

Abstract
The possibility of performing designed transition‐metal catalyzed reactions in biological and living contexts can open unprecedented opportunities to interrogate and interfere with biology. However, the task is far from obvious, in part because of the presumed incompatibly between organometallic chemistry and complex aqueous environments. Nonetheless, in the past decade there has been a steady progress in this research area, and several transition‐metal (TM)‐catalyzed bioorthogonal and biocompatible reactions have been developed. These reactions encompass a wide range of mechanistic profiles, which are very different from those used by natural metalloenzymes. Herein we present a summary of the latest progress in the field of TM‐catalyzed bioorthogonal reactions, with a special focus on those triggered by activation of multiple carbon‐carbon bonds.
Keywords: Biocatalysis, Bioorthogonal, Chemical biology, Cell biology, Organometallic catalysis
Transition‐metal catalyzed reactions are being increasingly used in biological contexts and even in living cells and organisms. Most of the processes so far developed rely on the ability of metal complexes to bind and activate unsaturated systems in a bioorthogonal way. The reactions are often tracked by fluorescence microscopy, by using reaction probes that emit light only after the reaction.
1. Introduction
Enzymes play essential roles in living organisms, promoting the chemical reactions that are needed to sustain life. [1] A substantial part of these enzymes feature metal cofactors at their active sites and are therefore coined as metalloenzymes. In most cases, these metals are first‐row transition elements, such as Zn, Mg, Fe or Mn, which participate in the catalytic process working as Lewis acids or redox centers. [2] In synthetic organic chemistry laboratories, the range of metals used for catalytic purposes is much higher than that used by Nature, and includes precious transition metals such as Pd, Ru, Rh, Ir or Au, which, when featuring appropriate ligands, can promote a wide variety of reactions that differ from those existing in nature. The impressive catalytic reactivity of complexes based on these metals stems mainly from two factors: on one hand, they can easily cyclize between oxidation states, which is essential from a mechanistic perspective and, on the other, they can bind and activate C−C multiple bonds, a feature not often seen in natural enzymes. [3]
Typical reactions catalyzed by transition metal complexes tend to involve mechanistic steps such as oxidative addition, reductive elimination, beta‐hydride elimination and/or transmetalations, among others. An illustrative example is the rhodium‐catalyzed hydrogenation of alkenes, which involves the oxidative addition of the catalyst to a hydrogen molecule, followed by migratory insertion of the double bond and subsequent reductive elimination leading to the reduced alkene (Scheme 1). [4]
Scheme 1.

Hydrogenation of alkenes depicting common mechanistic steps in transition‐metal‐catalyzed reactions.
Translating this type of organometallic processes to the same environments in which enzymes work is challenging, but very attractive, as it could considerably expand the possibilities for interrogating or interfering with living systems. However, the task is far from easy, in part because of the complexity of the environment of a living milieu, intrinsically aggressive for many of the typical intermediates of an organometallic reaction. In addition to water, the presence of thiols, amines and other nucleophiles that can coordinate to the metal, and of redox active species (e. g. glutathione, reactive oxygen species), can significantly alter the course of a standard organometallic process. Nonetheless, in recent years there has been an increasing number of reports that demonstrate the viability of performing designer transition‐metal promoted reactions in life‐like settings. [5] In many of the cases it is not clear whether the reactions are truly catalytic (exhibiting TON), but the simple fact of performing the abiotic transformations in living contexts is of high relevance and can have a profound impact in chemical and cell biology and in biomedicine.
In this minireview we present an updated summary of some of the more relevant and recent contributions in this emerging research field, with a special attention to reactions of unsaturated substrates, due to their orthogonal metal coordination and activation possibilities.
2. Transition Metal‐Catalyzed Bioorthogonal Reactions: Pioneering Examples
Owing to the scarcity of C−C multiple bonds in natural biomolecules, it is not surprising that the initial attempts to use transition metal catalysts in biological environments involved unsaturated reactants. Indeed, one of the first examples in this type of research is the ruthenium‐catalyzed hydrogenation of C−C double bonds in fatty acids within mesophyll protoplasts, using hydrogen gas. [6] However, the conditions needed to carry out the reduction were too drastic, and an extensive cell damage was observed even after only one hour of reaction. It was decades later when the field experimented a decisive impulse with the development of the famous Copper‐catalyzed Alkyne Azide cycloaddition (CuAAC), first reported by Sharpless and by Meldal (Scheme 2). [7] The reaction, which requires terminal alkynes, is very fast, compatible with physiological buffers, and highly chemoselective, thus avoiding interference with natural processes or with biological components. Therefore, it can be classified within the family of bioorthogonal reactions, and has been widely used for biological applications, as well as to modify biomolecules and biopolymers like carbohydrates, nucleic acids, lipids or proteins, among others. [8]
Scheme 2.

Copper‐catalyzed alkyne azide cycloaddition (CuAAC).
Nonetheless, the CuAAC still presents important drawbacks for their use in living contexts, in great part because of the toxicity of the copper (I) salts, and the usual requirement of reductants like ascorbate, that are not inoffensive from the biological perspective. [9] Therefore, along the years, there have been many efforts to develop alternative reaction conditions, as well as other type of bioorthogonal reactions that involve different organometallic intermediates. A key advance in the field was the work of Meggers and coworkers on the use of ruthenium catalysts to promote deallylation reactions. [10] In their pioneering article in 2006, the authors describe the ability of a Ru(II) complex to cleave an allylcarbamate protecting group under aqueous conditions. The reaction mechanism involves coordination of the C−C double bond to the ruthenium reagent and an ensuing cleavage of the alloc group with formation of a π‐allyl ruthenium complex. The ruthenium (II) catalyst is regenerated thanks to a nucleophilic addition of a thiol to this π‐allyl ligand. More importantly, the authors proved that the reaction can be carried out in the presence of HeLa cells using a non‐fluorescent caged rhodamine probe that recovers the fluorescence upon release of the alloc protecting group. This piece of research, which went relatively unnoticed for several years, along with the development of water compatible chemoselective methods to modify biopolymers, [11] is considered key in the emergence of this new area of research at the interface between chemistry and biology, dealing with the use of transition metal catalysts in living systems.
3. Reactions Initiated by the Activation of Alkynes
3.1. Cycloadditions
As mentioned above, one of the most widely used bioorthogonal reactions triggered by a transition metal catalyst is the CuAAC. The reaction is a metal‐catalyzed version of the thermal, dipolar cycloaddition between azides and alkynes reported by Huisgen in the 60s. [12] In presence of copper, these annulations take place via an organometallic mechanism, instead of through a classical concerted cycloaddition. Very importantly, the metal‐promoted version allows for control of the regiochemistry of the reaction. The currently accepted mechanism entails the formation of a Cu(I)‐acetylide species coordinated to a second copper complex, which in turn reacts with the azide leading to a dinuclear metalacyclic intermediate. A reductive elimination followed by protodemetalation then releases the triazole and the copper catalyst. [13]
The high rates, chemo and regioselectivity of the CuAAC, and the ease for attaching different type of functional groups to the reactants, has prompted its wide use in different biological applications, including bioconjugations, imaging, cell membrane modification, etc. as well as in material sciences. [8e] However, its use inside living cells or in vivo has been hampered by the toxicity of the copper, and by the relatively easy deactivation of the copper (I) reagent, due, in part, to its redox lability. [9] The use of external reductants is usually mandatory, but they can also affect the normal functioning of the cell. For instance, ascorbate can be readily converted into dehydroascorbate, which is highly reactive. [14]
One way to protect the copper (I) catalyst from deactivation, and enhance its orthogonal catalytic effectivity, consists of using azides containing chelating groups that can trap the catalyst and drive the desired cycloaddition. This approach was pioneered by Ting and coworkers, who designed an azide partner featuring a neighboring pyridine [15] (Scheme 3). Although effective for enhancing the orthogonality as well as the reaction rate, the strategy compromises the turnover as the product can sequester the copper complex.
Scheme 3.

