

Abstract
Background and Purpose
To investigate the diagnostic and prognostic value of axonal injury biomarkers in patients with inflammatory polyneuropathies.
Methods
Neurofilament light chain (NfL) and total tau (T‐tau) were measured in the cerebrospinal fluid (CSF) and plasma in 41 patients with Guillain–Barré syndrome (GBS), 32 patients with chronic inflammatory demyelinating polyneuropathy (CIDP), 10 with paraproteinemia‐related demyelinating polyneuropathy (PDN), and 8 with multifocal motor neuropathy (MMN), in comparison with 39 disease‐free controls and 59 other controls. Outcome was measured with the GBS‐disability score (GBS‐ds) or Inflammatory Neuropathy Cause and Treatment (INCAT) disability score.
Results
Neurofilament light chain levels in CSF and plasma were higher in GBS, CIDP, and PDN vs. disease‐free controls. Patients with MMN had higher NfL levels in plasma vs. disease‐free controls, but lower levels in CSF and plasma vs. patients with amyotrophic lateral sclerosis (ALS).
T‐tau levels in plasma were higher in GBS, CIDP, PDN, and MMN vs. all control groups.
Neurofilament light chain levels in CSF and plasma in patients with GBS correlated with GBS‐ds, as higher levels were associated with inability to run after 6 and 12 months. NfL levels in CSF and plasma in CIDP did not correlate significantly with outcome.
Conclusions
Acute and chronic inflammatory neuropathies are associated with an increase in levels of NfL in CSF and plasma, but NfL is validated as a prognostic biomarker only in GBS. NfL could be used in differentiating patients with MMN from ALS. T‐tau in plasma is a novel biomarker that could be used in a diagnostic assessment of patients with acute and chronic inflammatory polyneuropathies.
Keywords: chronic inflammatory demyelinating polyneuropathy (CIDP), CSF and plasma, diagnosis and prognosis, Guillain‐Barré syndrome (GBS), multifocal motor neuropathy (MMN), NfL, paraproteinemia‐related demyelinating polyneuropathy (PDN), T‐tau
Acute and chronic inflammatory neuropathies are associated with an increase in levels of neurofilament light chain (NfL) in cerebrospinal fluid (CSF) and plasma, but NfL is validated as a prognostic biomarker only in Guillain–Barré syndrome. NfL in CSF and plasma could be used in differentiating patients with multifocal motor neuropathy from ALS. T‐tau in plasma is a novel biomarker that could be used in a diagnostic assessment of patients with acute and chronic inflammatory polyneuropathies.
![]()
INTRODUCTION
Inflammatory polyneuropathies are usually divided into acute and chronic forms, but their presentation can be highly variable and, in some cases, difficult to differentiate from other non‐inflammatory neuropathies. Predicting disease course, prognosis, and response to immunomodulatory treatment is challenging due to the lack of reliable prognostic biomarkers [1, 2, 3]. Neurofilaments are the most abundant cytoskeletal component of mature neurons providing structural support, but they also control important cellular processes, such as axon conduction, distribution of organelles, and receptor recycling at the synapse [4]. Neurofilament light chain (NfL) is of particular interest, and it is considered to be a marker of axonal damage in a variety of different neurological disorders, including traumatic, inflammatory, and neurodegenerative diseases [5]. The first study on neurofilaments and inflammatory polyneuropathy dates back to 2006 showing high levels of neurofilament heavy chain (NfH) in cerebrospinal fluid (CSF) predicting worse motor and functional outcome in patients with Guillain–Barré syndrome (GBS) [6]. Interest into examining the role of NfL in acute and chronic inflammatory polyneuropathies has grown rapidly in the past 2 years [7, 8, 9, 10]. However, there is only one published study investigating NfL and comparing its levels in CSF in acute vs. chronic inflammatory polyneuropathies, though with a relatively low number of patients and without examining specificity by attempting comparison with other diseases [11].
Total tau (T‐tau) is a microtubule‐associated protein and has an important role in stabilizing neuronal microtubules and affects axonal transport [12]. Tau aggregation is a characteristic of several neurodegenerative diseases such as Alzheimer's disease and progressive supranuclear palsy [13, 14]. Besides NfL, T‐tau is one of the most established biomarkers of axonal injury in the central nervous system (CNS) [15, 16]. Tau in blood has also come to attention in recent years with studies showing high levels of T‐tau in serum in patients with traumatic brain injury (TBI) and hypoxic brain injury after cardiac arrest [17, 18]. Studies in patients with GBS regarding tau levels in CSF have shown inconsistent results [19, 20, 21], whereas tau has never been properly investigated in chronic inflammatory demyelinating polyneuropathy (CIDP). However, there is one study using not age‐matched patients with CIDP as a control to amyotrophic lateral sclerosis (ALS) showing higher levels of T‐tau in CSF in the ALS cohort [22]. This difference did not reach the level of statistical significance after adjusting for age. The relationship to healthy individuals has also not been investigated.
In this observational, single‐centre, retrospective study we investigated the usefulness of NfL and T‐tau in CSF and plasma in the differential diagnosis and prognostic evaluation of acute (GBS) and chronic inflammatory polyneuropathies, namely CIDP, paraproteinemic demyelinating neuropathy (PDN), and multifocal motor neuropathy (MMN).
METHODS
Patients
The patient group consisted of adult patients with acute‐ (GBS) and chronic inflammatory polyneuropathies (CIP) (i.e., CIDP, PDN, and MMN). The control group consisted of healthy controls (HC), patients with headache (HA), non‐inflammatory polyneuropathies (NIP), and ALS. All patients and controls, apart from the HC, had been recruited at the Department of Neurology at Karolinska University Hospital in Stockholm, Sweden between 2003 and 2017. The HC consisted of healthy volunteers recruited by the Department of Neurology at University Hospital of Umeå, Sweden. The HA group consisted of patients with sudden‐onset headache who had undergone diagnostic lumbar puncture (LP) to rule out subarachnoid haemorrhage and had no pathology in blood, CSF, brain imaging, and at follow‐up. The HA patients were considered as a second cohort of healthy controls, and together with the HC collectively constituted the disease‐free controls. Non‐inflammatory polyneuropathy (NIP) consisted of chronic idiopathic axonal polyneuropathies, hereditary demyelinating and axonal neuropathies, and diabetic axonal neuropathies.
The diagnosis of GBS was based on the Asbury Criteria from 1990, whereas the CIDP, PDN, and MMN diagnoses were based on the EFNS criteria from 2010 [23, 24, 25, 26]. The diagnosis of ALS was based on well‐recognized international criteria [27].
Clinical data were retrieved from the medical chart e‐health system TakeCare and the national quality register Swedish Neuro Registries.
