

ABSTRACT
Deubiquitination plays an important role in the regulation of the crosstalk between macroautophagy/autophagy and innate immune signaling, yet its regulatory mechanisms are not fully understood. Here we identify the deubiquitinase OTUD7B as a negative regulator of antiviral immunity by targeting IRF3 (interferon regulatory factor 3) for selective autophagic degradation. Mechanistically, OTUD7B interacts with IRF3, and activates IRF3-associated cargo receptor SQSTM1/p62 (sequestosome 1) by removing its K63-linked poly-ubiquitin chains at lysine 7 (K7) to enhance SQSTM1 oligomerization. Moreover, viral infection increased the expression of OTUD7B, which forms a negative feedback loop by promoting IRF3 degradation to balance type I interferon (IFN) signaling. Taken together, our study reveals a specific role of OTUD7B in mediating the activation of cargo receptors in a substrate-dependent manner, which could be a potential target against excessive immune responses.
Abbreviations: Baf A1: bafilomycin A1; CGAS: cyclic GMP-AMP synthase; DDX58/RIG-I: DExD/H-box helicase 58; DSS: dextran sodium sulfate; DUBs: deubiquitinating enzymes; GFP: green fluorescent protein; IFN: interferon; IKKi: IKBKB/IkappaB kinase inhibitor; IRF3: interferon regulatory factor 3; ISGs: interferon-stimulated genes; MAVS: mitochondrial antiviral signaling protein; MOI: multiplicity of infection; PAMPs: pathogen-associated molecular patterns; SeV: Sendai virus; siRNA: small interfering RNA; SQSTM1/p62: sequestosome 1; STING1: stimulator of interferon response cGAMP interactor 1; TBK1: TANK binding kinase 1; Ub: ubiquitin; WT: wild-type; VSV: vesicular stomatitis virus.
KEYWORDS: Antiviral immunity, cargo receptor, deubiquitination, selective autophagy, type I interferon signaling
Introduction
The innate antiviral immunity triggered by viral pathogen-associated molecular patterns (PAMPs) provides the first line of host defense against viral infection [1]. Host cells evoke initial antiviral responses by detecting PAMPs with pattern recognition receptors (PRRs), which trigger signaling cascades resulting in the induction of type I interferons (IFNs), pro-inflammatory cytokines, and chemokines [2]. Subsequently, type I IFNs induce the synthesis of downstream effector proteins to provoke comprehensive host defenses [3]. IRF3 (interferon regulatory factor 3) is an indispensable transcription factor for type I IFNs production in response to viral infection [4]. The activation of IRF3 is strictly orchestrated by multiple posttranscriptional mechanisms, including phosphorylation and ubiquitination [5,6]. In addition, the stability of IRF3 is also subtly controlled by proteasome-dependent system and autophagosome-dependent system to maintain its function [7–9].
Autophagy, a highly conserved process in eukaryotes, serves as a rheostat for immune reactions to maintain cell homeostasis [10]. Upon viral infection, autophagy can destroy viruses and viral components with the help of cargo receptors, by delivering the cargo to the autophagosome for autophagy-lysosomal degradation [11]. On the other hand, autophagy could also selectively target and degrade many critical components of immune factors, such as DDX58/RIG-I, CGAS, MAVS, STING1 and IRF3, to regulate antiviral immunity [9,12–15]. We recently found that ubiquitinated IRF3 could be recognized by the cargo receptor CALCOCO2/NDP52 and delivered for autophagic degradation upon viral infection [9]. However, the regulatory mechanisms of autophagosome-dependent degradation of IRF3 are not fully understood.
Ubiquitination and deubiquitination are the most prevalent post-translational modifications for numerous proteins and play important role in many physiological processes, such as innate immune responses and autophagy [16,17]. In our previous works, we performed a systematic function screening to investigate the functions of deubiquitinating enzymes (DUBs) in regulating antiviral immunity and autophagy, and found that deubiquitination plays an essential role in the regulation of the crosstalk between autophagy and antiviral immune signaling [18,19]. OTUD7B/Cezanne (OTU deubiquitinase 7B), a member of OTU deubiquitinases, controls many key cellular signaling pathways, such as MTORC2 and NFKB/NF-κB signaling pathway [20,21]. We recently identified OTUD7B as a negative regulator of autophagy through mediating the K63-K48 type ubiquitination type transition of PIK3C3/VPS34 [19]. Moreover, OTUD7B is reported to interact with hepatitis C virus (HCV) NS5A protein and its deubiquitinase activity could be enhanced by NS5A in human hepatoma cell [22], which implies that OTUD7B might play a critical role in antiviral pathway. However, whether OTUD7B orchestrates host antiviral immunity and its crosstalk with autophagy remains unclear.
Here, we found that OTUD7B serves as a negative regulator in antiviral immunity by targeting IRF3. Upon viral infection, OTUD7B is up-regulated and promotes cargo receptor SQSTM1/p62-mediated autophagic degradation of IRF3. Mechanistically, OTUD7B removes the K63-linked poly-ubiquitin chains from IRF3-associated SQSTM1 at lysine (K) 7, which enhances SQSTM1 activity through its oligomerization to mediate IRF3 degradation. Therefore, our findings demonstrate that OTUD7B plays an essential role in IRF3-centered antiviral immunity and prevents the host from excessive immune responses by specifically mediating the activation of cargo receptors in a substrate-dependent manner.
Results
OTUD7B negatively regulates type I IFN signaling
Mounting evidence demonstrated that OTUD7B regulates NFKB signaling and virus replication, which suggests that OTUD7B might also be involved in antiviral immunity [22–24]. To examine the effects of OTUD7B on type I IFN signaling as well as antiviral immunity, we performed luciferase reporter assay and found that ectopic expression of OTUD7B significantly suppressed the activation of IFN-stimulated response element (ISRE) by infection of Sendai virus (SeV), vesicular stomatitis virus (VSV) or intracellular (IC) poly (I:C) treatment (Figure 1A). To substantiate the role of OTUD7B under physiological conditions, we efficiently knocked out OTUD7B in 293 T cells (Figure 1B) and found that OTUD7B deficiency enhanced SeV, VSV or IC poly (I:C)-induced ISRE activation (Figure 1C). We next observed that OTUD7B deficiency inhibited viral infection and resulted in markedly less GFP+ (virus-infected) cells (Figure 1D, E). Additionally, we performed quantitative RT-PCR analysis and found that OTUD7B deficiency resulted in more IFIT2/ISG54, IFIT1/ISG56, ISG15 and IFNB1 mRNA than control cells during SeV infection (Figure 1F). This result is further confirmed in THP-1-derived macrophages infected with SeV (Figure 1G and S1). Collectively, these results suggest that OTUD7B negatively regulates type I IFN signaling as well as antiviral immunity.
Figure 1.
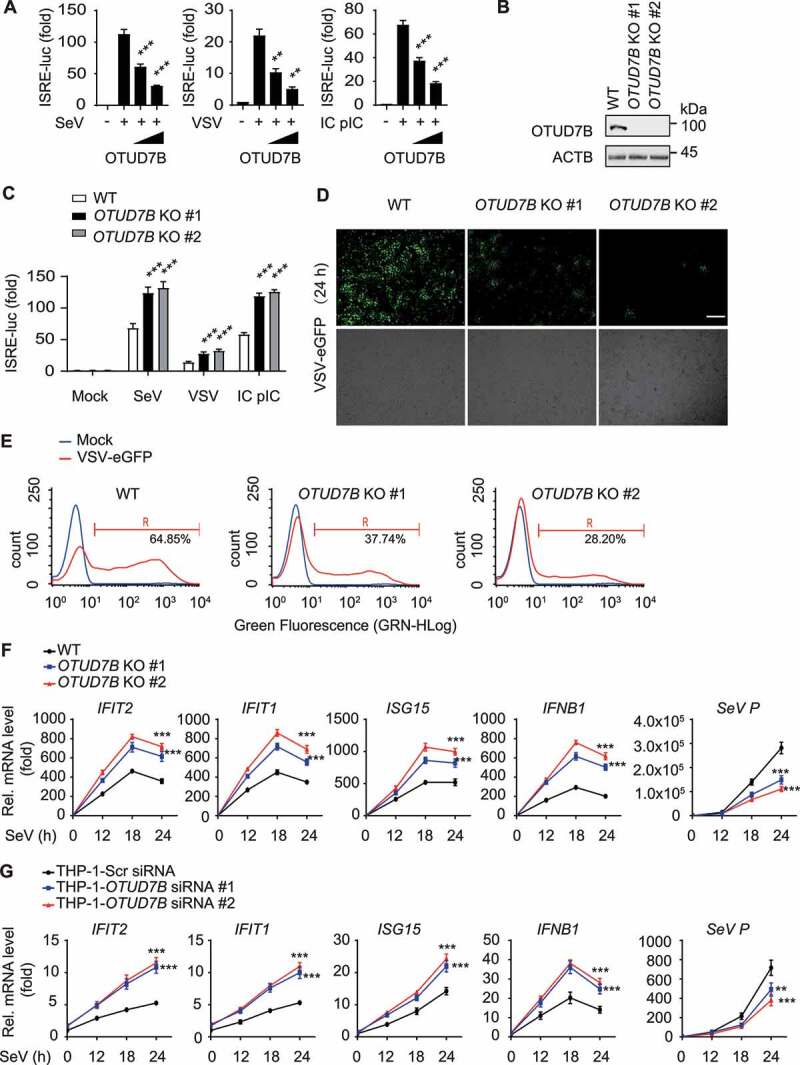
OTUD7B negatively regulates type I IFN signaling. (A) Luciferase activity in 293 T cells transfected with an ISRE luciferase reporter (ISRE-luc), together with the indicated plasmids along with empty vector (no wedge) or increasing amounts (wedge) of expression vector for HA-OTUD7B, followed by treatment with or without SeV (MOI = 0.1), VSV-eGFP (MOI = 0.1) or intracellular (IC) poly (I:C) (5 μg/mL) infection for 12 h respectively. (B) Immunoblot analysis of OTUD7B knockout (KO) 293 T cells. (C) Luciferase activity in wild-type (WT) or OTUD7B KO 293 T cells transfected with an ISRE-luc, then treated or untreated with SeV, VSV-eGFP or IC poly(I:C) for 12 h respectively. (D and E) Phase-contrast (PH) and fluorescence microscopy analysis (D) or flow cytometric analysis (E) of WT or OTUD7B KO 293 T cells treated with VSV-eGFP (MOI = 0.01) infection for 24 h. Scale bars: 200 μm. (F) Quantitative RT–PCR analysis of indicated gene expression in WT or OTUD7B KO 293 T cells treated with SeV (MOI = 0.1) infection at indicated time points. (G) Quantitative RT–PCR analysis of indicated gene expression in OTUD7B-depleted THP-1-derived macrophages transfected with scrambled (Scr) siRNA or OTUD7B siRNAs, followed by SeV (MOI = 0.1) infection at indicated time points. In (A, C, F and G), all error bars, mean values ± SEM, P-values were determined by unpaired two-tailed Student’s t test of n = 3 independent biological experiments, **P < 0.01 and *** P < 0.001. For (B, D and E), similar results are obtained for three independent biological experiments.