Pyridine directed CuAAC.
A more frequent approach to protect the Cu(I) complex while maintaining the reactivity consists of using bulky tristriazole‐like ligands (Scheme 4). The resulting complexes were used by Tirrell et al. to label the surface of mammalian cells by metabolically introducing azide moieties, which can participate in a CuAAC with a terminal alkyne bearing a fluorophore or an affinity tag like biotin [8d] (Scheme 4A). A related strategy was used by the group of Mascareñas to perform reactions inside mammalian cells, albeit with modest efficiencies. The group demonstrated that playing with this type of ligands it is possible to tune cell permeability, reactivity and side toxicity of the complexes, and even avoid the need for sodium ascorbate [16] (Scheme 4B).
Scheme 4.

Copper‐tristriazole complexes for CuAAC.
In recent years, there have been many efforts to develop alternatives to the CuAAC based on the use of other transition metals. A significant advance in this matter was the demonstration that thioalkynes undergo a relatively fast annulation with azides, in aqueous and complex biological buffers, under Ru(II) catalysis. [17] The reaction was also found to be orthogonal to the classic CuAAC, allowing to achieve mutually orthogonal transformations of substrates featuring both, alkynes and thioalkynes. In a more recent development, it has also been shown that it is possible to introduce an external control in the process, in the form of light (Scheme 5), by using a photoactivable ruthenium catalyst. [18] These reactions can be performed in the presence of HeLa cells, however it is likely that they are mostly taking place in the extracellular space.
Scheme 5.

Photoactivable ruthenium catalyst for thioalkyne‐azide coupling.
While alkynes (or thioalkynes) provide just one coordinating moiety for the metal, substrates exhibiting two alkyne pendants were reasoned to be good reactants for bioorthogonal metal‐promoted processes, owing to their bidentate coordination pattern and ensuing reactivity. Indeed, in 2020 the group of Mascareñas demonstrated that tailored diynes can engage in ruthenium‐catalyzed (2+2+2) formal cycloadditions with external alkynes, in aqueous buffers and even in the presence of cell lysates. While there was room for improvement in terms of yields, the increase of complexity achieved in these reactions is very notable. [19] More importantly, the authors showed that this multicomponent process can be performed inside mammalian cells. Remarkably, some the products resulting from the annulation of designed precursors do not internalize in cells, and tend to aggregate in the extracellular space, however, they can be generated inside the cells using the cycloaddition. These results highlight one of the potentials of the metal‐promoted reactions in biological chemistry, the generation of intracellular products that otherwise would not be possible to deliver to the cells (Scheme 6).
Scheme 6.

Ruthenium‐catalyzed (2+2+2) cycloaddition in live cells.
3.2. Cycloisomerizations
The ability of alkynes to partake in carbo‐ and hydrofunctionalization reactions, including cycloisomerizations, is well known and has been widely exploited in synthetic chemistry. Among the different metals that can catalyze these reactions, gold complexes have been especially attractive owing to their carbophilicity. [20] The tendency of gold salts to bind to alkynes has also been used for performing reactions in biological environments, initially in the context of developing gold sensors. For instance, Tae and coworkers used a gold induced cycloisomerization reaction of profluorogenic precursors to detect the presence of gold salts in cellular media [21] (Scheme 7). In their system, they use a caged rhodamine as reacting probe, whose fluorescence is switched off because the molecule is locked in a closed state [21a] (Scheme 7A). However the addition of Au(III) salts promotes a cyclization that unveils the fluorophore. A related approach entailing a cyclization to pro‐coumarin skeletons was reported by Kim and coworkers in 2010. [21b] The groups of Ahn and Patra[ 21c , 21d ] used a cyclization of 2‐ethynylbenzoic acid derivatives to isochromene derivatives with the concomitant release of fluorescent products to detect the presence of gold salts (Scheme 7B).
Scheme 7.

Bioorthogonal gold‐promoted cyclizations.
Other than in biosensing, gold catalysis has also been used to enable designed transformations in biological contexts. For instance, Tanaka and coworkers used a gold‐promoted activation of alkynes to unmask amines. [22] Another remarkable example on the application of gold catalysis in biological habitats was published by Mascareñas and coworkers, and entails an intramolecular hydroarylation reaction to form fluorescent aminocoumarins. [23] (Scheme 7C). The reaction was promoted by LAuCl type of complexes (specifically Au1), without the need to use chloride scavenging additives, as water can do the role as activating/solvating agent.
Importantly, the reaction works smoothly inside HeLa cells, without generating major side toxicity during the first hours. In this work, the authors were able to combine two different abiotic reactions promoted by either gold or ruthenium complexes, that can occur simultaneously inside the cells, opening the door to generating reaction circuits like the ones seen in living organisms. This area will likely receive much attention in the years to come.
In 2021, Tanaka and coworkers described an elegant intramolecular hydroamination of alkynes to yield the bioactive 5‐methylphenatrinium skeletons using gold (I) carbene reagents. [24] To protect the catalyst from the environment, and avoid its deactivation, the authors buried it within the protein HSA, using a albumin‐coumarin interaction (Scheme 8). [25] Under these conditions, the gold complex was significantly less toxic, and more active that in its free state. They also showed how the 5‐methylphenatrinium product leads to a decrease in the growth of cancer cells. These results highlight one of the more appealing avenues for applying this catalytic chemistry in the field of biomedicine: the controlled generation of drugs from inactive precursors.
Scheme 8.