Demographics for all groups and time between symptom onset and evaluation as well as treatment data for inflammatory polyneuropathies are presented in Table 1. In the case of the controls, CSF/blood sampling was done at the time of diagnostic assessment. All patients with inflammatory neuropathy had undergone a clinical assessment including a disease‐specific clinical disability scale, blood tests, LP, and electrophysiology testing. In the case of GBS and CIP, the CSF/blood sampling was in most cases done at time of diagnostic assessment, that is, in the acute progressive phase of disease prior to initiation of immunomodulatory treatment (pre‐treatment samples). However, some patients with GBS and CIP underwent their first CSF and blood sampling after treatment (post‐treatment samples) as presented in Table 1. The pre‐ and post‐treatment samples were paired in a few cases (4 patients with GBS, 2 patients with CIDP). Paired post‐treatment samples were obtained from those patients who had undergone a new LP if there was any diagnostic uncertainty such as the lack of elevated protein in CSF in patients with GBS or to re‐evaluate the levels of protein in CSF in patients with CIDP before changing treatment.
TABLE 1.
Demographics, time between symptom onset and evaluation, as well as treatment data
| Parameter | GBS | CIDP | PDN | MMN | HC + HA | NIP | ALS | Significant differences |
|---|---|---|---|---|---|---|---|---|
| N | 41 | 32 | 10 | 8 | 39 | 22 | 37 | |
| Male, N (%) | 23 (56) | 25 (78) | 10 (100) | 6 (75) | 17 (44) | 15 (68) | 17 (46) | NA |
| Age; years | ||||||||
| Mean (median) ‐ adjusted age group | 56 (56) | 60 (59) | 65 (69) | 49 (49) | 52 (51) | 58 (60) | 63 (64) | No |
| Range | 23–88 | 29–80 | 40–78 | 29–66 | 36–82 | 37–85 | 46–82 | |
| Pre‐treatment samples, N | 24 | 19 | 8 | 8 | NA | NA | NA | NA |
| Post‐treatment samples, N | 17 | 13 | 2 | 0 | NA | NA | NA | NA |
| Onset to CSF/blood sampling time point1 (GBS weeks; CIDP, PDN, MMN months) | ||||||||
| Mean (median) time to pre‐treatment sampling point | 1 (1) | 12 (8) | 49 (42) | 29 (24) | NA | NA | NA | GBS vs. CIDP, PDN, MMN p**** |
| Range: time to pre‐treatment sampling | 1–3 | 1–60 | 3–144 | 2–60 | ||||
| Mean (median) time to post‐treatment sampling point | 24 (8) | 38 (24) | 66 (66) | NA | GBS vs. CIDP, PDN p**** | |||
| Range: time to post‐treatment sampling point | 4–96 | 3–120 | 60–72 | NA | NA | |||
| Onset to final follow‐up (GBS months, CIP years) | ||||||||
| Mean (median) | 11.7 (12) | 6.9 (8) | 6.5 (5) | 7.1 (7.5) | NA | NA | NA | NA |
| Range | 1–12 | 2–10 | 2–10 | 2–10 | ||||
| Immunomodulatory therapy for inflammatory neuropathy, N (%) | 34 (83)3 | 32 (100)4 | 9 (90)5 | 8 (100)6 | NA | NA | NA | NA |
| Electrophysiological tests available, N (%) | 41 (100) | 29 (91) | 9 (90) | 8 (100) | NA | N | N | NA |
| Onset to electrophysiological test2 (GBS weeks, CIP months) | ||||||||
| Mean (median) time | 1.5 (1) | 15 (9) | 35 (36) | 27 (26) | GBS vs. CIDP, PDN, MMN p**** | |||
| Range | 1–3 | 2–54 | 3–60 | 2–60 | ||||
Note: 1Time from symptom onset to cerebrospinal fluid (CSF)/blood sampling; 2time from symptom onset to electrophysiological testing; 3 and 6intravenous (iv) immunoglobulins (IVIg); 4either IVIg of iv methylprednisolone (if the initial therapy was insufficient, other therapies were given: plasma exchange (4 patients), IVIg+iv methylprednisolone (7 patients), add‐on with oral prednisolone (6 patients), ciclosporin (5 patients), azathioprine (6 patients), cyclophosphamide (7 patients), mycophenolate mofetil (1 patient), autologous hematopoietic stem cell transplantation (1 patient); 5either IVIg of iv methylprednisolone (7 patients), rituximab (5 patients), add‐on with oral prednisolone (1 patient), cyclophosphamide (1 patient), plasma exchange (1 patient).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Abbreviations: ALS, amyotrophic lateral sclerosis; CIDP, chronic inflammatory demyelinating polyneuropathy; CSF, cerebrospinal fluid; GBS, Guillain–Barré syndrome; HA, headaches; HC, healthy controls; MMN, multifocal motor neuropathy; NA, not applicable; NIP, non‐inflammatory polyneuropathy; PDN, paraproteinaemic demyelinating neuropathy.
Electrophysiological examination was performed on all patients with inflammatory polyneuropathies included in this study. Duration of time between onset to CSF/blood sampling time point and onset to electrophysiology examination are presented in Table 1.
Electrophysiological examination of the 41 patients with GBS revealed 17 cases of acute inflammatory demyelinating polyneuropathy, 6 acute motor axonal neuropathy, 2 acute motor and sensory axonal neuropathy, and 12 equivocal cases. Furthermore, 2 of the 41 patients with GBS were classified as Miller Fisher syndrome (MFS), 2 as overlap MFS/GBS, and 2 as subacute inflammatory demyelinating polyneuropathy.
Within the CIDP group, 5 of the 32 patients had a variant presentation consistent with multifocal acquired demyelinating sensory and motor neuropathy (Lewis–Sumner syndrome), 2 with distal acquired demyelinating symmetric neuropathy (though without a paraprotein or MAG antibodies), 1 motor‐dominant CIDP, and 1 with acute‐onset CIDP. The remainder of the 23 patients had a typical CIDP.
Blood/CSF findings and comorbid conditions in patients and controls are presented in Table 2. All patients with GBS and CIP were tested for paraprotein. All 8 patients with MMN were tested for ganglioside antibodies (ab) (Table 2).
TABLE 2.
