

Abstract
Abstract
Prophylactic creatine treatment may reduce hypoxic brain injury due to its ability to sustain intracellular ATP levels thereby reducing oxidative and metabolic stress responses during oxygen deprivation. Using microdialysis, we investigated the real‐time in vivo effects of fetal creatine supplementation on cerebral metabolism following acute in utero hypoxia caused by umbilical cord occlusion (UCO). Fetal sheep (118 days’ gestational age (dGA)) were implanted with an inflatable Silastic cuff around the umbilical cord and a microdialysis probe inserted into the right cerebral hemisphere for interstitial fluid sampling. Creatine (6 mg kg−1 h−1) or saline was continuously infused intravenously from 122 dGA. At 131 dGA, a 10 min UCO was induced. Hourly microdialysis samples were obtained from −24 to 72 h post‐UCO and analysed for percentage change of hydroxyl radicals (•OH) and interstitial metabolites (lactate, pyruvate, glutamate, glycerol, glycine). Histochemical markers of protein and lipid oxidation were assessed at post‐mortem 72 h post‐UCO. Prior to UCO, creatine treatment reduced pyruvate and glycerol concentrations in the microdialysate outflow. Creatine treatment reduced interstitial cerebral •OH outflow 0 to 24 h post‐UCO. Fetuses with higher arterial creatine concentrations before UCO presented with reduced levels of hypoxaemia ( and ) during UCO which associated with reduced interstitial cerebral pyruvate, lactate and •OH accumulation. No effects of creatine treatment on immunohistochemical markers of oxidative stress were found. In conclusion, fetal creatine treatment decreased cerebral outflow of •OH and was associated with an improvement in cerebral bioenergetics following acute hypoxia.

Key points
Fetal hypoxia can cause persistent metabolic and oxidative stress responses that disturb energy homeostasis in the brain. Creatine in its phosphorylated form is an endogenous phosphagen; therefore, supplementation is a proposed prophylactic treatment for fetal hypoxia.
Fetal sheep instrumented with a cerebral microdialysis probe were continuously infused with or without creatine‐monohydrate for 10 days before induction of 10 min umbilical cord occlusion (UCO; 131 days’ gestation). Cerebral interstitial fluid was collected up to 72 h following UCO.
Prior to UCO, fetal creatine supplementation reduced interstitial cerebral pyruvate and glycerol concentrations.
Fetal creatine supplementation reduced cerebral hydroxyl radical efflux up to 24 h post‐UCO. Fetuses with higher arterial creatine concentrations before UCO and reduced levels of systemic hypoxaemia during UCO were associated with reduced cerebral interstitial pyruvate, lactate and •OH following UCO.
Creatine supplementation leads to some improvements in cerebral bioenergetics following in utero acute hypoxia.
Keywords: cerebral metabolism, creatine, hypoxia‐ischaemia, microdialysis, oxidative stress
Abstract figure legend Prophylactic fetal creatine supplementation is a proposed treatment strategy for acute transient asphyxia such as umbilical cord occlusion (UCO). Fetal sheep with higher circulating arterial creatine (Cr) concentrations before UCO and reduced levels of systemic hypoxaemia during UCO were associated with reduced accumulation of cerebral interstitial pyruvate (Pyr), lactate (Lac) and reactive oxygen species (specifically •OH) following UCO as measured by microdialysis. There are three proposed mechanisms, which include maintaining ATP turnover during hypoxia and therefore reducing the need for aerobic metabolism, anaerobic processes, and active reactive oxygen species scavenging. Created with BioRender.com.
Introduction
Perinatal asphyxia remains one of the most prominent causes of childhood morbidity and mortality, affecting up to 26 per 1000 live births around the globe each year (Kurinczuk et al., 2010; Lawn et al., 2010). Transient episodes of acute asphyxia such as those caused by nuchal cords, placental abruption or umbilical cord compression can range from <1 to 30 min or more (Anyaegbunam et al., 1986; Goldsmith, 2015; Nelson & Grether, 1998; Osak et al., 1997). The fetus can temporarily maintain adequate cellular oxidative metabolism through rapid, chemoreceptor‐mediated changes in the distribution of cardiovascular output, ensuring that blood flow and oxygen delivery to the brain and heart are preserved, or even increased (Giussani, 2016). However, as the acute asphyxic event progresses, hypoxia and acidosis worsen, eventually leading to a compromise of cellular oxidative metabolism and, a cascade of metabolic and oxidative stress responses that can ultimately result in cell death (Ferriero, 2004; Gunn & Bennet, 2009). Even when normal oxygen supply to the tissues is reinstated, cellular metabolism remains disrupted, resulting in further pathological consequences arising from reperfusion injury as well as secondary and tertiary energy failure (Azzopardi et al., 1989; Gunn & Bennet, 2009; Roth et al., 1997; Wyatt, 2002).
At a cellular level, acute hypoxia causes a progressive loss of ATP and a shift to anaerobic respiration, and the resulting metabolic stress can cause increased reactive oxygen species (ROS), accumulation of anaerobic by‐products, pyruvate and lactate, and depolarisation of the cellular membranes leading to the release of excitotoxic neurotransmitters such as glutamate (Ferriero, 2004; Volpe, 2008). While high‐energy phosphates are transiently restored during the immediate recovery phase of hypoxic–ischaemic (HI) brain injury, another phase of decreased high‐energy phosphate levels (i.e. ATP and phosphocreatine) occurs at about 8–24 h post‐injury, a phenomenon known as secondary energy failure (Vannucci et al., 2004). This later phase of energy failure is associated with poorer prognoses and long‐term neurodevelopmental impairment that characterises hypoxia–ischaemia encephalopathy (HIE) (Azzopardi et al., 1989; Hamilton et al., 1986; Hope, 1984; Kagan et al., 2021).
Hypoxia provokes changes in both neurons and glia that affect metabolite release and re‐uptake, and fundamentally affects aerobic cellular metabolism (Logica et al., 2016). Previous studies in fetal sheep have described delayed and persistent increases in ROS and multiple cellular metabolites, particularly pyruvate, lactate, glucose and glutamate, in brain interstitial fluid following acute global hypoxia (Miller et al., 2005; Yan et al., 2009). ROS, such as the superoxide radical (O2 •−), hydrogen peroxide (H2O2) and the hydroxyl radical (•OH), are particularly implicated in hypoxia‐mediated cellular injury due to their ability to directly damage cellular macromolecules such as proteins, lipids and nucleic acids (Ikeda et al., 1999; Myers, 1975; Rao et al., 2011; Yawno et al., 2017). The fetal brain is no exception and is highly vulnerable to ROS‐mediated oxidative stress due to its high oxygen consumption, abundant levels of free iron and unsaturated fatty acids, as well as relatively immature development of antioxidant systems (Castagne, 1999; Mishra & Delivoria‐Papadopoulos, 1988). Accordingly, increased ROS‐induced cellular and macromolecule by‐products have been reported in the plasma of newborns diagnosed with HIE (Chafer‐Pericas et al., 2016; Raicevic et al., 2010; Rogers et al., 1997), as well as in animal models of HI (Coimbra‐Costa et al., 2017; Lafemina et al., 2006; Rocha‐Ferreira et al., 2016; Yawno et al., 2017). The combination of perturbed cellular energy metabolism and oxidative stress responses following hypoxia and associated energy failure suggests that preventing or reducing these responses may be an effective approach to improving perinatal HI outcomes.
Creatine supplementation is proposed as a prophylactic treatment in pregnancy for preventing hypoxia‐mediated brain to the fetal brain during labour and birth (Ellery et al., 2016; Ireland et al., 2011; Muccini et al., 2021; Tran et al., 2021). Creatine in its phosphorylated form, phosphocreatine, acts physiologically in vertebrates as an ATP buffer through the creatine kinase circuit to maintain ATP‐dependent cellular metabolism (Wyss & Kaddurah‐Daouk, 2000). Creatine and phosphocreatine act as a spatial energy buffer and a cytosolic source of high‐energy phosphates for rapid re‐phosphorylation of ADP (temporal energy buffer), thus sustaining cellular ATP during processes of high metabolic demand such as during physical exertion (Clark, 1997; Muccini et al., 2021), and also during periods of tissue hypoxia (Alberti, 1977). Creatine and phosphocreatine's energy stabilising capacity has the potential to buffer cellular ATP depletion following HI, thereby helping to preserve metabolic and redox balance (Andres et al., 2005; Shen & Goldberg, 2012). In addition, creatine has been shown to have both direct and indirect antioxidant actions in vitro in cultured cells (Lawler et al., 2002; Sestili et al., 2006).
We have previously shown in fetal sheep that arterial creatine levels were significantly increase by 4 days of continuous intravenous creatine supplementation, and by 13 days there were increases in total creatine content (creatine plus phosphocreatine content) in several regions of the fetal brain (Tran et al., 2021). Therefore, in this study, we used cerebral microdialysis to investigate if intravenous creatine infusion could reduce the generation of oxidative and metabolic stress markers following transient (10 min) hypoxia induced by mechanical umbilical cord occlusion (UCO). We also investigated if any associations existed between available arterial creatine concentrations prior to UCO, the changes in blood gas measurements experienced during UCO, and the accumulation of metabolites and •OH in brain interstitial fluid following UCO.
Methods
Ethical approval
Twenty‐eight pregnant Border–Leicester/Merino cross ewes carrying singletons at 118 days’ gestational age (dGA; term is ∼145–147 dGA) were sourced from a private supplier who used induced ovulation and time mating procedures to allow calculation of gestation to ±1 day. The use of animals was approved by Monash Medical Centre Animal Ethics Committee (MMCA‐2017‐02) and was conducted in accordance with the Australian Code of Practice for the care and use of Animals for Scientific Purposes established by the National Health and Medical Research Council of Australia. The ewes were placed separately in individual pens at the Monash Medical Centre animal house holding facility, always in the company of other sheep, and allowed to acclimatise for 6 days prior to surgery. During this time they were maintained in a 12 h light–dark cycle (08.00–20.00 h) at an ambient temperature of ∼20°C. Ewes were fed twice daily with a lucerne chaff mixture with access to water ad libitum. The well‐being and food and water intake of each ewe was monitored daily throughout the entire experiment.