OTUD7B targets IRF3 and promotes its degradation
To identify the molecular target of OTUD7B in type I IFN signaling, we performed luciferase reporter assay and found that OTUD7B inhibited the activation of luciferase reporters induced by DDX58, CGAS and STING1, MAVS, TBK1, IKKi or IRF3 (5D) (a constitutively active mutant of IRF3) (Figure 2A). Further coimmunoprecipitation assay revealed that OTUD7B interacted with IRF3, but not DDX58, STING1, MAVS or TBK1, while it weakly interacted with IKKi (Figure 2B and S2A). Moreover, SeV infection only increased the association between OTUD7B and IRF3, but not IKKi, and the association between OTUD7B and IKKi was weak in both un-infected and virus-infected cells (Figure S2B,C). In addition, the endogenous association between OTUD7B and IRF3 was enhanced upon viral infection in A549 cells (Figure 2C and S2D). This result is further confirmed in THP-1-derived macrophages infected with SeV (Figure 2D). These data suggest that OTUD7B inhibits type I IFN signaling by targeting IRF3.
Figure 2.
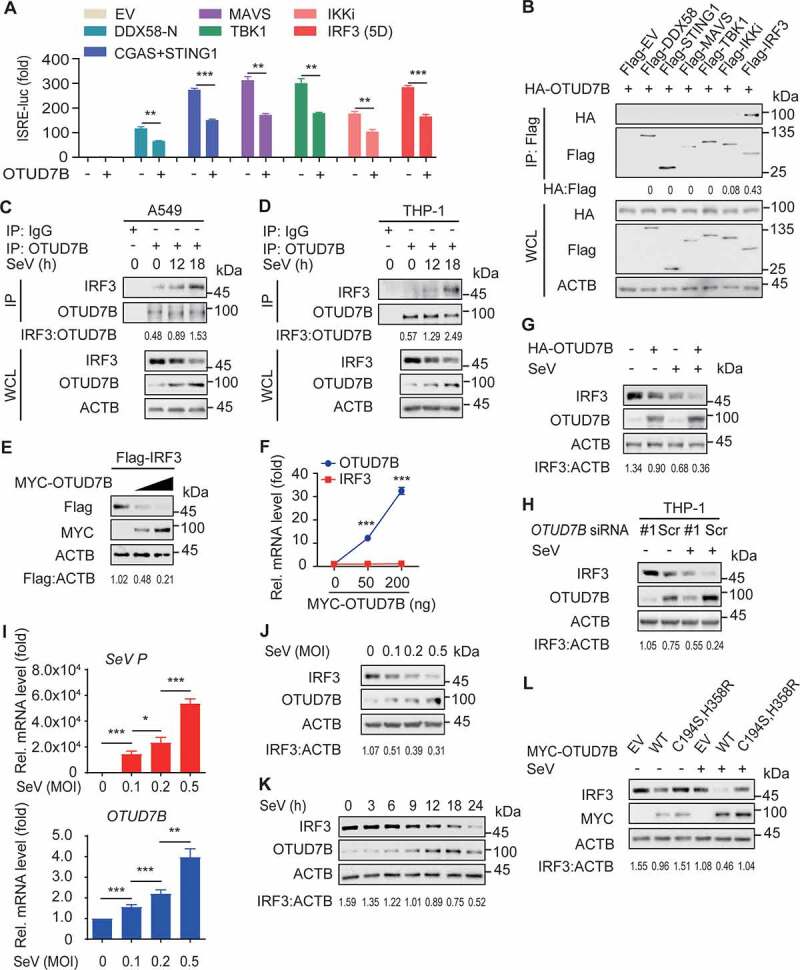
OTUD7B targets IRF3 and mediates the degradation of IRF3. (A) 293 T cells were transfected with the ISRE luciferase reporter, together with the indicated plasmids along with empty vector (EV) or expression vector for HA-OTUD7B. (B) Coimmunoprecipitation and immunoblot analysis of 293 T cells transfected with HA-OTUD7B together with Flag-tagged DDX58, STING1, MAVS, TBK1, IKKi or IRF3. (C) Protein lysates of A549 cells infected with SeV (MOI = 0.1) for indicated time points were immunoprecipitated with anti-OTUD7B antibody and immunoblot analysis with indicated antibodies. (D) Protein lysates of THP-1-derived macrophages infected with SeV (MOI = 0.1) for indicated time points were immunoprecipitated with anti-OTUD7B antibody and immunoblot analysis with indicated antibodies. (E) 293 T cells were transfected with Flag-IRF3, together with increasing amounts (wedge) of expression vector for MYC-OTUD7B, and the protein lysates were harvested for immunoblot analysis. (F) Quantitative RT-PCR analysis of OTUD7B mRNA and IRF3 mRNA in (E). (G) Protein lysates of 293 T cells transfected with plasmids encoding EV or HA-OTUD7B, followed by SeV (MOI = 0.1) infection for 18 h were immunoblotted with the indicated antibodies. (H) THP-1-derived macrophages were transfected with scrambled (Scr) siRNA or OTUD7B siRNA, followed by SeV (MOI = 0.1) infection for 18 h. Protein lysates were immunoblotted with indicated antibodies. (I) Quantitative RT-PCR analysis of OTUD7B mRNA and SeV P mRNA in A549 cells treated with indicated MOI of SeV for 12 h. (J) Protein lysates of A549 cells treated by SeV infection for 18 h with indicated MOI were immunoblotted with the indicated antibodies. (K) Protein lysates of A549 cells treated with SeV (MOI = 0.1) infection for indicated time points were immunoblotted with the indicated antibodies. (L) Protein lysates of 293 T cells transfected with wild-type (WT) OTUD7B or OTUD7BC194S,H358R mutant, following by treated with SeV (MOI = 0.1) infection for 18 h, were immunoblotted with the indicated antibodies. In (A, F and I), all error bars, mean values ± SEM, P-values were determined by unpaired two-tailed Student’s t test of n = 3 independent biological experiments, *P < 0.05, **P < 0.01 and *** P < 0.001. For (B-E, G, H and J-L), similar results are obtained for three independent biological experiments.
To dissect the inhibitory function of OTUD7B in type I IFN signaling through its interaction with IRF3, we examined the effect of OTUD7B on the stability of IRF3 and IKKi respectively. We found that increasing amount of OTUD7B significantly decreased the protein level of IRF3, but not IKKi (Figure 2E and S2E). Further quantitative RT-PCR analysis showed the abundance of IRF3 mRNA remained unchanged with increasing amount of OTUD7B mRNA (Figure 2F), suggesting that OTUD7B promotes IRF3 protein degradation. To further dissect the molecular mechanism of OTUD7B controlling IRF3 stability, we ectopically expressed OTUD7B in 293 T cells, and found that OTUD7B decreased the IRF3 protein abundance both in un-infected and SeV-infected cells, while viral infection resulted in increased OTUD7B protein abundance and the reduction of IRF3 protein abundance (Figure 2G and S2F). In addition, we knocked down OTUD7B in THP-1-derived macrophages and found that OTUD7B depletion resulted in increased level of IRF3 protein. Meanwhile, the endogenous expression of OTUD7B was increased by SeV infection (Figure 2H and S2G). Therefore, these results suggest that OTUD7B suppresses type I IFN signaling by mediating the degradation of IRF3.
To determine the mechanism of increased expression of OTUD7B during virus infection, we performed RT-PCR analysis of OTUD7B mRNA and SeV P mRNA in A549 cells treated with various titers of SeV. We found that increasing SeV load significantly resulted in increased OTUD7B mRNA level as well as OTUD7B protein abundance (Figure 2I,J and S2H). To further determine the dynamic pattern of OTUD7B protein after viral infection, the expression of OTUD7B and IRF3 were monitored at different time points after infection. We observed that the protein level of OTUD7B showed a remarkable increase upon viral infection, which resulted in more reduction of IRF3 protein (Figure 2K and S2I). Additionally, we found that OTUD7B-mediated IRF3 degradation required its DUB activity, as the catalytically inactive OTUD7B mutant, OTUD7BC194S,H358R failed to degrade IRF3 in both un-infected and SeV-infected cells (Figure 2L and S2J). Collectively, these results suggest that viral infection leads to the increase of OTUD7B which promotes the reduction of IRF3 protein in a virus load-dependent manner.