Blood and cerebrospinal fluid findings and comorbid conditions in patients with inflammatory neuropathies and controls
| Parameter | GBS | CIDP | PDN | MMN | HC | HA | NIP | ALS | ||
|---|---|---|---|---|---|---|---|---|---|---|
| GBSa | GBSb | CIDPa | CIDPb | |||||||
| CSF cells (×106/L) | ||||||||||
| Mean (median) | 3.8 (2) | 4.5 (2.5) | 3.4 (2.5) | 3.4 (2.5) | 2.3 (2.4) | 1.6 (1.4) | 1.7 (2.3) | 1.9 (2) | 1.9 (2) | 2.2 (1.3) |
| Range | 0–23 | 0–22 | 0–22 | 1–9.3 | 0.6–4 | 1.1–3 | 0–2.5 | 0–4 | 0–5.1 | 0–20 |
| Q‐albumin (CSF/plasma) | ||||||||||
| Mean (median) | 19.9 (15.8) | 21.6 (14.7) | 11.2 (10.2) | 18.2 (13.6) | 12.1 (12) | 4.8 (3.8) | 4.7 (4.3) | 5.2 (5) | 6.9 (7) | 6.3 (6.2) |
| Range | 5–50.2 | 3.5–62.4 | 3.2–21.2 | 2.6–59.8 | 5.6–19.8 | 2–9.2 | 2.9–7.6 | 2.2–8.5 | 1.1–15 | 2–11.9 |
| OCB in CSF, N (%) | 0 (0) | 0 (0) | 0 (0) | 1 (8) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 1 (5) | 1 (3) |
| Paraprotein, N (%) | 0 (0) | 0 (0) | 9 (47)1 | 3 (23) 1 | 10 (100) 2 | 0 | NA | NA | NA | NA |
| MAG‐antibodies2, N (%) | NA | NA | 0 (0) | 0 (0) | 9 (90) | 0 (0) | NA | NA | NA | NA |
| Ganglioside antibodies in patients tested, N (%) | 2 of 103 (20) | NA | 1 of 84 (13) | 3 of 55 (60) | 3 of 46 (75) | 3 of 87 (38) | NA | NA | NA | NA |
| Previous infections8, N (%) | 13 (54) | 7 (41) | NA | NA | NA | NA | NA | NA | NA | NA |
| Comorbid diseases: | ||||||||||
| Autoimmune, infectious | 2 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Diabetes (type 2) | 4 | 3 | 0 | 0 | 0 | 1 | 0 | 2 | 5 | 1 |
| Other diseases 10 | 13 | 8 | 20 | 11 | 8 | 4 | 1 | 8 | 11 | 20 |
Note: 1IgG or IgA; 2IgM; 3IgG‐GQ1b ab; 4IgM‐GM1, IgM‐GD1b ab; 5IgM‐GM1, IgM‐GM2, IgM‐GD1b, IgM‐GQ1b ab; 6IgM‐GM1, IgM‐GM2, IgM‐GD1a, IgM‐GD1b, IgM‐GQ1b ab; 7IgM anti‐GM1; 8within 6 weeks prior to onset; 9psoriasis and hepatitis C; 10cardiovascular (hypertension, atrial fibrillation, arrhythmia, hyperlipidemia); airway (chronic obstructive pulmonary disease [COPD], asthma); endocrine (hypothyroidism); orthopaedic (arthrosis); neurological (migraine, restless legs syndrome, essential tremor); others (rosacea, osteoporosis, anemia, nephrolithiasis).
Abbreviations: a, pre‐treatment; ALS, amyotrophic lateral sclerosis; b, post‐treatment; CIDP, chronic inflammatory demyelinating polyneuropathy; CSF, cerebrospinal fluid; GBS, Guillain–Barré syndrome; HA, headaches; HC, healthy controls; MMN, multifocal motor neuropathy; NA, not applicable; NIP, non‐inflammatory polyneuropathy; OCB, oligoclonal bands; PDN, paraproteinaemic demyelinating neuropathy; Q‐albumin, ratio of albumin in CSF (mg/L)/plasma (g/L) × 10−3 (reference range: 15–29 years <6.0 × 10−3; 30–49 years <7.0 × 10−3; ≥50 years <9.0 × 10−3).
Sixteen of the 32 patients with CIDP had undergone a neuroradiology examination of the brain and the spinal cord ruling out any pathology that could explain an increase of NfL and T‐tau in the CSF.
Clinical assessment
Patients with GBS had been assessed with the GBS‐disability scale (GBS‐ds) [28, 29] on the day of admission to hospital, at nadir, 3 months (±1 week), 6 months (±2 weeks), and 12 months (±4 weeks) later. Patients with CIP were assessed with the Inflammatory Neuropathy Cause and Treatment (INCAT) scale [30] at the abovementioned time points at admission to 12 months, and additionally at 18 months (±4 weeks), 24 months (±4 weeks), 5 years (±2 months), and 8–10 years post‐onset (the latter group combined). The scoring was done retrospectively by IK using data from medical charts and Swedish Neuro Registries when scoring was unavailable in the medical charts.
Biomaterial
Paired CSF and EDTA plasma from all patient groups was collected during routine clinical workup and saved at the biobank at the Department of Neurology, Karolinska University Hospital, Stockholm, Sweden between 2003 and 2017. CSF and plasma were centrifuged at 2000 g for 10 min and 400 μl of each was initially aliquoted into glass tubes and stored at −80°C. Before shipping, all samples were thawed and aliquoted into polypropylene tubes and sent to Neurochemistry Laboratory in Mölndal, Sweden in 2018 where NfL and T‐tau analysis were performed.
Analysis of NfL and T‐tau levels in CSF and plasma
In CSF, T‐tau was measured using the Lumipulse technology (Fujirebio), while NfL was measured using an in‐house enzyme‐linked immunosorbent assay (ELISA) method [31]. NfL and T‐tau measurements in plasma were performed using the NF‐Light and Tau 2.0 kits on a Simoa HD‐X Analyzer (Quanterix) according to the manufacturer's instructions.
Statistics
Statistical analyses were done using GraphPad Prism 8.0 (GraphPad Software). A logarithmic (log) transformation was used due to a non‐Gaussian distribution of data. Whenever distribution was normal, parametric tests were used to compare groups (t‐test if two groups or one‐way ANOVA if three or more groups). However, if the distribution was non‐Gaussian after log transformation, nonparametric tests were used (i.e., Mann–Whitney U test if two groups or Kruskal–Wallis if three or more groups). Multiple comparisons were corrected for with Holm‐Sidak or Dunn's test.
Correlation between different variables was tested with Pearson correlation or Spearman’s coefficient depending on distribution of data. A simple logistic regression model was used to investigate the association between NfL levels and clinical outcome at different time points, and multiple regression models (logistic and linear) were used to investigate the association between NfL, age, and clinical outcome. In multiple regression models, CSF and plasma levels of NfL were evaluated both as continuous and binary variables. NfL as a binary variable was stratified as low or high based on median pre‐treatment GBS, CIDP, PDN, or MMN group, respectively. NfL levels above the group median were considered as a cut‐off for high NfL. A further subanalysis was performed where NfL was stratified as low or high based on the 90th percentile HC + HA group.
This study was approved by Stockholm Ethical Review Board (EPN 2017/952–31/1 and 2018/832–32). The healthy controls were recruited by an approved study with the ethical permission number EPN 2011–39‐31 M. For this retrospective analysis of existing data, the need for written informed consent was waived.
RESULTS
All control groups were age‐matched with patients with inflammatory neuropathies (Table 1). NfL and T‐tau values in the patients with inflammatory neuropathies pertain to the combined pre‐ and post‐treatment samples, unless mentioned specifically as only pre‐ or post‐treatment values. The disease‐free control group was defined as the combined group of HC + HA.