Animal surgery
Prior to surgery and using a computer random number generator, the pregnant ewes were randomly assigned to either control or UCO group and then randomly assigned to either saline or creatine infusion (saline control, n = 6; creatine control, n = 7; saline UCO, n = 8; creatine UCO, n = 7). In this study, microdialysis probes were only inserted into the saline‐ and creatine‐treated UCO fetuses. Ewes and fetuses in this study were used in a previously published study detailing the experimental model and physiological outcomes of the creatine and cord occlusion protocols (Tran et al., 2021). Experimental protocols unique to this study are reported in detail below.
Briefly, at 118 dGA, after withdrawal of food for at least 18 h, each ewe was anaesthetised with intravenous sodium thiopentone (20 mg kg−1), intubated, and the anaesthesia then maintained by mechanical ventilation and inhalation of 1.5–3.5% isoflurane in oxygen:air (70:30). After sterile exteriorisation of the fetus during surgery and following the placement of an inflatable Silastic cuff around the umbilical cord, the fetuses were placed in the prone position and the head positioned for insertion of a microdialysis probe. An incision was made along the midline of the fetal head to expose the skull, and connective tissue cleared to reveal skull landmarks. A hole 1 mm in diameter was drilled into the skull using external co‐ordinates of 5 mm anterior of the coronal suture and 10 mm lateral to the midline of the right hemisphere. A microdialysis probe (CMA 20 Elite; 10 mm polyarylethersulfone membrane, 14 mm shaft length, 20 kDa cutoff; CMA Microdialysis, Kista, Sweden), connected to 1 m‐long inlet and outlet tubing (outer diameter: 2.5 mm, internal diameter: 1.7 mm; Microtube Extrusions, North Rocks, Australia), was inserted according to the manufacturer's instruction to a depth of 12 mm for sampling of interstitial fluid from parasagittal cortical grey matter and subcortical white matter. The probe was held in place using cyanoacrylate glue and dental acrylic. All fetal incisions were then closed using a continuous suture and all catheters, including a brachial artery and vein catheter and the umbilical cuff catheter, were exteriorised through the right flank of the ewe before closing all maternal incisions. Post‐surgery, a fentanyl patch (12.6 mg Durogesic 75, Sandoz, Boucherville, Canada) that provided 75 μg h−1 of analgesia was placed on the ewe's left inner thigh (Varcoe et al., 2019). The ewe was allowed 3 days of post‐operative recovery during which time antibiotics were administered daily (ewe: oxytetracycline, 50 mg in 5 ml, intravenous injection; fetus: ampicillin, 100 mg in 1 ml suspended in 25 IU ml−1 heparinised saline, intravenous injection; intra‐amniotic ampicillin, 400 mg in 4 ml suspended in 25 IU ml−1 heparinised saline) before the experimental procedures began.
Microdialysis experimental procedures
Ewes were housed in pens that restricted their sideways movement as soon as they had recovered from effects of post‐surgery anaesthesia and were moving freely, eating and drinking (usually, 3‐4 h). The microdialysis probe inlet and outlet were identified and both catheters extended with pre‐sterilised 0.5 m‐long vinyl tubing (outer diameter: 2.5 mm, internal diameter: 1.7 mm; Microtube Extrusions) using 100% ethanol‐soaked tubing adapters (CMA Microdialysis). The inlet catheter was connected to a syringe pump and the outlet connected to a fraction collector (BASi, West Lafayette, Indiana, USA). Freshly made artificial cerebrospinal fluid (aCSF; 148 mM NaCl, 4 mM KCl, 10 mM NaH2PO4, 40 mM Na2HPO4 in sterile water, pH 7.2–7.4) was passed through a 0.22 μm filter, and perfusion of the microdialysis probe at 1 μl min−1 was commenced. Care was taken to maintain patency within microdialysis probes by continually perfusing aCSF throughout the entire experimental protocol. Prior to starting the experiment, the probes were perfused at 1 μl min−1 and dialysate fluid was collected every 12 h and stored at −80°C. Terephthalic acid (5 mM TA; Sigma‐Aldrich, St Louis, MO, USA), a •OH trapping agent as used by our group previously (Yan et al., 2005), was added to the aCSF 42 h prior to commencing the experiment at 129 dGA to allow for equilibration with the interstitial fluid. Perfusion rate was then increased to 2 μl min−1 ∼50 h prior to performing the UCO at 130 dGA to increase dialysate volume collection to 120 μl h−1. Microdialysis fluid was collected hourly from 24 h prior to UCO and then continuously for 72 h after UCO. Dialysate was collected in capped borosilicate glass sample vials (BASi) using a fraction collector (BASi) and stored at −80°C until biochemical analysis was performed. Transit time from the probe tip to the outlet at the collection vial was determined to be 15 min, and therefore hourly samples were timed to account for this delay, i.e. an hourly sample collection began 15 min after the induction of UCO.
Creatine supplementation and UCO experimental procedures
The creatine supplementation and UCO protocol are described in detail elsewhere (Tran et al., 2021). Briefly, sterilised and filtered creatine monohydrate (Sigma‐Aldrich, St Louis, MO, USA) in saline was prepared at a concentration of 12 mg ml−1 and was delivered intravenously at 1.5 ml h−1 from 121 dGA at 09.00 h until 134 dGA; fetal weight was estimated to be 3 kg, thus, the infusion rate was ∼6 mg kg−1 h−1. Saline fetuses received an isovolumetric administration of 0.9% NaCl, pH 7.4 for the same duration. At 131 dGA, at 09.00 h, the Silastic cuff was inflated for 10 min using a predetermined volume of sterile water that would cause complete occlusion of the blood vessels in the umbilical cord; the internal cuff pressure was >100 mmHg. Successful UCO was determined by immediate hypertension and bradycardia (Tran et al., 2021). Control fetuses were not subjected to UCO.
Ewes were euthanised at 134 dGA with intravenous pentobarbital sodium (100 mg kg−1; Lethabarb; Virbac, Milperra, NSW, Australia) and allowing approximately a further 3 min for euthanasia of the fetus. The right hemisphere of the fetal brain was immediately immersion fixed in 4% paraformaldehyde (Merck) in 0.1 M phosphate buffer for 5 days and then coronally sectioned into 5 mm blocks for paraffin embedding.
2‐Hydroxy‐terephthalic acid HPLC
Terephthalic acid (TA) is an inert, non‐fluorescent molecule that forms a single stable isomer, 2‐hydroxy‐terephthalic acid (2‐OH‐TA) when oxidised with •OH. 2‐OH‐TA is highly fluorescent and can be detected at low concentrations at an excitation of 326 nm and emission at 432 nm (Yan et al., 2005), thereby providing a measurement of extracellular •OH concentration. Microdialysis samples were measured for 2‐OH‐TA efflux using HPLC as described previously (Yan et al., 2005). Due to technical issues, microdialysis samples from two animals from the saline UCO group and one animal from the creatine UCO group were not suitable for analysis owing to low and intermittent outflow; thus, the revised group numbers for successful microdialysis experiments are saline UCO, n = 6; creatine UCO, n = 6.
Briefly, a HPLC consisting of a solvent delivery system (LC‐10AT, Shimadzu, Kyoto, Japan), an automatic injector (SIL‐20AC, Shimadzu), an online degasser (DGU‐14A, Shimadzu), a C18 reverse‐phase column (5 μm particle size, 100 Å pore size, 250 mm × 4.6 mm; Luna, Phenomenex, Lane Cove, NSW, Australia), a fluorescent detector set at excitation 326 nm and emission 432 nm (RF‐20A, Shimadzu), and data collection software (LabSolutions, Shimadzu) was used. Microdialysis samples were diluted to 10% with mobile phase (v/v), and 10 μl of diluted sample was automatically injected into the HPLC system with a filtered mobile phase, 50 mM KH2PO4 in 30% (v/v) methanol (Sigma‐Aldrich); and pH adjusted to 3.2 with 1 M H3PO4 (Sigma‐Aldrich) at a flow rate of 0.7 ml min−1. Elution of 2‐OH‐TA occurred at 10.13 min, and 2‐OH‐TA concentration was determined from a standard curve generated from dissolved 2‐OH‐TA standards (Merck) after integration of the peak area under the curve (AUC). Assays of samples and standards were run with a calculated inter‐assay variability of 1.96% and intra‐assay variability of 0.58%.
GC‐MS
Targeted metabolomics was performed on microdialysis samples using GC‐MS. Due to low volumes caused again by inadequate outflow, one animal from each group was further removed from these analyses, i.e. revised group number for GC‐MS analysis is saline UCO, n = 5; creatine UCO, n = 5. In addition, not all of the hourly samples were analysed, but all time point samples that were measured are reported. An Agilent HP 6890N system equipped with a VF‐5 ms capillary column with 10 m Eziguard (J&W Scientific, Folsom, California, USA, 30 m, 250 μm inner diameter, 0.25 μm film thickness) and an Agilent 5975 MSD (Agilent Technologies, Santa Clara, CA, USA) in the electron ionisation mode with helium as the carrier gas were used. Using 1 ml glass vials (Verex, Phenomenex), 50 μl of sample, extraction blanks (MillQ water) or series of serially diluted external unlabelled standards (lactate, pyruvate, glutamate, glycine and glycerol) were mixed with 50 μl of analytical grade methanol (Fisher Scientific, Waltham, Massachusetts, USA) containing a mixture of stable isotope labelled internal standards (14.4 mM sodium [13C3]pyruvate (Sigma‐Aldrich), 23.7 mM sodium [13C3]l‐lactate (Sigma‐Aldrich), 10 mM [13C3]glycerol (Sigma‐Aldrich) and 2.5 mM 13C‐ and 15N‐labelled amino acid mix (MSK‐A2‐1.2; Cambridge Isotope Laboratories, USA, Tewksbury, Massachusetts, USA)). Samples were dried under vacuum for 40 min at 37°C. Following the drying procedure, which aided in precipitation of salt and small proteins, a second extraction step was performed which involved the addition of 100 μl of cold analytical grade ethanol (Fisher Chemicals) to all samples. The glass vials were then capped, vortexed and centrifuged for 5 min at 4°C 37,000 g. Approximately 100 μl of supernatant was transferred to 250 μl glass inserts in GC glass vials (Agilent) and again dried under vacuum for 30 min at 37°C. Samples were then first derivatised with 25 μl of pyridine containing 20 mg ml−1 methoxyamine hydrochloride (Sigma‐Aldrich), and then incubated for 1.5 h at 40°C. A second derivatisation step was performed by the addition of 25 μl of N‐methyl‐N‐(tert‐butyldimethylsilyl)‐trifluoroacetamide with 1% tert‐butyl‐dimethyl‐chlorosilane (Sigma‐Aldrich), followed by incubation for 30 min at 60°C. Two microlitres of the derivatised sample was injected in the splitless mode for a total GC run time of 18 min at a constant helium flow rate of 1 ml min−1. The tert‐butyl‐dimethylsilyl (TBDMS) derivatised samples were analysed by selected ion monitoring of lactate (261, 264 m/z), pyruvate (174, 177 m/z), glycerol (377, 380 m/z), glycine (246, 249 m/z) and glutamate (432, 438 m/z) ions. Absolute metabolite quantification was performed by integration of peak AUC for specific ions using Agilent Mass Hunter Quantitative analysis software (Agilent Technologies) and calculated from linear regression of the serially diluted external (unlabelled) standards using the isotope dilution technique. Assays of samples and standards were run with a calculated inter‐assay variability as follows: pyruvate, 8.4%; lactate, 31.6%; glycerol, 14.3%; glycine, 5.2%; and glutamate, 13.4%; and intra‐assay variability as follows: pyruvate, 1.3%; lactate, 0.5%; glycerol, 4.8%; glycine, 8.1%; and glutamate, 16.7%.
