

Abstract
We present herein the synthesis of a nearly square‐pyramidal chlorophosphorane supported by the tetradentate bis(amidophenolate) ligand, N,N′‐bis(3,5‐di‐tert‐butyl‐2‐phenoxy)‐1,2‐phenylenediamide. After chloride abstraction the resulting phosphonium cation efficiently promotes the disproportionation of 1,2‐diphenylhydrazine to aniline and azobenzene. Mechanistic studies, spectroscopic analyses and theoretical calculations suggest that this unprecedented reactivity mode for PV‐centres is induced by the high electrophilicity at the cationic PV‐center, which originates from the geometry constraints imposed by the rigid pincer ligand, combined with the ability of the o‐amidophenolate moieties to act as electron reservoir. This study illustrates the promising role of cooperativity between redox‐active ligands and phosphorus for the design of organocatalysts able to promote redox processes.
Keywords: Cooperative Effects, Lewis Acids, Non-Innocent Ligands, Organocatalysis, Redox Chemistry
An unprecedented P‐based catalyst able to promote hydrazine disproportionation has been synthesized. The available set of experimental results and theoretical calculations suggest that this reactivity is unlocked by the cooperation between a redox‐active bis(amidophenolate) ligand and the highly electrophilic central P‐atom.

Introduction
The redox chemistry of PIII species is defined by their reducing character and the high stability of the resulting PV derivatives; in particular when PV=O bonds are formed. [1] Textbook reactions such as the Wittig olefination, [2] the Staudinger reduction, [3] or the Mitsunobu inversion, [4] which are widely used even on industrial scale, make very productive use of this driving force. However, these classical protocols are low atom efficient due to the stoichiometric amounts of phosphine oxide waste that are typically produced. [5] The development of catalytic‐in‐phosphorus versions of these and other R3P‐triggered transformations is therefore highly desirable; [6] yet any design of a hypothetical catalytic cycle needs to face the challenge of shuttling back the catalyst from the oxidized PV=O form to the original PIII state. The recognition that phosphine oxides embedded in small rings are easier to deoxygenate than their acyclic congeners, therefore enabling faster catalyst turnovers and the use of milder reaction conditions, has been a breakthrough in this regard. [7] Phospholane oxides 1–3,[ 8 , 9 , 10 ] and phosphetane oxide 4, [11] are benchmark examples of the practical use of this guiding principle to provide operative PIII/PV=O catalytic cycles (Figure 1a).
Figure 1.
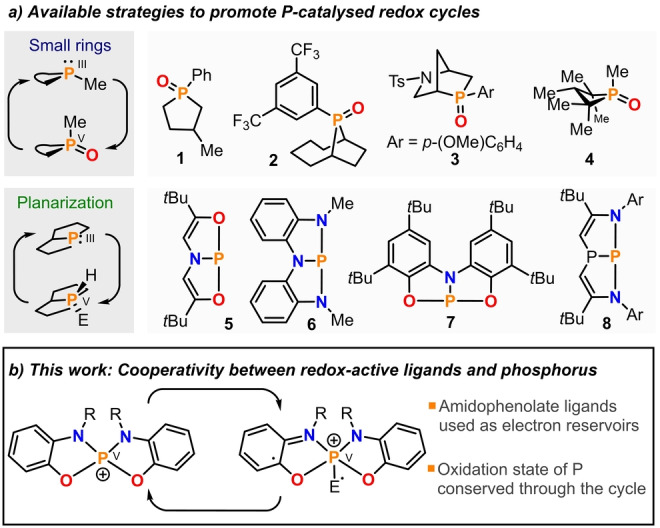
a) Strategies available for the construction of organophosphorus redox catalysts and selected examples. b) Design of P‐catalysts through redox cooperation between the ligand and a central PV atom.
T‐shaped PIII species such as 5–8 have also been intensively evaluated for the development of P‐based catalysts (Figure 1a). [12] Upon planarization of the P‐atom the original HOMO–LUMO energy gap gets significantly reduced and this facilitates the formal oxidative addition of polar H−E molecules over the P‐atom.[ 13 , 14 ] Theoretical studies suggest however that such reactions are unlikely to proceed in a concerted fashion at the P‐center, but rather through cooperation with the ligand. [15] A catalytic cycle based on the use of these PIII/PV couples has been engineered consisting on the transfer hydrogenation between ammonia borane and azobenzene. [16] Interestingly, although the two strategies just showcased seem to be unrelated, they actually rely on the same principle: the promotion of biphilic behavior at the central P‐atom by imposing non‐trigonal geometries. In P‐catalysts thus constructed the donor and acceptor characters are both localized at P, and the redox processes occurring thoroughout the catalytic cycle are P‐centered.
Being aware of the progress achieved in the catalysis arena by leveraging metal‐ligand redox cooperativity, [17] we hypothesized that the design of redox‐active P‐catalysts might also benefit from the use of this fundamentally different approach (Figure 1b). [18] Specifically, our attention was attracted by the work of Heyduk, which illustrates the participation of ZrIV and other d0 centers in redox processes employing amidophenolate and bis(amidophenolate) ligands as electron reservoirs. [19] These reports evidence that the central metal only provides a Lewis acidic position for the coordination of the substrate, while the electrons are given and taken on demand by the ligand. Thus, we speculated that phosphonium cations derived from similar ligand frameworks should also contain all necessary ingredients to act as redox‐active catalysts. Namely, electrophilic reactivity at the electronically unsaturated PV center, [20] and a supply of electrons provided by the cooperating amidophenolate substituents. Note that any hypothetical P‐catalyst based on this design maintains the PV oxidation state through the entire catalytic cycle.