OTUD7B promotes the autophagic degradation of IRF3
To investigate which degradation system substantially involves in OTUD7B-mediated IRF3 degradation, we used ubiquitin-proteasome inhibitor and autophagy inhibitors to find that the degradation of IRF3 induced by OTUD7B was blocked by 3-methyladenine (3 MA), chloroquine (CQ) and bafilomycin A1 (Baf A1), but not the proteasome inhibitor MG132 (Figure 3A). In addition, OTUD7B could dramatically promote the degradation of IRF3 under autophagy-induced condition (Figure 3B, C), while OTUD7B depletion showed the opposite effect (Figure 3D,E). Coimmunoprecipitation analysis showed that IRF3 interacted with LC3B, and OTUD7B could significantly enhance this association (Figure 3F). In contrast, OTUD7B depletion decreased the endogenous association between IRF3 and LC3 in virus-infected THP-1-derived macrophages (Figure 3G). Further confocal microscopy analysis revealed that SeV infection resulted in more numbers of GFP-LC3B puncta and enhanced the colocalization between IRF3 and LC3B. In consistent with these results, OTUD7B depletion decreased the colocalization between IRF3 and LC3B (Figure 3H,I). These results suggest that OTUD7B promotes the autophagic degradation of IRF3.
Figure 3.
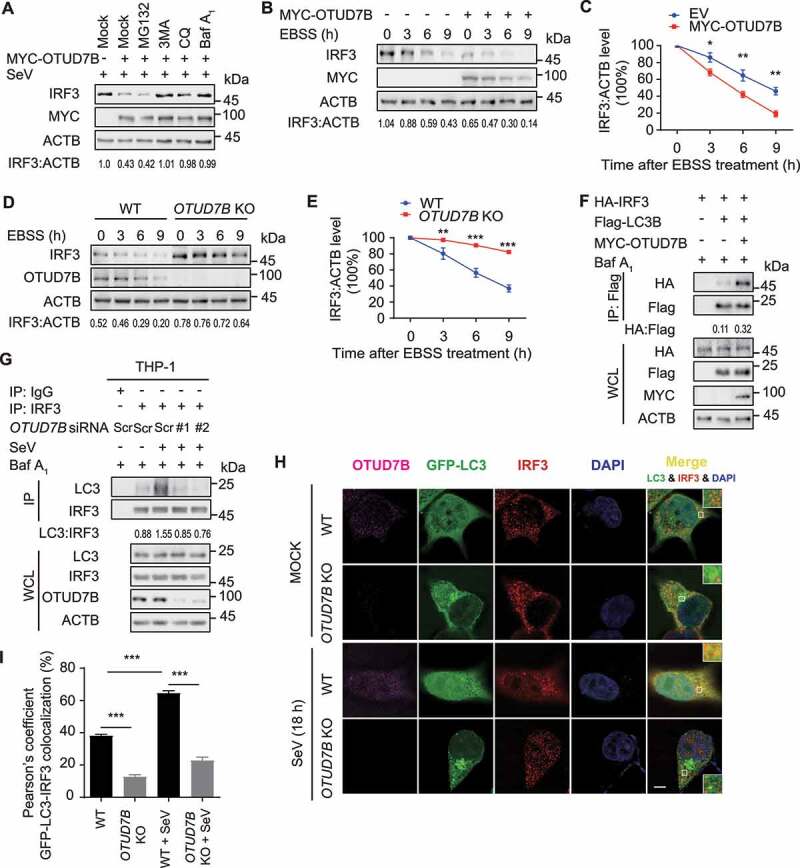
OTUD7B promotes the autophagic degradation of IRF3. (A) 293 T cells were transfected with plasmids encoding empty vector (EV) or MYC-OTUD7B, followed by SeV (MOI = 0.1) infection for 12 h. Protein lysates of the cells treated with MG132 (10 μM), 3 MA (10 mM), CQ (50 μM) and bafilomycin A1 (Baf A1) (0.2 μM) for 6 h respectively, were immunoblotted with the indicated antibodies. (B) Immunoblot analysis of protein extracts of 293 T cells transfected with plasmids encoding EV or MYC-OTUD7B, followed by Earle’s balanced salt solution (EBSS) treatment for the indicated time points. (C) Quantification of the expression levels of IRF3 shown in (B). (D) Wild-type (WT) and OTUD7B knockout (KO) 293 T cells were cultured in EBSS for the indicated time points. Protein extracts were analyzed with indicated antibodies. (E) Quantification of the expression levels of IRF3 shown in (D). (F) Coimmunoprecipitation and immunoblot analysis of protein lysates of 293 T cells transfected with vectors expressing HA-IRF3, Flag-LC3B and MYC-OTUD7B, followed by treatments of Baf A1 (0.2 μM) for 6 h. (G) THP-1-derived macrophages were transfected with scrambled (Scr) siRNA or OTUD7B siRNAs, followed by SeV (MOI = 0.1) infection for 18 h and Baf A1 (0.2 μM) treatment for 6 h. Protein lysates were immunoprecipitated and immunoblotted with indicated antibodies. (H and I) WT and OTUD7B KO 293 T cells were transfected with plasmid encoding GFP-LC3B, followed by SeV (MOI = 0.1) infection for 18 h. Confocal microscopy (H) of by labeling of GFP-LC3, OTUD7B and IRF3 with specific primary antibody and Alexa Fluor 633 goat anti-Mouse IgG (H + L) (pink) or CF568 Goat anti-Rabbit IgG (H + L) (red). Scale bars: 20 μm. Quantitative analysis (I) of the colocalization (30 cells per group). Data are expressed as means ± SD of 30 cells. In (C, E and I), all error bars, mean values ± SEM, P-values were determined by unpaired two-tailed Student’s t test of n = 3 independent biological experiments, *P < 0.05, **P < 0.01 and *** P < 0.001. For (A, B, D, F-G), similar results are obtained for three independent biological experiments.
OTUD7B promotes the SQSTM1-mediated IRF3 degradation
Accumulating evidence unravels the pivotal roles of cargo receptors in delivering substrates for selective autophagic degradation [13,15,25,26]. We have found that IRF3 could interact with cargo receptors, including SQSTM1 and CALCOCO2 [9], which was further validated in a exogenous experimental system under SeV infection (Figure 4A). To testify whether OTUD7B controls the IRF3 degradation via these cargo receptors, we performed coimmunoprecipitation analysis and found that OTUD7B enhanced the association of IRF3-SQSTM1, but not IRF3-CALCOCO2 (Figure 4B and S3A,B). Likewise, we showed that OTUD7B deficiency remarkably attenuated the association between endogenous IRF3 and SQSTM1 (Figure 4C). In addition, we observed that OTUD7B could interact with SQSTM1 (Figure 4D). To further confirm the role of OTUD7B in the association between IRF3 and SQSTM1, we performed immunoprecipitation and found that OTUD7B interacted with IRF3 at 12 h post-SeV infection, while the apparent interaction between OTUD7B-IRF3 and SQSTM1 could be detected at a later time point (around 18 h post infection), which revealed the dynamic assembly of the OTUD7B-IRF3-SQSTM1 complex (Figure 4E and S3C). To further substantiate the association between SQSTM1, IRF3 and OTUD7B, we applied two-step coimmunoprecipitation analysis to examine the associated proteins of IRF3 (IP-1) and IRF3-associated OTUD7B (IP-2). Sequential immunoprecipitation analysis result showed that IRF3, OTUD7B and SQSTM1 are present in the same trimolecular complex (Figure S3D). Taken together, these results suggest that OTUD7B firstly targets to IRF3, and then promotes the association between IRF3 and SQSTM1 at the later stage of viral infection.
Figure 4.