Immunohistochemistry
Immunohistochemistry was carried out on 8 μm paraffin‐embedded sections. Two sections from the parietal–temporal lobe were sampled corresponding to section 720 and section 1120 according to the Michigan State University Sheep Atlas (Johnson et al., 2013). Tissue slides were dewaxed in xylene, rehydrated in decreasing concentrations of ethanol, and antigen retrieval was conducted by heating in citrate buffer (10 mM tri‐sodium citrate in dH2O, pH 6.0; Sigma‐Aldrich). Sections were then rinsed with phosphate buffer saline (PBS), blocked for endogenous peroxidases with 0.3% hydrogen peroxide in methanol for 30 min at room temperature, washed in PBS again then blocked for non‐specific binding with 4% BSA in PBS for 4‐hydroxynonenal (4‐HNE) immunohistochemistry, or 10% normal rabbit serum in PBS for 3‐nitrotyrosine (3‐NT) immunohistochemistry for 1 h. Sections were then incubated overnight at 4°C with primary rabbit anti‐4‐HNE antibody (1:500, Merck Millipore, Burlington, Massachusetts, USA), or anti‐3NT antibody (1:200, Merck Millipore) in antibody diluent. Negative controls (no primary antibody) were included in all runs and demonstrated no positive staining. The sections were washed and incubated in secondary biotinylated IgG antibody (1:200 in PBS raised in corresponding animal). Antigen–antibody complex was visualised with avidin–biotin complex (ABC) (Vectastain, Vector Laboratories, Burlingame, CA, USA) and 3,3′‐diamniobenzidine (MP Biomedicals, Seven Hills, NSW, Australia). Immunohistochemistry stains were counterstained with haematoxylin accordingly.
Slides were scanned at ×20 magnification using an Aperio Scanscope AT Turbo (Leica Biosystems, Nußloch, Baden‐Wurttemberg, Germany). Regions of interest included the cortical grey matter (GM), subcortical white matter (SCWM), periventricular white matter (PVWM), corpus callosum (CC), striatum, putamen, internal capsule, thalamic nuclei and dorsal hippocampus (CA1–CA3 and dentate gyrus). Regions were outlined using Aperio Image Scope (Leica Biosystems) and positive‐stained antigens were manually counted for cell density (cells per μm2) analysis in 6–10 fields of view of 200 × 200 μm (except for hippocampal CA1–CA3 and CC, where it was 100 × 100 μm). All analyses were completed blinded to experimental treatment groups.
Data processing and statistical analyses
To determine the stability of basal microdialysis 2‐OH‐TA and cerebral metabolite efflux over the repeated microdialysis sampling period, baseline collections were compared using repeated measures two‐way ANOVA to assess for time or treatment main effects. No significant time effect on efflux levels was found, thereby demonstrating that basal efflux for each parameter was stable across the baseline period (−24 to −1 h). Thus, for each fetus, the means of baseline collections were calculated, and data presented as a percentage change from average basal levels to allow for inter‐individual and group comparisons.
To assess changes across time periods, the experimental timeline was separated a priori into specific time blocks: early recovery (0–8 h post‐UCO), delayed recovery (9–24 h post‐UCO; to account for secondary energy failure (Gunn & Bennet, 2009)), and late recovery (24–72 h post‐UCO); these time blocks correspond to that used previously (Tran et al., 2021). Each time block includes a pre‐UCO time point (−2 h post‐UCO) to assess significant changes from baseline. For each microdialysis analyte, a two‐way repeated measures ANOVA was conducted to assess treatment (P TREAT) and time following UCO (P TIME) effects and interaction (P INT) across each of these time blocks. Any significant main effect for time or interaction was followed up using Šidák's multiple comparisons post hoc testing.
For immunohistochemistry analyses, two sections per animal were analysed for greater sampling across the fetal brain. Regions that were analysed in both sections (i.e. SCWM, PVWM, GM, CC) were averaged across both sections if no significant sampling effect was found. Data sets were assessed for main effects of UCO (P UCO), main effects of creatine treatment (P TREAT) and interactions between the main effects (P INT) by two‐way ANOVA. Where a significant interaction was observed, post hoc analysis was performed using Tukey's multiple comparison test.
To determine the correlation between the plasma creatine concentration, blood gas measurements obtained during UCO, and the resulting changes in oxidative and metabolic cerebral metabolites, multiple linear regression modelling was conducted. The methodology and results of plasma creatine concentration and blood gas measurements have been reported elsewhere (Tran et al., 2021). The percentage change from baseline of blood gas parameters was calculated from the value obtained −30 min prior to the start of the UCO and the value at the 9 min time point of the 10 min duration of UCO, and was used to establish the degree of hypoxaemia, hypercapnia and lactic acidosis each fetus experienced. Raw arterial blood gases and metabolites (−30 min, +9 min and +72 h relative to UCO) are reported in Supplementary Data S2. The oxidative and metabolic cerebral metabolite changes were calculated as the AUC of the relative (percentage) change from average basal levels from 0 to 72 h after UCO. This approach was taken to represent the total impact of the hypoxia on the specific metabolite as a result of UCO. The concentration of arterial creatine prior to UCO was calculated as the average creatine plasma concentration across days 4–8 of creatine infusion, i.e. days 6–0 before the UCO. Within this analysis, the saline UCO and creatine UCO groups were combined to assess the effect of arterial creatine concentrations alone irrespective of treatment group. The assumption of little to no multi‐collinearity was confirmed if the variance inflation factor was <3.
Data are presented as means ± SD. Data were assessed for normality using the Shapiro–Wilk test. Statistical significance was accepted for P ≤ 0.05. ANOVA statistical analysis and regression modelling was computed using GraphPad Prism (Version 8.4.3 for Windows, GraphPad Software, San Diego, CA, USA). 2‐OH‐TA and metabolite efflux concentrations are reported uncorrected for probe membrane efficiency.
Results
Effects of creatine supplementation and UCO on interstitial •OH concentrations
Basal levels of 2‐OH‐TA
Basal cerebral levels of 2‐OH‐TA efflux were stable across the 24 h prior to commencement of the UCO (Table 1). The mean basal concentration of 2‐OH‐TA (uncorrected for probe membrane efficiency) for saline‐ and creatine‐treated fetuses was 0.40 ± 0.02 and 0.39 ± 0.03 μmol l−1, respectively (Table 1).
Table 1.
Two‐way ANOVA and combined average baseline concentrations of cerebral interstitial 2‐OH‐TA and metabolites in saline (n = 5–6)‐ or creatine (n = 5–6)‐treated fetuses in dialysate (uncorrected for dialysate recovery efficiency) during the 24 h prior to UCO
| Group | Two‐way ANOVA | ||||
|---|---|---|---|---|---|
| Saline | Creatine | P TREAT | P TIME | P INT | |
| 2‐OH‐TA (μmol l−1) | 0.40 ± 0.02 | 0.39 ± 0.03 | 0.893 | 0.261 | 0.474 |
| Pyruvate (μmol l−1) | 31.32 ± 16.98 | 21.70 ± 12.34 | 0.049 * | 0.659 | 0.767 |
| Lactate (μmol l−1) | 671.00 ± 164.70 | 748.10 ± 181.00 | 0.597 | 0.201 | 0.636 |
| Glycerol (μmol l−1) | 8.52 ± 2.74 | 6.52 ± 3.29 | 0.049 * | 0.979 | 0.866 |
| Glycine (μmol l−1) | 29.05 ± 1.76 | 29.55 ± 6.65 | 0.922 | 0.135 | 0.060 |
| Glutamate (μmol l−1) | 9.75 ± 2.63 | 7.55 ± 2.66 | 0.470 | 0.678 | 0.921 |
Data are means ± SD. * P ≤ 0.05: statistically significant creatine treatment effects.
Effects of UCO on 2‐OH‐TA
There were no time or interaction effects for 2‐OH‐TA concentrations in the dialysate outflow across any of the post‐UCO time intervals (Supplementary Data S1). However, creatine treatment led to a decrease in the 2‐OH‐TA concentration compared to saline fetuses during the early recovery phase (0–8 h post‐UCO; F(1, 10) = 4.96, P TREAT = 0.050; Fig. 1; Supplementary Data S1). A similar effect for creatine treatment to reduce 2‐OH‐TA concentrations below saline‐treated fetuses occurred in the delayed recovery phase (9–24 h post‐UCO; F(1, 10) = 4.90, P TREAT = 0.050). By late recovery, 24–72 h after UCO, there were no differences of 2‐OH‐TA levels between the treatment groups (F(1, 10) = 1.29, P TREAT = 0.283).
Figure 1. Percentage change from baseline 2‐OH‐TA microdialysis efflux in the fetal subcortical white matter and cortical grey matter of saline‐treated (filled circles; n = 6) and creatine‐treated (open circles; n = 6) fetuses following 10 min UCO occurring at 0 h (grey bar).

First data point represents 1 h pre‐UCO. Data calculated as percentage change from pre‐UCO values and presented as means ± SD (saline‐UCO, n = 6; creatine‐UCO, n = 6). * P ≤ 0.05 indicates statistically significant main effect of creatine treatment.