Results and Discussion
Synthesis and Reactivity of Square Pyramidal Phosphoranes
To put this idea into practice, bis(amidophenolate) ligand 9, first reported by Wieghardt was chosen as the most promising candidate; [21] three are the main reasons for this election: i) Its tetradentate nature should avoid ligand decoordination upon oxidation; ii) the rigidity of the tricyclic ligand imposes a distorted geometry to the PV‐atom, which is expected to increase its Lewis acidity; and finally, iii) having the two amidophenolate moieties connected by an o‐phenylene linker imparts additional stability upon two‐electron oxidation because a relatively robust diiminoquinone will be formed in the backbone of 9ox2 (Scheme 1a). [19]
Scheme 1.
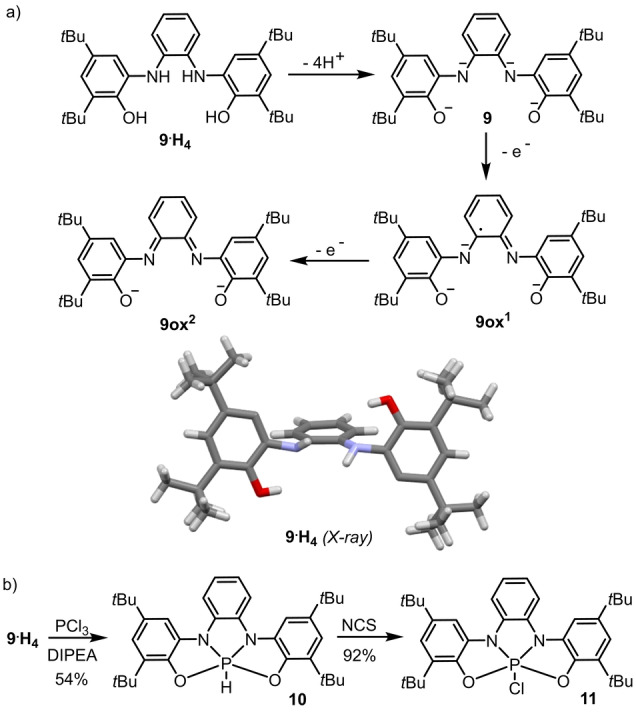
Relevant oxidation levels and labels for the bis(amidophenolate) ligand 9 and synthesis of of hydrophosphorane 10 and chlorophosphorane 11.
Mixing protonated 9⋅H4 and PCl3 in the presence of 3.15 equivalents of DIPEA affords a relatively clean reaction mixture from which a white solid was isolated in 54 % yield after purification by flash chromatography (Scheme 1b). [22] The ESI mass spectrum of the sample displays a [M+1]+ molecular ion peak at 545.3 Da, which indicates a 1 : 1 P/ligand ratio. Moreover, the newly obtained compound is characterized by a 1H NMR signal at 8.81 ppm. (J P−H=782.3 Hz) and a 31P NMR signal at −39.8 ppm. with the same coupling constant. These data are consistent with the formation of hydridophosphorane 10. Diffusion of MeOH into toluene solutions of 10 led to the formation of crystals suitable for X‐ray diffraction, which confirmed the proposed connectivity (Figure 2a). A bond length analysis of the obtained structure suggests that the ligand is in its reduced form (N1−C15, 1.394(1) Å; N2−C16, 1.397(1) Å; C15−C16, 1.410(1) Å) [21] and as expected from the geometric constraints imposed by 9, the τ‐value of only 0.02 bears out a strongly pronounced square pyramidal character with the hydride moiety located at the apical site. [23] 10‐P‐5 species depicting square or nearly perfect rectangular pyramid geometry have been already described; [24] however, to the best of our knowledge, this is the first hydridophosphorane sharing this spatial distribution. [25]
Figure 2.