HSA‐gold metalloenzyme for the generation of 5‐methylphenantrenium cores.
3.3. Bond cleavage reactions
Since the seminal work of Meggers and coworkers mentioned above, [10] uncaging reactions promoted by transition metal catalysts have gained a lot of attraction. Indeed, most biological applications of organometallic catalysis involve the removal of protecting groups by some type of bond‐cleaving reaction. [26]
Palladium is one more profusely used metals for this type of reactions. This is somewhat surprising, as palladium reagents can be readily deactivated under the stringent conditions of a biological milieu. However, the tuneability and enormous transformative potential of palladium‐based reagents is very attractive. Therefore, in 2014, the group of Chen, reported a depropargylation reaction catalyzed by homogeneous palladium catalysts. [27] In their work, the authors describe the use of a variety of palladium salts and complexes with oxidation states ranging from (0) to (IV) to perform deallylation and depropargylation processes. However, they didn′t establish a clear relationship between the structure or oxidation state of the palladium catalysts and the reactivity. Importantly, the authors demonstrated the utility of this uncaging for controlling the activity of an enzyme. In particular, they modified the lysine 134 of a phosphorthreonine lyase using biosynthetic procedures to incorporate a propargyloxycarbonil (Poc)‐capped lysine, observing that this modified protein lacked activity (Scheme 9A). The addition of suitable palladium salts promoted the depropargylation and allowed to recover the enzymatic activity. Although the efficiency of the catalytic processes is probably low, the method proved useful for biological interrogation.
Scheme 9.

Palladium‐catalyzed depropargylations in cells and in vivo. (Dppf)=diphenylphosphinoferrocoene.
Related depropargylations of designed probes were also described by the group of Bradley, using a Pd‐carbene catalyst tethered to a cell penetrating peptide which facilitated the internalization of the metal complex. [28]
As commented above, homogeneous Pd reagents are not particular efficient catalysts in living contexts, however, by choosing appropriate ligands it is possible to obtain better results. In fact, tailored phosphine ligands allow to improve the catalytic efficiency of deallylation and depropargylation reactions in living mammalian cells [29] (Scheme 9B). The incorporation of the phosphine ligands also allows to modulate other properties, like targeting and transport.
Using specifically designed substrates, palladium‐catalyzed decaging reactions can also be used for theranostic applications, as demonstrated by Wu et al. with a prodrug featuring a caged coumarin fused to nitrogen mustard (NCI). [30] Palladium‐catalyzed depropargylation of the coumarin precursor triggers the release of NCI, obtaining both a fluorescent molecule and a bioactive drug. Upon loading both components (catalyst and precursor) in different liposomes, it is possible to target them specifically to tumors within live mice observing both fluorescence and reduction in the tumor growth (Scheme 9C).
The above examples use small‐sized palladium complexes as catalytic reagents. However, it is possible to use catalysts with a larger size. Indeed, a pallado‐miniprotein, which is competent for depropagylation reactions in live mammalian cells has recently been disclosed to work in intracellular settings. [31] By equipping a bZIP basic region peptide with two histidine residues at i and i+4 positions, it is possible to pinch Pd(II) salts so that the resulting peptide becomes more helicoidal (Scheme 10A). This conformational change facilitates the cellular penetration of the stapled peptide, which can now work as an intracellular catalytic reagent to promote the depropargylation of appropriately designed fluorogenic probes (Scheme 10B). [32] The bis‐histidine chelating strategy proved to be quite versatile and could be integrated in a peptide containing an Arginine‐Glycine‐Asparagine (RGD) targeting group, which allows for preferential internalization in cell lines presenting high levels of integrin receptors. [33]
Scheme 10.

Palladopeptide catalyzed uncaging of alkyne‐containing molecules.
Using this work as reference, the group of Zou developed a way to deliver gold complexes into living cells. In their report, the authors leveraged the ability of gold to transmetalate to palladium and release an active gold catalyst from a gold acetylide precursor. The resulting gold chloride can be used as a catalyst to carry out the cyclization of phenyl propiolates. Moreover, by fusing a palladopeptide with the tripeptide RGD targeting motif, [34] the authors were able to deliver the cytotoxic gold complex to cancer cell lines overexpressing integrin receptors (Scheme 10C).
Designed depropargylation reactions can also be performed by copper complexes, as demonstrated by Chen in 2019, using Cu(I)BTAA as catalytic reagent, a reagent which had been originally developed for CuAAC. [35] By using this methodology, the authors were able to deliver the cytotoxic drug etoposide at the membrane of cancer cells through tethering it to an affibody that binds with high affinity to the HER2 receptor of the SKBR‐3 cancer cell line (Scheme 11A).
Scheme 11.

Cleavage of substituted alkynes. (Cod)=cyclooctadiene.
The use of internal propargylic thioethers as dimerizing linkers can allow the release of two different bioactive molecules, as shown by Bernardes and coworkers [36] (Scheme 11B). This method is also appropriate to build drug‐antibody conjugates using stimuli responsible linkers. This feature was used for tethering doxorubicin (Dox) to anti‐HER2 nanobodies, conjugates that can be directed towards the HER2 receptor of specific cancer cell lines. Then, a palladium‐catalyzed reaction allows for the controlled release of the drug in the vicinity of the target cells. Later they also showed that the activation of internal alkynes to release secondary amines could be performed by reaction with platinum salts like KPtCl4 or cisplatin. [37] They also used this technology for the release of the anticancer drug monomethylauristatin (MMAE) in the surface of HeLa cells (Scheme 11C). Moreover, the authors prove the viability of carrying out a similar cleavage reaction in a colorectal zebrafish xenograft model, for uncaging cytotoxic fluorouracil.
The group of Tanaka used the gold‐promoted reactivity of alkynes to label organs within living mice. Specifically, they used the coumarin‐BSA interaction to locate gold reagents into specific organs, which can then promote the electrophilic activation of an alkynyl ester in a Proc‐protected profluorophore, favoring an amidation reaction with nearby lysines [25] (Scheme 12). The BSA can be modified with different sugars to target either the liver or the intestine when injected into live mice. This work confirms that gold catalysis can be used within living mammals, opening the door for future developments in the field.
Scheme 12.
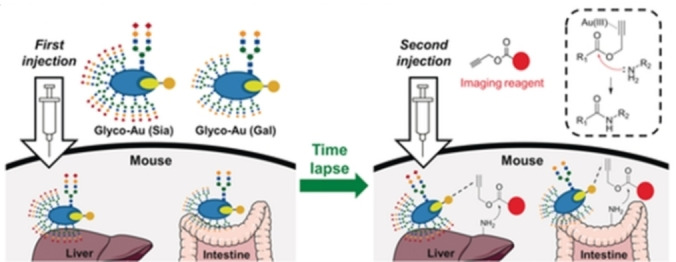
Gold‐catalyzed labelling of organs. Reproduced from ref. 25 with permission from Wiley‐VCH GmbH, © 2017.
3.4. Sonogashira coupling
Alkynes can also be used for cross‐coupling processes, especially carbon‐carbon bond forming reactions. [38] This is the case of the well‐known Sonogashira coupling, a palladium promoted reaction between terminal alkynes and haloarenes. [39] The group of Lin demonstrated that this reaction can be performed in the presence of live cells. Using an adaptation of a catalytic system developed by Davis for protein labeling, [40] they were able to carry out a copper‐free Sonogashira coupling between an alkyne‐modified ubiquitin and substituted iodoarenes to label proteins inside cells [41] (Scheme 13A). The use of this reaction was expanded by Chen and coworkers to label GFP and other proteins inside living enteric pathogens (Scheme 13B). [42]
Scheme 13.