Effects of creatine supplementation and UCO on cerebral metabolism
Basal concentration of metabolites
Basal interstitial concentrations of lactate, glycine and glutamate during the 24 h prior to UCO (i.e. at −24, −18 −12, −6, −2 h prior to UCO) did not differ between the saline or creatine treatment groups (P TREAT > 0.05; Table 1). In contrast, basal levels of pyruvate and glycerol efflux during the 24 h prior to UCO were significantly lower in creatine‐treated fetuses than in saline‐treated fetuses (both P TREAT = 0.049; Table 1). As no significant fluctuations with time during the baseline microdialysis sampling period were found for any of the metabolites (−24 to −2 h; P TIME > 0.05; Table 1), the pre‐UCO data were used to calculate the relative percentage change from the average baseline levels for each animal (Fig. 2A–E ).
Figure 2. Metabolite efflux from microdialysis sampling in the fetal subcortical white matter and cortical grey matter following UCO.

Saline‐treated (filled circles; n = 5) and creatine‐treated (open circles; n = 5) fetuses underwent a 10 min UCO occurring at 0 h (grey bar). Levels of pyruvate (A), lactate (B), glycerol (C), glycine (D), and glutamate (E) efflux are presented as percentage change from baseline. There were no significant interaction effects of time and treatment. #P ≤ 0.05, ##P ≤ 0.01 refer to a time main effect and indicate statistically significant differences compared to baseline in both saline‐ and creatine‐treated fetuses. Data are means ± SD.
Effects of UCO on interstitial metabolite concentrations
There were no interaction or creatine treatment effects for any of the metabolite concentrations across any recovery time period following UCO (Supplementary Data S1). There were significant time effects for pyruvate, lactate and glycerol occurring mainly during the early recovery period. The UCO induced a rise in pyruvate, lactate and glycerol concentrations in the cerebral interstitial fluid with a similar temporal pattern identified in both saline‐ and creatine‐treated fetuses during the early recovery stage (0–8 h post‐UCO; F(1.72, 12.45) = 7.39, P TIME = 0.009; F(2.05, 14.35) = 4.79, P TIME = 0.025 and F(2.26, 16.38) = 6.24; P TIME = 0.008 respectively, Fig. 2A–C , Supplementary Data S1). Pyruvate concentrations were significantly increased up to ∼69% compared to baseline and remained elevated up to 8 h post‐UCO in both treatment groups (Fig. 2A ). Lactate and glycerol concentrations were significantly increased up to 6 h post‐UCO with efflux reaching peak increases of ∼34% and ∼201%, respectively (Fig. 2B and C ). By the delayed recovery stage (9–24 h post‐UCO), both pyruvate and lactate concentrations had returned to near baseline in both treatment groups (P TIME > 0.05). Glycerol concentrations remained affected 9–24 h following UCO irrespective of treatment (F(3.55, 27.49) = 2.94, P TIME = 0.043; Supplementary Data S1), and returned by late recovery (>24 h post‐UCO). Levels of glycine and glutamate were not affected by UCO, though there was a near‐to‐significant interaction between time and creatine treatment during the late recovery period for glycine concentrations (F(8, 63) = 1.98, P INT = 0.064) (Fig. 2D and E , Supplementary Data S1).
Relationship between arterial creatine concentration and blood gas measurements during UCO on metabolic changes within the fetal brain
Potential relationships between the circulating arterial creatine concentrations prior to UCO, the degree of hypoxaemia, hypercapnia and lactic acidosis experienced by each fetus, and changes in cerebral 2‐OH‐TA and metabolites following UCO were then assessed with multivariate regression analyses. Overall, there were significant associations between fetal arterial creatine concentrations, arterial and , and the cumulative percentage change of cerebral interstitial pyruvate, lactate and 2‐OH‐TA (Fig. 3; Supplementary Data S3). The level of arterial creatine was positively correlated with the percentage change of and , which were then negatively associated with percentage change in cerebral pyruvate accumulation, suggesting that higher levels of arterial creatine and reduced changes in arterial and during UCO resulted in lower levels of pyruvate accumulation (P = 0.006 and P < 0.0001, respectively; Fig. 3; Supplementary Data S3). A similar relationship between higher levels of creatine and reduced changes in arterial during UCO resulting in lower levels of cerebral lactate accumulation was also found (P = 0.017; Fig. 3; Supplementary Data S3). In addition, a significant relationship between arterial creatine, arterial and cerebral 2‐OH‐TA was observed, in which higher arterial creatine and reduced changes in during UCO was associated with lower cerebral 2‐OH‐TA levels (P = 0.050; Fig. 3; Supplementary Data S3).
Figure 3. Significant relationships between arterial creatine concentration prior to UCO and percentage change of arterial (A) and arterial (B) during UCO to predict cerebral pyruvate accumulation following UCO; arterial to predict cerebral lactate levels (C); and arterial to predict cerebral 2‐OH‐TA levels (D).

Each data point represents a fetus; note there is no measurement of for one fetus due to technical error. n (number of cases analysed) = 9–11. [Colour figure can be viewed at wileyonlinelibrary.com]
Effects of creatine supplementation and UCO on histochemical markers of oxidative stress
4‐Hydroxynonenal
Lipid peroxidation (4‐HNE immunoreactivity) was found in the cytoplasm in all of the fetal brain regions examined, with some cells displaying intense positive staining within the nucleus (Fig. 4). The level of 4‐HNE immunoreactivity was increased in both saline‐ and creatine‐treated fetuses following UCO in the SCWM and thalamus (F(1, 24) = 6.06, P UCO = 0.021; F(1, 24) = 4.84, P UCO = 0.038, respectively; Table 2 and Fig. 4). Creatine treatment did not alter the intensity of 4‐HNE immunoreactivity or the cell density in any of these regions in either control or UCO fetuses.
Figure 4. Representative immunohistochemical staining of 4‐hydroxynonenal (4‐HNE) in subcortical white matter (SCWM) and thalamus.

Cell nuclei were counterstained with haematoxylin. Black arrows indicate positive staining of 4‐HNE within cells. Bar corresponds to 100 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 2.
Cell density counts (cells mm−2) and two‐way ANOVA of main effects and interaction of immunohistochemical staining of 4‐hydroxynonenal (4‐HNE) and 3‐nitrotyrosine (3‐NT)
| Control | UCO | Statistics | |||||
|---|---|---|---|---|---|---|---|
| Brain region | Saline (n = 6) | Creatine (n = 7) | Saline (n = 8) | Creatine (n = 7) | P UCO | P TREAT | P INT |
| 4‐HNE | |||||||
| SCWM | 118.75 ± 64.65 | 138.39 ± 48.13 | 186.88 ± 78.93 | 194.46 ± 67.58 | 0.021 * | 0.594 | 0.813 |
| GM | 114.38 ± 37.89 | 74.11 ± 34.68 | 121.09 ± 62.81 | 137.50 ± 42.53 | 0.061 | 0.510 | 0.125 |
| PVWM | 143.13 ± 99.72 | 171.61 ± 86.86 | 203.75 ± 86.78 | 174.64 ± 98.71 | 0.375 | 0.993 | 0.422 |
| Hipp DG | 50.00 ± 32.17 | 86.31 ± 58.67 | 74.48 ± 32.77 | 102.98 ± 43.88 | 0.223 | 0.060 | 0.814 |
| Hipp CA1–3 | 222.22 ± 223.03 | 264.29 ± 220.57 | 420.83 ± 277.42 | 390.48 ± 218.34 | 0.086 | 0.949 | 0.693 |
| Thalamus | 63.89 ± 37.79 | 64.29 ± 26.34 | 92.19 ± 47.37 | 112.5 ± 62.27 | 0.038 * | 0.557 | 0.572 |
| CC | 125.00 ± 133.65 | 95.24 ± 93.15 | 102.08 ± 65.12 | 85.71 ± 74.80 | 0.648 | 0.517 | 0.850 |
| Internal capsule | 14.58 ± 14.61 | 88.10 ± 70.85 | 43.75 ± 65.16 | 51.19 ± 70.99 | 0.870 | 0.100 | 0.171 |
| Putamen | 36.81 ± 41.95 | 56.55 ± 43.56 | 38.54 ± 37.38 | 35.71 ± 33.67 | 0.527 | 0.575 | 0.455 |
| Striatum | 18.06 ± 13.35 | 26.77 ± 13.36 | 25.00 ± 11.36 | 29.76 ± 16.91 | 0.354 | 0.211 | 0.709 |
| 3‐NT | |||||||
| SCWM | 45.83 ± 40.73 | 9.64 ± 11.40 | 61.72 ± 67.98 | 53.93 ± 32.03 | 0.088 | 0.206 | 0.410 |
| GM | 31.88 ± 14.81 | 20.89 ± 12.54 | 35.16 ± 18.76 | 45.36 ± 18.01 | 0.036* | 0.951 | 0.102 |
| PVWM | 24.58 ± 28.89 | 18.04 ± 28.87 | 43.28 ± 57.71 | 37.68 ± 28.74 | 0.214 | 0.689 | 0.975 |
| Hipp DG | 12.50 ± 22.82 | 0.00 ± 0.00 | 9.90 ± 6.66 | 23.81 ± 24.26 | 0.102 | 0.911 | 0.045* |
| Hipp CA1–3 | 2.78 ± 6.81 | 0.00 ± 0.00 | 10.42 ± 17.68 | 33.33 ± 60.09 | 0.102 | 0.411 | 0.296 |
| Thalamus | 21.53 ± 19.62 | 4.17 ± 4.81 | 38.02 ± 30.41 | 23.81 ± 23.04 | 0.042* | 0.072 | 0.853 |
| CC | 5.56 ± 8.61 | 9.52 ± 16.26 | 43.75 ± 63.58 | 11.11 ± 13.61 | 0.177 | 0.326 | 0.213 |
| Internal capsule | 2.78 ± 5.05 | 8.33 ± 11.54 | 3.65 ± 4.69 | 8.33 ± 8.74 | 0.890 | 0.111 | 0.889 |
| Putamen | 9.72 ± 18.38 | 2.98 ± 3.96 | 10.42 ± 13.36 | 3.47 ± 4.87 | 0.900 | 0.145 | 0.983 |
| Striatum | 2.78 ± 5.05 | 10.12 ± 7.93 | 17.19 ± 25.63 | 16.07 ± 11.64 | 0.100 | 0.605 | 0.484 |
Data are means ± SD. Two‐way ANOVA: * P ≤ 0.05 indicates statistically significant main effects or interaction. CC: corpus collosum; GM: grey matter; Hipp CA1–3: hippocampus cornu ammonis; Hipp DG: hippocampus dentate gyrus; PVWM: periventricular white matter; SCWM: subcortical white matter.