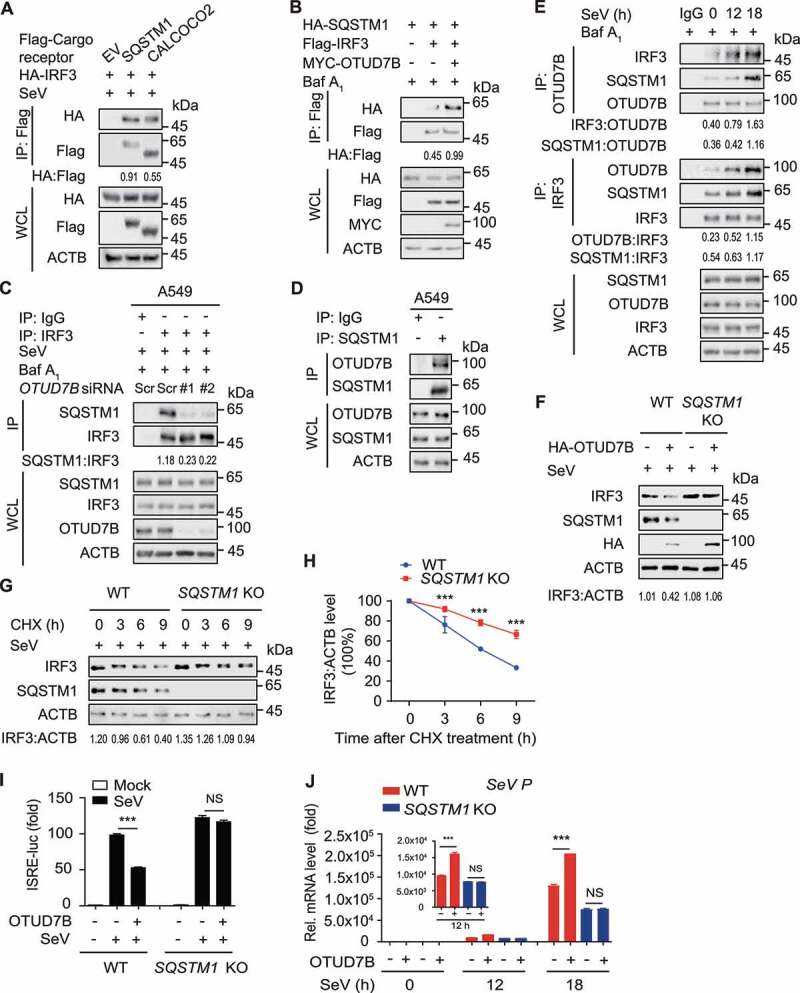
OTUD7B promotes the SQSTM1-mediated IRF3 degradation. (A) 293 T cells were transfected with vectors encoding HA-IRF3, together with Flag-tagged SQSTM1 and CALCOCO2, followed by SeV (MOI = 0.1) infection for 18 h. Protein lysates were harvested for immunoprecipitation with anti-Flag beads and immunoblot analysis with anti-HA. (B) 293 T cells were transfected with vectors encoding HA-SQSTM1 and Flag-IRF3, together with MYC-OTUD7B. Protein lysates were harvested after bafilomycin A1 (Baf A1) (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblot analysis with anti-HA. (C) A549 cells were transfected with scrambled (Scr) siRNA or OTUD7B siRNAs, followed by SeV (MOI = 0.1) infection for 12 h. Protein lysates were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation and immunoblot using indicated antibodies. (D) Protein lysates of A549 cells were immunoprecipitated with anti-SQSTM1 antibody and immunoblot analysis with indicated antibodies. (E) A549 cells were treated with SeV (MOI = 0.1) infection for indicated time points. Protein lysates were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation and immunoblot using indicated antibodies. (F) Protein lysates of wild-type (WT) and SQSTM1 knockout (KO) 293 T cells transfected with plasmids encoding empty vector (EV) or HA-OTUD7B, followed by SeV (MOI = 0.1) infection for 12 h were immunoblotted with the indicated antibodies. (G) Protein lysates of WT and SQSTM1 KO 293 T cells infected with SeV (MOI = 0.1) for 18 h, followed by cycloheximide (CHX) (100 μg/mL) treatment for the indicated time points, were immunoblotted with the indicated antibodies. (H) Quantification of the expression levels of IRF3 shown in (G). (I) Luciferase activity in WT or SQSTM1 KO 293 T cells transfected with an ISRE promoter-driven luciferase reporter, together with EV or expression vector for HA-OTUD7B, followed by SeV (MOI = 0.1) infection for 12 h. (J) Quantitative RT-PCR analysis of indicated gene expression in WT or SQSTM1 KO 293 T cells transfected with plasmids encoding EV or HA-OTUD7B, followed by SeV (MOI = 0.1) infection for indicated time points. In (H-J), all error bars, mean values ± SEM, P-values were determined by unpaired two-tailed Student’s t test of n = 3 independent biological experiments, *** P < 0.001. NS: not significant. For (A-G), similar results are obtained for three independent biological experiments.
We next sought to substantiate whether SQSTM1 involves in IRF3 autophagic degradation. Indeed, OTUD7B failed to promote the degradation of IRF3 in SQSTM1 KO cells (Figure 4F). Similarly, SQSTM1 depletion slowed the clearance of IRF3 in cycloheximide (CHX)-treated cells (Figure 4G,H and S3E). To address the functional importance of SQSTM1 in IRF3-centered type I IFN signaling, we performed luciferase reporter assay and found that OTUD7B could not inhibit the activation of luciferase reporters in SQSTM1 KO cells (Figure 4I). Consistently, OTUD7B failed to promote the replication of SeV upon viral infection in SQSTM1 KO cells (Figure 4J). These data suggest that OTUD7B inhibits antiviral immunity via SQSTM1-mediated autophagic degradation of IRF3.
OTUD7B inhibits the K63-linked poly-ubiquitination of SQSTM1 at K7
OTUD7B plays a key role in many cellular processes via deubiquitinating its substrates, including ZAP70, TRAF3, TRAF6, RIP1 and LSD1 [21,27–30]. We also observed that OTUD7B mediates IRF3 degradation in a DUB activity-dependent manner (Figure 2L). Thus, we sought to examine whether OTUD7B modulates the ubiquitination of IRF3. Unexpectedly, the ubiquitination of IRF3 was unchanged in OTUD7B deficient cells (Figure S4A). However, OTUD7B depletion remarkably increased the poly-ubiquitination of SQSTM1 (Figure 5A). Structural and biochemical studies suggested that OTUD7B could bind Lys (K)11, K48, K63 and Met-1 linked ubiquitin chains [31,32]. We next co-expressed the indicated ubiquitin mutants with SQSTM1 in the absence or presence of OTUD7B to investigate OTUD7B-mediated deubiquitination of SQSTM1. Immunoprecipitation assays showed that OTUD7B predominantly inhibited the K63-linked poly-ubiquitination of SQSTM1 (Figure 5B and S4B). Consistently, viral infection decreased K63-linked poly-ubiquitination of SQSTM1, while depletion of OTUD7B abrogated this process (Figure 5C). Likewise, OTUD7BC194S,H358R mutant failed to cleave the K63-linked poly-ubiquitin chains of SQSTM1 (Figure 5D), indicating that OTUD7B regulates the deubiquitination of SQSTM1 in a DUB activity-dependent manner. We further investigated the sites at which OTUD7B cleaves the poly-ubiquitin chains from SQSTM1. It has been reported that lysine 7 residue (K7) is critical for SQSTM1 dimerization, ubiquitin binding, as well as aggregate formation [33]. We found that SQSTM1K7R (K7 to arginine (R) point mutation of SQSTM1) mutant could hardly undergo the K63-linked poly-ubiquitination and OTUD7B failed to inhibit the K63-linked poly-ubiquitination of SQSTM1K7R mutant (Figure 5E,F and S4C). These results suggest that OTUD7B inhibits the K63-linked poly-ubiquitination of SQSTM1.
Figure 5.
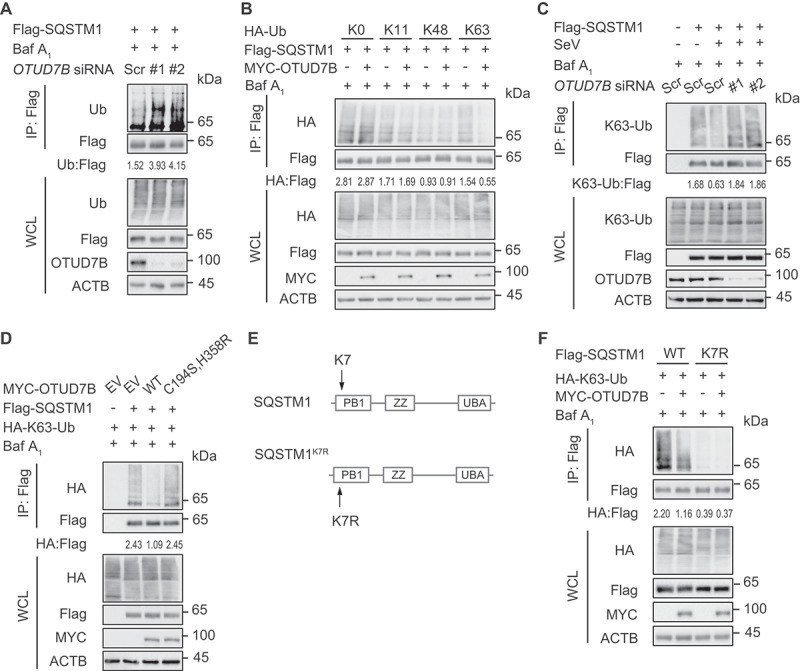
OTUD7B inhibits the K63-linked poly-ubiquitination of SQSTM1 at residue K7. (A) 293 T cells were transfected with scrambled (Scr) siRNA or OTUD7B siRNAs, together with expression vector for Flag-SQSTM1. Protein lysates were harvested after bafilomycin A1 (Baf A1) (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with indicated antibodies. (B) Protein lysates of 293 T cells transfected with plasmids expressing Flag-SQSTM1 and indicated HA-tagged ubiquitin (Ub) mutants, together with empty vector (EV) or expression vector of MYC-OTUD7B, were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with anti-HA antibody. The HA-K0-Ub indicates the ubiquitin mutant in which all the K are substituted with R. The K11, K48 and K63 mutants of HA-Ub indicate the ubiquitin mutant in which the specific K is intact while other K are substituted with R. Different amounts of plasmids for various mutants of Ub were transfected into cells for equal expression with Flag-SQSTM1 overexpression. (C) 293 T cells were transfected with Scr siRNA or OTUD7B siRNAs, together with plasmids expressing Flag-SQSTM1, followed by SeV (MOI = 0.1) infection for 12 h. Protein lysates were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation and immunoblot using indicated antibodies. (D) Protein lysates of 293 T cells transfected with plasmids expressing Flag-SQSTM1 and HA-K63-Ub, together with expression vector for WT MYC-OTUD7B and OTUD7BC194S,H358R mutant, were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with anti-HA antibody. (E) Schematic diagram of SQSTM1K7R mutant. (F) Protein lysates of 293 T cells transfected with plasmids expressing WT Flag-SQSTM1 and SQSTM1K7R mutant, together with expression vector of HA-K63-Ub and MYC-OTUD7B, were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with anti-HA antibody. Samples in (A-D and F) were incubated for 5 min with 1% SDS before immunoprecipitation. For (A-D and F), similar results are obtained for three independent biological experiments.