CSF NfL in inflammatory polyneuropathies versus controls
The median NfL levels in patients and controls are shown in Figure 1a.
FIGURE 1.
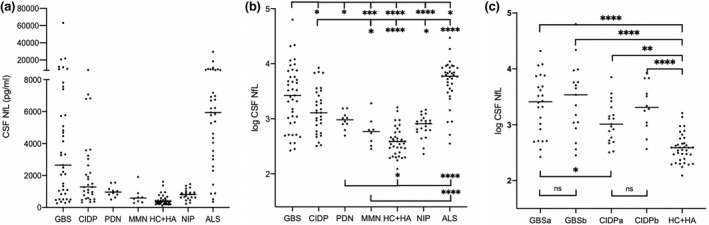
Neurofilament light chain (NfL) in the cerebrospinal fluid (CSF) in patients with inflammatory polyneuropathies and controls. (a) Absolute concentration of NfL in CSF. (b) Log transformed NfL values in Guillain–Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), paraproteinaemic demyelinating neuropathy (PDN), and multifocal motor neuropathy (MMN) vs. controls. (c) Log transformed NfL values in GBS and CIDP patients prior to and following immunomodulatory treatment. ALS, amyotrophic lateral sclerosis; CIDP, chronic inflammatory demyelinating polyneuropathy; GBS, Guillain–Barré syndrome; HA, headaches; HC, healthy controls; MMN, multifocal motor neuropathy; NIP, non‐inflammatory polyneuropathy; PDN, paraproteinaemic demyelinating neuropathy. a, pre‐treatment; b, post‐treatment; horizontal lines represent median values. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Median NfL levels were higher in patients with GBS compared to other inflammatory neuropathies, HC + HA, and NIP, but lower compared to ALS. When comparing only pre‐treatment NfL levels, patients with GBS had higher levels vs. those with CIDP (Figure 1c).
Neurofilament light chain levels in patients with CIDP were higher compared to MMN, HC + HA, and NIP, but lower compared to patients with ALS. NfL levels in patients with PDN were higher than HC + HA, but lower than ALS. There was no difference between patients with MMN and HC + HA, but NfL levels were lower than in patients with ALS (Figure 1b).
Plasma NfL in inflammatory polyneuropathies vs. controls
The median NfL levels in patients and controls are shown in Figure 2a.
FIGURE 2.
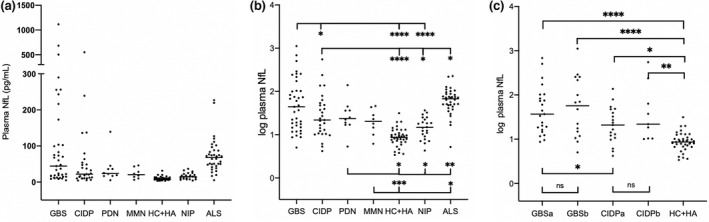
Neurofilament light chain (NfL) in plasma in patients with inflammatory polyneuropathies and controls. (a) Absolute concentration of NfL in plasma. (b) Log transformed NfL values in GBS, Guillain–Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), paraproteinaemic demyelinating neuropathy (PDN), and multifocal motor neuropathy (MMN) vs. controls. (c) Log transformed NfL values in GBS and CIDP patients prior to and following immunomodulatory treatment. ALS, amyotrophic lateral sclerosis; CIDP, chronic inflammatory demyelinating polyneuropathy; GBS, Guillain–Barré syndrome; HA, headaches; HC, healthy controls; MMN, multifocal motor neuropathy; NIP, non‐inflammatory polyneuropathy; PDN, paraproteinaemic demyelinating neuropathy. a, pre‐treatment; b, post‐treatment; horizontal lines represent median values; ns, not significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Median NfL levels in patients with GBS were higher compared to CIDP, HC + HA, and NIP. Patients with GBS had higher pre‐treatment NfL levels than those with CIDP (Figure 2c).
Neurofilament light chain levels in patients with CIDP, PDN, and MMN were higher compared to HC + HA, but lower than ALS. In addition, NfL levels were higher in CIDP and PDN compared to NIP (Figure 2b).
CSF and plasma T‐tau in inflammatory polyneuropathies vs. controls
Median T‐tau levels in patients and controls are shown in Figures 3a and 4a. The median T‐tau levels in CSF were higher in patients with CIDP compared to HC + HA.
FIGURE 3.
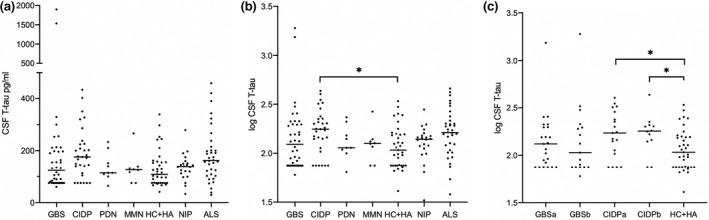
Total tau (T‐tau) in the cerebrospinal fluid (CSF) in patients with inflammatory polyneuropathies and controls. (a) Absolute concentration of T‐tau in CSF. (b) Log transformed NfL values in GBS, Guillain–Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), paraproteinaemic demyelinating neuropathy (PDN), and multifocal motor neuropathy (MMN) vs. controls. (c) Log transformed NfL values in GBS and CIDP patients prior to and following immunomodulatory treatment. ALS, amyotrophic lateral sclerosis; CIDP, chronic inflammatory demyelinating polyneuropathy; GBS, Guillain–Barré syndrome; HA, headaches; HC, healthy controls; MMN, multifocal motor neuropathy; NIP, non‐inflammatory polyneuropathy; PDN, paraproteinaemic demyelinating neuropathy. a, pre‐treatment; b, post‐treatment; horizontal lines represent median values. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
FIGURE 4.
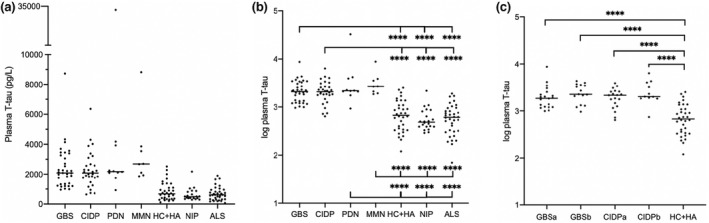
Total tau (T‐tau) in plasma in patients with inflammatory polyneuropathies and controls. (a) Absolute concentration of T‐tau in plasma. (b) Log transformed NfL values in Guillain–Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), paraproteinaemic demyelinating neuropathy (PDN), and multifocal motor neuropathy (MMN) vs. controls. (c) Log transformed NfL values in GBS and CIDP patients prior to‐ and following immunomodulatory treatment. ALS, amyotrophic lateral sclerosis; CIDP, chronic inflammatory demyelinating polyneuropathy; GBS, Guillain–Barré syndrome; HA, headaches; HC, healthy controls; MMN, multifocal motor neuropathy; NIP, non‐inflammatory polyneuropathy; PDN, paraproteinaemic demyelinating neuropathy. a, pre‐treatment; b, post‐treatment; horizontal lines represent median values. ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
The median t‐tau levels in plasma were higher in all subgroups of patients with inflammatory neuropathy compared to all controls (Figure 4a,b).