Sonogashira couplings in live cells.
Overall, alkynes, owing to their metal coordination ability, and in some cases, the acidity of terminal derivatives, represent excellent functional groups to carry out bioorthogonal metal‐promoted reactions, even in living contexts.
4. Reactions Initiated by the Activation of Alkenes
4.1. C−O bond cleavage
Appropriately designed alkenes can also coordinate to transition metals and undergo chemoselective transformations. Most of the applications of TM‐mediated reactions of alkenyl containing precursors deal with the removal of allyl or alloc protecting groups. This is the case of the previously mentioned ruthenium‐promoted deallyloxylation developed by Meggers, that can be considered as a kickstarting step of this field of TM‐catalyzed reactions in biological habitats. [10] In their work, they demonstrated the ability of a Ru(II) complex to catalyze the cleavage of allylcarbamates in the presence of mammalian cells (Scheme 14A). Notably, the reaction required the presence of external thiols to regenerate the ruthenium catalyst. This issue was solved by the same group by developing Kitamura‐type of Ru(IV) precatalysts, [43] that showed a 60‐fold improvement in the turnover number for the in vitro reactions, and avoids the need for extra thiol addition. [44] The system was further improved in 2017 by a third‐generation catalyst that boosted both the reaction rate and the TON of the previous iterations. [45] Recently, iridium catalysts have also been shown to catalyze the removal of alloc groups. [46]
Scheme 14.

Ruthenium‐catalyzed deallyloxylation.
The group of Mascareñas developed in 2014 a Ru(II)‐catalyzed uncaging of the DNA binding agents DAPI and Ethidium Bromide. [47] These compounds when capped as allylcarbamates are distributed throughout the cell but, after the deprotection with a ruthenium (II) reagent, they recover their DNA binding ability and accumulate in the cell nuclei. The lower cytotoxicity of the protected binding agents in comparison with their uncaged counterparts means that this strategy could also be used for the controlled activation of cytotoxic agents.
Using the same type of Ru catalysts, the group of Rotello has developed a variety of deallylation platforms based on the encapsulation of catalysts in different type of gold nanoparticles. [48] This approach protects the ruthenium complex from the cellular environment and allows for controlling the activity of the resulting nanozymes by different stimuli (Scheme 14B).
Recently, the group of Tanaka developed a clever strategy to leverage the deallylation reaction for a proximity‐dependent labeling of specific cells. Using a Kitamura‐type ruthenium catalyst with a tag capable of binding to HSA, they demonstrated that the resulting hybrid can cleave the allyl group of a fluoromethylene‐bearing alloc‐protected anilide, to give a highly reactive ortho quinone methide that is trapped by nearby nucleophiles (Scheme 15). This strategy can be used to label cells with proapoptotic peptides leading to cell death. [49] The whole process mimic related strategies that traditionally rely on enzymes (such as APEX or BioID) [50] further hinting at the ability of metal catalysts to replace or complement enzymes in biological settings.
Scheme 15.

Cell tagging through Ru‐catalyzed deallylation.
The deallylation strategy has also been implemented in living organisms that require the presence of an external metal catalyst to survive (auxotrophs). In this regard, the group of Balskus used an E. Coli. strain unable to synthesize paraaminobenzoic acid (PABA), a biosynthetic precursor of folic acid. The lack of PABA renders the organism unable to grow due to a defect in the nucleotide metabolism (Scheme 16A). [51] The addition of N‐allylated PABA to the bacteria does not lead to growth of the bacteria due to the inability of the organism to metabolize that compound. However, when N‐allyl PABA is supplied along with a simple Ru(II) precatalyst, the growth was recovered, proving that the TM‐catalyzed deallylation reaction can be rendered essential to living organisms.
Scheme 16.

Deallylation as a mean for bacterial cell survival.
A similar strategy was used by the group of Mayer, devising a system consisting of an E. Coli strain that requires the non‐canonical amino acids (nCAAs) 3‐iodo‐L‐tyrosine (3iY) or 3‐nitro‐L‐tyrosine (3nY) to survive in a medium containing ampicillin. When these cells are cultivated in a medium rich in ampicillin and the alloc‐protected versions of 3iY or 3nY, they cannot survive. However, if an external Ru catalyst is added, the deprotection reaction releases the natural amino acids therefore allowing the survival of the bacteria (Scheme 16B). These systems could be used for an efficient screening of bioorthogonal reactions as well as a mean for controlling the growth of genetically modified organisms (GMOs). [52]
The possibility of manipulating the quinoline ligand in the Kitamura‐type of ruthenium precatalysts was leveraged by Mascareñas and coworkers to target the reagents to the mitochondria of living cells (Scheme 17A). [53] This strategy allowed the localized generation of bioactive compounds in that organelle, which can be advantageous over the production in the cytosol or other parts of the cell. For instance, the mitochondrial uncoupler 1,4‐dinitrophenol (DNP) needs to be added in large amounts to the cells, over 500 μM, to induce the depolarization of the mitochondria. However, when using a combination of an inactive allylDNP derivative and a mitochondria‐located ruthenium catalyst capable of promoting the deallylation reaction, the biological response is much more effective, and only 150 μM of the caged drug is needed. This localized, catalytic generation of an active product in a specific organelle represents a promising therapeutic strategy. A similar mitochondria targeting approach was used by the same group to perform palladium‐catalyzed deallylations. [29]
Scheme 17.

Applications of ruthenium catalysts with designed ligands for target‐selective deallylations.
Ward and coworkers have extensively worked on the building of artificial metalloenzymes based on the interaction between streptavidin (Sav) and biotin. One of these systems was also based on ruthenium quinoline scaffolds and used for deallylation reactions. [54] After several processes of directed evolution, the authors were able to build ruthenium‐protein complexes that present better activities than the isolated ruthenium reagents. They used the artificial metalloenzyme to catalyze the deallylation of allyloxycarbonyl‐triiodothyronine. The released triiodothyronine can then activate a gene switch leading to the production of a luciferase that acts as optical readout (Scheme 17B). A similar strategy albeit using an encapsulated HaloTag instead of streptavidin‐biotin interactions was developed by Jessen‐Trefzer, allowing the localized deallylation of fluorophores within live cells. [55]
4.2. C−C bond formation
The activation of alkenes by coordination to transition metals can also be used for other type of C−C bond‐forming reactions, like metathesis. [56] At first sight, it doesn′t seem to be a reaction that could be easily translated to biological settings, mostly due to the sensitivity of the required ruthenium complexes towards thiols. This issue has been smartly approached by the group of Ward, by designing ruthenium‐containing metalloenzymes based on the SAV‐biotin interaction, which can catalyze a metathesis reaction in the bacterial periplasm. They could track the process by running a ring‐closing metathesis that releases a fluorescent coumarin (Scheme 18A). [57] A similar metathesis reaction was also cleverly employed by the same group to carry out the uncaging of carboxylic acids, alcohols and amines. [58] The deprotection reaction proceeds through the generation of an intermediate dihydroanthracene that evolves through aromatization releasing the product (Scheme 18B).
Scheme 18.