OTUD7B enhances the oligomerization of SQSTM1
The failure of deubiquitination on SQSTM1K7R mutant suggests that OTUD7B might modulate SQSTM1 oligomerization activity to suppress type I IFN signaling. To investigate whether OTUD7B regulates the dimerization of SQSTM1, we performed the binding assay and found that SQSTM1 underwent auto-interaction and this association remarkably increased in the presence of OTUD7B (Figure 6A and S5A). We next used disuccinimidyl suberate (DSS) cross-linking assay to assess the change of SQSTM1 oligomerization and observed that OTUD7B considerably promoted the oligomerization of SQSTM1 (Figure 6B and S5B). In contrast, OTUD7B deficiency reduced the oligomerization of SQSTM1 upon viral infection (Figure 6C and S5C). Likewise, we knocked down OTUD7B in THP-1-derived macrophages and found that OTUD7B depletion considerably decreased the oligomerization of SQSTM1 (Figure 6D). Additionally, SQSTM1 underwent oligomerization under autophagy-induced conditions and OTUD7B deficiency attenuated this oligomerization (Figure 6E and S5D). Since K7 is critical for SQSTM1 oligomerization, we next examined whether OTUD7B affected the oligomerization of SQSTM1K7R mutant. As expected, SQSTM1K7R mutant could hardly undergo the oligomerization and OTUD7B failed to promote the oligomerization of SQSTM1K7R mutant (Figure S5E). These results indicate that OTUD7B regulates SQSTM1 oligomerization activity via the deubiquitination of SQSTM1 at K7.
Figure 6.
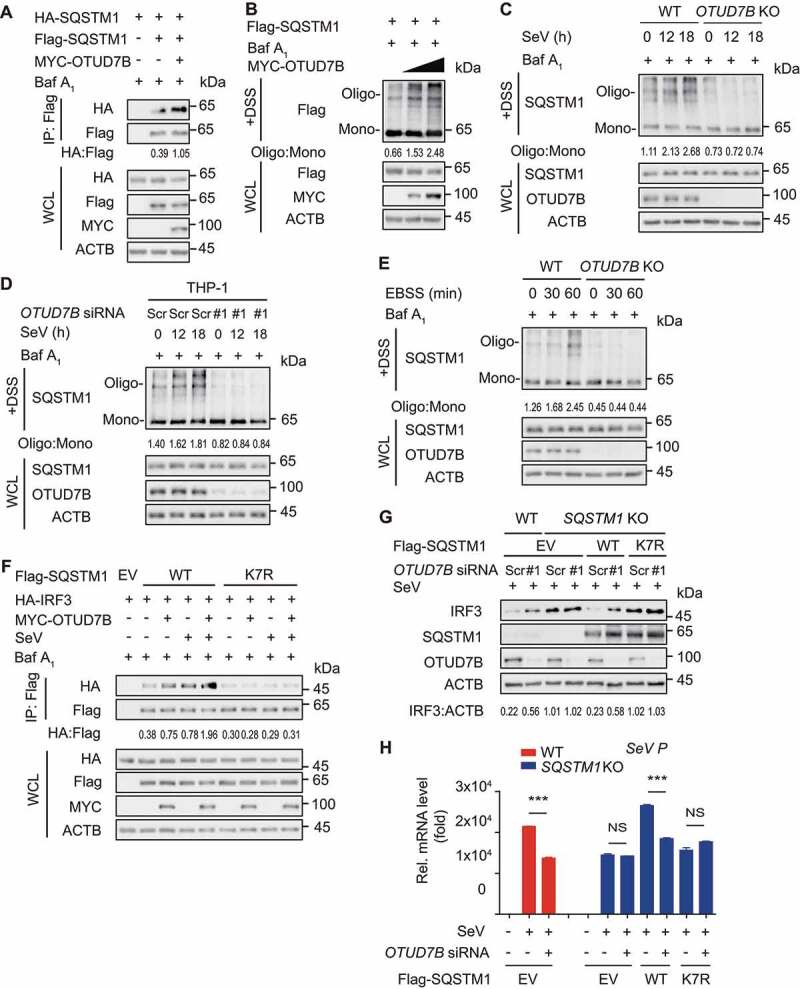
OTUD7B regulates SQSTM1 oligomerization activity. (A) 293 T cells were transfected with vectors encoding HA-SQSTM1 and Flag-SQSTM1, together with MYC-OTUD7B. Protein lysates were harvested after bafilomycin A1 (Baf A1) (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with anti-HA antibody. (B) 293 T cells transfected with plasmids encoding Flag-SQSTM1 and increasing amounts (wedge) of expression vector for MYC-OTUD7B were harvested after Baf A1 (0.2 μM) treatment (6 h). Disuccinimidyl suberate (DSS) was used to cross-link the protein and immunoblot analysis was used to measure the polymer content of Flag-SQSTM1. (C) Wild-type (WT) and OTUD7B knockout (KO) 293 T cells were treated with SeV (MOI = 0.1) infection for indicated time points, followed by Baf A1 (0.2 μM) treatment (6 h). DSS was used to cross-link the protein and immunoblot analysis was used to measure the polymer content of SQSTM1. (D) THP-1-derived macrophages were transfected with scrambled (Scr) siRNA or OTUD7B siRNA, followed by SeV (MOI = 0.1) infection at indicated time points and Baf A1 (0.2 μM) treatment for 6 h. DSS was used to cross-link the protein and immunoblot analysis was used to measure the polymer content of SQSTM1. (E) WT and OTUD7B KO 293 T cells were treated with balanced salt solution (EBSS) for indicated time points, followed by Baf A1 (0.2 μM) treatment (6 h). DSS was used to cross-link the protein and immunoblot analysis was used to measure the polymer content of SQSTM1. (F) 293 T cells were transfected with plasmids encoding empty vector (EV) or MYC-OTUD7B, together with WT SQSTM1 or SQSTM1K7R mutant and HA-IRF3, following by SeV (MOI = 0.1) infection for 18 h. Protein lysates were harvested after bafilomycin A1 (Baf A1) (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with anti-HA antibody. (G) WT and SQSTM1 KO 293 T cells were transfected with Scr siRNA or OTUD7B siRNA, together with expression vector for Flag-SQSTM1. Protein lysates were harvested after SeV (MOI = 0.1) infection for 12 h and immunoblotted with indicated antibody. (H) Quantitative RT-PCR analysis of indicated gene expression in WT or SQSTM1 KO 293 T cells transfected with Scr siRNA or OTUD7B siRNA, together with WT SQSTM1 or SQSTM1K7R mutant, and followed by SeV (MOI = 0.1) infection for 12 h. In (H), all error bars, mean values ± SEM, P-values were determined by unpaired two-tailed Student’s t test of n = 3 independent biological experiments, *** P < 0.001. NS: not significant. For (A-G), similar results are obtained for three independent biological experiments.
To further substantiate the functional importance of deubiquitination on K7 of SQSTM1 in IRF3-centered type I IFN signaling, we overexpressed OTUD7B with wild type (WT) SQSTM1 or SQSTM1K7R mutant and found that OTUD7B markedly increased the association of IRF3 and WT SQSTM1, but not its K7R mutant (Figure 6F and S5F). In addition, OTUD7B lost its ability to degrade IRF3 after virus infection when restored with the SQSTM1K7R mutant in SQSTM1 KO cells (Figure 6G). Consistently, the OTUD7B deficiency-mediated decrease of SeV replication, was abrogated in SQSTM1K7R mutant-reconstituted cells (Figure 6H). These results indicate that OTUD7B enhances SQSTM1 oligomerization activity to promote IRF3 degradation.
OTUD7B specifically promotes the activation of IRF3-associated SQSTM1 to prevent excessive type I IFN activation
Previously we and other group demonstrated that SQSTM1 targets and degrades many critical components of immune factors, such as DDX58, CGAS and MAVS [13,16,34]. In the current study, we identified OTUD7B as a positive regulator to enhance the activation of SQSTM1. Thus, we sought to investigate whether OTUD7B modulates the degradation of other SQSTM1-associated immune factors during viral infection. However, OTUD7B could hardly promote the degradation of DDX58, CGAS or MAVS, while it markedly promotes IRF3 degradation (Figure S6A). Meanwhile, we knocked down OTUD7B in THP-1-derived macrophages and found that OTUD7B depletion considerably upregulated the protein level of IRF3, but not DDX58, MAVS or CGAS in both un-infected and SeV-infected cells (Figure S6B). In addition, we found that depletion of OTUD7B in THP-1-derived macrophages resulted in decreased association between SQSTM1 and IRF3, but not DDX58, MAVS, or CGAS (Figure S6C). However, we observed that IRF3 depletion significantly attenuated the association of OTUD7B and SQSTM1 (Figure 7A). These results suggest that OTUD7B might specifically mediate the activation of IRF3-associated SQSTM1.
Figure 7.