Pre‐ and post‐treatment biomarker levels in non‐paired samples
Post‐treatment NfL and T‐tau levels in CSF and plasma did not differ from pre‐treatment levels in patients with GBS and CIDP (Figures 1, 4).
NfL and T‐tau levels in CSF vs. plasma and relationship to level of the blood–nerve barrier damage
There was a correlation between the levels of NfL in CSF and plasma in the combined groups of patients and controls (Spearman’s r = 0.8, p < 0.0001), as well as in all groups (except for PDN and MMN) separately (GBS Spearman’s r = 0.7, p < 0.0001; CIDP r = 0.7, p < 0.0001; HC + HA r = 0.6, p < 0.0001; NIP r = 0.6, p = 0.003; ALS r = 0.7, p < 0.0001).
Correlation between NfL in CSF and albumin quotient (CSF/plasma quotient of albumin, Q.alb) was seen in patients with GBS (r = 0.6, p < 0.001), but not CIDP, PDN, MMN, or HC + HA. Correlation between NfL in plasma and Q.alb was seen in patients with GBS (r = 0.5, p < 0.003) and CIDP (r = 0.5, p < 0.004), but not PDN, MMN, and HC + HA.
There was no correlation between T‐tau in CSF and plasma. Nor did T‐tau in CSF or plasma correlate with Q.alb in any of the patient groups (Table S1 ).
NfL and T‐tau levels vs. clinical and electrophysiological outcome measures
In patients with GBS, NfL levels in CSF correlated with GBS‐ds at all time points, that is, the time of diagnostic LP and all time points of follow‐up, but in plasma only with the time points of follow‐up (Table 3).
TABLE 3.
Correlation between neurofilament light chain values and clinical scales in patients with Guillain–Barré syndrome, chronic inflammatory demyelinating polyneuropathy, and multifocal motor neuropathy at different time points
| GBS | CIDP | MMN | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CSF | CSF | CSF | ||||||||
| Spearman’s r | GBSds 0 m | GBSds 3 m | GBSds 6 m | GBSds 12 m | Spearman’s r | INCAT 3 m | INCAT 18 m | INCAT 8–10 y | Spearman’s r | INCAT‐arm 0 m |
| r | 0.43 | 0.51 | 0.48 | 0.46 | r | 0.48 | 0.69 | 0.7 | r | 0.54 |
| p value | 0.03 | 0.01 | 0.02 | 0.03 | p value | 0.04 | 0.001 | 0.01 | p value | 0.21 |
| p value summary | * | * | * | * | p value summary | * | ** | * | p value summary | ns |
| Plasma | Plasma | Plasma | ||||||||
| Spearman’s r | GBSds 0 m | GBSds 3 m | GBSds 6 m | GBSds 12 m | Spearman’s r | INCAT 3 m | INCAT 18 m | INCAT 8–10 y | Spearman’s r | INCAT‐arm 0 m |
| r | 0.31 | 0.48 | 0.46 | 0.46 | r | 0.39 | 0.57 | 0.28 | r | 0.81 |
| p value | 0.16 | 0.03 | 0.03 | 0.04 | p value | 0.09 | 0.01 | 0.39 | p value | 0.01 |
| p value summary | ns | * | * | * | p value summary | ns | * | ns | p value summary | * |
Abbreviations: 0 m, the time of diagnostic lumbar puncture; 3 m, 3 months after symptom onset; 6 m, 6 months after symptom onset, etc.; CIDP, chronic inflammatory demyelinating polyneuropathy; CSF, cerebrospinal fluid; GBS, Guillain–Barré syndrome; GBSds, GBS disability scale; INCAT, Inflammatory Neuropathy Cause and Treatment disability score; m, months; MMN, multifocal motor neuropathy; ns, not significant; r, Spearman'`s correlation coefficient; y, year.
* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
In patients with CIDP, NfL levels in CSF correlated with INCAT at 3 and 18 months, and 8–10 years, but in plasma only with INCAT at 18 months. There was no correlation between NfL levels and INCAT in patients with PDN. Due to the predominant upper limb involvement in MMN, we did a subanalysis of correlation of the INCAT‐arm score (0–5 points) with NfL and found that higher NfL levels in plasma correlated with a higher level of hand impairment in patients with MMN at the time of diagnostic investigation (Spearman’s r = 0.81, p = 0.01) (Table 3).
No relationship measured with the correlation coefficient was found between clinical outcome and pre‐treatment T‐tau levels in CSF and plasma in any of the subgroups of inflammatory polyneuropathy.
Low compound muscle action potential (CMAP) values in the tibial nerve correlated with high NfL levels in CSF (Spearman’s r = −0.5, p = 0.03), high NfL in plasma (r = −0.5, p = 0.02), and high T‐tau levels in CSF (r = −0.6, p = 0.003) in patients with GBS, but not those with CIDP (Table S1).
NfL as predictors of outcome
To further investigate the prognostic value of NfL and the association between NfL levels and outcome, we performed regression analyses (linear and logistic). Within the logistic model we divided outcome into two groups (i.e., favourable vs. unfavourable outcome). Favourable outcome in patients with GBS was defined as ability to run, corresponding to GBS‐ds 0–1 points (0 = healthy, 1 = minor symptoms and capable of running), and unfavourable outcome as inability to run (i.e., GBS‐ds of 2 or more points). Patients with MFS were excluded in regression analyses.
Favourable outcome in patients with CIDP was defined as a maximum of 2 out of 10 points on the INCAT scale, and unfavourable outcome as 3 or more points.
When pre‐treatment NfL was analysed as a binary variable and median values of the respective group were taken as the cut‐off for high NfL, the following results were found.
GBS
In CSF and plasma, NfL levels higher than group median were associated with the inability to run after 3, 6, and 12 months (Table 4 , data for 3 months not shown in the table). When taking age into consideration, the multiple logistic model showed the same result for outcome at 6 and 12 months. A similar association was found when analysing NfL in CSF and outcome as continuous variables, and NfL as continuous and outcome as binary variable (data not shown).
TABLE 4.
