cutoffs predicted for sequence identification at the associated taxonomic level are given to the right of the parentheses. The numbers above the curves are the highest confidence measures obtained for the global similarity cutoffs
TABLE 4.
Global similarity cutoffs and confidence measures predicted for the CBSITScomplete, CBSITS1, CBSITS2, and CBS5.8S data sets
| Level | Prediction | CBSITScomplete | CBSITS1 | CBSITS2 | CBS5.8S |
|---|---|---|---|---|---|
| Species | Global cutoff | 0.994 | 0.994 | 0.994 | 0.994 |
| confidence | 0.8553 | 0.8803 | 0.8992 | 0.8589 | |
| Genus | Global cutoff | 0.951 | 0.969 | 0.973 | 0.994 |
| confidence | 0.6605 | 0.5708 | 0.5692 | 0.434 | |
| Family | Global cutoff | 0.894 | 0.951 | 0.95 | 0.987 |
| confidence | 0.7208 | 0.6347 | 0.6374 | 0.3484 | |
| Order | Global cutoff | 0.847 | 0.931 | 0.926 | 0.987 |
| confidence | 0.8223 | 0.6595 | 0.7363 | 0.3264 | |
| Class | Global cutoff | 0.795 | 0.852 | 0.891 | 0.987 |
| confidence | 0.7765 | 0.7103 | 0.785 | 0.3726 |
Similar to the ITS barcodes (partial 18S, complete ITS, partial LSU), the confidence measures obtained for species identification by complete ITS, ITS1, and ITS2 were high (>0.8553). CBSITS2 had the highest global confidence measure of 0.8947, followed by CBSITS1 (0.8873), CBSITScomplete (0.8553), and CBS5.8S (0.8168). At the genus and higher taxonomic levels, they were low, in particular for ITS1 and ITS2 at the genus and family levels, and for 5.8S at all taxonomic levels. The CBSITScomplete data set had the highest confidence measures for genus, family, order, and class identification, suggesting that it might be better to use the complete ITS region for sequence identification at higher taxonomic levels. The high number of indistinguishable species by the 5.8S gene and the low confidence measures obtained indicate that the 5.8S gene is not useful as a biomarker for fungal identification at any taxonomic level.
Tables S3–S5 show the similarity cutoffs and the associated confidence measures obtained for the clades of the CBSITScomplete, CBSITS1, and CBSITS2 data sets. They are also given in the Supporting Information files CBSITScomplete.cutoffs.json, CBSITS1.cutoffs.json, and CBSITS2.cutoffs.json.
The similarity cutoffs and associated confidence measures obtained by complete ITS, ITS1, and ITS2 at all taxonomic levels also varied significantly between different clades of the data sets (see Table 5). Most of the taxa (86.19% for complete ITS, 84.56% for complete ITS1, and 78.06% for complete ITS2) had a local confidence measure higher than the global confidence measure predicted for the whole data sets. In addition, the median local confidence measures obtained at all taxonomic levels were significantly higher than the global confidence measure, suggesting that it is better to use local similarity cutoffs rather than using one single similarity cutoff for fungal identification.
TABLE 5.
