

Abstract
During the early phase of the pandemic (20 February–4 April 2020), we have investigated the temporal and geographical evolution of the virus in Lombardy showing the circulation of at least seven lineages distributed differently in the Region. In the present study, the molecular epidemiology of SARS‐CoV‐2 was monitored in a period between two pandemic waves in order to track the circulation of new variants (April‐August 2020). A great majority of SARS‐CoV‐2 strains (70.8%) belonged to lineages B, B.1, B.1.1 and B.1.1.1, and five strains belonging to four lineages were already reported in Italy (B.1.1.148, B.1.1.162, B.1.1.71, and B.1.425). In addition, 21 SARS‐CoV‐2 strains belonged to six lineages not previously observed in Italy were detected. No variants of concern were observed. A total of 152/1274 (11.3%) amino acid changes were observed among spike gene sequences and only 26/152 (17.1%) occurred in the receptor‐binding domain region of the spike protein. Results of this study are indicative of ongoing transmission throughout the lockdown period, rather than re‐introduction of novel lineages past lockdown. The use of molecular epidemiology in Italy should be promoted in order to provide additional understanding of the transmission of the disease and to have major effect on controlling the spread of disease.
Keywords: COVID‐19, molecular epidemiology, NGS, SARS‐CoV‐2, whole genome sequencing
Key Message.
Surveillance of SARS‐CoV‐2 circulating strains is crucial to provide important information on viral evolution and epidemiology. Tracking SARS‐CoV‐2 evolution is fundamental for the assessment of vaccine‐induced immunity and antiviral therapy efficacy against emerging variants.
1. INTRODUCTION
Since coronavirus disease 2019 (COVID‐19) was initially reported in China on 30th December 2019,1, 2 SARS‐CoV‐2 has been spreading worldwide. As of 5 February 2021, there have been 104 million confirmed infections and more than 2 million deaths have been reported worldwide. 1 Lombardy with a population of 10 million is the most densely populated and affected Region in Italy during the first wave with more than 90,000 cases at the end of May 2020.3, 4 The population density coupled with the high level of transportation links to Europe creates the conditions to host and favor the spread of a highly transmissible viruses such as SARS‐CoV‐2. During the early phase of the pandemic (20 February–4 April 2020), we have investigated the temporal and geographical evolution of the virus in Lombardy. 5 This study has documented the circulation of at least seven lineages distributed differently in the Region. The first pandemic wave showed as sharp down at the end of May 2020. However, a steading number of cases was reported (more than 10,000 June‐August 2020) and international road transport to and from several EU states was re‐established after 1 June 2020.4, 6 In the present study, the molecular epidemiology of SARS‐CoV‐2 was monitored in a period between two pandemic waves in order to track the circulation of new variants.
2. MATERIAL AND METHODS
A total of 89 respiratory samples with SARS‐CoV‐2 cycle threshold values <24 were selected for sequencing. Clinical samples were collected between 15 April and 20 August 2020 and tested positive for SARS‐CoV‐2 as previously described.5, 7 Total RNAs were extracted from nasopharyngeal swabs by using QIAamp Viral RNA Mini Kit, followed by purification with Agencourt RNA Clean XP beads. Virus genomes (GISAID EPI_ISL_1133145‐1133202 and 1166095–1166108) were generated by using a multiplex approach, using version 1 of the CleanPlex SARS‐CoV‐2 Research and Surveillance Panel (Paragon Genomics, Inc.), according to the manufacturer's protocol starting with 50 ng of total RNA and followed by Illumina sequencing on a MiSeq platform. NGS data were also analyzed with an in‐house pipeline. Lineages were assigned from alignment file using the Phylogenetic Assignment of Named Global Outbreak LINeages tool PANGOLIN v1.07 (https://github.com/hCoV‐2019/pangolin). 8 The study protocol was approved by the local Research Ethics Committee of Fondazione IRCCS Policlinico San Matteo (P_20200085574). This study was conducted in accordance with the principles of the 1964 Declaration of Helsinki. Informed consent was waived in accordance with Italian governmental regulations on observational retrospective studies.