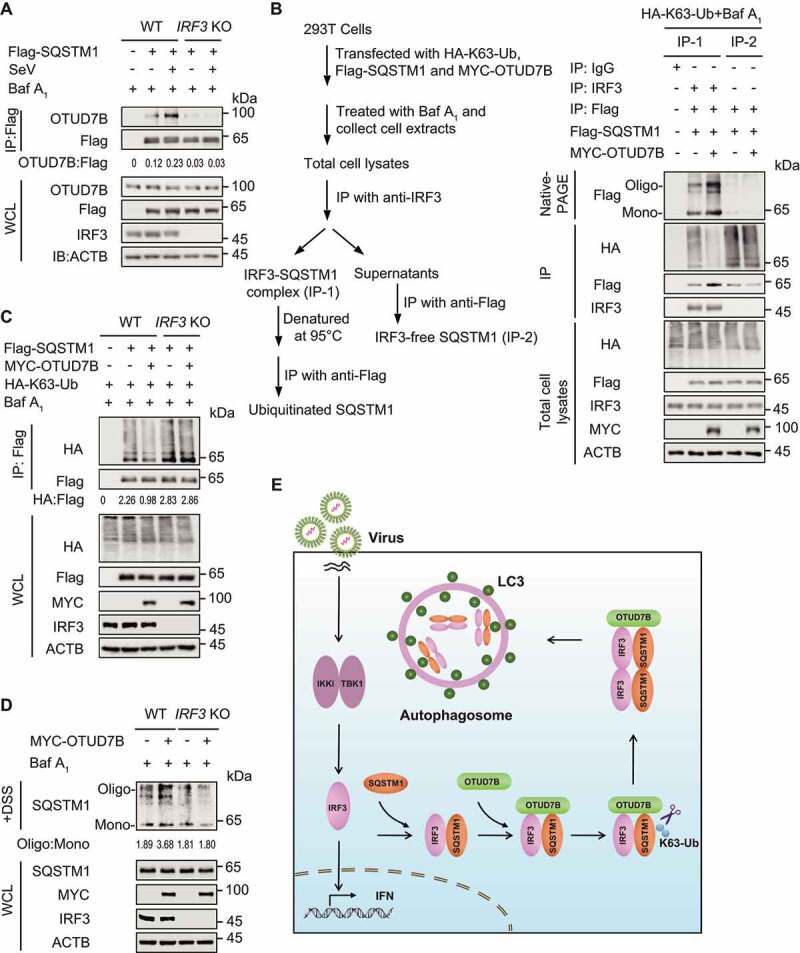
OTUD7B specifically promotes the activation of IRF3-associated SQSTM1 to prevent excessive type I IFN activation. (A) Protein lysates of wild-type (WT) or IRF3 knockout (KO) 293 T cells transfected with Flag-SQSTM1, following by SeV (MOI = 0.1) infection for 18 h, were harvested after bafilomycin A1 (Baf A1) (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with indicated antibody. (B) Protein lysates of 293 T cells transfected with plasmids expressing Flag-SQSTM1 and HA-K63-ubiquitin (Ub), together with empty vector (EV) or expression vector of MYC-OTUD7B, were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation and immunoblot analysis. Protein lysates were firstly immunoprecipitated with IRF3 antibody (IP-1), followed by stripping and re-immunoprecipitation with anti-Flag to get IRF3-associated SQSTM1. The supernatants of IP-1 were further used for immunoprecipitation with anti-Flag beads to get IRF3-free SQSTM1 (IP-2). Since TBS buffer containing DSS is not suitable for immunoprecipitation analysis, 10% native PAGE was used to examine the oligomerization of SQSTM1 in the whole-cell extracts and IRF3-free supernatants. (C) Protein lysates of WT or IRF3 KO 293 T cells transfected with Flag-SQSTM1 and HA-K63-Ub, together with EV or expression vector of MYC-OTUD7B, were harvested after Baf A1 (0.2 μM) treatment (6 h) for immunoprecipitation with anti-Flag beads and immunoblotted with indicated antibody. (D) Protein lysates of WT or IRF3 KO 293 T cells transfected with EV or expression vector of MYC-OTUD7B, were harvested after Baf A1 (0.2 μM) treatment (6 h). DSS was used to cross-link the protein and immunoblot analysis was used to measure the polymer content of SQSTM1. (E) Work model to illustrate how OTUD7B negatively regulates type I IFN signaling. Samples in (C) were incubated for 5 min with 1% SDS before immunoprecipitation. For (A-D), similar results are obtained for three independent biological experiments.
To substantiate the role of IRF3 in the activation of SQSTM1 triggered by OTUD7B, we applied two-step coimmunoprecipitation analysis to assess the ubiquitination and oligomerization of IRF3-associated SQSTM1 (IP-1) and IRF3-unassociated SQSTM1 (IRF3-free SQSTM1, IP-2). Since TBS buffer with DSS is not suitable for immunoprecipitation analysis, the oligomerization of SQSTM1 in the whole cell lysates and the supernatants of IP-1 was detected by native PAGE. We observed that OTUD7B failed to modulate the K63-linked poly-ubiquitination of IRF3-free SQSTM1, as well as its oligomerization (Figure 7B). We further examined the ubiquitination of SQSTM1 in IRF3 KO cells and found that OTUD7B failed to decrease the K63-linked poly-ubiquitination of SQSTM1 in the absence of IRF3 (Figure 7C). Meanwhile, in IRF3 KO cells, OTUD7B could not enhance SQSTM1 oligomerization (Figure 7D). These results suggest that OTUD7B specifically mediates the activation of SQSTM1 in a substrate-dependent manner.
Discussion
Autophagy, an evolutionary conserved degradative process, has been reported to regulate type I IFN signaling through mediating the degradation of IRF3 [8,9]. IRF3 is an essential transcription factor in innate immune signaling. We have found IRF3 could be ubiquitinated and specifically degraded through autophagy with the help of cargo receptor CALCOCO2 [9]. Here, we identified another cargo receptor SQSTM1, which selectively sequesters aggregates into inclusion bodies and delivers them to autophagosome, also mediates the degradation of IRF3. By removing K63-linked poly-ubiquitination of SQSTM1 at K7 residue, OTUD7B plays a critical role in controlling the activation of SQSTM1 to degrade IRF3. Thus, our studies illustrate that distant cargo receptors may coordinate the stability of IRF3 via diverse regulatory mechanisms. Not only ubiquitinated IRF3 could be recognized by cargo receptors and delivered to autophagosome for degradation [9], but also the ability of cargo receptor, such as SQSTM1, is regulated by ubiquitination to co-aggregate with IRF3. Such multiple regulatory mechanisms ensure the stringent control of the stability of IRF3 to maintain host antiviral immunity and homeostasis.
Selective autophagy is regulated at diverse levels, including the selection of components and autophagy receptors [35]. As a selective autophagy receptor, SQSTM1 recognizes and delivers the ubiquitinated proteins to the autophagosome for degradation [36]. The oligomerization of SQSTM1 plays critical roles in mediating its activity in autophagy [37]. However, the regulatory mechanisms of SQSTM1 oligomerization are not fully uncovered. Ubiquitination has been reported to modulate the activity and function of SQSTM1 in autophagy. Recent studies showed that K63-linked poly-ubiquitination inhibits the oligomerization and sequestration function of SQSTM1 in response to oxidative stress, to block autophagy-lysosomal degradation of protein aggregates [33,38]. Thus, dynamic ubiquitination of SQSTM1 should be subtly regulated and plays an important role in autophagy-lysosomal degradation system [38,39]. Ubiquitination can be reversed by DUBs, which remove ubiquitin chains from the target protein [40]. In this study, we identified deubiquitinase OTUD7B as a negative regulator for the K63-linked poly-ubiquitination of SQSTM1. OTUD7B binds to SQSTM1 and removes the K63-linked poly-ubiquitin chains from SQSTM1 at K7, which is critical for SQSTM1 oligomerization and ubiquitin binding [33]. OTUD7B-mediated deubiquitination of SQSTM1 enhances its oligomerization and accelerates IRF3 degradation. Additionally, our study showed that viral infection and nutrition stress increased the oligomerization of SQSTM1, and OTUD7B deficiency remarkably attenuated SQSTM1 oligomerization. Our findings suggest that OTUD7B regulates the activation of SQSTM1 to mediate IRF3 degradation.
Moreover, IRF3 is indispensable for OTUD7B to regulate the activity of SQSTM1. IRF3 serves as a specific bridge in OTUD7B-SQSTM1 association. IRF3 depletion significantly attenuates the association of OTUD7B and SQSTM1, as well as SQSTM1 oligomerization. In addition, OTUD7B fails to modulate the K63-linked poly-ubiquitination and oligomerization of IRF3-free SQSTM1. These lines of evidence raise the possibility that OTUD7B specifically promotes the activation of SQSTM1 in a substrate-dependent manner. We further demonstrate that OTUD7B could not enhance the activation of SQSTM1 in IRF3-depleted cells. Intriguingly, the substrate IRF3 modulates its own degradation by mediating the activation of its cargo receptor SQSTM1. However, the regulatory mechanism of IRF3 in controlling the activation of its cargo receptor SQSTM1 via OTUD7B is far from clear. Dissecting the downstream factors of IRF3 may shed light on this question.
OTUD7B was reported as a specific DUB to hydrolyze the K63-linked poly-ubiquitin chains [32]. In a recent study, we found OTUD7B participates in autophagy via reducing the K63-linked poly-ubiquitination of PIK3C3 [19]. Moreover, OTUD7B reduces the K63-linked poly-ubiquitination of MLST8 to regulate MTORC2 signaling [20]. In addition, OTUD7B is reported to modulate the binding partner specificity of LSD1 by removing its K63-linked poly-ubiquitin chains [30]. Here, we found OTUD7B removed the K63-linked poly-ubiquitin chains form SQSTM1 and governed its binding partner specificity to IRF3. Apart from K63-linked poly-ubiquitin chains, OTUD7B also shows the specificity to interact with K11, K48 and Met1-linked ubiquitin chains [32]. Specificity for particular ubiquitin chain architectures leads to the generation of specific cleavage products and specific function of DUB [41]. Thus, K63-linked poly-ubiquitin chains might be a specific signal for OTUD7B-mediated deubiquitination in regulating antiviral immunity.