3‐Nitrotyrosine
Free radical‐mediated cell damage can result in 3‐NT modification of proteins (Fig. 5). The cellular density of 3‐NT positive immunoreactivity increased following UCO in the cortical GM and thalamus in both saline‐ and creatine‐treated fetuses (F(1, 24) = 4.96, P UCO = 0.036; F(1, 24) = 4.63, P UCO = 0.042, respectively; Table 2). 3‐NT immunoreactivity was predominately within the cytosol, with some positive staining also observable along axons, especially in the cortical GM (Fig. 5). In the hippocampus dentate gyrus (DG), there was a significant interaction between UCO and creatine treatment on 3‐NT (F(1, 24) = 4.50, P INT = 0.045), with 3‐NT immunoreactivity almost entirely absent in creatine‐treated controls, but elevated in creatine‐treated fetuses subjected to UCO (P = 0.055), but not compared to the saline‐treated fetuses after UCO. There were no correlations between the total magnitude of change of 2‐OH‐TA following UCO on the cell density of 4‐HNE and 3‐NT positive immunostaining within the SCWM and GM (Supplementary Data S4).
Figure 5. Representative immunohistochemical staining of 3‐nitrotyrosine (3‐NT) in cortical grey matter (GM), hippocampus dentate gyrus (Hipp DG) and thalamus.
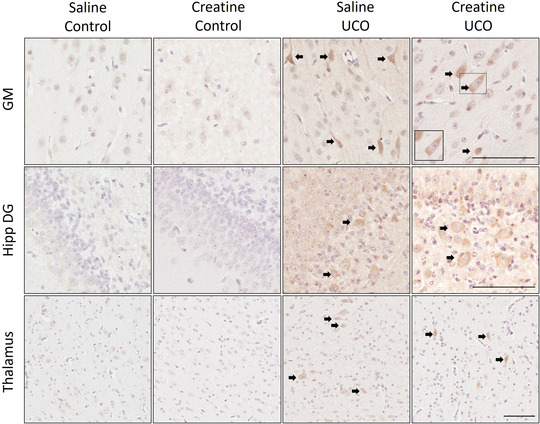
Cell nuclei were counterstained with haematoxylin. Black arrows indicate positive staining of 3‐NT within cells. Bar corresponds to 100 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
In this study we used microdialysis probes inserted into the near‐term fetal sheep brain to capture shifts in interstitial fluid metabolites following acute in utero hypoxia. The key findings of this study were, firstly, that creatine supplementation reduced basal pyruvate and glycerol levels within the cerebral interstitial fluid in the absence of hypoxia; secondly, that creatine treatment reduced hydroxyl radical (·OH) efflux up to 24 h following UCO. However, creatine supplementation did not alter lipid and protein oxidation that was observable in specific brain regions at 72 h following UCO. Lastly, we report a relationship where fetuses with higher arterial creatine concentrations displayed smaller (reduced) changes in arterial and during UCO and reduced pyruvate, lactate and 2‐OH‐TA accumulation within the fetal cerebral interstitial fluid following UCO.
Creatine and oxidative stress
This is the first study to demonstrate an in vivo reduction in •OH in the fetal brain following creatine treatment and UCO. Further, multivariate regression analysis revealed that fetuses with higher circulating creatine levels were associated with reduced 2‐OH‐TA and a reduced degree of hypoxaemia during the UCO. We hypothesise that this relationship results from a decrease in fetal tissue aerobic metabolism due to the increased creatine levels maintaining ATP turnover for longer under hypoxic conditions, thereby reducing the need for oxidative phosphorylation (Scopes, 1973). The relationship between reduced hypoxaemia during UCO and the smaller changes of cerebral pyruvate and lactate (discussed further below) also supports the hypothesis that there is a reduced reliance on mitochondrial bioenergetics during UCO conferred by increased creatine levels (Bouillaud et al., 2021). We have previously reported from the same cohort of fetuses that creatine supplementation had no effect on state I, IV or III mitochondrial respiration in the cerebral grey or white matter 72 h after UCO (Muccini et al., 2022). While the relationship between oxygen deprivation and ROS production in the brain is well established (Abramov et al., 2007; Niatsetskaya et al., 2012), our results suggest there is now a need to ascertain in real time if creatine preserves mitochondrial bioenergetics during acute hypoxia and investigate the actual cellular and mitochondrial oxygen flux and subsequent ROS rates (Tran et al., 2021).
Improved mitochondrial bioenergetics alone would not explain why 2‐OH‐TA was consistently below baseline during the immediate and early recovery period in the creatine‐treated fetuses. This may not be due to increased •OH scavenging by intracellular antioxidant defence systems, given that creatine treatment does not enhance the expression and activity of the main antioxidant enzymes such as superoxide dismutase, glutathione peroxidase and catalases in skeletal muscle (Guimarães‐Ferreira et al., 2012; Lawler et al., 2002). A possibility is that creatine can directly scavenge ROS moieties. In a cell‐free paradigm, Lawler et al. (2002) and Guimarães‐Ferreira et al. (2012) both showed that creatine had direct scavenger effects on the superoxide radical (O2 •−) and the peroxynitrite (OONO−) anion, but not on the generation of hydrogen peroxide (H2O2), although Sestili et al. (2006) was able to demonstrate this capacity for direct scavenging of H2O2 in cultured cells. The effects of creatine treatment on the efflux of these other ROS were not assessed in this study, however, as •OH is formed by successive reduction of oxygen (Chen & Schopfer, 1999), the potentially reduced production of these oxidative metabolites may explain why the average percentage change of •OH was consistently below zero.
Another potential mechanism for direct reduction of •OH could involve creatinine, the non‐enzymatic breakdown product of creatine, and its capacity to also scavenge •OH radicals (Ienaga & Yokozawa, 2011; Nakamura et al., 1991). Given we have shown total creatine content is increased in the fetal brain with supplementation (Tran et al., 2021), there may also be a relative increase in the presence of creatinine within the cerebral parenchyma. This would require direct measurement as it remains unknown if creatine supplementation increases brain creatinine levels, and there are conflicting studies on whether serum creatinine levels increase following creatine supplementation (Taes, 2003; Williamson & New, 2014). While it has been established that creatine and creatinine have a lower direct scavenging capacity compared to other enzymatic antioxidants (Lawler et al., 2002), our findings suggest it could support active scavenging of excess ROS in the fetal brain given the limited antioxidant capacity of fetal tissues (Gitto et al., 2009; Saugstad, 1996).
Finally, the known vasodilator effects of increased creatine could have contributed to lower 2‐OH‐TA following UCO with creatine treatment (Prass et al., 2006). Vasodilatation may have resulted in an effective washing out of •OH from the extracellular space, but not before this ROS had been able to oxidatively modify cell proteins and lipids. Clearly, a study investigating the effects of increased circulating creatine on cerebral blood flow in the fetal brain should be conducted, perhaps using microspheres so that regional effects on blood flow can be revealed.
Despite the implied antioxidant effects discussed above, and our observed changes in brain metabolites, there was no indication that creatine treatment had ameliorated the increased lipid (4‐HNE) and protein (3‐NT) oxidation in the SCWM, GM and thalamus observed at the end of the experiment, at 72 h after UCO. The dynamic changes in •OH efflux found in the current study were not different between groups after 24 h post UCO, i.e. 2 days before histochemical assessments were made. This suggests that despite the seeming return to normal oxidative metabolism, there are possibly other persistent cellular perturbations, or remnants of cellular damage that were not ameliorated by the creatine treatment.
Creatine and cellular metabolites
The efflux of cellular metabolites such as pyruvate, lactate and glycerol occurred in clearly defined phases. Pyruvate and lactate efflux increased soon after the UCO and had returned to baseline levels by 8 h post‐UCO, while glycerol levels remained increased from UCO induction up to 24 h post‐UCO. The time to metabolite concentration recovery to baseline is markedly slower compared to other studies in rats that report recovery within 1 h following reoxygenation (Roehl et al., 2012; Zoremba et al., 2007). This slow recovery in the fetal sheep brain might be due to a longer reliance on glycolysis after oxygenation had been restored in this in utero ovine model.
The increased pyruvate changes following UCO in the fetuses that experienced greater hypoxaemia during UCO is consistent with the fact that the greater the hypoxia, the greater the need for anaerobic glycolysis with associated increases in pyruvate and lactate production (Siesjö & Nilsson, 1971). Fetuses with higher arterial creatine concentrations and reduced decreases in and during the UCO were associated with lower cerebral pyruvate and lactate accumulation suggesting a reduced need for this metabolic shift during and early in recovery following hypoxia. These observations are consistent with studies in intensely contracting skeletal muscle which together support that elevated creatine reduced the shift to glycogenolysis and anaerobic glycolysis during hypoxia and thus reduced changes in pyruvate and lactate flux (Balsom et al., 1995; Scopes, 1973).
Another interesting outcome of this study was an observed reduction in basal pyruvate and glycerol levels in creatine‐treated fetuses, prior to UCO. Pyruvate is generated via glycolysis and other cytosolic sources but serves ultimately as a substrate for the mitochondrial citric acid cycle for oxidative phosphorylation and ATP generation. Flux between intracellular and extracellular pyruvate is rapid, with levels between the compartments demonstrated to be closely comparable, at least in vitro (O'donnell‐Tormey et al., 1987; Quek et al., 2016). Accordingly, the decrease in extracellular pyruvate observed with creatine treatment most likely reflects reduced intracellular pyruvate production. Indeed, increased intracellular phosphocreatine inhibits key glycolytic enzymes – phosphofructokinase and pyruvate kinase – which would subsequently reduce pyruvate production (Kemp, 1973; Storey & Hochachka, 1974). This feedback control is integral to the buffering capability of the creatine kinase phosphagen system as depletion of phosphocreatine and ATP following increased metabolic demand would then lead to the activation of glycolysis to maintain cellular bioenergetics. The decrease in basal cerebral interstitial pyruvate concentration occurred in the absence of changes to basal lactate levels with creatine treatment, suggesting no adverse changes to basal cellular metabolism (Kuhr & Korf, 1988) and maintenance of lactate shuttling between astrocytes and neurons (Pellerin et al., 1998).