3. RESULTS
Based on the GISAID EpiCoV™ database (https://www.epicov.org/epi3/) as of 5 February 2021, a total of 3212 SARS‐CoV Italian sequences were available and a comparison between them was performed and presented in Table 1 SARS‐CoV‐strains. A SARS‐CoV‐2 lineages analysis performed using the Pangolin web application suggested that the B lineages were the most common in Lombardy (Figure 1). In detail, a great majority of SARS‐CoV‐2 strains (63/89; 70.8%) belonged to lineages B, B.1, B.1.1 and B.1.1.1. Five strains grouped into clade A including sequences from China and many from South East Asia, Japan, South Korea, Australia, and the USA. The distribution of lineages in the early phase of pandemic, a similar distribution of lineages also continued until August. 5 This may suggest that the initial multiple lineage introduction in Lombardy was followed by local transmission events during the lockdown period. In addition, five strains belonging to four lineages, already reported in Italy (B.1.1.148, B.1.1.162, B.1.1.71, and B.1.425) were detected. Finally, 21 SARS‐CoV‐2 strains belonged to six lineages not previously observed in Italy were detected (Table 1). This finding showed the introduction of additional lineages in the context of a predominant circulation of previously introduced lineages.
TABLE 1.
Lineages identified in this study
| Lineages | This study (no.) | Italian sequences (no.) | Most common countries | Earliest date | Count | Description a |
|---|---|---|---|---|---|---|
| A | 5 | 16 | United_Arab_Emirates 19.0%, Germany 13.0%, China 13.0%, United States of America 6.0%, Japan 5.0% | 30–12–2019 | 1312 | Root of the pandemic lies within lineage A |
| B | 9 | 21 | United Kingdom 36.0%, United States of America 16.0%, China 13.0%, Spain 4.0%, Singapore 3.0% | 24–12–2019 | 4976 | Base of this lineage also lies in China, with many global exports, two distinct SNPs `8782TC` and `28144CT` define this lineage |
| B.1 | 45 | 743 | United States of America 46.0%, United Kingdom 13.0%, Canada 5.0%, Spain 4.0%, France 3.0% | 24–01–2020 | 50516 | A large European lineage that corresponds to the Italian outbreak. |
| B.1.1 | 1 | 29 | United States of America 27.0%, United Kingdom 23.0%, Canada 6.0%, Germany 6.0%, Netherlands 5.0% | 24–02–2020 | 2766 | European lineage |
| B.1.1.1 | 3 | 150 | United Kingdom 76.0%, Denmark 2.0%, Italy 2.0%, United States of America 2.0%, Switzerland 2.0% | 02–03–2020 | 7604 | UK/ Europe lineage |
| B.1.1.148 | 1 | 1 | USA 89.0%, Canada 5.0%, Germany 2.0%, UK 2.0%, Mexico 1.0% | 16–05–2020 | 87 | USA lineage |
| B.1.1.161 | 10 | 0 | Saudi_Arabia 24.0%, UK 21.0%, Switzerland 10.0%, Czech_republic 10.0%, Denmark 4.0% | 09–03–2020 | 202 | Saudi Arabian lineage |
| B.1.1.162 | 1 | 57 | United Kingdom 23.0%, United States of America 15.0%, Japan 10.0%, Canada 9.0%, Spain 6.0% | 26–02–2020 | 1927 | Australian/UK lineage |
| B.1.1.38 | 1 | 0 | United Kingdom 23.0%, United States of America 15.0%, Japan 10.0%, Canada 9.0%, Spain 6.0% | 24–03–2020 | 260 | UK lineage |
| B.1.1.44 | 1 | 0 | United Kingdom 56.0%, Spain 20.0%, United States of America 11.0%, Denmark 2.0%, Switzerland 1.0% | 23–03–2020 | 630 | UK/Spain lineage |
| B.1.1.71 | 2 | 4 | Netherlands 27.0%, United_Arab_Emirates 21.0%, UK 17.0%, Australia 8.0%, Belgium 7.0% | 06–03–2020 | 146 | The Netherlands lineage |
| B.1.1.74 | 7 | 0 | United Kingdom 87.0%, United States of America 5.0%, Peru 3.0%, Ireland 1.0%, Russia 1.0% | 23–05–2020 | 268 | Northern Irish lineage |
| B.1.243 | 1 | 0 | United States of America 98.0%, Mexico 1.0%, Canada 0.0%, Switzerland 0.0%, South_Korea 0.0% | 23–03–2020 | 5256 | USA lineage |
| B.1.379 | 1 | 0 | United Kingdom 91.0%, France 3.0%, United States of America 1.0%, Sierra_Leone 1.0%, Germany 1.0% | 16–03–2020 | 67 | UK lineage, previously B.1.5.30 |
| B.1.425 | 1 | 1 | United States of America 90.0%, Turkey 10.0%, Italy 0.0% | 296 | USA lineage (UT), reassigned from part of B.1.370 |
https://cov‐lineages.org/lineages.html.