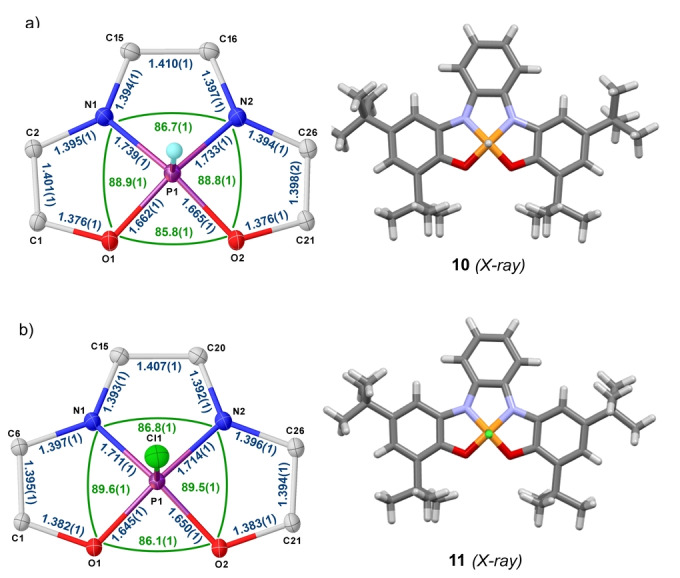
X ray structures of 10 and 11. Ellipsoids are set at 50 % probability; solvent molecules were removed for clarity. [27]
Treatment of 10 with N‐chlorosuccinimide delivers the corresponding chlorophosphorane 11, in which the square pyramidal geometry is preserved although the pyramidalization degree at P decreases (Sum of the basal angles O1−P1−N1, N1−P1−N2, O2−P1−N1 and O1−P1−O2 in 11=352.0° vs. 350.5° in 10; Figure 2b). This is rationalized by a diminished s‐orbital character in the apical bond in accordance with the Bent's rule. [26]
Compound 11 serves as precursor for a series of phosphoranes 12–15 via exchange of the chloride by cyanide, fluoride, amino or aryl anions, respectively (Scheme 2). The connectivity of 12 and 14 were also confirmed crystallographically (see Figure S16 and S17). To evaluate the ability of ligand 9 to participate in redox processes after coordination to a PV‐center, a dichloromethane solution of 14 (0.1 m in NBu4PF6) was prepared and investigated by cyclic voltammetry. A reversible one‐electron oxidation event at E 1/2 ox=0.55 V (vs. Fc/Fc+) was observed, which can only be assigned as a ligand‐centered one‐electron process (9→9ox1 ) since the central atom is already a PV center (Figure 3a). In agreement with that oxidation potential, the addition of one equivalent of AgSbF6 to a solution of 14 in CH2Cl2 at −78 °C resulted in the development of an intense blue coloration. The EPR spectrum of the newly formed species, which we assumed to be 16 shows coupling with the phosphorus, α (31)P=0.8 G; two identical nitrogen nuclei, α (14)N=3.5 G and two pairs of protons α (1)H=2.4 and 2.1 G (Figure 3a). The Mulliken spin density distribution calculated at the UB3LYP‐D3(BJ)/def2‐TZVP level is also shown in Figure 3a. Large‐spin populations are located on the nitrogen atoms (ca. 15 % each), with significant contributions from C4, C9, C10 and C17 (9 % each). A rather small contribution is located at the P‐center (ca. 4 %). The reversible formation of 16 confirms that the ligand scaffold can act as electron reservoir once coordinated to a PV‐center and thereby may assist the P atom by transferring electrons in a hypothetical catalytic cycle.
Scheme 2.
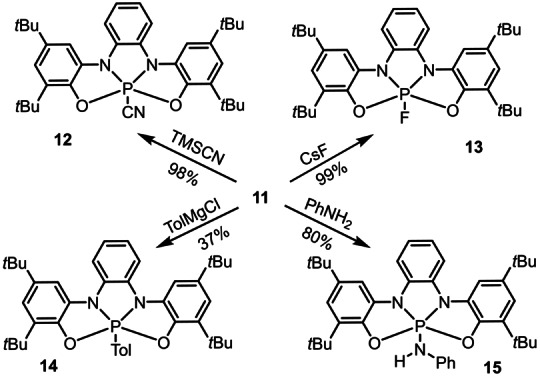
Chloride ligand exchange at chlorophosphorane 11.
Figure 3.
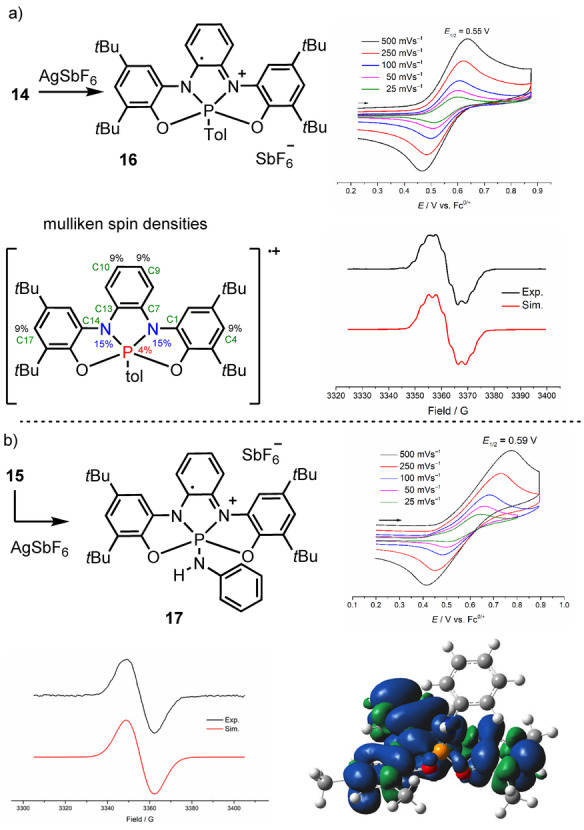
One electron oxidation of 14 and 15. The X band EPR spectra of 16 and 17 were recorded at 200 K in CH2Cl2 (0.5 mM). Mulliken spin density distributions calculated at the UB3LYP‐D3(BJ)/def2‐TZVP level and contour plot of spin‐density distribution depicted at iso‐density value of 0.0004 au.
Cyclic voltammetry of 15 showed a quasi‐reversible oxidative wave at E 1/2 ox=0.59 V (vs. Fc/Fc+). In agreement with this result, reaction of 15 with 1 equiv of AgSbF6 at −78 °C produced a violet solution, whose EPR spectrum in dichloromethane at 200 K exhibited an isotropic signal at g=2.003. Unfortunately, the lack of hyperfine structure of the spectrum prevents further characterization of this species (Figure 3b).