Ruthenium metalloenzymes for metathesis.
In 2019 Tanaka leveraged his BSA‐embedding strategy to carry out a metathesis reaction inside SW620 cells. [59]
In 2017, the group of Weissleder reported the translation of another powerful metal‐catalyzed C−C bond forming transformation, the Heck reaction, into living cells. [60] To do so, they encapsulated a palladium catalyst inside a biocompatible polymer which avoids a rapid deactivation of the catalyst under the stressing atmosphere of a living cell. This strategy allowed to synthesize a fluorescent aminocoumarin through an intramolecular Heck reaction, as indicated in Scheme 19. In this case, the success of the reaction heavily relies on the protective environment of the polymeric nanostructure.
Scheme 19.

Palladium‐catalyzed Heck reaction inside live cells.
4.3. Isomerization
In 2019 the group of Mascareñas demonstrated the viability of performing ruthenium‐catalyzed isomerizations of allylic alcohols to ethyl ketones under biologically relevant conditions and even in live mammalian cells (Scheme 20). [61] The reaction mechanism involves ruthenium hydride intermediates, that are able of surviving, at least in part, inside the stringent milieu of a living cell. To track the reaction, the authors synthesize an amine‐bearing naphthalene equipped with an allylic alcohol that, when isomerized to the corresponding ketone, provides a push‐pull fluorescent product (Scheme 20A). Using a designed diallylic alcohol precursor, the reaction can also be used to promote the formation of alpha‐beta unsaturated products which are known glutathione(GSH)‐depleting agents (Scheme 20B).
Scheme 20.

Ruthenium‐catalyzed isomerization in living cells.
5. Reactions Initiated by the Activation of Allenes
Allenes are very versatile motifs for TM‐catalyzed reactions due to their intrinsic strain and improved coordination abilities to metal complexes. [62] However, there has not been significant progress in the translation of reactions involving allenes into biological settings. The only example so far published was developed by Chen and coworkers, and consists of the palladium triggered decaging of allenyltyrosines (Scheme 21). This reaction can be used for chemical rescuing of caged tyrosines relevant for protein function. [63] This feature is demonstrated by the expression of non‐functional caged proteins through genetic code expansion, which, upon treatment with a deallenylation palladium catalyst, can recover their activity. Specifically, they expressed a Src Kinase, with its phosphorylation site blocked by an allenyl motif. The protein function is recovered after palladium‐catalyzed decaging.
Scheme 21.

Palladium‐catalyzed decaging of a tyrosine exposing the OH motif to kinase activity.
6. Other Reactions
Although the main focus of this review consists in transformations that involve some form of activation of C−C multiple bonds, there have been also significant developments in other type of TM‐catalyzed bioorthogonal reactions.
The most studied reaction in this category involves the reduction of azides, usually under iron catalysis (Scheme 22), [64] although other reduction methods, such as those based on ruthenium‐based photocatalysts, have also been successfully used. [65] The cellular application of this reaction was first described by the group of Meggers and collaborators in 2012 by using an iron porphyrin system (Scheme 22A), [64a] which seemed to be capable of driving the process, harnessing the reducing properties of the interior of the cells.
Scheme 22.

Bioorthogonal iron‐catalyzed azide reduction.
Other examples include the cleavage of a self‐immolative group after reduction of the azide, which then releases a fluorophore or active drug, as seen in an example by Rotello and coworkers. [64b] In this report, the authors use the aforementioned iron catalyst in a polymeric network to uncage antibiotics in bacterial biofilms (Scheme 22B). Despite the many applications of this method, the interpretation of results can be difficulted by the fact that the reducing environment of cells may trigger the reduction of the azide even in the absence of the metal catalyst.[ 64a , 65 ]
The Suzuki‐Miyaura cross coupling has also been translated into bacterial cells by the group of Davis (Scheme 23). Using a water‐soluble palladium catalytic system, they were able to carry out the coupling between a p‐iodophenylalanine‐containing transmembrane protein and a fluorescent boronic acid in the surface of E. Coli. [66]
Scheme 23.

Suzuki‐Miyaura coupling in the surface of E. Coli.
The carbonyl reduction is a textbook reaction that can transform aldehydes or ketones into alcohols. This reaction is carried out in living organisms using enzymes such as carbonylreductase, [67] which often use NADH or NADPH as the reducing agent. Remarkably, NADH can also work as reductant in an iridium catalyzed reduction of aldehydes in biological settings (Scheme 24). The reaction was tested by using an aldehyde‐containing BODIPY probe which is five times more emissive when reduced than in its oxidized form; although in the cellular environment, the alcohol form only showed twice the fluorescence of the aldehyde counterpart. The addition of Ir1 to the cells incubated with Bodipy‐CHO increased the fluorescent by 1.6‐fold in comparison with the starting material. To demonstrate that the NADH was the ultimate responsible for the reduction of the carbonyl group, the authors added pyruvate, a known NADH depleting agent to the cells. Under these conditions, they did not observe a fluorescent increase, strongly suggesting that indeed the NADH in the reducing agent, probably by forming an Iridium‐hydride species, a well‐studied process. [68]
Scheme 24.

Iridium‐catalyzed reduction of aldehydes.
Another relevant and versatile class of organometallic intermediates are metal carbenes. It is worth, therefore, commenting a recent report demonstrating the viability of exporting metal‐carbene chemistry to mammalian cells. [69] Metal carbenes are well known, powerful reagents in organometallic chemistry, that can participate as intermediates in many relevant organic transformations, ranging from the cyclopropanation of alkenes to C−H or N−H insertions. Their high reactivity suggest that they might not survive in biological soups, however, in this report the authors demonstrate that an appropriate engineering of the probes allows for copper‐catalyzed N−H insertions of diazo precursors, even in cellular habitats. Specifically, the reaction consists of a Cu(II) catalyzed annulation between 1,2‐diamino naphthalene and an α‐diazo‐β‐keto esters (Scheme 25). The reaction proceeds through an N−H insertion of the carbene followed by cyclization and oxidative aromatization. The methodology was also used for the in‐situ preparation of mitochondria fragmenting agents. Moreover, the authors use a copper tristriazole complex coupled with an RGD motif to target the reaction towards cells with high levels of integrin receptors.
Scheme 25.

Copper‐catalyzed carbene insertions.
Finally, and despite falling outside the mechanistic aspects of reactions discussed in this review, mostly involving traditional organometallic steps, the use of photocatalysis in live settings should be mentioned. This kind of reactivity has been mainly explored for the distance‐dependent labeling of proteins, pioneered by the group of Nakamura [70] and recently expanded by Macmillan and coworkers to living cells. [71] This strategy consists of the photocatalytic generation of a highly reactive species (such as carbene from a diazirine) which can travel short distances before reacting with the solvent. However, if, during that time, this intermediate finds a suitable biomolecular partner, it can form a covalent bond with it, therefore allowing for the proximity‐dependent labeling (Scheme 26) [71a] This is an area of Chemical Biology that is clearly expanding.
Scheme 26.