FIGURE 1.
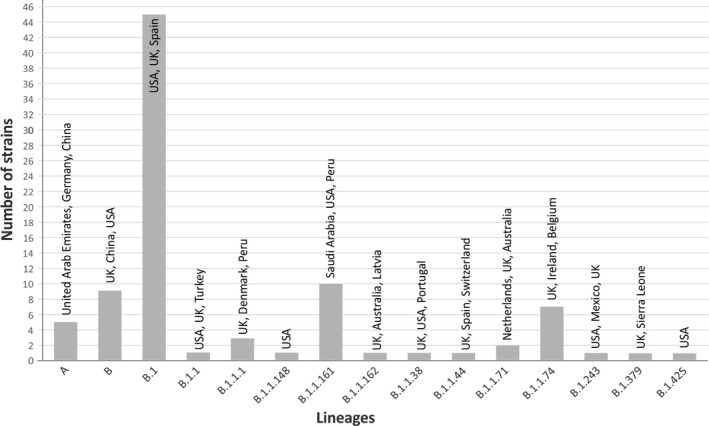
Distribution of SARS‐CoV‐2 strains according to lineages assigned by Pangolin COVID‐19 Lineage Assigner online tool (https://github.com/hCoV‐2019/pangolin). The most common countries in which each lineage was identified are reported above the bars
Since summer 2020, a series of SARS‐CoV‐2 variants of concern (VOC) (eg, VOC 202012/01, and 501Y.V2) harboring several amino acidic changes were highlighted by ECDC as of potential increased pathogenicity. 9 Some of them were associated with increased infection cases in the UK (VOC 202012/01) and later in other countries and other few (VOC501Y.V2 and Brasil) were associated with a reduced neutralization by plasma. In the period from 15 April to 20 August 2020 of this analysis, none of these variants were observed.
A total of 152/1274 (11.3%) amino acid (aa) changes were observed among spike gene sequences; however, only 10/152 (6.6%, 0.8% of total aa) changes (T29I, E281G, Q564R, F565S, D614G, S640A, N641H, K964R) were observed in at least two SARS‐CoV‐2 strains. Of note, only 26/152 (17.1%, 2.0% of total) changes occurred in the receptor‐binding domain (RBD) region of the spike protein.
4. DISCUSSION
The phylogenetic analysis performed on the viral sequences collected during the first period of the pandemic in Italy, mainly originated in Lombardy, suggested a clear circulation of at least seven SARS‐CoV‐2 lineages. 5 Similar results were also observed in a subsequent study including 460 Italian strains previously reported 10 and in a more extended study aiming to describe SARS‐CoV‐2 circulation in European region. 11 The generated genomes in the present study provide additional insight into the SARS‐CoV‐2 lineages and variants circulating in Lombardy in the period between the first and the before second pandemic wave. Results of the analyses seem to indicate that additional lineages were introduced in Lombardy during the summer but these introductions did not lead to further transmission of the virus in the community, or a limited transmission has occurred. The segregation of specific lineages was observed probably as a result of the strict lockdown measures applied during the three‐month state of emergency (March‐May 2020). Several lineages mainly circulating in other countries have been identified, and few of them were not previously reported in Italy. None of “high risk” variants have been observed to circulate in Lombardy in the study period. However, the recently circulation of UK variants (B.1.1.7) associated with a more transmissible virus raise the attention on global changing of SARS‐CoV‐2 circulation. The ongoing vaccine campaign should be supported with real‐time surveillance of circulating variants in order to monitor the emergence of mutations associated with poorly antibody recognition.
Finally, the great majority of mutations within the spike gene region were observed in single strain and only D614G changes seem to be fixed in the SARS‐CoV‐2 population analyzed. In addition, most of them located outside the RBD region with therefore a limited impact on antibodies recognition.