In an attempt to isolate the conceivable planar phosphonium cation resulting from the chloride abstraction in 11, dichloromethane solutions of this compound were initially treated with NaSbF6 and Na[B(C6H3(CF3)2)4]. No reaction was observed under these conditions. Conversely, addition of Et3Si(C6H6)[B(C6F5)4] immediately produced dark brown solutions from which no discrete species could be isolated. Identical result was obtained by reaction of 10 with (Ph3C)2[B12Cl12]. Finally, addition of NaSbF6 to an equimolar mixture of 11 and dimethylaminopyridine (DMAP) allowed the isolation of the phosphonium‐DMAP adduct 18 in 55 % yield, whose connectivity was confirmed by X‐ray diffraction (Figure 4a). Due to the cationic nature in 18 the P‐atom gets deeper embedded in the ligand cavity (Ʃang at P(18)=352.8°) diminishing its pyramidalization if compared with 11 and 12.
Figure 4.
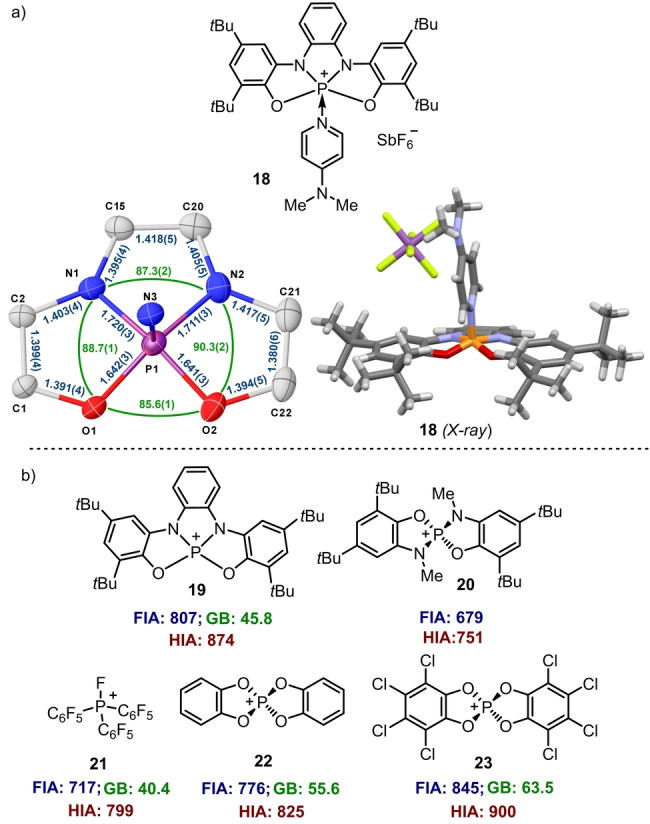
Synthesis and structure of phosphonium‐DMAP adduct 18 and estimated Lewis acidity for 19–23. Ellipsoids are set at 50 % probability; solvent molecules were removed for clarity. [27] Estimated Lewis acidity for 19–23 by the Gutmann–Beckett method and computed fluoride and hydride ion affinities (kJ mol−1) at the PW6B95‐D3(BJ)/def2‐QZVPP//B3LYP‐D3(BJ)/def2‐TZVP level of theory. Values for 21–23 obtained from reference [20d].
The effective Lewis acidity of phosphonium ion 19 has been determined by the Gutmann–Beckett method (Δδ31P=45.8 ppm.) to be lower than the one of structurally similar hitherto isolated catecholato‐based phosphonium ions 22 and 23, but stronger than that of [(C6F5)3PF]+ 21 (Figure 4b; see the Supporting Information for experimental details). [28] This value fits the expected trend since oxygen‐based catecholate substituents are more electron withdrawing than amidophenolate ones. In contrast, the isodesmically computed fluoride ion affinity (FIA) and hydride ion affinity (HIA) for 19 are higher than that reported for 21 or 22, and only slightly surpassed by that of octa‐chlorinated 23. Note that the FIA and HIA values include the reagent deformation penalty to form the corresponding fluoride and hydride adducts. [29] This energy is not negligible for 22 and 23 because their geometries substantially change from tetrahedral to trigonal bipyramidal to form the corresponding adduct, while 19 only requires a minimum displacement of the central P‐atom outside of the ONNO plane. For comparison purposes we also calculated the FIA and HIA values for quasi tetrahedral 20. The values are notably lower than those of 19 even though both 19 and 20 are amidophenolate substituted phosphonium ions, confirming the analysis (Figure 4b). Hence, in 19 coexist a planar Lewis super‐acidic P‐center and an easy to oxidize ligand framework.
Catalytic Disproportionation of 1,2‐Diphenylhydrazine
At this stage we set out to investigate the catalytic activity of transient phosphonium cation 19 taking as model transformation the disproportionation of 1,2‐diphenylhydrazine, a reaction already known to occur at d0‐centers through redox cooperation between the metal and the assisting ligand. [19c] Chlorophosphorane 11 was used as precatalyst in combination with Na[B(C6H3(CF3)2)4] as redox inert chloride abstractor agent. Thus, a chloroform solution containing 1,2‐diphenylhydrazine (10 equiv), 11 (1 equiv) and Na[B(C6H3(CF3)2)4] (1 equiv) was prepared in a Young tube under N2 atmosphere at room temperature. This resulted in the immediate development of a dark green solution, which slowly turned dark brown upon the course of the reaction. Quantitative 1H NMR analysis of the final solution employing hexamethylbenzene as internal standard revealed that after 16 h. the initial hydrazine had been completely consumed and 4.5 equivalents of azobenzene (84 % isolated yield) had been produced together with slightly more than 8 equivalents of free aniline (71 % isolated yield as N‐phenylbenzamide). The 31P NMR spectrum of the same reaction mixture features a single signal at δ=−34.6 ppm., which coincides with that of anilido compound 15, although it is slightly broader. Compound 15 is probably formed by deprotonation of an aniline moiety coordinated to phosphonium cation 19 under the operating reaction conditions, and it is not catalytically active in the model disproportionation. However, in situ protonation of 15 with [H(OEt2)2][B(C6H3(CF3)2)4] generates a catalytically competent species. These experiments make us believe that 15 is involved in the formation of an off‐cycle resting state of the catalyst.