Local similarity cutoffs and confidence measures predicted for the reference CBSITScomplete, CBSITS1, and CBSITS2 data sets
| Level | Prediction | CBSITScomplete | CBSITS1 | CBSITS2 |
|---|---|---|---|---|
| Species | Cutoffs | 0.906–0.999 | 0.931–0.996 | 0.919–0.996 |
| Median cutoff | 0.991 | 0.988 | 0.99 | |
| Confidences | 0.788–1 | 0.7855–1 | 0.8165–1 | |
| Median confidence | 0.9165 | 0.9233 | 0.9185 | |
| Genus | Cutoff | 0.825–0.987 | 0.7–0.993 | 0.621–0.995 |
| Median cutoff | 0.939 | 0.958 | 0.959 | |
| Confidence | 0.5913–0.9725 | 0.504–0.9326 | 0.5115–0.9512 | |
| Median confidence | 0.7735 | 0.6835 | 0.6795 | |
| Family | Cutoff | 0.825–0.92 | 0.702–0.966 | 0.502–0.966 |
| Median cutoff | 0.8845 | 0.9255 | 0.9245 | |
| Confidence | 0.6592–0.8325 | 0.5408–0.9266 | 0.5772–0.9266 | |
| Median confidence | 0.7509 | 0.6813 | 0.6688 | |
| Order | Cutoff in Sordariomycetes | 0.848 | 0.946 | 0.925 |
| Confidence in Sordariomycetes | 0.8887 | 0.7166 | 0.8136 | |
| Cutoff in Agaricomycetes | 0.837 | 0.86 | 0.891 | |
| Confidence in Agaricomycetes | 0.7925 | 0.6877 | 0.6264 | |
| Cutoff in Dothideomycetes | 0.84 | 0.802 | 0.913 | |
| Confidence in Dothideomycetes | 0.8399 | 0.806 | 0.819 | |
| Cutoff in Ascomycota | 0.848 | 0.931 | 0.926 | |
| Confidence in Ascomycota | 0.8405 | 0.6827 | 0.7893 | |
| Cutoff in Basidiomycota | 0.837 | 0.86 | 0.891 | |
| Confidence in Basidiomycota | 0.8015 | 0.7039 | 0.6478 | |
| Class | Cutoff in Ascomycota | 0.831 | 0.852 | 0.891 |
| Confidence in Ascomycota | 0.8252 | 0.7485 | 0.8108 | |
| Cutoff in Basidiomycota | 0.501 | 0.482 | ||
| Confidence in Basidiomycota | 0.9448 | 0.9552 |
Notes: Only clades with more than 30 sequences and 10 groups were included in the prediction.
Overall, the local confidence measures obtained for species identification were higher than 0.85 except for some genera and their higher classifications such as Colletotrichum, Penicillium, Talaromyces, Aspergillus, and Fusarium for ITS complete, Oidiodendron, Talaromyces, and Colletotrichum for ITS1, and Talaromyces, Microascus, and Coprinellus for ITS2. At the higher more inclusive taxonomic levels, local confidence measures were getting lower, in particular for ITS1 and ITS2 at the genus and family levels (with a median value of less than 0.7).
When comparing the resolving powers of the complete ITS with the ITS barcodes (partial 18S, complete ITS, partial LSU), out of 202 taxa having both predictions, 161 (79.7%) taxa had a confidence measure obtained by complete ITS higher than the confidence measure obtained by the ITS barcodes, suggesting that it might be better to extract the ITS region from the ITS barcodes to have an optimal identification.
When comparing the resolving powers of the complete ITS with ITS1 (ITS2) at the genus and higher taxonomic levels, out of 94 taxa that had both ITS and ITS1 (ITS2) predictions, 93.62% of the taxa had an ITS confidence measure higher than the ITS1 (ITS2) confidence measure. At the species level, the resolving powers of the complete ITS and ITS1 (ITS2) were about the same. Out of 118 (119) taxa that had both ITS and ITS1 (ITS2) predictions, 54.2% (53.8%) of the taxa had a confidence measure obtained by ITS greater than the confidence measure obtained by ITS1 (ITS2).
When comparing ITS1 and ITS2, their resolving powers were about the same. Out of 252 taxa having both predictions, 126 taxa had a higher ITS1 confidence measure and 127 taxa had a higher ITS2 confidence measure. For species identification, ITS1 was more divergent (25.6% indistinguishable species) than ITS2 (26.36% indistinguishable species). ITS1 had either a significantly lower percentage of indistinguishable species and/or higher confidence measure in genera such as Agaricus, Alternaria, Aspergillus, Bipolaris, Chaetomium, Penicillium, Sarocladium, and Talaromyces and their higher classifications, whereas ITS2 performed better for clades such as Mortierella, Ophiostoma, Humicola, Cordycipitaceae, Peniophoraceae, and Exobasidiomycetes.
3.4. Diversity and community structure of the global soil samples based on the CBS collection
3.4.1. Taxonomic classification of the soil data set
Out of 42,626 OTUs of the soil data set, a total of 24,659 (57.85%) OTUs were classified from species to class rank using the reference filamentous fungal CBSITS2 data set with the ITS2 similarity cutoffs predicted in the previous section. The classification result is given in the Supporting Information file globalsoil.xlsx and visualized in the Supporting Information file globalsoil.CBSITS2_BLAST.krona.html in which 1493 (3.5%), 5908 (13.86%), 8078 (18.95%), 10,908 (25.59%), and 24,451 (57.36%) OTUs were classified at the species, genus, family, order, and class level, respectively including 1146 (2.67%), 1686 (3.96%), 1649 (3.89%), 742 (1.74%), and 112 (0.26%) newly identified sequences (Figure 8). Among the 347, 4222, 6429, 10,166, and 24,339 OTUs successfully identified by both UNITE+INSDC and CBS data sets at the species, genus, family, order, and class level, respectively, 122, 2007, 4123, 8485, and 22,298 had the same name. For OTUs classified with different names, 195 (86%), 1532 (69%), 1571 (68%), 1143 (67%), and 957 (47%) were updated with a new name as their similarity scores to the best matches were higher than the scores obtained previously in Tedersoo et al. (2014), thus showing a significant improvement for the classification of the OTUs of the global soil samples based on the CBS collection.