Iridium‐photocatalyzed proximity labeling using diazirines.
Photocatalysts have also been used in biological contexts for the reduction of azides [65] and for the uncaging of pyridinium immolative linkers. [72] In one notable example by the group of Winssinger, it was shown how a nucleic acid template can accelerate photocatalytic uncaging processes (Scheme 27). [72a]
Scheme 27.

Nucleic acid‐templated photocatalytic uncaging of a Rhodamine.
7. Summary and Outlook
In the past decade, the number of bio‐compatible and bioorthogonal reactions catalyzed by homogeneous transition metals has notably expanded, providing chemists with mechanistically diverse alternatives to carry out reactions in biological and living settings. What started as a mere curiosity and very specific reactions, has become a whole research field that promises to impact chemical and cell biology and biomedicine. Indeed, a wide number of very relevant applications for biological sensing, localized activation of drugs or the formation of gene circuits have been already described. Major challenges in the field are related with the low turnover numbers, and the ready deactivation of the active catalysts in the biological soups, especially under live settings. Some groups have found ways to protect the metal complex from the cellular environment through encapsulation in polymers, conjugation to proteins or embedding them in nanoparticles. Overall, the development of very active, low toxic catalysts that presents new to nature reactivities and a higher scope and promiscuity that native enzymes represents a challenging and appealing task for the near future.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Andrés Seoane obtained his PhD from the University of Santiago de Compostela (USC) in 2016 under the supervision of Prof. J. L. Mascareñas and Prof. Moisés Gulías. After a postdoctoral stay in the University of California San Diego (UCSD) with Prof. N. K. Devaraj, he returned to USC where he is working on the development of biologically relevant metal‐catalyzed reactions.

Biographical Information
José Luis Mascareñas completed his PhD at the University of Santiago and postdoctoral studies at Stanford University (USA) under the supervision of Prof. Paul Wender. He became professor in 1993 and full professor in 2005, at the University of Santiago de Compostela. In 2014 he was awarded with an ERC Advanced Grant (http://metbiocat.eu/) and, more recently, an ERC Proof of Concept (2020). He has been scientific director of CIQUS since 2014. In 2015 he received the gold medal of the Spanish Society of Chemistry, and in 2016, was appointed as member of the European Academy of Sciences. His current research splits between a synthetic program aimed at discovering novel methods based on metal catalysis, and a chemical biology program focused on the development of synthetic tools for biological intervention.