Collectively, our findings provide new insights into how OTUD7B regulates innate antiviral immunity and its crosstalk with autophagy. Upon virus infection, OTUD7B interacts with IRF3, and then OTUD7B activates IRF3-associated SQSTM1 by removing its K63-linked poly-ubiquitin chains at K7, leading to the increased activation of SQSTM1 to promote IRF3 degradation, which prevents the host from excessive type I IFN activation. Moreover, viral infection increased the expression of OTUD7B, which forms a negative feedback loop by promoting IRF3 degradation to balance type I IFN signaling (Figure 7E). Collectively, we demonstrate that OTUD7B plays a specific role in mediating the activation of cargo receptors in a substrate-dependent manner and it might be a potential target against excessive immune responses.
Materials and methods
Cell lines and culture conditions
HEK293 T (GNHu17), A549 (TCHu150) and THP-1 (TCHu57) cells were obtained from the Cell Bank of the Chinese Academy of Sciences. HEK293 T and A549 cells were maintained in DMEM medium (Corning, 10–013-CVR) with 10% (vol:vol) fetal bovine serum (Gibco, 10099141) and 1% L-glutamine (Gibco, 35050061). THP-1 cells were maintained in RPMI 1640 medium (Gibco, C22400500BT) with 10% (vol:vol) fetal bovine serum and 1% L-glutamine. THP-1 cells were differentiated into macrophages cultured with RPMI 1640 containing 100 ng/ml phorbol-12-myristate-13-acetate (PMA; Sigma, P8139) for 12 h, and then the macrophages had a resting period of 24 h before being stimulated. All cells were incubated at 37°C incubator with 5% CO2.
Plasmids and transfection
Plasmids for OTUD7B and its mutant were cloned into the pcDNA3.1 vector (provided by Rong-Fu Wang laboratory, Houston Methodist Research Institute) for transient expression. HEK293 T transfection was performed using Lipofectamine 2000 (Invitrogen, 11668019) according to procedures recommended by the manufacturer. Chemically synthesized 21-nucleotide siRNA duplexes were obtained from TranSheepBio and transfected using Lipofectamine RNAiMAX (Invitrogen, 13778150) according to the manufacturer’s instructions. The sequences of target siRNAs are as follows:
SiOTUD7B-1: 5’-GGAUGACAUCGUUCAAGAATT-3’;
SiOTUD7B-2: 5’-CCCAUCCCUUGGAACGUAATT-3’;
Scrambled siRNA: 5’-UUCUCCGAACGUGUCACGUTT-3’.
Antibodies and reagents
Monoclonal anti-Flag M2-peroxidase (A8592), monoclonal anti-ACTB/β-actin antibody produced in mouse AC-74 (A2228), anti-Flag M2 affinity gel (A2220) and anti-MB21D1/CGAS antibody (HPA031700) were purchased from Sigma. Anti-MYC-horseradish peroxidase (11814150001) and anti-HA-peroxidase (high affinity from rat immunoglobulin G1) (12013819001) were purchased from Roche. DDX58/RIG-I (D14 G6) rabbit monoclonal antibody (mAb) (3743S), K63-linkage specific polyubiquitin (D7A11) antibody (5621S) and LC3B (D11) XP® Rabbit mAb (3868S) were purchased from Cell Signaling Technology. Anti-ubiquitin (sc-8017), IRF3 antibody (FL-425) (sc-9082), IRF3 antibody (SL-12) (sc-33641) and mouse monoclonal anti-MAVS (sc-166583) were purchased from Santa Cruz Biotechnology. OTUD7B monoclonal antibody (66276-1-Ig) and SQSTM1/p62 polyclonal antibody (18420-1-AP) were purchased from Proteintech Group. Goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 568 (A-11011) and goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 633 (A-21050) were purchased from Invitrogen.
Generation of knockout cell lines
For OTUD7B knockout cells, lentiviral particles were produced by transfecting HEK293 T cells with target sequences cloned into pLentiCRISPRv2 (Addgene, 52961; deposited by Jun Cui). The medium was changed the following day and the viral containing supernatant was collected 48 h after transfection, filtered through a 0.45-μm filter and subsequently used to infect cells with polybrene (8 μg/mL; Sigma, 107689). 293 T cells were infected by incubation with lentivirus-containing supernatant for 48 h. To generate OTUD7B knockout cells, the target sequences (OTUD7B guide #1: 5′-GAGGAGATCTCGCGCTAGCCC-3′; OTUD7B guide #2: 5′-GTCAGATTTTGTCCGTTCCAC-3′) were cloned into pLentiCRISPRv2 by cutting with BsmBI. Infected cells were treated with 1 μg/mL puromycin for 48 h to enrich transfected cells, which were then diluted and placed into 96-well plates for single colonies. The gRNAs for SQSTM1 and IRF3 were previously reported [15,42].
Immunoprecipitation and immunoblot analysis
For immunoprecipitation, whole-cell extracts were prepared after transfection or stimulation with appropriate ligands, followed by incubation overnight with the appropriate antibodies plus protein A/G beads (Pierce, 20423) or anti-Flag (Sigma, A2220). Beads were then washed five times with low-salt lysis buffer (50 mM HEPES, pH 7.5 [Gibco, 15630080], 150 mM NaCl [Sigma, S5886], 1 mM EDTA [Vetec, 60–00-4], 10% glycerol [Vetec, V900122], 1.5 mM MgCl2 [Vetec, V900020], 1% Triton X-100 [Sigma, T9284]), and immunoprecipitates were eluted with 2× SDS Loading Buffer (FD Biotechnology, FD003) and resolved by SDS-PAGE. For sequential immunoprecipitation assays, Flag-IRF3 was immunoprecipitated from the cell extracts with anti-Flag beads. After extensive washing with the low-salt lysis buffer, the proteins were eluted with 3 × FLAG Peptide (APEX Bio, A6001) and then centrifuged at 10,000 × g at 4°C for 1 min to collect the supernatant. The supernatant was incubated overnight with anti-OTUD7B antibodies plus protein A/G beads. Beads were then washed five times with low-salt lysis buffer and immunoprecipitates were eluted with 2× SDS Loading Buffer and resolved by SDS-PAGE. For deubiquitination assays in cultured cells, the cells were lysed with low-salt lysis buffer and the supernatants were denatured at 95°C for 5 min in the presence of 1% SDS. The denatured lysates were diluted with lysis buffer to reduce the concentration of SDS below 0.1% followed by immunoprecipitation (denature-IP) with the indicated antibodies. Proteins were transferred to PVDF membranes (Bio-Rad, 1620177) and further incubated with the appropriate antibodies. Immobilon Western Chemiluminescent HRP Substrate (Millipore, WBKLS0500) was used for protein detection.
Fluorescence microscopy
Cells were cultured on glass bottom culture dishes (Nest Scientific, 801002) and directly observed as previously described [13]. For examination by immunofluorescence microscopy, cells were fixed with 4% paraformaldehyde for 15 min, and then permeabilized in methyl alcohol for 10 min at −20°C. After washing with PBS for three times, cells were blocked in 5% fetal goat serum (Boster Biological, AR1009) for 1 h, and then incubated with primary antibodies diluted in 10% bull serum albumin (Sigma, A1933) overnight. The cells were washed, and followed by a fluorescently labeled secondary antibody (Alexa Fluor 633 goat anti-mouse IgG (H + L) (pink) or CF568 goat anti-rabbit IgG (H + L) (red). Confocal images were examined using a Leica TCS-SP8 confocal microscope (TCS-SP8, Leica) equipped with a × 100 NA oil-immersion objective.
Virus infection
SeV and VSV-eGFP were kindly provided by Dr. Xiaofeng Qin (Suzhou Institute of Systems Medicine). SeV and VSV-eGFP were titered on Vero cells. Virus titers were measured by means of 50% of the tissue culture’s infectious dose (TCID50).
Quantitative RT-PCR
Total RNA was extracted from cells using the Trizol reagent (Invitrogen, 10296010) according to the manufacturer’s instructions. For RT-PCR analysis, cDNA was generated with HiScript® II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme, R223-01) and was analyzed by quantitative real-time PCR using the 2× RealStar SYBR Mixture (GenStar, A311). The sequences of primers are as follows:
IFNB1: Forward: 5′-GATGAACTTTGACATCCCTGAG-3′,
Reverse: 5′-TCAACAATAGTCTCATTCCAGC-3′;
ISG15: Forward: 5′- CGCAGATCACCCAGAAGATCG-3′,
Reverse: 5′- TTCGTCGCATTTGTCCACCA-3′;
IFIT2: Forward: 5′-TATTGGTGGCAGAAGAGGAAGA-3′,
Reverse: 5′-CAGGTGAAATGGCATTTTAGTT-3′;
IFIT1: Forward: 5′-TCAGGTCAAGGATAGTCTGGAG-3′,
Reverse: 5′-AGGTTGTGTATTCCCACACTGTA-3′;
SeV P: Forward: 5′-TGTTATCGGATTCCTCGACGCAGTC-3′,
Reverse:5′-TACTCTCCTCACCTGATCGATTATC-3′.