FIGURE 8.
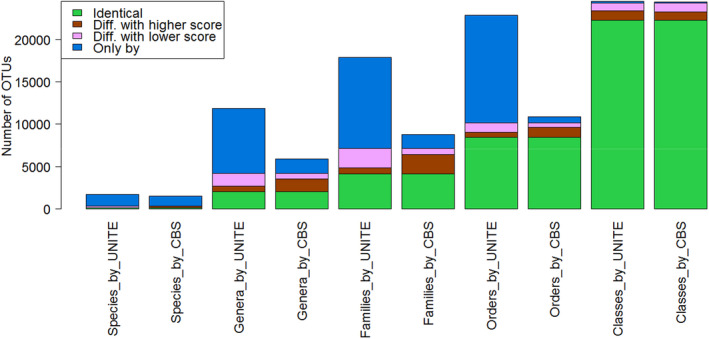
Comparisons of the OTUs identifications of the soil samples studied in Tedersoo et al. (2014) by the UNITE+INSDC and CBS data sets. The green colour shows the number of OTUs that were identified with the same name by both data sets. The brown colour shows the number of OTUs that were identified by the associated data set with a different name and higher score than the other data set. The pink colour shows the number of OTUs that were identified by the associated data set with a different name and lower score than the other data set. The blue colour shows the number of the OTUs that were only identified by the associated data set
3.4.2. Taxon diversity and community structure
To avoid the problem of suboptimal identification due to the lack of CBSITS2 reference sequences, all OTUs that were newly identified with a lower score were removed. In the end, 23,958 OTUs were classified up to the class level using the CBSITS2 barcodes in which 1463, 5225, 7343, 10,367, and 23,367 were revealed at the species, genus, family, order, and class level, respectively. Based on the obtained classification, the taxon diversity and community structure of the global soil samples were studied. Figure 9 shows the relative proportion of the OTUs at the genus level in all and different biomes and sampling sites. The relative proportion of the OTUs at the higher taxonomic levels can be found in Figure S6. It can be seen that the relative proportion of the classified sequences at all taxonomic levels varied across different biomes. Even though they were distributed across all or most sites, the majority of them were still restricted to the type of the biome as also observed in Tedersoo et al. (2014).
FIGURE 9.
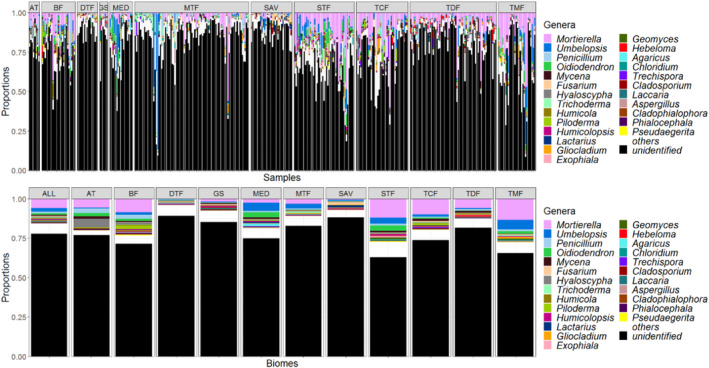
The diversity of filamentous fungi at the genus level in all biomes Arctic tundra (AT), boreal forests (BF), dry tropical forests (DTF), grassland and shrubland (GS), moist tropical forests (MTF) Mediterranean (MED), savannas (SAV), southern temperate forests (STF), temperate coniferous forests (TCF), temperate deciduous forests (TDF), and tropical montane forests (TMF) studied by Tedersoo et al. (2014). The identification of the OTUs was done based on the reference filamentous fungal CBSITS2 barcodes (Vu et al., 2019) with the predicted ITS2 similarity cutoffs
Mortierella (8%), Umbelopsis (3%), Penicillium (1.4%), and Oidiodendron (1.3%) were the four largest genera in the soil samples revealed by the CBS collection. The proportions of the remaining genera were less than 1%. Next to these four genera, Hyaloscypha was also revealed with a relatively high proportion in AT (9.33%), Piloderma in BF (3%), Fusarium in DTF (1.1%), Humicolopsis in GS (1.56%), Agaricus in MED (2%), Trichoderma in MTF (1.2%), Fusarium in SAV (2.3%), Geomyces in STF (1.47%), Mycena in TCF (1.56%), Mycena in TDF (1.6%), and Gliocladium in TMF (1.3%).