The decrease in the basal cerebral interstitial levels of glycerol with creatine supplementation was an unexpected finding, particularly as 2‐OH‐TA levels were not changed, nor were those of any other marker suggestive of oxidative cell damage. The reduced glycerol concentrations within the interstitial fluid could have occurred because of reduced lipolysis or increased neuronal glycerol utilisation via phosphorylation or oxidation (Nguyen et al., 2003). Indeed, in vitro and in vivo studies of creatine supplementation have observed reduced accumulation of triacylglycerol in the liver (Da Silva et al., 2014; Da Silva et al., 2017; Earnest et al., 1996), suggesting that creatine might reduce circulating fatty acids and/or increase fatty acid oxidation. Whether similar mechanisms due to creatine occur in the brain is not known. Fatty acids are actively catabolised for energy production in the fetal brain and are important for fetal brain development as well as during the perinatal transition (Clandinin et al., 1980; Desoye & Herrera, 2021; Steiner, 2019). Therefore, the effect of creatine supplementation during pregnancy on fetal cerebral cellular energy metabolism and fatty acid requirements, even in the absence of hypoxia, needs to be investigated further.
Methodological considerations
The key methodological consideration of this study is the use of microdialysis itself. The insertion of the microdialysis probe into tissue causes mechanical injury and after a time will induce gliosis, ultimately reducing the efficiency and recovery rate of dialysates (Bungay et al., 2003). While there are mathematical modelling approaches to determine microdialysis efficiency, they do not take into account endogenous microvasculature remodelling which must occur after such wounding by implantation of a probe, degree of brain tissue sensitivity, blood flow, tissue resistance and metabolism (Chen et al., 2002; Justice, 1993; Morrison et al., 1991), and also in the circumstance of the fetal brain which continues to develop over time. However, the long duration of the probe implantation in the current study (16 days) is less of a confounding factor because a degree of wound repair must have occurred, as shown by the relative stability of the efflux measurement in the days before the UCO was undertaken. Thus, while probe efficiency and therefore actual metabolite concentrations could not be calculated, the relative changes in metabolite flux allowed us to examine transient and dynamic changes over time.
It is important to note that in vivo recovery of microdialysis dialysate is influenced by local blood flow which may have been affected by the UCO itself. As mentioned above, increases in local blood flow may lower in vivo dialysate recovery due to increased clearance of molecules from the tissue bed, thereby underestimating measurements within interstitial fluid. This is of particular importance as changes in cerebral blood flow have been identified up to 4 days post asphyxia, corresponding also to the severity of hypoxic injury (Bennet et al., 1998; Van Bel et al., 1987), as well as an impairment of cerebral vasoreactivity caused by oedema or vascular endothelial injury (Rosenberg, 1988). Whether creatine supplementation affects cerebral blood flow in the fetal brain is unknown but, as mentioned, creatine is known to have vasodilatory effects in the adult brain (Prass et al., 2007). Therefore, relative changes between treatment groups, i.e. saline UCO vs. creatine UCO, may be under‐ or overestimated. Nonetheless, the data from this study demonstrate that the consequences of altered cerebral metabolism can be measured robustly by the efflux of specific cellular metabolites in the fetal brain using microdialysis.
We report here, as well as in our previous study of the same animals (Tran et al., 2021), that average arterial plasma creatine concentrations were significantly higher in fetuses supplemented with creatine for 10 days, though there remained a range of initial arterial plasma creatine concentrations irrespective of creatine treatment as well as a range of arterial blood gas and metabolite absolute values during UCO. Thus, a strength of conducting multiple variable regression modelling is that it accounts for subject variability, such as that often observed clinically. Furthermore, the multivariate analysis revealed that denoting the degree of hypoxaemia by the relative changes of blood gases, rather than from the absolute values, was predictive of cerebral metabolic changes that followed the UCO. While measurement of relative or absolute changes of fetal oxygenation is unlikely to be clinically feasible, the potential relationships we have identified warrant further investigation.
Conclusion
This is the first study to investigate the in vivo temporal metabolic and oxidative milieu of the fetal cerebral interstitial space following creatine supplementation and acute mild hypoxia arising from UCO. While histological assessments 72 h after UCO may not have been able to fully capture beneficial changes afforded by fetal creatine treatment within the brain, our results demonstrate that fetuses with increased levels of arterial creatine prior to UCO displayed a lower degree of hypoxaemia and were likely to have improved cellular bioenergetics, i.e. reduced cerebral pyruvate, lactate and 2‐OH‐TA accumulation. Further studies are warranted to investigate the effects of these reduced metabolic and oxidative stress responses at different time points relating to the peak metabolic and oxidative stress response and long‐term postnatal effects. Overall, the findings of this study provides evidence for creatine supplementation to the fetus to improve cellular metabolic stability as well as reduce reactive oxygen species efflux, specifically •OH, within the fetal brain following an acute asphyxic event.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
Conceptualisation: N.T.T., R.J.S., D.W.W. and S.J.E.; methodology: N.T.T., G.M.K., I.N., N.H., R.J.S., D.W.W. and S.J.E.; data curation: N.T.T. and G.M.K.; data analysis: N.T.T.; writing – original draft preparation: N.T.T.; writing – review and editing: N.T.T., G.M.K., A.M.M., I.N., R.J.S., D.W.W. and S.J.E. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
N.T.T. was supported by a PhD scholarship from the School of Health & Biomedical Sciences, RMIT University. This research was supported by a grant to D.W.W., R.J.S. and S.J.E. from the National Health & Medical Research Council of Australia (1124493), and to D.W.W. and S.J.E. from the Victorian Government Infrastructure Support Scheme. S.J.E. was supported by Australian National Health and Medical Research Council (NHMRC) Early Career Fellowship (1125539).
Supporting information
Peer Review History
Statistical Summary Document
Supplementary Data S1.
Supplementary Data S2.
Supplementary Data S3.
Supplementary Data S4.
Acknowledgements
The authors acknowledge the technical support provided by Peter Boklund from CMA Microdialysis for microdialysis probe design.
Open access publishing facilitated by RMIT University, as part of the Wiley – RMIT University agreement via the Council of Australian University Librarians.
Biography
Nhi Thao Tran is a Post‐Doctoral Scientist in the Perinatal Transition Research Group at the Ritchie Centre, The Hudson Institute of Medical Research, Australia. She obtained her BBiomed (Hons) from the University of Melbourne in 2016. In 2018−2022, she completed her PhD studies in fetal physiology and neurodevelopment at the School of Health & Biomedical Sciences, RMIT University, Australia, and the Ritchie Centre focusing on evaluating the prenatal administration of creatine to protect the fetal/neonatal brain. Her research interests include improving the neurological outcomes of newborns following complications during pregnancy and at the time of birth.

Edited by: Laura Bennet & Justin Dean
Linked articles: This article is highlighted in a Perspective article by Davidson et al. To read this article, visit https://doi.org/10.1113/JP283330.
The peer review history is available in the Supporting Information section of this article (https://doi.org/10.1113/JP282840#support‐information‐section).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Abramov, A. Y. , Scorziello, A. , & Duchen, M. R. (2007). Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. Journal of Neuroscience, 27(5), 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti, K. G. (1977). The biochemical consequences of hypoxia. Journal of Clinical Pathology Supplement, s3‐11(1), 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres, R. H. , Huber, A. W. , Schlattner, U. , Pérez‐Bouza, A. , Krebs, S. H. , Seiler, R. W. , Wallimann, T. , & Widmer, H. R. (2005). Effects of creatine treatment on the survival of dopaminergic neurons in cultured fetal ventral mesencephalic tissue. Neuroscience, 133(3), 701–713. [DOI] [PubMed] [Google Scholar]
- Anyaegbunam, A. , Brustman, L. , Divon, M. , & Langer, O. (1986). The significance of antepartum variable decelerations. American Journal of Obstetrics and Gynecology, 155(4), 707–710. [DOI] [PubMed] [Google Scholar]
- Azzopardi, D. , Wyatt, J. S. , Cady, E. B. , Delpy, D. T. , Baudin, J. , Stewart, A. L. , Hope, P. L. , Hamilton, P. A. , & Reynolds, E. O. R. (1989). Prognosis of newborn infants with hypoxic‐ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatric Research, 25(5), 445–451. [DOI] [PubMed] [Google Scholar]
- Balsom, P. D. , Söderlund, K. , Sjödin, B. , & Ekblom, B. (1995). Skeletal muscle metabolism during short duration high‐intensity exercise: Influence of creatine supplementation. Acta Physiologica Scandinavica, 154(3), 303–310. [DOI] [PubMed] [Google Scholar]
- Bennet, L. , Peebles, D. M. , Edwards, A. D. , Rios, A. , & Hanson, M. A. (1998). The cerebral hemodynamic response to asphyxia and hypoxia in the near‐term fetal sheep as measured by near infrared spectroscopy. Pediatric Research, 44(6), 951–957. [DOI] [PubMed] [Google Scholar]