Association between neurofilament light chain levels and outcome in patients with Guillain–Barré syndrome and chronic inflammatory demyelinating polyneuropathy
| GBS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Simple logistic regression | Multiple logistic regression | ||||||||||
| CSF | CSF | ||||||||||
| Outcome1 | Variable | OR | CI 95% | p value | p value summary | Outcome1 | Variable | OR | CI 95% | p value | p value summary |
| 6 months | NfL2 | 15.8 | 1.9–351.4 | 0.02 | * | 6 months | Age | 0.9 | 0.8–1.1 | 0.9 | ns |
| NfL2 | 22.6 | 1.8–917.4 | 0.03 | * | |||||||
| 12 months | NfL2 | 31.5 | 3.2–817.9 | 0.009 | ** | 12 months | Age | 1.03 | 0.9–1.2 | 0.57 | ns |
| NfL2 | 20.8 | 1.7–713.5 | 0.03 | * | |||||||
| PLASMA | PLASMA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Outcome1 | Variable | OR | CI 95% | p value | p value summary | Outcome1 | Variable | OR | CI 95% | p value | p value summary |
| 6 months | NfL2 | 31.5 | 3.3–817.9 | 0.009 | ** | 6 months | Age | 1.1 | 0.9–1.3 | 0.39 | ns |
| NfL2 | 31.2 | 2.02–1357 | 0.02 | * | |||||||
| 12 months | NfL2 | 13.8 | 1.6–311.5 | 0.03 | * | 12 months | Age | 1.01 | 0.8–1.2 | 0.9 | ns |
| NfL2 | 59.5 | 2.4–9756 | 0.03 | * | |||||||
| CIDP | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Simple logistic regression | Multiple logistic regression | ||||||||||
| CSF | CSF | ||||||||||
| Outcome3 | Variable | OR | CI 95% | p value | p value summary | Outcome3 | Variable | OR | CI 95% | p value | p value summary |
| 6 months | NfL4 | 8.2 | 1.2–83.4 | 0.04 | * | 6 months | Age | 1.05 | 0.95–1.18 | 0.40 | ns |
| NfL4 | 3.7 | 0.30–59.2 | 0.31 | ns | |||||||
| 12 months | NfL4 | 8.0 | 1.4–81.2 | 0.04 | * | 12 months | Age | 1.03 | 0.94–1.14 | 0.50 | ns |
| NfL4 | 4.73 | 0.42–77.53 | 0.22 | ns | |||||||
| 18 months | NfL4 | 18.0 | 2.1–425.3 | 0.02 | * | 18 months | Age | 1.10 | 0.98–1.28 | 0.14 | ns |
| NfL4 | 5.27 | 0.33–151.5 | 0.25 | ns | |||||||
| 24 months | NfL4 | 15.0 | 1.6–359.4 | 0.03 | * | 24 months | Age | 1.01 | 0.91–1.11 | 0.89 | ns |
| NfL4 | 10.03 | 0.66–376.2 | 0.13 | ns | |||||||
| PLASMA | PLASMA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Outcome3 | Variable | OR | CI 95% | p value | p value summary | Outcome3 | Variable | OR | CI 95% | p value | p value summary |
| 6 months | NfL4 | 3.0 | 0.5–22.1 | 0.25 | ns | 6 months | Age | 1.09 | 0.99–1.22 | 0.11 | ns |
| NfL4 | 0.84 | 0.06–9.39 | 0.89 | ns | |||||||
| 12 months | NfL4 | 2.9 | 0.4–21.7 | 0.26 | ns | 12 months | Age | 1.07 | 0.98–1.19 | 0.19 | ns |
| NfL4 | 0.97 | 0.07–11.64 | 0.98 | ns | |||||||
| 18 months | NfL4 | 18.7 | 2.1–439.3 | 0.02 | * | 18 months | Age | 1.15 | 0.99–1.52 | 0.13 | ns |
| NfL4 | 1.13 | 1.02–1.38 | 0.07 | ns | |||||||
| 24 months | NfL4 | 16.7 | 1.8–397.4 | 0.02 | * | 24 months | Age | 1.01 | 0.91–1.11 | 0.89 | ns |
| NfL4 | 15.1 | 1.15–540.2 | 0.06 | ns | |||||||
Note: 1Binary outcome favourable vs. unfavourable (unfavourable outcome = inability to run, GBSds 2 points or higher); 2binary variable low vs. high (based on median GBS cohort CSF NfL or plasma NfL); 3binary outcome favourable vs. unfavourable (unfavourable = INCAT 3 points or higher); 4binary variable low vs. high (based on median CIDP cohort CSF NfL or plasma NfL); age in years as a continuous variable.
Abbreviations: CI 95%, 95% confidence interval; CIDP, chronic inflammatory demyelinating polyneuropathy; CSF, cerebrospinal fluid; GBS, Guillain–Barré syndrome; GBSds, GBS disability scale; INCAT, Inflammatory Neuropathy Cause and Treatment disability score; NfL, neurofilamaent light chain; ns, not significant; OR, odds ratio.
* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
CIDP, PDN, and MMN
In patients with CIDP, the simple logistic model showed that pre‐treatment NfL levels higher than group median at 6 (CSF), 12 (CSF), 18 (CSF and plasma), and 24 months (CSF and plasma) were associated with an unfavourable outcome. However, the multiple logistic model including age did not show any statistically significant results (Table 4).
Logistic regression was not performed in patients with PDN and MMN due to small sample size.
NfL and T‐tau pre‐treatment levels in differentiating inflammatory neuropathies from each other and from non‐inflammatory neuromuscular disorders
Patient groups were compared using the receiver‐operating characteristic (ROC) analysis measuring the area under the curve (AUC). Median T‐tau levels in plasma were significantly higher in all inflammatory neuropathy subgroups vs. all controls. Median NfL levels in plasma and CSF were lower in patients with MMN vs. ALS (see Table S2 for more details, AUC values, 95% confidence intervals and p values).
DISCUSSION
In this study, we investigated NfL and T‐tau in both CSF and plasma in patients with GBS and different subtypes of chronic inflammatory neuropathies in comparison with controls, to evaluate the roll of these axonal injury biomarkers as diagnostic and prognostic markers.
We confirmed the finding of high NfL levels in CSF and plasma in the acute phase of GBS and CIDP, and also the usefulness of NfL in both CSF and plasma as a prognostic biomarker in GBS, including the correlation between NfL values and electrophysiological findings in the acute phase of GBS. However, we found no robust prognostic capability of NfL in CIDP, since NfL did not correlate with outcome in patients with CIDP when age was taken into account. This may be due to the heterogeneity with regards to long‐term immunomodulatory treatments in the CIDP cohort. Nor did NfL levels correlate with outcome in PDN and MMN, mainly due to assessment using the INCAT scale, which is not optimal for these neuropathies. Of interest though is the correlation between pre‐treatment NfL levels in plasma and the INCAT‐arm score at the time of sampling in patients with MMN, indicating a greater extent of nerve damage in patients with higher plasma NfL. The lack of correlation between NfL levels in plasma and the INCAT‐arm score at later time points could suggest reversible nerve damage in treated patients with MMN.
The source of NfL released into the CSF in patients with GBS and CIDP is likely damaged proximal nerve roots which are surrounded by CSF in the subarachnoid space of the spinal cord. The lack of correlation between NfL (in CSF and plasma) with Q.alb in the patients with PDN and MMN suggests that high plasma NfL levels in these patients has its origin in the peripheral (PNS) and not the central nervous system (CNS).