When considering only the OTUs that were newly identified or given a new name with a higher similarity score by the CBS collection, 478, 393, 55, 989, 2076, and 229 OTUs were assigned to a functional group of animal‐ and mycoparasites, animal pathogens, ectomycorrhizal fungi, plant pathogens, saprotrophs, and others, respectively, using FUNGuild (Nguyen et al., 2016), and 2281 OTUs remained unassigned. The CBS collection clearly contributed to the identification of plant pathogenic fungi in the global soil samples as the number of plant pathogenic OTUs newly assigned by the CBS collection was 35.65% of the plant pathogenic OTUs (2774) assigned based on the classification obtained previously in Tedersoo et al. (2014) (see the Supporting Information file globalsoil.xlsx).
3.5. Run‐time performance
The benchmarks were performed on a Linux x86‐64 platform of a high‐performance computing (HPC) cluster (16 GB RAM, 2 cores) at the Dutch national e‐infrastructure SURFsara. To predict a global similarity cutoff, dnabarcoder needed ~78 min at the species level and ~ 138 min at the genus and higher taxonomic levels. It took dnabarcoder 2, 4, 10, 22, and 48 min to predict local similarity cutoffs for species identification for the genera, families, orders, classes, and phyla of the data set, respectively. For genus identification, it took 10, 21, 43, and 97 min to predict similarity cutoffs for the families, orders, classes, and phyla of the data set. For family identification, it took 19, 38, and 79 min to predict similarity cutoffs for the orders, classes, and phyla of the data set. For order identification, it took 39 and 82 min to predict similarity cutoffs for the classes and phyla of the data set. Finally, it took 85 min to predict similarity cutoffs for class identification in Ascomycota and Basidiomycota. For the classification of the UNITE and global soil data sets, it took dnabarcoder ~35 minutes. For the verification, it took dnabarcoder ~176 min to verify 5421 classification results at the species level.
4. DISCUSSION
The accuracy and prediction of fungal sequence identification have recently been addressed in Vu et al. (2019) and Lücking et al. (2020, 2021). A number of strategies were suggested in Lücking et al. (2020) to improve the accuracy and precision of fungal sequence identification, including implementing secondary DNA barcodes for groups where ITS does not provide sufficient precision and using multiple sequence alignments based phylogenetic approaches as more accurate alternatives. These suggestions are currently impractical for metabarcoding studies, given the large amount of data containing up to millions of sequences from environmental samples to be analysed. The ITS region will probably remain the marker of choice for fungal metabarcoding studies for the foreseeable future.
Computational methods for classifying sequences have primarily been developed for bacteria, after which they were adopted for fungi. However, the verification of the classification results of these methods was often neglected (Lücking et al., 2020). In our recent study in Vu et al. (2020), we compared four different classification methods for fungal classification including BLAST, the Ribosomal Database Project (RDP) Bayesian classifier (Wang et al., 2007), and two deep learning‐based classifiers, viz. a convolutional neural network (CNN, LeCun et al., 2015) and a deep belief network (DBN, Hinton & Salakhutdinov, 2006). We found that when classifying a data set whose sequences were not present in the training data set, BLAST was the tool that performed the best in terms of taxonomic identification.