IRF3: Forward: 5′- TCCCACTCCCTTCCCAAACCT-3′,
Reverse: 5′- AGCGTCCTGTCTCCCACTTCG-3′;
OTUD7B: Forward: 5′- GACAGAGAGCCTACTCGCC-3′,
Reverse: 5′- CACAGATGGGCATTTCCAGG-3′;
RPL13A: Forward: 5′-GCCATCGTGGCTAAACAGGTA-3′,
Reverse: 5′-GTTGGTGTTCATCCGCTTGC-3′.
Luciferase and reporter assays
Cells were plated in 24-well plates and transfected with plasmids encoding the ISRE luciferase reporter (firefly luciferase, 30 ng) and pRL-TK (Renilla luciferase, 10 ng), which were kindly provided by Dr. Rong-Fu Wang, together with different plasmids (100 ng). Cells treated with SeV, VSV or IC poly(I:C) stimulation for the indicated times were collected and luciferase activity was measured with Dual-Luciferase Assay (Promega, E1910) with a Luminoskan Ascent luminometer (Thermo Fisher Scientific). Reporter gene activity was determined by normalization of the firefly luciferase activity to Renilla luciferase activity. The values were means ± SD of 3 independent transfections performed in parallel.
Detection of SQSTM1 oligomerization
293 T cells were seeded in 12-well plates and treated as indicated. The cell pellets were collected into 1.5-ml microcentrifuge tubes and lysed in TBS buffer (50 mM Tris-HCl pH 7.9, 150 mM NaCl, 0.5% Triton X-100, pH 7.4) with phosphatase inhibitor (Roche, 04906837001) and EDTA-free protease inhibitor (Bimake, B14011) on a rocker for 30 min on ice, and then centrifuged at 6000 × g at 4°C for 15 min to discard the supernatants. The pellets were washed twice with TBS buffer and resuspend in TBS buffer containing 2 mM fresh disuccinimidyl suberate (DSS; Thermo Fisher Scientific, 21655) cross-linker at 37°C for 30 min to crosslink with flipping the tubes every 10 min, and then spun at 6000 × g at 4°C for 15 min. The crosslinked pellets were resuspended in 25 μl 2× SDS loading buffer and boiled at 100°C for 5 min, and analyzed by immunoblotting of anti-SQSTM1/p62 antibody.
To analyze the SQSTM1/p62 oligomerization in IRF3-associated material and IRF3-free supernatants respectively, sequential immunoprecipitation is performed. Since TBS buffer containing DSS is not suitable for immunoprecipitation analysis, whole-cell extracts were prepared with low-salt lysis buffer and 10% native PAGE was used to examine the oligomerization of SQSTM1/p62 in the whole-cell extracts and IRF3-free supernatants.
Statistical analysis
Data are represented as mean ± SEM unless otherwise indicated, and Student’s t-test was used for all statistical analysis with the GraphPad Prism 8 software. Differences between two groups were considered significant when P value was less than 0.05.
Supplementary Material
Funding Statement
This work was supported by the National Natural Science Foundation of China [92042303, 31870862, 31970700 and 32170876], Science and Technology Planning Project of Guangzhou, China [201907010038], Guangdong Basic and Applied Basic Research Foundation [2020B1515120090], and China Postdoctoral Science Foundation [2020TQ0388 and 2020M683036].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Wang J, Flavell RA, Li HB.. Antiviral immunity: a link to bile acids. Cell Res. 2019;29(3):177–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Takeuchi O, Akira S. Recognition of viruses by innate immunity. Immunol Rev. 2007;220:214–224. [DOI] [PubMed] [Google Scholar]
- [3].Lian H, Zang R, Wei J, et al. The zinc-finger protein ZCCHC3 binds RNA and facilitates viral RNA sensing and activation of the RIG-I-like receptors. Immunity. 2018;49(3):438–448.e5. [DOI] [PubMed] [Google Scholar]
- [4].Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4(5):491–496. [DOI] [PubMed] [Google Scholar]
- [5].Qin BY, Liu C, Lam SS, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10(11):913–921. [DOI] [PubMed] [Google Scholar]
- [6].Zhang Z, Wang D, Wang P, et al. OTUD1 negatively regulates type I IFN induction by disrupting noncanonical ubiquitination of IRF3. J Immunol. 2020;204(7):1904–1918. [DOI] [PubMed] [Google Scholar]
- [7].Higgs R, Gabhann JN, Larbi NB, et al. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. 2008;181(3):1780–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jiang LQ, Xia T, Hu YH, et al. IFITM3 inhibits virus-triggered induction of type I interferon by mediating autophagosome-dependent degradation of IRF3. Cell Mol Immunol. 2018;15(9):858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu Y, Jin S, Liu Q, et al. Selective autophagy controls the stability of transcription factor IRF3 to balance type I interferon production and immune suppression. Autophagy. 2021;17(6):1379–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Martin PK, Marchiando A, Xu R, et al. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol. 2018;3(10):1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kumsta C, Chang JT, Lee R, et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat Commun. 2019;10(1):5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Du Y, Duan T, Feng Y, et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018;37(3):351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen M, Meng Q, Qin Y, et al. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell. 2016;64(1):105–119. [DOI] [PubMed] [Google Scholar]
- [14].Jin S, Tian S, Luo M, et al. Tetherin suppresses Type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell. 2017;68(2):308–322. [DOI] [PubMed] [Google Scholar]
- [15].Prabakaran T, Bodda C, Krapp C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 2018;37(8):e97858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res. 2016;26(4):457–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yin Z, Popelka H, Lei Y, et al. The roles of ubiquitin in mediating autophagy. Cells. 2020;9(9):2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu Q, Wu Y, Qin Y, et al. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci Adv. 2018;4(5):eaar2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tian S, Jin S, Wu Y, et al. High-throughput screening of functional deubiquitinating enzymes in autophagy. Autophagy. 2021;17(6):1367–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang B, Jie Z, Joo D, et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature. 2017;545(7654):365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ji Y, Cao L, Zeng L, et al. The N-terminal ubiquitin-associated domain of Cezanne is crucial for its function to suppress NF-κB pathway. J Cell Biochem. 2018;119(2):1979–1991. [DOI] [PubMed] [Google Scholar]
- [22].Sianipar IR, Matsui C, Minami N, et al. Physical and functional interaction between hepatitis C virus NS5A protein and ovarian tumor protein deubiquitinase 7B. Microbiol Immunol. 2015;59(8):466–476. [DOI] [PubMed] [Google Scholar]
- [23].Hu H, Brittain GC, Chang JH, et al. OTUD7B controls non-canonical NF-kappa B activation through deubiquitination of TRAF3. Nature. 2013;494(7437):371–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang ZC, Chen Q, Wang J, et al. Sulforaphane mitigates LPS-induced neuroinflammation through modulation of Cezanne/NF-κB signalling. Life Sci. 2020;262:118519. [DOI] [PubMed] [Google Scholar]
- [25].He X, Zhu Y, Zhang Y, et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 2019;38(14):e100978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xian H, Yang S, Jin S, et al. LRRC59 modulates type I interferon signaling by restraining the SQSTM1/p62-mediated autophagic degradation of pattern recognition receptor DDX58/RIG-I. Autophagy. 2020;16(3):408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hu H, Wang H, Xiao Y, et al. Otud7b facilitates T cell activation and inflammatory responses by regulating Zap70 ubiquitination. J Exp Med. 2016;213(3):399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang B, Yang C, Wang R, et al. OTUD7B suppresses Smac mimetic-induced lung cancer cell invasion and migration via deubiquitinating TRAF3. J Exp Clin Cancer Res. 2020;39(1):244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Luong LA, Fragiadaki M, Smith J, et al. Cezanne regulates inflammatory responses to hypoxia in endothelial cells by targeting TRAF6 for deubiquitination. Circ Res. 2013;112(12):1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gong Z, Li A, Ding J, et al. OTUD7B deubiquitinates LSD1 to govern its binding partner specificity, homeostasis, and breast cancer metastasis. Adv Sci (Weinh). 2021;8(15):e2004504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bonacci T, Suzuki A, Grant GD, et al. Cezanne/OTUD7B is a cell cycle-regulated deubiquitinase that antagonizes the degradation of APC/C substrates. EMBO J. 2018;37(16):e98701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mader J, Huber J, Bonn F, et al. Oxygen-dependent asparagine hydroxylation of the ubiquitin-associated (UBA) domain in Cezanne regulates ubiquitin binding. J Biol Chem. 2020;295(8):2160–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pan JA, Sun Y, Jiang YP, et al. TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell. 2016;61(5):720–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu J, Wu X, Wang H, et al. HFE inhibits type I IFNs signaling by targeting the SQSTM1-mediated MAVS autophagic degradation. Autophagy. 2021;17(8):1962–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61(6):609–624. [DOI] [PubMed] [Google Scholar]
- [36].Ciuffa R, Lamark T, Tarafder AK, et al. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015;11(5):748–758. [DOI] [PubMed] [Google Scholar]
- [37].Lee Y, Chou TF, Pittman SK, et al. Keap1/Cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 2017;19(1):188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guo C, Zhang Y, Nie Q, et al. SQSTM1/ p62 oligomerization contributes to Aβ-induced inhibition of Nrf2 signaling. Neurobiol Aging. 2021;98:10–20. [DOI] [PubMed] [Google Scholar]
- [39].Lim D, Lee HS, Ku B, et al. Oligomer model of PB1 domain of p62/SQSTM1 based on crystal structure of homo-dimer and calculation of helical characteristics. Mol Cells. 2019;42(10):729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26(4):399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. 2019;20(6):338–352. [DOI] [PubMed] [Google Scholar]
- [42].Vargas JNS, Wang C, Bunker E, et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell. 2019;74(2):347–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.