Acknowledgements
This work has received financial support from Spanish grant PID2019‐108624RB‐I00 and Juan de la Cierva‐Incorporación fellowship (IJC2018‐036705‐I to A.S.), the Consellería de Cultura, Educación e Ordenación Universitaria (ED431C‐2021/25 and Centro Singular de Investigación de Galicia accreditation2019‐2022, ED431G 2019/03), the European Regional Development Fund (ERDF), and the European Research Council (Advanced Grant No. 340055).
A. Seoane, J. L. Mascareñas, Eur. J. Org. Chem. 2022, e202200118.
References
- 1.D. L. Nelson, M. M. Cox in Lehninger Principles of Biochemistry, 7th ed., W. H. Freeman, New York, 2017.
- 2. Sigel R. K. O., Pyle A. M., Chem. Rev. 2007, 107, 97–113. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Rabinovich D. in Compr. Organomet. Chem. III (Eds. Mingos M. P., Crabtre R. H.). Elsevier, 2007, 1, 93–117; [Google Scholar]
- 3b. Grate J. W., Frye G. C. in Sensors Update, Vol. 2 (Eds.: Baltes H., Göpel W., Hesse J.), Wiley-VCH, Weinheim, 1996, pp. 10–20. [Google Scholar]
- 4. Daniel C., Koga N., Fu X. Y., Morokuma K., J. Am. Chem. Soc. 1988, 110, 3773–3787. [Google Scholar]
- 5.
- 5a. Martínez-Calvo M., Mascareñas J. L., Coord. Chem. Rev. 2018, 359, 57–79; [Google Scholar]
- 5b. Destito P., Vidal C., López F., Mascareñas J. L., Chem. Eur. J. 2021, 27, 4789–4816. [DOI] [PubMed] [Google Scholar]
- 6. Vigh L., Joó F., Csépló Á., Eur. J. Biochem. 1985, 146, 241–244. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2708–2711; [Google Scholar]
- 7b. Tornøe C. W., Christensen C., Meldal M., J. Org. Chem. 2002, 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. del Amo D. S., Wang W., Jiang H., Besanceney C., Yan A. C., Levy M., Liu Y., Marlow F. L., Wu P., J. Am. Chem. Soc. 2010, 132, 16893–16899; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Salic A., Mitchison T. J., Proc. Nat. Acad. Sci. 2008, 105, 2415–2420; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Jao C. Y., Roth M., Welti R., Salic A., Proc. Nat. Acad. Sci. 2009, 106, 15332–15337; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. and A. J. L., Tirrell D. A., J. Am. Chem. Soc. 2003, 125, 11164–11165; [DOI] [PubMed] [Google Scholar]
- 8e. Li L., Zhang Z., Molecules 2016, 21, 1393–1415. [Google Scholar]
- 9. Kennedy D. C., McKay C. S., Legault M. C. B., Danielson D. C., Blake J. A., Pegoraro A. F., Stolow A., Mester Z., Pezacki J. P., J. Am. Chem. Soc. 2011, 133, 17993–18001. [DOI] [PubMed] [Google Scholar]
- 10. Streu C., Meggers E., Angew. Chem. Int. Ed. 2006, 45, 5645–5648; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5773–5776. [Google Scholar]
- 11.
- 11a. Antos J. M., Francis M. B., Curr. Opin. Chem. Biol. 2006, 10, 253–262; [DOI] [PubMed] [Google Scholar]
- 11b. Chalker J. M., Wood C. S. C., Davis B. G., J. Am. Chem. Soc. 2009, 131, 16346–16347; [DOI] [PubMed] [Google Scholar]
- 11c. Schiemann O., Piton N., Plackmeyer J., Bode B. E., Prisner T. F., Engels J. W., Nat. Protoc. 2007, 2, 904–923. [DOI] [PubMed] [Google Scholar]
- 12. Breugst M., Reissig H.-U., Angew. Chem. Int. Ed. 2020, 59, 12293–12307; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12389–12404. [Google Scholar]
- 13.
- 13a. Worrell B. T., Malik J. A., Fokin V. V., Science. 2013, 340, 457–460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Fang Y., Bao K., Zhang P., Sheng H., Yun Y., Hu S.-X., Astruc D., Zhu M., J. Am. Chem. Soc. 2021, 143, 1768–1772. [DOI] [PubMed] [Google Scholar]
- 14. McKay C. S., Finn M. G., Chem. Biol. 2014, 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Uttamapinant C., Tangpeerachaikul A., Grecian S., Clarke S., Singh U., Slade P., Gee K. R., Ting A. Y., Angew. Chem. Int. Ed. 2012, 51, 5852–5856; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5954–5958. [Google Scholar]
- 16. Miguel-Ávila J., Tomás-Gamasa M., Olmos A., Pérez P. J., Mascareñas J. L., Chem. Sci. 2018, 9, 1947–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Destito P., Couceiro J. R., Faustino H., López F., Mascareñas J. L., Angew. Chem. Int. Ed. 2017, 56, 10766–10770; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10906–10910. [Google Scholar]
- 18. Gutiérrez-González A., Destito P., Couceiro J. R., Pérez-González C., López F., Mascareñas J. L., Angew. Chem. Int. Ed. 2021, 60, 16059–16066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miguel-Ávila J., Tomás-Gamasa M., Mascareñas J. L., Angew. Chem. Int. Ed. 2020, 59, 17628–17633; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 17781–17786. [Google Scholar]
- 20. Pflästerer D., Rudolph M., Hashmi A. S. K., Isr. J. Chem. 2018, 58, 622–638. [Google Scholar]
- 21.
- 21a. Yang Y. K., Lee S., Tae J., Org. Lett. 2009, 11, 5610–5613; [DOI] [PubMed] [Google Scholar]
- 21b. Do J. H., Kim H. N., Yoon J., Kim J. S., Kim H. J., Org. Lett. 2010, 12, 932–934; [DOI] [PubMed] [Google Scholar]
- 21c. Seo H., Jun M. E., Egorova O. A., Lee K. H., Kim K. T., Ahn K. H., Org. Lett. 2012, 14, 5062–5065; [DOI] [PubMed] [Google Scholar]
- 21d. Patil N. T., Shinde V. S., Thakare M. S., Kumar P. H., Bangal P. R., Barui A. K., Patra C. R., Chem. Commun. 2012, 48, 11229–11231. [DOI] [PubMed] [Google Scholar]
- 22. Vong K., Yamamoto T., Chang T., Tanaka K., Chem. Sci. 2020, 11, 10928–10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vidal C., Tomás-Gamasa M., Destito P., López F., Mascareñas J. L., Nat. Commun. 2018, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang T. C., Vong K., Yamamoto T., Tanaka K., Angew. Chem. Int. Ed. 2021, 60, 12446–12454. [DOI] [PubMed] [Google Scholar]
- 25. Tsubokura K., Vong K. K. H., Pradipta A. R., Ogura A., Urano S., Tahara T., Nozaki S., Onoe H., Nakao Y., Sibgatullina R., Kurbangalieva A., Watanabe Y., Tanaka K. Angew. Chem. Int. Ed. 2017, 56, 3579–3584; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3633–3638. [Google Scholar]
- 26. Sabatino V., Unnikrishnan V. B., Bernardes G. J. L., Chem. Catalysis. 2022, 2, 39–51. [Google Scholar]
- 27. Li J., Yu J., Zhao J., Wang J., Zheng S., Lin S., Chen L., Yang M., Jia S., Zhang X., Chen P. R., Nat. Chem. 2014, 6, 352–361. [DOI] [PubMed] [Google Scholar]
- 28. Indrigo E., Clavadetscher J., Chankeshwara S. V., Megia-Fernandez A., Lilienkampf A., Bradley M., Chem. Commun. 2017, 53, 6712–6715. [DOI] [PubMed] [Google Scholar]
- 29. Martínez-Calvo M., Couceiro J. R., Destito P., Rodríguez J., Mosquera J., Mascareñas J. L., ACS Catal. 2018, 8, 6055–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li B., Liu P., Wu H., Xie X., Chen Z., Zeng F., Wu S., Biomaterials 2017, 138, 57–68. [DOI] [PubMed] [Google Scholar]
- 31. Learte-Aymamí S., Curado N., Rodríguez J., Vázquez M. E., Mascareñas J. L., J. Am. Chem. Soc. 2017, 139, 16188–16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Learte-Aymamí S., Vidal C., Gutiérrez-González A., Mascareñas J. L., Angew. Chem. Int. Ed. 2020, 59, 9149–9154; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 9234–9239. [Google Scholar]
- 33. Ruoslahti E., Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [DOI] [PubMed] [Google Scholar]
- 34. Long Y., Cao B., Xiong X., Chan A. S. C., Sun R. W. Y., Zou T., Angew. Chem. Int. Ed. 2021, 60, 4133–4141; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4179–4187. [Google Scholar]
- 35. Wang X., Liu Y., Fan X., Wang J., Ngai W. S. C., Zhang H., Li J., Zhang G., Lin J., Chen P. R., J. Am. Chem. Soc. 2019, 141, 17133–17141. [DOI] [PubMed] [Google Scholar]