Up to this point, in most metabarcoding studies, a single similarity cutoff has been used for sequence identification, and its accuracy and precision were typically not assessed in those studies. To the best of our knowledge, dnabarcoder is the first tool that allows the users to study extensively local similarity cutoffs for sequence identification for different clades of fungi. For a predicted similarity cutoff for sequence identification in a clade, a confidence measure is computed. This confidence measure helps the user to evaluate the resolving power of the DNA marker in that clade. Our results showed that the similarity cutoffs predicted for the clades of the filamentous fungal ITS barcode data set of Vu et al. (2019) varied significantly, and most of the taxa had a prediction confidence measure significantly higher than the confidence predicted for the whole data set, indicating that it is better to use different similarity cutoffs predicted for different fungal clades rather than using one single similarity cutoff for fungal identification. When comparing the predicted local similarity cutoffs with the traditional similarity cutoffs used in metabarcoding studies, the local similarity cutoffs assigned fewer sequences of the UNITE general FASTA release data set. However, the obtained accuracy and precision values were significantly higher. We also showed that the resolving powers of the complete ITS, ITS1, and ITS2 were similar for fungal species identification. At higher taxonomic levels, the complete ITS region had a better resolving power than ITS1 and ITS2. Using dnabarcoder, we were able to show that the CBS collection clearly improved fungal identification in the global soil samples collected in Tedersoo et al. (2014), in particular for plant pathogenic fungi.
Dnabarcoder allows the users to assess the classification results based on multiple sequence alignments and phylogenetic trees. These advanced analyses are available for the classification results with more than two reference sequences present in the reference data set. In addition, it is more practical for small data sets as it took almost three hours to verify the classification results of ~5500 sequences.
We would like to emphasize that dnabarcoder can be used to predict similarity cut‐offs for any biomarker. Although dnabarcoder was initially developed for fungi, it is applicable to any other organisms where DNA barcodes are used for identification. This study used the full fungal ITS region, the ITS1 spacer, the 5.8S gene, and the ITS2 spacer of the CBS collection. It would be relevant to study similarity cutoffs for other fungal groups that were not present in the CBS barcode data sets, for the groups where alternative DNA barcodes are used, and for other similarity search‐based programs such as DIAMOND (Buchfink et al., 2021) and mmseqs2 (Mirdita et al., 2021).
Metabarcoding seeks to capture authentic patterns and processes in nature—in other words, to get as close to biological reality as possible (Burian et al., 2021). However, the present study suggests that the field of metabarcoding loses significant resolution and scientific explanatory power by relying on a single sequence similarity threshold value for taxonomic assignment. We introduce a software tool that seeks to reflect actual biological patterns more accurately. While our tool clearly is not free from shortcomings and complications, it still represents a significant improvement over singular, static threshold value‐solutions for taxonomic assignments. Our work emphasizes the problems of using similarity‐based operational taxonomic units (OTUs; Blaxter et al., 2005) in metabarcoding studies. After all, if sequence variation is averaged away already in an initial OTU clustering step, then that would clearly cap the full potential of our tool in the subsequent sequence identification step. In a metabarcoding context, the full power of the approach presented in the present study will be unleashed only in association with the use of amplicon sequence variants (ASVs; Callahan et al., 2017).
Appropriate use of amplicon sequence variants is tightly coupled to the existence of richly populated and well‐annotated reference sequence databases (Callahan et al., 2017). Mycology, sadly, does not enjoy such databases. A few percent of the estimated millions of extant fungal species have been described, and type specimen‐derived DNA sequence data are available for less than 10% of the number of described species. Clearly, the heavily funded field of metabarcoding relies on fields and undertakings that do not enjoy the same level of funding: traditional taxonomic work, the sequencing of legacy type specimens in herbaria (Bieker et al., 2020), and the pursuit of the many novel “dark taxa” fungal lineages that are being unearthed by environmental sequencing efforts (e.g., Khan et al., 2020; Tedersoo et al., 2017). Ironically, in breaking new technological ground, the present authors find themselves pleading for the funding of traditional taxonomical endeavours.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supinfo
ACKNOWLEDGEMENTS
The DNA sequence data set used in this study was created with support by the “Fonds Economische Structuurver‐sterking (FES)”, Dutch Minister of Education, Culture and Science grant BEK/BPR‐2009/137964‐U, “Making the Tree of Life Work”, and the Royal Academy of Arts and Sciences in the Netherlands (KNAW). We would like to thank the Westerdijk Institute barcoding team for generating the DNA barcode data, and Konstanze Bensch and the Mycobank team for managing and curating all the fungal taxa. We also thank SURFsara (surf.nl/), the Dutch national e‐infrastructure, for the support in using the HPC cluster for all the experiments done in the manuscript.