Theoretical Calculations on the Operating Mechanism
In an effort to shed light on the mechanism governing this transformation, electronic structure calculations were carried out. The reaction intermediates and selected transition states were optimized at the PBE‐D3(BJ)/def2‐SVP level of theory, [30] making use of the D3 Grimme dispersion correction with Becke–Johnson damping. [31] The nature of the stationary points was confirmed by harmonic frequency calculations, which were also used for the thermodynamical corrections to the free energy. In order to account for solvation effects, single point corrections were computed with the C‐PCM model with chloroform as solvent at the B3LYP‐D3(BJ)/def2‐TZVP level. [32] Standard state corrections for solution were also applied to the free energy values at 298.15 K. The Gaussian 16A.03 program package was utilized in these calculations (see the Supporting Information for computational details). [33]
As shown in Figure 5, our studies start already with 1,2‐diphenylhydrazine coordinated to the phosphonium cation Int I. A relay is required to move a proton from the P‐coordinated nitrogen of the hydrazine moiety (N3) to the neighbor nitrogen atom (N4). We suggest an intramolecular stepwise mechanism, by which the bis(amidophenolate) ligand cooperates to the transfer via initial protonation of one of its amidophenolate nitrogen atoms Int IIa. The unhindered rotation of the hydrazine moiety around the P−N axis allows the formation of a hydrogen bond between the hydrazine outer nitrogen and the just formed N−H unit of bis(amidophenolate) ligand Int IIb; thus, facilitating the formation of intermediate Int III via transition state TSII. This ligand‐assisted pathway for proton transfer requires a moderate Gibbs activation energy (ΔG ≠) of 21.6 kcal mol−1. The latter is an upper bond, given that proton tunneling could lower the effective barrier. [34] Additionally, an external aniline molecule may also serve as proton shuttle connecting Int I with Int III through an energetically favored path. Subsequent elimination of aniline follows with a low barrier ΔG ≠=8.9 kcal mol−1 delivering the key nitrenoide Int IV.
Figure 5.

Free energy profile for the disproportionation of 1,2‐diphenylhydrazine catalyzed by 19 calculated at the B3LYP‐D3(BJ)/def2‐TZVP(C‐PCM)//PBE‐D3(BJ)/def2‐SVP level of theory.
Our calculations suggest that Int IV may exist in various spin states such as singlet IV or as diradical Int IV′. A stability analysis of the wave function of the closed shell singlet reveals an UKS/RKS instability. Comparison of the energetics for the singlet states (closed shell and broken symmetry diradical) and the triplet IV′ reveals that the latter is the most stable electronic state for the intermediate; 3.3 kcal mol−1 lower than the closed shell singlet. These observations were also confirmed with the range separated functional ωB97XD. [35] The spin density plot of the open shell triplet IV′ is shown in the Supporting Information (Figure S20). Thermodynamically favorable hydrogen atom transfer (HAT) from 1,2‐diphenylhydrazine to Int IV′ delivers Int V′, which is identical to compound 17, and 1,2‐diphenylhydrazinyl radical. Abstraction of a second hydrogen atom presumably from the last species generates Int VI. Departure of azobenzene releases Int VII, which corresponds to an aniline moiety coordinated to the phosphonium cation 19. Exchange of aniline by 1,2‐diphenylhydrazine regenerates Int I and closes the catalytic cycle (Figure 5). The suggested reaction pathway singles out TSI as the TOF‐determining transition state; note however that the barriers for the exothermic hydrogen atom transfer processes have not been determined.
Kinetics and Trapping Experiments
To gain further insight into the reaction mechanism, kinetic studies were undertaken. The conversion of 1,2‐diphenylhydrazine into aniline and azobenzene was monitored by 1H NMR spectroscopy in CDCl3 using C6Me6 as internal standard. The temperature was kept constant at 25 °C, 15 (5–10 mol %) was used as precatalyst, and an aniline/anilinium buffer (0.1 M) was added to all control experiments to avoid slow poisoning of the catalyst upon reaction progression.
By use of the variable time normalization analysis (VTNA), [36] the order in substrate has been determined to be 1.0 (Figure S4), as expected from the substantial Int IV/Int VI energy difference (Figure 5 and S19). In addition, the reaction rate is proportional to the square root of the total amount of catalyst added (order 0.5 in catalyst; Figure S5). This suggests that the majority of the P‐catalysts exists outside the catalytic cycle as an inactive dimer in equilibrium with the active species. Moreover, this dimerization should occur before the turnover limiting step TS1. Hence, diffusion ordered NMR (DOSY) experiments were undertaken to determine if 15 forms aggregates in solution under the working conditions (Figures S6–S8 and Table S2). [37] From these experiments a diffusion coefficient D=6.56×10−10 m2 s−1 was determined for pure 15, and its volume predicted to be 971 Å3. This compares well with that of structurally related 14, which has been determined by X‐ray crystallography (946 Å3) and provides direct evidence for existence of pure 15 as monomer. Interestingly, the diffusion coefficient estimated for 15 in the presence of an aniline/anilinium buffer (5.0 equiv of each, as used in our kinetic experiments) is 5.06×10−10 m2 s−1 , which corresponds to an aggregate volume of 2114 Å3. This value is slightly higher than twice the volume expected for 15 and it is a clear indication pointing to the dimerization of 15. While the actual connectivity of the dimer remains unknown to us, making use of theoretical calculations we were able to find a dimeric structure in which two molecules of 15 are bridged through an anilinium moiety (Figure S9). Its dissociation free energy has been estimated in 15.6 kcal mol−1. An extensive conformational search has not been performed; therefore, it is likely that other dimer structures are energetically available. Nonetheless, the evidence of a single structure would already explain the observed kinetics and the NMR diffusion results.