- Bouillaud, F. , Hammad, N. , & Schwartz, L. (2021). Warburg effect, glutamine, succinate, alanine, when oxygen matters. Biology, 10(10), 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bungay, P. M. , Newton‐Vinson, P. , Isele, W. , Garris, P. A. , & Justice, J. B. (2003). Microdialysis of dopamine interpreted with quantitative model incorporating probe implantation trauma. Journal of Neurochemistry, 86(4), 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagne, V. (1999). Relationships between neuronal death and the cellular redox status. Focus on the developing nervous system. Progress in Neurobiology, 59(4), 397–423. [DOI] [PubMed] [Google Scholar]
- Chafer‐Pericas, C. , Cernada, M. , Rahkonen, L. , Stefanovic, V. , Andersson, S. , & Vento, M. (2016). Preliminary case control study to establish the correlation between novel peroxidation biomarkers in cord serum and the severity of hypoxic ischemic encephalopathy. Free Radical Biology & Medicine, 97, 244–249. [DOI] [PubMed] [Google Scholar]
- Chen, K. C. , Höistad, M. , Kehr, J. , Fuxe, K. , & Nicholson, C. (2002). Theory relating in vitro and in vivo microdialysis with one or two probes. Journal of Neurochemistry, 81(1), 108–121. [DOI] [PubMed] [Google Scholar]
- Chen, S.‐X. , & Schopfer, P. (1999). Hydroxyl‐radical production in physiological reactions. European Journal of Biochemistry, 260(3), 726–735. [DOI] [PubMed] [Google Scholar]
- Clandinin, M. T. , Chappell, J. E. , Leong, S. , Heim, T. , Swyer, P. R. , & Chance, G. W. (1980). Intrauterine fatty acid accretion rates in human brain: Implications for fatty acid requirements. Early Human Development, 4(2), 121–129. [DOI] [PubMed] [Google Scholar]
- Clark, J. F. (1997). Creatine and phosphocreatine: A review of their use in exercise and sport. Journal of Athletic Training, 32(1), 45–51. [PMC free article] [PubMed] [Google Scholar]
- Coimbra‐Costa, D. , Alva, N. , Duran, M. , Carbonell, T. , & Rama, R. (2017). Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biology, 12, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva, R. P. , Kelly, K. B. , Leonard, K. ‐. A. , & Jacobs, R. L. (2014). Creatine reduces hepatic TG accumulation in hepatocytes by stimulating fatty acid oxidation. Biochimica et Biophysica Acta – Molecular and Cell Biology of Lipids, 1841(11), 1639–1646. [DOI] [PubMed] [Google Scholar]
- Da Silva, R. P. , Leonard, K.‐A. , & Jacobs, R. L. (2017). Dietary creatine supplementation lowers hepatic triacylglycerol by increasing lipoprotein secretion in rats fed high‐fat diet. Journal of Nutritional Biochemistry, 50, 46–53. [DOI] [PubMed] [Google Scholar]
- Desoye, G. , & Herrera, E. (2021). Adipose tissue development and lipid metabolism in the human fetus: The 2020 perspective focusing on maternal diabetes and obesity. Progress in Lipid Research, 81, 101082. [DOI] [PubMed] [Google Scholar]
- Earnest, C. P. , Almada, A. L. , & Mitchell, T. L. (1996). High‐performance capillary electrophoresis‐pure creatine monohydrate reduces blood lipids in men and women. Clinical Science, 91(1), 113–118. [DOI] [PubMed] [Google Scholar]
- Ellery, S. J. , Dickinson, H. , Mckenzie, M. , & Walker, D. W. (2016). Dietary interventions designed to protect the perinatal brain from hypoxic‐ischemic encephalopathy–Creatine prophylaxis and the need for multi‐organ protection. Neurochemistry International, 95, 15–23. [DOI] [PubMed] [Google Scholar]
- Ferriero, D. M. (2004). Neonatal brain injury. New England Journal of Medicine, 351(19), 1985–1995. [DOI] [PubMed] [Google Scholar]
- Gitto, E. , Pellegrino, S. , Gitto, P. , Barberi, I. , & Reiter, R. J. (2009). Oxidative stress of the newborn in the pre‐ and postnatal period and the clinical utility of melatonin. Journal of Pineal Research, 46(2), 128–139. [DOI] [PubMed] [Google Scholar]
- Giussani, D. A. (2016). The fetal brain sparing response to hypoxia: Physiological mechanisms. Journal of Physiology, 594(5), 1215–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith, J. (2015). Overview and initial management of delivery room resuscitation. Neonatal‐perinatal medicine (10th ed., pp. 460–463). Ohio: Elsevier Saunders. [Google Scholar]
- Guimarães‐Ferreira, L. , Pinheiro, C. H. J. , Gerlinger‐Romero, F. , Vitzel, K. F. , Nachbar, R. T. , Curi, R. , & Nunes, M. T. (2012). Short‐term creatine supplementation decreases reactive oxygen species content with no changes in expression and activity of antioxidant enzymes in skeletal muscle. European Journal of Applied Physiology, 112(11), 3905–3911. [DOI] [PubMed] [Google Scholar]
- Gunn, A. J. , & Bennet, L. (2009). Fetal hypoxia insults and patterns of brain injury: Insights from animal models. Clinics in Perinatology, 36(3), 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, P. , Cady, E. B. , Wyatt, J. S. , Hope, P. L. , Delpy, D. T. , & Reynolds, E. O. R. (1986). Impaired energy metabolism in brains of newborn infants with increased cerebral echodensities. Lancet, 327(8492), 1242–1246. [DOI] [PubMed] [Google Scholar]
- Hope, P. (1984). Cerebral energy metabolism studied with phosphorus NMR spectroscopy in normal and birth‐asphyxiated infants. Lancet, 324(8399), 366–370. [DOI] [PubMed] [Google Scholar]
- Ienaga, K. , & Yokozawa, T. (2011). Creatinine and HMH (5‐hydroxy‐1‐methylhydantoin, NZ‐419) as intrinsic hydroxyl radical scavengers. Drug Discoveries & Therapeutics, 5(4), 162–175. [DOI] [PubMed] [Google Scholar]
- Ikeda, T. , Choi, B. H. , Yee, S. , Murata, Y. , & Quilligan, E. J. (1999). Oxidative stress, brain white matter damage andintrauterine asphyxia in fetal lambs. International Journal of Developmental Neuroscience, 17(1), 1–14. [DOI] [PubMed] [Google Scholar]
- Ireland, Z. , Castillo‐Melendez, M. , Dickinson, H. , Snow, R. , & Walker, D. W. (2011). A maternal diet supplemented with creatine from mid‐pregnancy protects the newborn spiny mouse brain from birth hypoxia. Neuroscience, 194, 372–379. [DOI] [PubMed] [Google Scholar]
- Johnson, J. K. , Sudheimer, K. D. , Davis, K. K. , Kerndt, G. M. , & Winn, B. M. (2013). The Sheep Brain Atlas. https://brains.anatomy.msu.edu/brains/sheep/index.html.
- Justice, J. B. (1993). Quantitative microdialysis of neurotransmitters. Journal of Neuroscience Methods, 48(3), 263–276. [DOI] [PubMed] [Google Scholar]
- Kagan, B. J. , Ermine, C. M. , Frausin, S. , Parish, C. L. , Nithianantharajah, J. , & Thompson, L. H. (2021). Focal ischemic injury to the early neonatal rat brain models cognitive and motor deficits with associated histopathological outcomes relevant to human neonatal brain injury. International Journal of Molecular Sciences, 22(9), 4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp, R. G. (1973). Inhibition of muscle pyruvate kinase by creatine phosphate. Journal of Biological Chemistry, 248(11), 3963–3967. [PubMed] [Google Scholar]
- Kuhr, W. G. , & Korf, J. (1988). Extracellular lactic acid as an indicator of brain metabolism: Continuous on‐line measurement in conscious, freely moving rats with intrastriatal dialysis. Journal of Cerebral Blood Flow and Metabolism, 8(1), 130–137. [DOI] [PubMed] [Google Scholar]
- Kurinczuk, J. J. , White‐Koning, M. , & Badawi, N. (2010). Epidemiology of neonatal encephalopathy and hypoxic‐ischaemic encephalopathy. Early Human Development, 86(6), 329–338. [DOI] [PubMed] [Google Scholar]
- Lafemina, M. J. , Sheldon, R. A. , & Ferriero, D. M. (2006). Acute hypoxia‐ischemia results in hydrogen peroxide accumulation in neonatal but not adult mouse brain. Pediatric Research, 59(5), 680–683. [DOI] [PubMed] [Google Scholar]
- Lawler, J. M. , Barnes, W. S. , Wu, G. , Song, W. , & Demaree, S. (2002). Direct antioxidant properties of creatine. Biochemical and Biophysical Research Communications, 290(1), 47–52. [DOI] [PubMed] [Google Scholar]
- Lawn, J. E. , Kerber, K. , Enweronu‐Laryea, C. , & Cousens, S. (2010). 3.6 million neonatal deaths–what is progressing and what is not? Seminars in Perinatology, 34(6), 371–386. [DOI] [PubMed] [Google Scholar]
- Logica, T. , Riviere, S. , Holubiec, M. I. , Castilla, R. , Barreto, G. E. , & Capani, F. (2016). Metabolic changes following perinatal asphyxia: Role of astrocytes and their interaction with neurons. Frontiers in Aging Neuroscience, 8, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S. L. , Yan, E. B. , Castillo‐Meléndez, M. , Jenkin, G. , & Walker, D. W. (2005). Melatonin provides neuroprotection in the late‐gestation fetal sheep brain in response to umbilical cord occlusion. Developmental Neuroscience, 27(2–4), 200–210. [DOI] [PubMed] [Google Scholar]
- Mishra, Om. P , & Delivoria‐Papadopoulos, M. (1988). Anti‐oxidant enzymes in fetal guinea pig brain during development and the effect of maternal hypoxia. Brain Research, 42(2), 173–179. [DOI] [PubMed] [Google Scholar]
- Morrison, P. F. , Bungay, P. M. , Hsiao, J. K. , Ball, B. A. , Mefford, I. N. , & Dedrick, R. L. (1991). Quantitative microdialysis: Analysis of transients and application to pharmacokinetics in brain. Journal of Neurochemistry, 57(1), 103–119. [DOI] [PubMed] [Google Scholar]
- Muccini, A. M. , Tran, N. T. , De Guingand, D. L. , Philip, M. , Della Gatta, P. A. , Galinsky, R. , Sherman, L. S. , Kelleher, M. A. , Palmer, K. R. , Berry, M. J. , Walker, D. W. , Snow, R. J. , & Ellery, S. J. (2021). Creatine metabolism in female reproduction, pregnancy and newborn health. Nutrients, 13(2), 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccini, A. M. , Tran, N. T. , Hale, N. , McKenzie, M. , Snow, R. J. , Walker, D. W. , & Ellery, S. J. (2022). The effects of in utero fetal hypoxia and creatine treatment on mitochondrial function in the late gestation fetal sheep brain. Oxidative Medicine and Cellular Longevity, 2022, 3255296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, R. E. (1975). Four patterns of perinatal brain damage and their conditions of occurrence in primates. Advances in Neurology, 10, 223–234. [PubMed] [Google Scholar]
- Nakamura, K. O. , Ienaga, K. , Yokozawa, T. , Fujitsuka, N. , & Oura, H. (1991). Production of methylguanidine from creatinine via creatol by active oxygen species: Analyses of the catabolism in vitro. Nephron, 58(1), 42–46. [DOI] [PubMed] [Google Scholar]