Vu, D. , Nilsson, R. H. , & Verkley, G. J. M. (2022). Dnabarcoder: An open‐source software package for analysing and predicting DNA sequence similarity cutoffs for fungal sequence identification. Molecular Ecology Resources, 22, 2793–2809. 10.1111/1755-0998.13651
Handling Editor: Kin‐Ming (Clement) Tsui
DATA AVAILABILITY STATEMENT
The filamentous fungal ITS barcode data set (Vu et al., 2019) was deposited in GenBank under the BioProject number PRJNA422523 and can be downloaded via the link https://www.ncbi.nlm.nih.gov/bioproject/PRJNA422523. The general FASTA release of the UNITE database (Abarenkov et al., 2020; Nilsson et al., 2019) can be downloaded via the link https://doi.org/10.15156/BIO/786368. The global soil data set was given in Tedersoo et al. (2014). The current taxa of the sequences of both data sets, downloaded from Mycobank, are given in the Supporting Information file MBclassification.xlsx. The source code is available under the Apache Licence version 2.0 software licence at https://github.com/vuthuyduong/dnabarcoder.
REFERENCES
- Abarenkov, K. , Adams, R. I. , Laszlo, I. , Agan, A. , Ambrosio, E. , Antonelli, A. , Bahram, M. , Bengtsson‐Palme, J. , Bok, G. , Cangren, P. , & Coimbra, V. (2016). Annotating public fungal ITS sequences from the built environment according to the MIxS‐built environment standard—A report from a may 23–24, 2016 workshop (Gothenburg, Sweden). MycoKeys, 16, 1–15. [Google Scholar]
- Abarenkov, K. , Zirk, A. , Piirmann, T. , Pöhönen, R. , Ivanov, F. , Nilsson, R. H. , & Kõljalg, U. (2020). UNITE general FASTA release for fungi. UNITE Community. 10.15156/BIO/786368 [DOI] [Google Scholar]
- Altschul, S. F. , Madden, T. L. , Schäffer, A. A. , Zhang, J. , Zhang, Z. , Miller, W. , & Lipman, D. J. (1997). Gapped BLAST and PSI‐BLAST: A new generation protein database search programs. Nucleic Acids Research, 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldrian, P. , Větrovský, T. , Lepinay, C. , & Kohout, P. (2021). High‐throughput sequencing view on the magnitude of global fungal diversity. Fungal Diversity. 10.1007/s13225-021-00472-y [DOI] [Google Scholar]
- Bensch, S. , Inumaru, M. , Sato, Y. , Lee Cruz, L. , Cunningham, A. A. , Goodman, S. J. , Levin, I. I. , Parker, P. G. , Casanueva, P. , Hernández, M. A. , & Moreno‐Rueda, G. (2020). Contaminations contaminate common databases. Molecular Ecology Resources, 21, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieker, V. C. , Barreiro, F. S. , Rasmussen, J. A. , Brunier, M. , Wales, N. , & Martin, M. D. (2020). Metagenomic analysis of historical herbarium specimens reveals a postmortem microbial community. Molecular Ecology Resources, 20, 1206–1219. [DOI] [PubMed] [Google Scholar]
- Blaxter, M. , Mann, J. , Chapman, T. , Thomas, F. , Whitton, C. , Floyd, R. , & Abebe, E. (2005). Defining operational taxonomic units using DNA barcode data. Philosophical Transactions B, 360, 1935–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink, B. , Reuter, K. , & Drost, H. G. (2021). Sensitive protein alignments at tree‐of‐life scale using DIAMOND. Nature Methods, 18, 366–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burian, A. , Mauvisseau, Q. , Bulling, M. , Domisch, S. , Qian, S. , & Sweet, M. (2021). Improving the reliability of eDNA data interpretation. Molecular Ecology Resources, 21, 1422–1433. [DOI] [PubMed] [Google Scholar]
- Callahan, B. , McMurdie, P. , & Holmes, S. (2017). Exact sequence variants should replace operational taxonomic units in marker‐gene data analysis. The ISME Journal: Multidisciplinary Journal of Microbial Ecology, 11, 2639–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton, G. E. , & Salakhutdinov, R. R. (2006). Reducing the dimensionality of data with neural networks. Science, 313, 504–507. [DOI] [PubMed] [Google Scholar]
- Hofstetter, V. , Buyck, B. , Eyssartier, G. , Schnee, S. , & Gindro, K. (2019). The unbearable lightness of sequenced‐based identification. Fungal Diversity, 96, 243–284. [Google Scholar]