We also found higher T‐tau levels mainly in plasma in patients with inflammatory polyneuropathies. Of importance is the fact that tau is not present only in the CNS, but also in the PNS [32, 33, 34]. T‐tau is considered to be a marker of a neuronal cell body damage whereas NfL is a marker of axonal damage [35, 36]. Our observation of increased T‐tau levels in plasma but not CSF in patients with GBS, PDN, and MMN and lack of correlation between CSF and plasma T‐tau and Q.alb, suggests that T‐tau in plasma originates from damaged distal segments of peripheral nerves in inflammatory polyneuropathies. This may also be the case in CIDP whose T‐tau levels in plasma are increased, but a more proximal source might specifically be involved in CIDP, since T‐tau levels are increased even in CSF in these patients. Whether the source of T‐tau in CSF in CIDP are the lower motor neuron cell bodies, dorsal root ganglia, or maybe subclinically the CNS, remains unclear.
With regard to post‐treatment NfL and T‐tau levels, the long half‐life of NfL in CSF (2–3 months) [37] and the short half‐life of T‐tau in CSF (3 weeks) [38] may explain the continuation of high NfL levels in CSF but a decrease in T‐tau in post‐treatment samples obtained within a relatively short time from pre‐treatment samples.
Limitations of our study include its retrospective design, lack of a larger proportion of paired samples before and after the treatment, low number of patients with PDN and MMN, lack of clinical assessment with other disability scales (such as I‐RODS) or by measuring grip strength (especially in patients with MMN), lack of data on antibodies against nodal/paranodal epitopes (e.g., anti‐neurofascin 155), and heterogeneity of patients with NIP.
In conclusion, our systematic retrospective study verifies an increase in NfL levels in not only CSF but also in plasma in the acute phase of GBS and CIDP, and also provides evidence for a prognostic value of NfL in CSF and plasma in the evaluation of both short‐ and long‐term outcome of patients with GBS, but not in CIDP. Furthermore, we show high NfL levels in plasma in MMN and its use in the short‐term evaluation of upper extremity function. We also demonstrate that NfL in CSF and plasma can be used as a biomarker to differentiate MMN from ALS, which could be useful in rare cases when late‐stage MMN cannot be electrophysiologically differentiated from progressive spinal muscular atrophy. In addition, we provide a novel finding suggesting a diagnostic value of T‐tau in plasma, an easily accessible blood biomarker, in differentiating inflammatory neuropathies from non‐inflammatory neuromuscular disorders. Finally, our results suggest that elevated NfL and T‐tau levels in CSF and plasma are released from damaged peripheral nerves and nerve roots, though we cannot rule out a subclinical CNS origin.
The predictive value of NfL and T‐tau for outcome as well as their possible capability to determine the short‐term efficacy of immunomodulatory treatments in inflammatory neuropathies needs to be verified in prospective controlled studies.
AUTHOR CONTRIBUTIONS
Ivan Kmezic: Conceptualization (equal); data curation (lead); formal analysis (equal); investigation (lead); methodology (equal); project administration (lead); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Kristin Samuelsson: Supervision (equal); visualization (equal); writing – review and editing (equal). Anja Finn: Methodology (equal); supervision (equal); writing – review and editing (equal). Zane Upate: Supervision (supporting); validation (equal); writing – review and editing (supporting). Kaj Blennow: Data curation (equal); investigation (equal); resources (equal); writing – review and editing (equal). Henrik Zetterberg: Data curation (equal); investigation (equal); resources (equal); writing – review and editing (equal). Rayomand Press: Conceptualization (lead); data curation (equal); formal analysis (equal); funding acquisition (lead); methodology (equal); supervision (lead); visualization (lead); writing – original draft (equal); writing – review and editing (lead).
CONFLICT OF INTEREST
Ivan Kmezic declares no conflicts of interest. Kristin Samuelsson has served on scientific advisory boards and/or as a consultant for Akcea Theurapeutics, Inc. and Alnylam Pharmaceuticals. Anja Finn declares no conflict of interest. Zane Upate declares no conflicts of interest. Kaj Blennow has served as a consultant, on advisory boards, or on data monitoring committees for Abcam, Axon, BioArctic, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Pharmatrophix, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the work presented in this article. Kaj Blennow is supported by the Swedish Research Council (#2017–00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB‐201809‐2016615), the Swedish Alzheimer Foundation (#AF‐742881), Hjärnfonden, Sweden (#FO2017‐0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF‐agreement (#ALFGBG‐715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019‐466‐236), the National Institutes of Health (NIH), USA (grant #1R01AG068398–01), and the Alzheimer's Association 2021 Zenith Award (ZEN‐21‐848495). Henrik Zetterberg has served on scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures at symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside the submitted work). Henrik Zetterberg is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018–02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809–2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF‐21‐831376‐C, #ADSF‐21‐831381‐C, and #ADSF‐21‐831377‐C), the Olav Thon Foundation, the Erling‐Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019‐0228), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No. 860197 (MIRIADE), European Union Joint Program for Neurodegenerative Disorders (JPND2021‐00694), and the UK Dementia Research Institute at UCL. Rayomand Press declares no conflicts of interest.
Supporting information
Table S1‐S2
ACKNOWLEDGEMENTS
Ivan Kmezic and Rayomand Press have received a research grant for this study from the Swedish patient national association for neurological diseases Neuro Sweden (Neuroförbundet). Ivan Kmezic has received a research grant for this study from the Stockholm patient national association for neurological diseases Neuro Stockholm. The authors of this paper would like to especially acknowledge the contribution of Prof. Anders Svenningsson who provided the healthy control samples from the Department of Neurology, University Hospital of Umeå, Sweden. Open access funding enabled and organized by ProjektDEAL.
Kmezic I, Samuelsson K, Finn A, et al. Neurofilament light chain and total tau in the differential diagnosis and prognostic evaluation of acute and chronic inflammatory polyneuropathies. Eur J Neurol. 2022;29:2810‐2822. doi: 10.1111/ene.15428
DATA AVAILABILITY STATEMENT
Anonymized and raw data supporting the findings of this study are availabel on resonable request within the current regulations.