Subsequently, the ability of Int IV to act as H‐atom acceptor was also evaluated. For this purpose, we conducted the reaction of 11 and TolN3 in the presence of a stoichiometric amount of 9,10‐dihydroanthracene as benchmark HAT reagent with a C−H bond dissociation free energy (BDFE) of 76 kcal mol−1. Inspection of the 1H NMR spectrum of the crude reaction mixture confirmed the formation of toluidine and anthracene albeit in only moderate yield (Scheme 3a).
Scheme 3.
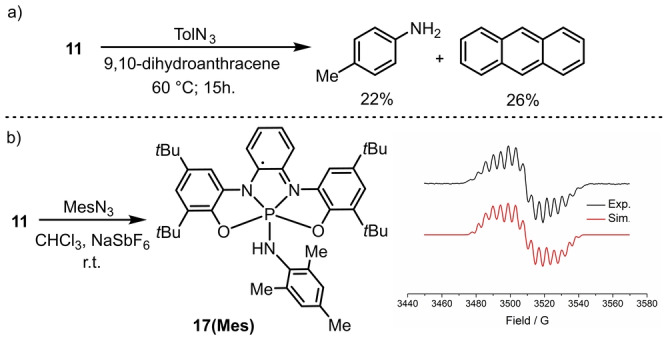
a) Trapping of the nitrenoide intermediate. b) Detection of 17(Mes) through X band EPR. Spectra recorded at 298 K in CH2Cl2 (0.5 mM).
In a final attempt to trap Int IV, a significantly bulkier azide, 2,4,6‐(Me)3C6H2N3 (MesN3), was made react with 11 and the blue‐green solution thus obtained analyzed by EPR. Neither the spectrum pattern obtained is characteristic for a triplet state (s=1) nor the distinct half‐field signal for the forbidden (Δms =±2) were observed. We recorded instead the 16‐line spectrum of a monoradical centered at g=2.003, which can be well simulated considering the hyperfine coupling of an electron with a phosphorus nucleus α (31)P=13.1 G; three inequivalent N nuclei with α (14)N=8.6, 3.7 and 2.8 G, respectively; and the four protons in p‐position to the nitrogen atoms, α (1)H=5.1, 4.6, 3.5 and 3.5 G. These hyperfine couplings are in good agreement with those computed for 17(Mes) (Scheme 3b and Figure S3). Therefore, we assume that driven by the favorable thermodynamics, Int IV(Mes) abstracts a hydrogen atom from the reaction medium delivering 17(Mes) (Scheme 3b). A detailed mechanistic proposal and kinetic model that summarize all the information available can be found in the Supporting Information.
Conclusion
In summary, we report the use of a rigid redox‐active tetradentate scaffold as supporting ligand for the synthesis of a series of phosphoranes featuring a nearly square pyramidal coordination environment. After activation of these compounds by abstraction of the apical substituent, the resulting phosphonium cation is able to catalyze the disproportionation of 1,2‐diphenylhydrazine into aniline and azobenzene. The available set of experimental results and theoretical data suggest that it is the synergy between strong electrophilic properties at the phosphorus center and the electron‐reservoir function of the ligand what unlocks this reactivity. This work expands the concept of metal–ligand redox cooperativity to a typical non‐metallic element such phosphorus, and doing so, it offers a new tool for the design of unprecedented redox‐active organocatalysts of promising reactivity.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (projects INST 186/1237‐1 and 405832858) is gratefully acknowledged. Technical assistance from the NMR and MS services at the Faculty of Chemistry (University of Göttingen) are gratefully acknowledged. We also thank Prof. Marina Bennati and Andreas Meyer (MPI for Biophysical Chemistry, Göttingen) for providing generous access to EPR facilities and Anne‐Kathrin Kreyenschmidt (University of Göttingen) for assistance during the measurement of DOSY NMR experiments. Open Access funding enabled and organized by Projekt DEAL.
S. B. H. Karnbrock, C. Golz, R. A. Mata, M. Alcarazo, Angew. Chem. Int. Ed. 2022, 61, e202207450; Angew. Chem. 2022, 134, e202207450.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐17trc).
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Cadogan J. I. G., Mackie R. K., Chem. Soc. Rev. 1974, 3, 87–137. [Google Scholar]
- 2. Wittig G., Geissler G., Justus Liebigs Ann. Chem. 1953, 580, 44–57. [Google Scholar]
- 3. Staudinger H., Meyer J., Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar]
- 4.
- 4a. Mitsunobu O., Yamada Y., Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382; [Google Scholar]
- 4b. Mitsunobu O., Synthesis 1981, 1–28. [Google Scholar]
- 5.