- Nelson, K. B. , & Grether, J. K. (1998). Potentially asphyxiating conditions and spastic cerebral palsy in infants of normal birth weight. American Journal of Obstetrics and Gynecology, 179(2), 507–513. [DOI] [PubMed] [Google Scholar]
- Nguyen, N. H. T. , Bråthe, A. , & Hassel, B. (2003). Neuronal uptake and metabolism of glycerol and the neuronal expression of mitochondrial glycerol‐3‐phosphate dehydrogenase. Journal of Neurochemistry, 85(4), 831–842. [DOI] [PubMed] [Google Scholar]
- Niatsetskaya, Z. V. , Sosunov, S. A. , Matsiukevich, D. , Utkina‐Sosunova, I. V. , Ratner, V. I. , Starkov, A. A. , & Ten, V. S. (2012). The oxygen free radicals originating from mitochondrial complex i contribute to oxidative brain injury following hypoxia–ischemia in neonatal mice. The Journal of Neuroscience, 32(9), 3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'donnell‐Tormey, J. , Nathan, C. F. , Lanks, K. , Deboer, C. J. , & De La Harpe, J. (1987). Secretion of pyruvate. An antioxidant defense of mammalian cells. Journal of Experimental Medicine, 165(2), 500–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osak, R. , Webster, K. M. , Bocking, A. D. , Campbell, M. K , & Richardson, B. S. (1997). Nuchal cord evident at birth impacts on fetal size relative to that of the placenta. Early Human Development, 49(3), 193–202. [DOI] [PubMed] [Google Scholar]
- Pellerin, L. , Pellegri, G. , Bittar, P. G. , Charnay, Y. , Bouras, C. , Martin, J.‐L. , Stella, N. , & Magistretti, P. J. (1998). Evidence supporting the existence of an activity‐dependent astrocyte‐neuron lactate shuttle. Developmental Neuroscience, 20(4–5), 291–299. [DOI] [PubMed] [Google Scholar]
- Prass, K. , Royl, G. , Lindauer, U. , Freyer, D. , Megow, D. , Dirnagl, U. , Stöckler‐Ipsiroglu, G. , Wallimann, T. , & Priller, J. (2006). Improved reperfusion and neuroprotection by creatine in a mouse model of stroke. Journal of Cerebral Blood Flow & Metabolism, 27(3), 452–459. [DOI] [PubMed] [Google Scholar]
- Prass, K. , Royl, G. , Lindauer, U. , Freyer, D. , Megow, D. , Dirnagl, U. , Stöckler‐Ipsiroglu, G. , Wallimann, T. , & Priller, J. (2007). Improved reperfusion and neuroprotection by creatine in a mouse model of stroke. Journal of Cerebral Blood Flow and Metabolism, 27(3), 452–459. [DOI] [PubMed] [Google Scholar]
- Quek, L.‐E. , Liu, M. , Joshi, S. , & Turner, N. (2016). Fast exchange fluxes around the pyruvate node: A leaky cell model to explain the gain and loss of unlabelled and labelled metabolites in a tracer experiment. Cancer & Metabolism, 4(1), 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raicevic, S. , Cubrilo, D. , Arsenijevic, S. , Vukcevic, G. , Živkovic, V. , Vuletic, M. , Barudžic, N. , Andjelkovic, N. , Antonovic, O. , & Jakovljevic, V. (2010). Oxidative stress in fetal distress: potential prospects for diagnosis. Oxidative Medicine and Cellular Longevity, 3(3), 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. , Lin, Z. , Drobyshevsky, A. , Chen, L. , Ji, X. , Ji, H. , Yang, Y. , Yu, L. , Derrick, M. , Silverman, R. B. , & Tan, S. (2011). Involvement of neuronal nitric oxide synthase in ongoing fetal brain injury following near‐term rabbit hypoxia‐ischemia. Developmental Neuroscience, 33(3–4), 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha‐Ferreira, E. , Rudge, B. , Hughes, M. P. , Rahim, A. A. , Hristova, M. , & Robertson, N. J. (2016). Immediate remote ischemic postconditioning reduces brain nitrotyrosine formation in a piglet asphyxia model. Oxidative Medicine and Cellular Longevity, 2016, 5763743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehl, A. B. , Zoremba, N. , Kipp, M. , Schiefer, J. , Goetzenich, A. , Bleilevens, C. , Kuehn‐Velten, N. , Tolba, R. , Rossaint, R. , & Hein, M. (2012). The effects of levosimendan on brain metabolism during initial recovery from global transient ischaemia/hypoxia. BMC Neurology, 12, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, M. S. , Wang, W. , Mongelli, M. , Pang, C. P. , Duley, J. A. , & Chang, A. M. Z. (1997). Lipid peroxidation in cord blood at birth: A marker of fetal hypoxia during labour. Gynecologic and Obstetric Investigation, 44(4), 229–233. [DOI] [PubMed] [Google Scholar]
- Rosenberg, A. A. (1988). Regulation of cerebral blood flow after asphyxia in neonatal lambs. Stroke, 19(2), 239–244. [DOI] [PubMed] [Google Scholar]
- Roth, Sc , Baudin, J. , Cady, E. , Johal, K. , Townsend, Jp , Wyatt, J. S. , Reynolds, E. O. R. , & Stewart, A. L. (1997). Relation of deranged neonatal cerebral oxidative metabolism with neurodevelopmental outcome and head circumference at 4 years. Developmental Medicine and Child Neurology, 39(11), 718–725. [DOI] [PubMed] [Google Scholar]
- Saugstad, O. D. (1996). Mechanisms of tissue injury by oxygen radicals: Implications for neonatal disease. Acta Paediatrica, 85(1), 1–4. [DOI] [PubMed] [Google Scholar]
- Scopes, R. K. (1973). Studies with a reconstituted muscle glycolytic system. The rate and extent of creatine phosphorylation by anaerobic glycolysis. Biochemical Journal, 134(1), 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sestili, P. , Martinelli, C. , Bravi, G. , Piccoli, G. , Curci, R. , Battistelli, M. , Falcieri, E. , Agostini, D. , Gioacchini, A. M. , & Stocchi, V. (2006). Creatine supplementation affords cytoprotection in oxidatively injured cultured mammalian cells via direct antioxidant activity. Free Radical Biology & Medicine, 40(5), 837–849. [DOI] [PubMed] [Google Scholar]
- Shen, H. , & Goldberg, M. P. (2012). Creatine pretreatment protects cortical axons from energy depletion in vitro. Neurobiology of Disease, 47(2), 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjö, B. K. , & Nilsson, L. (1971). The influence of arterial hypoxemia upon labile phosphates and upon extracellular and intracellular lactate and pyruvate concentrations in the rat brain. Scandinavian Journal of Clinical and Laboratory Investigation, 27(1), 83–96. [DOI] [PubMed] [Google Scholar]
- Steiner, P. (2019). Brain fuel utilization in the developing brain. Annals of Nutrition and Metabolism, 75(1), 8–18. [DOI] [PubMed] [Google Scholar]
- Storey, K. B. , & Hochachka, P. W. (1974). Activation of muscle glycolysis: A role for creatine phosphate in phosphofructokinase regulation. FEBS Letters, 46(1–2), 337–339. [DOI] [PubMed] [Google Scholar]
- Taes, Y. E. C. (2003). Creatine supplementation does not affect kidney function in an animal model with pre‐existing renal failure. Nephrology, Dialysis, Transplantation, 18(2), 258–264. [DOI] [PubMed] [Google Scholar]
- Tran, N. T. , Kelly, S. B. , Snow, R. J. , Walker, D. W. , Ellery, S. J. , & Galinsky, R. (2021). Assessing creatine supplementation for neuroprotection against perinatal hypoxic‐ischaemic encephalopathy: A systematic review of perinatal and adult pre‐clinical studies. Cells. 10(11), 2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, N. T. , Muccini, A. M. , Snow, R. J. , Nitsos, I. , Hale, N. , Walker, D. W. , & Ellery, S. J. (2021). The physiological effects of creatine supplementation in fetal sheep before, during, and after umbilical cord occlusion and global hypoxia. Journal of Applied Physiology, 131(3), 1088–1099. [DOI] [PubMed] [Google Scholar]
- Van Bel, F. , Van De Bor, M. , Stijnen, T. , Baan, J. , & Ruys, J. H. (1987). Cerebral blood flow velocity pattern in healthy and asphyxiated newborns: A controlled study. European Journal of Pediatrics, 146(5), 461–467. [DOI] [PubMed] [Google Scholar]
- Vannucci, R. C. , Towfighi, J. , & Vannucci, S. J. (2004). Secondary energy failure after cerebral hypoxia–ischemia in the immature rat. Journal of Cerebral Blood Flow & Metabolism, 24(10), 1090–7. [DOI] [PubMed] [Google Scholar]
- Varcoe, T. J. , Darby, J. R. T. , Gatford, K. L. , Holman, S. L. , Cheung, P. , Berry, M. J. , Wiese, M. D. , & Morrison, J. L. (2019). Considerations in selecting postoperative analgesia for pregnant sheep following fetal instrumentation surgery. Anime Fronts, 9(3), 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe, J. J. (2008). Neurology of the newborn. E‐book: Elsevier Health Sciences. [Google Scholar]
- Williamson, L. , & New, D. (2014). How the use of creatine supplements can elevate serum creatinine in the absence of underlying kidney pathology. BMJ Case Reports, 2014(1), bcr2014204754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt, J. (2002). Applied physiology: Brain metabolism following perinatal asphyxia. Current Paediatrics, 12(3), 227–231. [Google Scholar]
- Wyss, M. , & Kaddurah‐Daouk, R. (2000). Creatine and creatinine metabolism. Physiological Reviews, 80(3), 1107–1213. [DOI] [PubMed] [Google Scholar]
- Yan, E. B. , Baburamani, A. A. , Walker, A. M. , & Walker, D. W. (2009). Changes in cerebral blood flow, cerebral metabolites, and breathing movements in the sheep fetus following asphyxia produced by occlusion of the umbilical cord. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 297(1), R60‐R69. [DOI] [PubMed] [Google Scholar]
- Yan, E. B. , Unthank, J. K. , Castillo‐Melendez, M. , Miller, S. L. , Langford, S. J. , & Walker, D. W. (2005). Novel method for in vivo hydroxyl radical measurement by microdialysis in fetal sheep brain in utero. Journal of Applied Physiology, 98(6), 2304–2310. [DOI] [PubMed] [Google Scholar]
- Yawno, T. , Mahen, M. , Li, J. , Fahey, M. C. , Jenkin, G. , & Miller, S. L. (2017). The beneficial effects of melatonin administration following hypoxia‐ischemia in preterm fetal sheep. Frontiers in Cellular Neuroscience, 11, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoremba, N. , Homola, A. , Rossaint, R. , & Syková, E. (2007). Brain metabolism and extracellular space diffusion parameters during and after transient global hypoxia in the rat cortex. Experimental Neurology, 203(1), 34–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peer Review History
Statistical Summary Document
Supplementary Data S1.
Supplementary Data S2.
Supplementary Data S3.
Supplementary Data S4.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.