- 36. Stenton B. J., Oliveira B. L., Matos M. J., Sinatra L., Bernardes G. J. L., Chem. Sci. 2018, 9, 4185–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oliveira B. L., Stenton B. J., Unnikrishnan V. B., de Almeida C. R., Conde J., Negrão M., Schneider F. S. S., Cordeiro C., Ferreira M. G., Caramori G. F., Domingos J. B., Fior R., Bernardes G. L. J. Am. Chem. Soc. 2020, 142, 10869–10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Biffis A., Centomo P., Del Zotto A., Zecca M., Chem. Rev. 2018, 118, 2249–2295. [DOI] [PubMed] [Google Scholar]
- 39. Chinchilla R., Nájera C., Chem. Rev. 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]
- 40. Chalker J. M., Wood C. S. C., Davis B. G., J. Am. Chem. Soc. 2009, 131, 16346–16347. [DOI] [PubMed] [Google Scholar]
- 41. Li N., Lim R. K. V., Edwardraja S., Lin Q., J. Am. Chem. Soc. 2011, 133, 15316–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li J., Lin S., Wang J., Jia S., Yang M., Hao Z., Zhang X., Chen P. R., J. Am. Chem. Soc. 2013, 135, 7330–7338. [DOI] [PubMed] [Google Scholar]
- 43. Tanaka S., Saburi H., Murase T., Yoshimura M., Kitamura M., J. Org. Chem. 2006, 71, 4682–4684. [DOI] [PubMed] [Google Scholar]
- 44. Völker T., Dempwolff F., Graumann P. L., Meggers E., Angew. Chem. Int. Ed. 2014, 53, 10536–10540; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10705–10710. [Google Scholar]
- 45. Völker T., Meggers E., ChemBioChem 2017, 18, 1083–1086. [DOI] [PubMed] [Google Scholar]
- 46. Singh N., Gupta A., Prasad P., Mahawar P., Gupta S., Sasmal P. K., Inorg. Chem. 2021, 60, 12644–12650. [DOI] [PubMed] [Google Scholar]
- 47.
- 47a. Sánchez M. I., Penas C., Vázquez M. E., Mascareñas J. L., Chem. Sci. 2014, 5, 1901–1907; [PMC free article] [PubMed] [Google Scholar]
- 47b. Vázquez O., Sánchez M. I., Martinez-Costas J., Vázquez M. E., Mascareñas J. L., Org. Lett. 2010, 12, 216–219; [DOI] [PubMed] [Google Scholar]
- 47c. Sánchez M. I., Vázquez O., Vázquez M. E., Mascareñas J. L., Chem. Commun. 2011, 47, 11107–11109. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Tonga G. Y., Jeong Y., Duncan B., Mizuhara T., Mout R., Das R., Kim S. T., Yeh Y. C., Yan B., Hou S., Rotello V. M., Nat. Chem. 2015, 7, 597–603; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48b. Zhang X., Liu Y., Gopalakrishnan S., Castellanos-Garcia L., Li G., Malassiné M., Uddin I., Huang R., Luther D. C., Vachet R. W., Rotello V. M., ACS Nano 2020, 14, 4767–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ahmadi P., Muguruma K., Chang T. C., Tamura S., Tsubokura K., Egawa Y., Suzuki T., Dohmae N., Nakao Y., Tanaka K., Chem. Sci. 2021, 12, 12266–12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qin YW., Cho K. F., Cavanagh P. E., Ting A. Y., Nat. Methods 182 2021, 18, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee Y., Umeano A., Balskus E. P., Angew. Chem. Int. Ed. 2013, 52, 11800–11803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12016–12019. [Google Scholar]
- 52. Rubini R., Mayer C., ACS Chem. Biol. 2020, 15, 3093–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tomás-Gamasa M., Martínez-Calvo M., Couceiro J. R., Mascarenãs J. L., Nat. Commun. 2016, 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Okamoto Y., Kojima R., Schwizer F., Bartolami E., Heinisch T., Matile S., Fussenegger M., Ward T. R., Nat. Commun. 2018, 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lohner P., Zmyslia M., Thurn J., Pape J. K., Gerasimaitė R., Keller-Findeisen J., Groeer S., Deuringer B., Süss R., Walther A., Hell S. W., Lukinavičius G., Hugel T., Jessen-Trefzer C., Angew. Chem. Int. Ed. 2021, 60, 23835–23841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.
- 56a. Schuster M., Blechert S., Angew. Chem. Int. Ed. 1997, 36, 2036–2056; [Google Scholar]; Angew. Chem. 1997, 109, 2124–2144; [Google Scholar]
- 56b. Ogba O. M., Warner N. C., O'Leary D. J., Grubbs R. H., Chem. Soc. Rev. 2018, 47, 4510–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jeschek M., Reuter R., Heinisch T., Trindler C., Klehr J., Panke S., Ward T. R., Nature. 2016, 537, 661–665. [DOI] [PubMed] [Google Scholar]
- 58. Sabatino V., Rebelein J. G., Ward T. R., J. Am. Chem. Soc. 2019, 141, 17048–17052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eda S., Nasibullin I., Vong K., Kudo N., Yoshida M., Kurbangalieva A., Tanaka K., Nat. Catal. 2019, 2, 780–792. [Google Scholar]
- 60. Miller M. A., Askevold B., Mikula H., Kohler R. H., Pirovich D., Weissleder R., Nat. Commun. 2017, 8, 15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vidal C., Tomás-Gamasa M., Gutiérrez-González A., Mascareñas J. L., J. Am. Chem. Soc. 2019, 141, 5125–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.
- 62a. Mascareñas J. L., Varela I., López F., Acc. Chem. Res. 2019, 52, 465–479; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62b. Faustino H., Varela I., Mascareñas J. L., López F., Chem. Sci. 2015, 6, 2903–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang J., Zheng S., Liu Y., Zhang Z., Lin Z., Li J., Zhang G., Wang X., Li J., Chen P. R., J. Am. Chem. Soc. 2016, 138, 15118–15121. [DOI] [PubMed] [Google Scholar]
- 64.
- 64a. Sasmal P. K., Carregal-Romero S., Han A. A., Streu C. N., Lin Z., Namikawa K., Elliott S. L., Köster R. W., Parak W. J., Meggers E., ChemBioChem 2012, 13, 1116–1120; [DOI] [PubMed] [Google Scholar]
- 64b. Huang R., Li C. H., Cao-Milán R., He L. D., Makabenta J. M., Zhang X., Yu E., Rotello V. M., J. Am. Chem. Soc. 2020, 142, 10723–10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.
- 65a. Chen Y., Kamlet A. S., Steinman J. B., Liu D. R., Nat. Chem. 2011, 3, 146–153; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65b. Holtzer L., Oleinich I., Anzola M., Lindberg E., Sadhu K. K., Gonzalez-Gaitan M., Winssinger N., ACS Cent. Sci. 2016, 2, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Spicer C. D., Triemer T., Davis B. G., J. Am. Chem. Soc. 2011, 134, 800–803. [DOI] [PubMed] [Google Scholar]
- 67. Bose S., Ngo A. H., Do L. H., J. Am. Chem. Soc. 2017, 139, 8792–8795. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a. Liu Z., Romero-Canelón I., Qamar B., Hearn J. M., Habtemariam A., Barry N. P. E., Pizarro A. M., Clarkson G. J., Sadler P. J., Angew. Chem. Int. Ed. 2014, 53, 3941–3946; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4022–4027; [Google Scholar]
- 68b. Yang L., Bose S., Ngo A. H., Do L. H., ChemMedChem 2017, 12, 292–299. [DOI] [PubMed] [Google Scholar]
- 69. Gutiérrez S., Tomás-Gamasa M., Mascareñas J. L., Angew. Chem. Int. Ed. 2021, 60, 22017–22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.
- 70a. Sato S., Nakamura H., Angew. Chem. Int. Ed. 2013, 52, 8681–8684; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8843–8846; [Google Scholar]
- 70b. Nakane K., Sato S., Niwa T., Tsushima M., Tomoshige S., Tagushi H., Ishikawa M., Nakamura H., J. Am. Chem. Soc. 2021, 143, 7726–7731. [DOI] [PubMed] [Google Scholar]
- 71.
- 71a. Geri J. B., Oakley J. V., Reyes-Robles T., Wang T., McCarver S. J., White C. H., Rodriguez-Rivera F. P., D. L. Parker Jr. , Hett E. C., Fadeyi O. O., Oslund R. C., MacMillan D. W. C., Science. 2020, 367, 1091–1097; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71b. Buksh B. F., Knutson S. D., Oakley J. V., Bissonnette N. B., Oblinsky D. G., Schwoerer M. P., Seath C. P., Geri J. B., Rodriguez-Rivera F. P., Parker D. L., Scholes G. D., Ploss A., MacMillan D. W. C., J. Am. Chem. Soc. 2022, 144, 6154–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.
- 72a. Saarbach J., Lindberg E., Folliet S., Georgeon S., Hantschel O., Winssinger N., Chem. Sci. 2017, 8, 5119–5125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72b. Lindberg E., Angerani S., Anzola M., Winssinger N., Nat. Commun. 2018, 9, 1–9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72c. Angerani S. S., Winssinger N., J. Am. Chem. Soc. 2020, 142, 12333–12340. [DOI] [PubMed] [Google Scholar]