- Khan, F. K. , Kluting, K. , Tångrot, J. , Urbina, H. , Ammunet, T. , Eshghi Sahraei, S. , Rydén, M. , Ryberg, M. , & Rosling, A. (2020). Naming the untouchable—Environmental sequences and niche partitioning as taxonomical evidence in fungi. IMA Fungus, 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koljalg, U. , Nilsson, R. H. , Abarenkov, K. , Tedersoo, L. , Taylor, A. F. , Bahram, M. , Bates, S. T. , Bruns, T. D. , Bengtsson‐Palme, J. , Callaghan, T. M. , & Douglas, B. (2013). Towards a unified paradigm for sequence‐based identification of fungi. Molecular Ecology, 22, 5271–5277. [DOI] [PubMed] [Google Scholar]
- LeCun, Y. , Bengio, Y. , & Hinton, G. (2015). Deep learning. Nature, 521, 436–444. [DOI] [PubMed] [Google Scholar]
- Lücking, R. , Aime, M. C. , Robbertse, B. , Miller, A. N. , Aoki, T. , Ariyawansa, H. A. , Cardinali, G. , Crous, P. W. , Druzhinina, I. S. , Geiser, D. M. , & Hawksworth, D. L. (2021). Fungal taxonomy and sequence‐based nomenclature. Nature Microbiology, 6, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking, R. , Aime, M. C. , Robbertse, B. , Miller, A. N. , Ariyawansa, H. A. , Aoki, T. , Cardinali, G. , Crous, P. W. , Druzhinina, I. S. , Geiser, D. M. , & Hawksworth, D. L. (2020). Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus, 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita, M. , Steinegger, M. , Breitwieser, F. , Söding, J. , & Levy Karin, E. (2021). Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics, 37, 3029–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L. T. , Schmidt, H. A. , von Haeseler, A. , & Minh, B. Q. (2015). IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, N. H. , Song, Z. , Bates, S. T. , Branco, S. , Tedersoo, L. , Menke, J. , Schilling, J. S. , & Kennedy, P. G. (2016). FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecology, 20, 241–248. [Google Scholar]
- Nilsson, R. H. , Kristiansson, E. , Ryberg, M. , Hallenberg, N. , & Larsson, K. H. (2008). Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and ITS implications for molecular species identification. Evolutionary Bioinformatics, 4, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson, R. H. , Larsson, K. H. , Taylor, A. F. S. , Bengtsson‐Palme, J. , Jeppesen, T. S. , Schigel, D. , Kennedy, P. , Picard, K. , Glöckner, F. O. , Tedersoo, L. , & Saar, I. (2019). The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Research, 47, D259–D264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondov, B. D. , Bergman, N. H. , & Phillippy, A. M. (2011). Interactive metagenomic visualization in a web browser. BMC Bioinformatics, 12, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paccanaro, P. , Casbon, J. A. , & Saqi, M. A. (2006). Spectral clustering of proteins sequences. Nucleic Acids Research, 34, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, W. R. , & Lipman, D. J. (1988). Improved tools for biological sequence comparison. Proceedings of the National Academy of Sciences, 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa, F. , Varoquaux, G. , Gramfort, A. , Michel, V. , Thirion, B. , Grisel, O. , Blondel, M. , Prettenhofer, P. , Weiss, R. , Dubourg, V. , & Vanderplas, J. (2011). Scikit‐learn: Machine learning in python. The Journal of Machine Learning Research, 12, 2825–2830. [Google Scholar]
- Robert, V. , Vu, D. , Amor, A. B. H. , van de Wiele, N. , Brouwer, C. , Jabas, B. , Szoke, S. , Dridi, A. , Triki, M. , Daoud, S. B. , & Chouchen, O. (2013). MycoBank gearing up for new horizons. IMA Fungus, 4, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch, C. L. , Robbertse, B. , Robert, V. , Vu, D. , Cardinali, G. , Irinyi, L. , Meyer, W. , Nilsson, R. H. , Hughes, K. , Miller, A. N. , & Kirk, P. M. (2014). Finding needles in haystacks: Linking scientific names, reference specimens and molecular data for fungi. Database, 2014, bau061. 10.1093/database/bau061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch, C. L. , Seifert, K. A. , Huhndorf, S. , Robert, V. , Spouge, J. L. , Levesque, C. A. , Chen, W. , Fungal Barcoding Consortium , Fungal Barcoding Consortium Author List , Bolchacova, E. , & Voigt, K. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proceedings of the National Academy of Sciences, 109, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers, F. , Wilm, A. , Dineen, D. , Gibson, T. J. , Karplus, K. , Li, W. , Lopez, R. , McWilliam, H. , Remmert, M. , Söding, J. , & Thompson, J. D. (2011). Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal omega. Molecular Systems Biology, 11(7), 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich, E. J. , Berbee, L. M. , Blackwell, M. , Hibbett, D. S. , James, T. Y. , Spatafora, J. W. , & Taylor, J. W. (2009). The fungi. Current Biology, 19, R840–R845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stielow, J. B. , Levesque, C. A. , Seifert, K. A. , Meyer, W. , Irinyi, L. , Smits, D. , Renfurm, R. , Verkley, G. J. M. , Groenewald, M. , Chaduli, D. , & Lomascolo, A. (2015). One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia, 35, 242–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlet, P. , Coissac, E. , Pompanon, F. , Brochmann, C. , & Willerslev, E. (2012). Towards next‐generation biodiversity assessment using DNA metabarcoding. Molecular Ecology, 21, 2045–2050. [DOI] [PubMed] [Google Scholar]
- Tang, J. , Liu, J. , Zhang, M. , & Mei, Q. (2016). Visualizing large‐scale and high‐dimensional data. In WWW’16 (pp. 287–297). 10.1145/2872427.2883041 [DOI] [Google Scholar]
- Tedersoo, L. , Bahram, M. , Polme, S. , Kõljalg, U. , Yorou, N. S. , Wijesundera, R. , Ruiz, L. V. , Vasco‐Palacios, A. M. , Thu, P. Q. , Suija, A. , & Smith, M. E. (2014). Global diversity and geography of soil fungi. Science, 346, 1256688. [DOI] [PubMed] [Google Scholar]
- Tedersoo, L. , Bahram, M. , Puusepp, R. , Nilsson, R. H. , & James, T. Y. (2017). Novel soil‐inhabiting clades fill gaps in the fungal tree of life. Microbiome, 5, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu, D. , Eberhardt, U. , Szöke, S. , Groenewald, M. , & Robert, V. (2012). A laboratory information management system for DNA barcoding workflows. Integrative Biology, 4, 744–755. [DOI] [PubMed] [Google Scholar]
- Vu, D. , Georgievska, S. , Szöke, S. , Kuzniar, A. , & Robert, V. (2018). fMLC: Fast multi‐level clustering and visualization of large molecular datasets. Bioinformatics, 34, 1577–1579. [DOI] [PubMed] [Google Scholar]
- Vu, D. , Groenewald, M. , de Vries, M. , Gehrmann, T. , Stielow, B. , Eberhardt, U. , Al‐Hatmi, A. , Groenewald, J. Z. , Cardinali, G. , Houbraken, J. , & Boekhout, T. (2019). Large‐scale analysis of filamentous fungal DNA barcodes reveals thresholds for species and higher taxon delimitation. Studies in Mycology, 92, 135–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu, D. , Groenewald, M. , Szöke, S. , Cardinali, G. , Eberhardt, U. , Stielow, B. , De Vries, M. , Verkleij, G. J. , Crous, P. W. , Boekhout, T. J. , & Robert, V. (2016). DNA barcoding analysis of more than 9000 yeast isolates contributes to quantitative thresholds for yeast species and genera delimitation. Studies in Mycology, 85, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu, D. , Groenewald, M. , & Verkley, G. (2020). Convolutional neural networks improve fungal classification. Scientific Reports, 10, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu, D. , Szöke, S. , Wiwie, C. , Cardinali, G. , Röttger, R. , & Robert, V. (2014). Massive fungal biodiversity data re‐annotation with multi‐level clustering. Scientific Reports, 4, 6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo
Data Availability Statement
The filamentous fungal ITS barcode data set (Vu et al., 2019) was deposited in GenBank under the BioProject number PRJNA422523 and can be downloaded via the link https://www.ncbi.nlm.nih.gov/bioproject/PRJNA422523. The general FASTA release of the UNITE database (Abarenkov et al., 2020; Nilsson et al., 2019) can be downloaded via the link https://doi.org/10.15156/BIO/786368. The global soil data set was given in Tedersoo et al. (2014). The current taxa of the sequences of both data sets, downloaded from Mycobank, are given in the Supporting Information file MBclassification.xlsx. The source code is available under the Apache Licence version 2.0 software licence at https://github.com/vuthuyduong/dnabarcoder.