REFERENCES
- 1. Shahrizaila N, Lehmann HC, Satoshi Kuwabara S. Guillain‐Barré syndrome. Lancet. 2021;397(10280):1214‐1228. [DOI] [PubMed] [Google Scholar]
- 2. Bunschoten C, Jacobs BC, Van den Bergh PYK, et al. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol. 2019;18(8):784‐794. [DOI] [PubMed] [Google Scholar]
- 3. Eftimov F, Lucke IM, Querol LA, et al. Diagnostic challenges in chronic inflammatory demyelinating polyradiculoneuropathy. Brain. 2020;143(11):3214‐3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bomont P. The dazzling rise of neurofilaments: physiological functions and roles as biomarkers. Curr Opin Cell Bio. 2021;68:181‐191. [DOI] [PubMed] [Google Scholar]
- 5. Gaetani L, Blennow K, Calabresi P, et al. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90(8):870‐881. [DOI] [PubMed] [Google Scholar]
- 6. Petzold A, Hinds N, Murray NMF, et al. CSF neurofilament levels: a potential prognostic marker in Guillain‐Barré syndrome. Neurology. 2006;67(6):1071‐1073. [DOI] [PubMed] [Google Scholar]
- 7. Axelsson M, Sjögren M, Andersen O, et al. Neurofilament light protein levels in cerebrospinal fluid predict long‐term disability of Guillain‐Barré syndrome: a pilot study. Acta Neurol Scand. 2018;138(2):143‐150. [DOI] [PubMed] [Google Scholar]
- 8. Van Lieverloo GGA, Wieske L, Verhamme C, et al. Serum neurofilament light chain in chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Sys. 2019;24(2):187‐194. [DOI] [PubMed] [Google Scholar]
- 9. Godelaine J, De Schaepdryver M, Bossuyt X, et al. Prognostic value of neurofilament light chain in chronic inflammatory demyelinating polyneuropathy. Brain Commun. 2021;3(1):fcab018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martín‐Aguilar L, Camps‐Renom P, Lleixà C, et al. Serum neurofilament light chain predicts long‐term prognosis in Guillain‐Barré syndrome patients. J Neurol Neurosurg Psychiatry. 2021;92:70‐77. [DOI] [PubMed] [Google Scholar]
- 11. Mariotto S, Farinazzo A, Magliozzi R, et al. Serum and cerebrospinal neurofilament light chain levels in patients with acquired peripheral neuropathies. J Peripher Nerv Syst. 2018;23(3):174‐177. [DOI] [PubMed] [Google Scholar]
- 12. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5‐21. [DOI] [PubMed] [Google Scholar]
- 14. Scheltens P, Kaj Blennow K, Breteler MMB, et al. Alzheimer's disease. Lancet. 2016;388(10043):505‐517. [DOI] [PubMed] [Google Scholar]
- 15. Blennow K, Brody DL, Kochanek PM, et al. Traumatic brain injuries. Nat Rev Dis Primers. 2016;2:16084. [DOI] [PubMed] [Google Scholar]
- 16. Staffaroni AM, Kramer AO, Casey M, et al. Association of blood and cerebrospinal fluid tau level and other biomarkers with survival time in sporadic Creutzfeldt‐Jakob disease. JAMA Neurol. 2019;76(8):969‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rubenstein R, Chang B, Davies P, et al. A novel, ultrasensitive assay for tau: potential for assessing traumatic brain injury in tissues and biofluids. J Neurotrauma. 2015;32(5):342‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Randall J, Mörtberg E, Provuncher GK, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation. 2013;84(3):351‐356. [DOI] [PubMed] [Google Scholar]
- 19. Jin K, Takeda A, Shiga Y, Sato S, et al. CSF tau protein: a new prognostic marker for Guillain‐Barré syndrome. Neurology. 2006;67(8):1470‐1472. [DOI] [PubMed] [Google Scholar]
- 20. Petzold A, Brettschneider J, Jin K, et al. CSF protein biomarkers for proximal axonal damage improve prognostic accuracy in the acute phase of Guillain‐Barré syndrome. Muscle Nerve. 2009;40(1):42‐49. [DOI] [PubMed] [Google Scholar]
- 21. Wang XK, Zhang HL, Meng FH, et al. Elevated levels of S100B, tau and pNFH in cerebrospinal fluid are correlated with subtypes of Guillain‐Barré syndrome. Neurol Sci. 2013;34(5):655‐661. [DOI] [PubMed] [Google Scholar]
- 22. Kasai T, Kojima Y, Ohmichi T, et al. Combined use of CSF NfL and CSF TDP‐43 improves diagnostic performance in ALS. Ann Clin Transl Neurol. 2019;6(12):2489‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain‐Barré syndrome. Ann Neuro. 1990;27:S21‐S24. [DOI] [PubMed] [Google Scholar]
- 24. Van den Bergh PYK, Hadden RDM, Bouche P, et al. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society ‐ first revision. Eur J Neurol. 2010;17(3):356‐363. [DOI] [PubMed] [Google Scholar]
- 25. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies . Report of a joint task force of the European federation of neurological societies and the peripheral nerve society‐‐first revision. J Peripher Nerv Syst. 2010;15(3):185‐195. [DOI] [PubMed] [Google Scholar]
- 26. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy . Report of a joint task force of the European federation of neurological societies and the peripheral nerve society‐‐first revision. J Peripher Nerv Syst. 2010;15(4):295‐301. [DOI] [PubMed] [Google Scholar]
- 27. Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293‐299. [DOI] [PubMed] [Google Scholar]
- 28. Hughes RA, Newsom‐Davis JM, Perkin GD, Pierce JM. Controlled trial prednisolone in acute polyneuropathy. Lancet. 1978;2(8093):750‐753. [DOI] [PubMed] [Google Scholar]
- 29. Van Koningsveld R, Schmitz PIM, Avander Meché FG, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain‐Barré syndrome: randomized trial. Lancet. 2004;363(9404):192‐196. [DOI] [PubMed] [Google Scholar]
- 30. Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50(2):195‐201. [DOI] [PubMed] [Google Scholar]
- 31. Gaetani L, Höglund K, Parnetti L, et al. A new enzyme‐linked immunosorbent assay for neurofilament light in cerebrospinal fluid: analytical validation and clinical evaluation. Alzheimer’s Res Ther. 2018;10(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trojanowski JQ, Schuck T, Schmidt ML, et al. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37(2):209‐215. [DOI] [PubMed] [Google Scholar]
- 33. Georgieff IS, Liem RK, Mellado W, et al. High molecular weight tau: preferential localization in the peripheral nervous system. J Cell Sci. 1991;100(Pt 1):55‐60. [DOI] [PubMed] [Google Scholar]
- 34. Fischer I, Baas PW. Resurrecting the mysteries of Big tau. Trends Neurosci. 2020;43(7):493‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Day GS, Yarbrough MY, Körtvelyessy P, et al. Prospective quantification of CSF biomarkers in antibody‐mediated encephalitis. Neurology. 2021;96(20):e2546‐e2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wieske L, Smyth D, Lunn MP, et al. Fluid biomarkers for monitoring structural changes in polyneuropathies: their use in clinical practice and trials. Neurotherapeutics. 2021;18(4):2351‐2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bergman J, Dring A, Zetterberg H, et al. Neurofilament light in CSF and serum is a sensitive marker for axonal white matter injury in MS. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97(6):1284‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2
Data Availability Statement
Anonymized and raw data supporting the findings of this study are availabel on resonable request within the current regulations.