- 5a. Trost B. M., Angew. Chem. Int. Ed. Engl. 1995, 34, 259–281; [Google Scholar]; Angew. Chem. 1995, 107, 285–307; [Google Scholar]
- 5b. Sheldon R. A., Pure Appl. Chem. 2000, 72, 1233–1246. [Google Scholar]
- 6.For recent reviews, see:
- 6a. Lao Z., Toy P. H., Beilstein J. Org. Chem. 2016, 12, 2577–2587; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Longwitz L., Werner T., Pure Appl. Chem. 2019, 91, 95–102; [Google Scholar]
- 6c. Lipshultz J. M., Li G., Radosevich A. T., J. Am. Chem. Soc. 2021, 143, 1699–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Kalkeren H. A., Leenders S. H. A. M., Hommersom C. R. A., Rutjes F. P. T. J., van Delft F. L., Chem. Eur. J. 2011, 17, 11290–11295. [DOI] [PubMed] [Google Scholar]
- 8.Selected references for the use of 1:
- 8a. O'Brien C. J., Tellez J. L., Nixon Z. S., Kang L. J., Carter A. L., Kunkel S. R., Przeworski K. C., Chass G. A., Angew. Chem. Int. Ed. 2009, 48, 6836–6839; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6968–6971; [Google Scholar]
- 8b. O'Brien C. J., US Patent US8901365, 2014;
- 8c. Tsai Y.-L., Lin W., Asian J. Org. Chem. 2015, 4, 1040–1043. [Google Scholar]
- 9.For the use of 2 see: Coyle E. E., Doonan B. J., Holohan A. J., Walsh K. A., Lavigne F., Krenske E. H., O'Brien C. J., Angew. Chem. Int. Ed. 2014, 53, 12907–12911; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13121–13125. [Google Scholar]
- 10.References for the use of 3 and other members of the HypPhos family:
- 10a. Lorton C., Castanheiro T., Voituriez A., J. Am. Chem. Soc. 2019, 141, 10142–10147; [DOI] [PubMed] [Google Scholar]
- 10b. Cai L., Zhang K., Chen S., Lepge R. J., Houk K. N., Krenske E. H., Kwon O., J. Am. Chem. Soc. 2019, 141, 9537–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For selected examples of the use of phospholane 4, see:
- 11a. Nykaza T. V., Harrison T. S., Ghosh A., Putnik R. A., Radosevich A. T., J. Am. Chem. Soc. 2017, 139, 6839–6842; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Longwitz L., Spannenberg A., Werner T., ACS Catal. 2019, 9, 9237–9244; [Google Scholar]
- 11c. Lecomte M., Lipshultz J. M., Kim-Lee S. H., Li G., Radosevich A. T., J. Am. Chem. Soc. 2019, 141, 12507–12512; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Li G., Miller S. P., Radosevich A. T., J. Am. Chem. Soc. 2021, 143, 14464–14469; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Lipshultz J. M., Radosevich A. T., J. Am. Chem. Soc. 2021, 143, 14487–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Culley S. A., Arduengo A. J., J. Am. Chem. Soc. 1984, 106, 1164–1165; [Google Scholar]
- 12b. Arduengo A. J., Steward C. A., Davidson F., Dixon D. A., Becker J. Y., Culley S. A., Mizen M. B., J. Am. Chem. Soc. 1987, 109, 627–647. [Google Scholar]
- 13.For seminal publications, see:
- 13a. McCarthy S. M., Lin Y.-C., Devarajan D., Chang J. W., Yennawar H. P., Rioux R. M., Ess D. H., Radosevich A. T., J. Am. Chem. Soc. 2014, 136, 4640–4650; [DOI] [PubMed] [Google Scholar]
- 13b. Zhao W., McCarthy S. M., Lai T. Y., Yennawar H. P., Radosevich A. T., J. Am. Chem. Soc. 2014, 136, 17634–17644; [DOI] [PubMed] [Google Scholar]
- 13c. Cui J., Li Y., Ganguly R., Inthirarajah A., Hirao H., Kinjo R., J. Am. Chem. Soc. 2014, 136, 16764–16767; [DOI] [PubMed] [Google Scholar]
- 13d. Robinson T. P., De Rosa D. M., Aldridge S., Goicoechea J. M., Angew. Chem. Int. Ed. 2015, 54, 13758–13763; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13962–13967. [Google Scholar]
- 14.For recent reviews, see:
- 14a. Brand A., Uhl W., Chem. Eur. J. 2019, 25, 1391–1404; [DOI] [PubMed] [Google Scholar]
- 14b. Abbenseth J., Goicoechea J. M., Chem. Sci. 2020, 11, 9728–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Zeng G., Maeda S., Taketsugu T., Sakaki S., Angew. Chem. Int. Ed. 2014, 53, 4633–4637; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4721–4725; [Google Scholar]
- 15b. Pal A., Vanka K., Inorg. Chem. 2016, 55, 558–565. [DOI] [PubMed] [Google Scholar]
- 16. Dunn N. L., Ha M., Radosevich A. T., J. Am. Chem. Soc. 2012, 134, 11330–11333. [DOI] [PubMed] [Google Scholar]
- 17.For selected review articles on metal-ligand cooperation, see:
- 17a. Lyaskovskyy V., de Bruin B., ACS Catal. 2012, 2, 270–279; [Google Scholar]
- 17b. Luca O. R., Crabtree R. H., Chem. Soc. Rev. 2013, 42, 1440–59; [DOI] [PubMed] [Google Scholar]
- 17c. Khusnutdinova J. R., Milstein D., Angew. Chem. Int. Ed. 2015, 54, 12236–12273; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12406–12445; [Google Scholar]
- 17d. Broere D. L. J., Plessius R., van der Vlugt J. I., Chem. Soc. Rev. 2015, 44, 6886–6915. [DOI] [PubMed] [Google Scholar]
- 18.For a recent review on main group-ligand cooperation, see: Greb L., Ebner F., Ginzburg Y., Sigmund L. M., Eur. J. Inorg. Chem. 2020, 3030–3047. [Google Scholar]
- 19.