5. CONCLUSION
We reported on the limited but ongoing within‐region transmission during the first wave of SARS‐CoV‐2 in Lombardy. These features are indicative of ongoing transmission throughout the lockdown period, rather than re‐introduction of novel lineages past lockdown. However, it is critical to acknowledge the extremely limited amount of genomic data from Italy compared with many other localities that clearly impacted the strength of the conclusions that can be drawn here. It is therefore vital that Italy build better structures for effective genomic epidemiology prior to any future major outbreaks of emerging infectious disease.
CONFLICT OF INTERESTS
All authors have no conflicts of interest to disclose.
AUTHORS’ CONTRIBUTION
Monica Tallarita: Methodology (equal), Writing‐review & editing (equal). Federica Giardina: Methodology (equal), Writing‐review & editing (equal). Federica Novazzi: Methodology (equal), Writing‐review & editing (equal). Stefano Gaiarsa: Methodology (equal), Writing‐review & editing (equal). Gherard Batisti Biffignandi: Methodology (equal), Writing‐review & editing (equal). Stefania Paolucci: Supervision (equal); Methodology (equal), Writing‐review & editing (equal). Francesca Rovida: Supervision (equal); Writing‐review & editing (equal). Antonio Piralla: Conceptualization (equal); Supervision (lead); Writing‐review & editing (equal). Fausto Baldanti: Conceptualization (equal); Supervision (equal); Writing‐review & editing (equal).
ACKNOWLEDGEMENTS
We thank Daniela Sartori for manuscript editing.
Tallarita M, Giardina F, Novazzi F, et al. Spread of multiple SARS‐CoV‐2 lineages April‐August 2020 anticipated the second pandemic wave in Lombardy (Italy). Pediatr Allergy Immunol. 2022;33 (Suppl. 27):89–92. 10.1111/pai.13641
Monica Tallarita, Federica Giardina and Federica Novazzi contributed equally to the work and share first authorship.
Funding information
AP and FB have received funding from the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No 101003650. This study was supported by “Ricerca Finalizzata” from Ministry of Health, Italy (grants no. GR‐2013‐02358399 and COVID‐2020‐12371817).
REFERENCES
- 1. Dong E, Du H, Gardner L. An interactive web‐based dashboard to track COVID‐19 in real time. Lancet Infect Dis. 2020;20:533‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eurostat . The statistical office of the European Union statistics. https://ec.europa.eu/Eurostat.web/cities/data/database (Accessed 4 April 2021)
- 4. Regione Lombardia ‐ Dashboard COVID 19 update of cases. https://www.regione.lombardia.it/wps/portal/istituzionale/HP/servizi‐e‐informazioni/cittadini/salute‐e‐prevenzione/coronavirus/dashboard‐covid19 (Accessed 4 April 2021)
- 5. Alteri C, Cento V, Piralla A, et al. Genomic epidemiology of SARS‐CoV‐2 reveals multiple lineages and early spread of SARS‐CoV‐2 infections in Lombardy, Italy. Nat Commun. 2021;12:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. European Union . https://ec.europa.eu/info/live‐work‐travel‐eu/coronavirus‐response/travel‐during‐coronavirus‐pandemic_en. (accessed 4 Apr 2021).
- 7. Piralla A, Ricchi M, Cusi MG, et al. Residual SARS‐CoV‐2 RNA in nasal swabs of convalescent COVID‐19 patients: Is prolonged quarantine always justified? Int J Infect Dis. 2021;102:299‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rambaut A, Holmes EC, O'Toole Á, et al. A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. European Centre for Disease Prevention and Control . Risk Related to Spread of New SARS‐CoV‐2 Variants of Concern in the EU/EEA, First Update – 21 January 2021. ECDC; 2021. https://www.ecdc.europa.eu/en/publications‐data/covid‐19‐risk‐assessment‐spread‐new‐variants‐concern‐eueea‐first‐update (Accessed 4 April 2021). [Google Scholar]
- 10. Di Giallonardo F, Duchene S, Puglia I, et al. Genomic epidemiology of the first wave of SARS‐CoV‐2 in Italy. Viruses. 2020;12:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alm E, Broberg EK, Connor T, et al. Geographical and temporal distribution of SARS‐CoV‐2 clades in the WHO European Region, January to June 2020. Euro Surveill. 2020;25:2001410. [DOI] [PMC free article] [PubMed] [Google Scholar]