- 19a. Blackmore K. J., Ziller J. W., Heyduk A. F., Inorg. Chem. 2005, 44, 5559–5561; [DOI] [PubMed] [Google Scholar]
- 19b. Haneline M. R., Heyduk A. F., J. Am. Chem. Soc. 2006, 128, 8410–8411; [DOI] [PubMed] [Google Scholar]
- 19c. Blackmore K. J., Lal M., Ziller J. W., Heyduk A. F., J. Am. Chem. Soc. 2008, 130, 2728–2729; [DOI] [PubMed] [Google Scholar]
- 19d. Blackmore K. J., Lal M., Ziller J. W., Heyduk A. F., Eur. J. Inorg. Chem. 2009, 735–743; [Google Scholar]
- 19e. Heyduk A. F., Zarkesh R. A., Hguyen A. I., Inorg. Chem. 2011, 50, 9849–9863. [DOI] [PubMed] [Google Scholar]
- 20.Selected examples of highly Lewis acidic P-centers:
- 20a. Holthausen M. H., Mehta M., Stephan D. W., Angew. Chem. Int. Ed. 2014, 53, 6538–6541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6656–6659; [Google Scholar]
- 20b. Zhou J., Liu L. L., Cao L. L., Stephan D. W., Chem 2018, 4, 2699–2708; [Google Scholar]
- 20c. Mehlmann P., Witteler T., Wilm L. F. B., Dielmann F., Nat. Chem. 2019, 11, 1139–1143; [DOI] [PubMed] [Google Scholar]
- 20d. Roht D., Stirn J., Stephan D. W., Greb L., J. Am. Chem. Soc. 2021, 143, 15845–15851. [DOI] [PubMed] [Google Scholar]
- 21. Chaudhuri P., Hess M., Müller J., Hildenbrand K., Bill E., Weyhermüller T., Wieghardt K., J. Am. Chem. Soc. 1999, 121, 9599–9610. [Google Scholar]
- 22.
- 22a. Burgada R., Laurenço C., J. Organomet. Chem. 1974, 66, 255–270; [Google Scholar]
- 22b. Tlahuextl M., Martínez-Martínez F. J., Rosalez-Hoz M. J., Contreras R., Phosphorus Sulfur Silicon 1997, 123, 5–19; [Google Scholar]
- 22c. Montgomery C. D., Boeré R. T., Dawn A., Jelier B. J., J. Chem. Crystallogr. 2013, 43, 127–133. [Google Scholar]
- 23. Addison A. W., Rao T. N., Reedijk J., van Rijn J., Verschoor G. C., J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar]
- 24.
- 24a. Clark T. E., Day R. O., Holmes R. R., Inorg. Chem. 1979, 18, 1668–1674; [Google Scholar]
- 24b. Sharma R., Ravikanth M., J. Porphyrins Phthalocyanines 2016, 20, 895–917; [Google Scholar]
- 24c. Gilhula J. C., Radosevich A. T., Chem. Sci. 2019, 10, 7177–7182; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24d. Eickhoff L., Ohms L., Bresien J., Villinger A., Michalik D., Schulz A., Chem. Eur. J. 2022, 28, e202103983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.While preparing this manuscript the synthesis of compound 10 was independently published: Volodarsky S., Malahov I., Bawari D., Diab M., Malik N., Tumanskii B., Dobrovetsky R., Chem. Sci. 2022, 13, 5957–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bent H. A., Chem. Rev. 1961, 61, 275–311. [Google Scholar]
- 27.Deposition Numbers 2153553 (for 9H 4 ), 2153554 (for 10), 2153556 (for 11), 2153555 (for 12), 2169268 (for 14), and 2153557 (for 18) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 28.
- 28a. Mayer U., Gutmann V., Gerger W., Monatsh. Chem. 1975, 106, 1235–1257; [Google Scholar]
- 28b. Beckett M. A., Strickland G. C., Holland J. R., Sukumar Varma K., Polymer 1996, 37, 4629–4631. [Google Scholar]
- 29. Erdmann P., Greb L., Angew. Chem. Int. Ed. 2022, 61, e202114550; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202114550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868; [DOI] [PubMed] [Google Scholar]
- 30b. Weigend F., Ahlrichts R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 31. Grimme S., Ehrlich S., Goerigk L., J. Comb. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 32. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- 33.Gaussian 16, Revision A.03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 34. Kästner J., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 158–168. [Google Scholar]
- 35. Chai J.-D., Head-Gordon M., Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Burés J., Angew. Chem. Int. Ed. 2016, 55, 2028–2031; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2068–2071; [Google Scholar]
- 36b. Burés J., Angew. Chem. Int. Ed. 2016, 55, 16084–16087; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16318–16321; [Google Scholar]
- 36c. Nielsen C. D.-T., Burés J., Chem. Sci. 2019, 10, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.
- 37a. Jerschow A., Müller N., J. Magn. Reson. 1996, 123, 222–225; [Google Scholar]
- 37b. Jerschow A., Müller N., J. Magn. Reson. 1997, 125, 372–375. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.