

ABSTRACT
Atherosclerosis, the leading cause of cardiovascular death, is driven by hyperlipidemia, inflammation and aggravated by aging. As chaperone-mediated autophagy (CMA), a selective type of lysosomal degradation for intracellular proteins, diminishes with age and is inhibited by lipid excess, we studied if the decline in CMA could contribute to atherosclerosis pathogenesis. We found that CMA declines in human and murine vasculature with disease progression. Inhibition and reactivation of CMA using transgenic mouse models establishes a protective effect of CMA against atherogenesis. CMA upregulation ameliorates both systemic metabolic parameters, and vascular cell function. Our work suggests CMA reactivation could be a viable therapeutic strategy to prevent and reduce cardiovascular disease.
KEYWORDS: Cardiovascular disease, cholesterol, inflammation, insulin, lysosomes, macrophages, smooth muscle cells
Atherosclerosis, the leading cause of cardiovascular death, is driven by hyperlipidemia, inflammation and aggravated by aging. As chaperone-mediated autophagy (CMA), a selective type of lysosomal degradation for intracellular proteins, diminishes with age and is inhibited by lipid excess, we studied if the decline in CMA could contribute to atherosclerosis pathogenesis. We found that CMA declines in human and murine vasculature with disease progression. Inhibition and reactivation of CMA using transgenic mouse models establishes a protective effect of CMA against atherogenesis. CMA upregulation ameliorates both systemic metabolic parameters, and vascular cell function. Our work suggests CMA reactivation could be a viable therapeutic strategy to prevent and reduce cardiovascular disease.
Patients with cardiovascular disease (CVD), even those treated with lipid-lowering or other medication, remain at a high residual risk of stroke or myocardial infarction following thrombotic occlusions. Thus, research into pathogenesis of atherosclerosis, the main underlying cause of CVD, is still much needed.
Risk factors for atherosclerosis development and progression include systemic parameters such as hyperlipidemia, hypertension, insulin resistance and age. At the local vascular level, atherosclerosis involves a complex interplay between macrophage infiltration and pro-inflammatory activation, smooth muscle cells dedifferentiation into a secretory and inflammatory phenotype, and induction of cell death. Together these events promote formation of a large necrotic core and enhance matrix degradation, which weakens the atherogenic plaque and increases the risk of its rupture and subsequent thrombotic occlusions.
In this work [1], we recognized the potential impact that CMA could have on the main risk factors for atherosclerosis and set to investigate the consequences of changes in CMA activity on vascular disease progression. CMA regulates cellular lipid and glucose metabolism through timely degradation of limiting enzymes and other metabolism-related proteins. CMA activity declines with age and upon persistent lipid overload, two main risk factors for atherosclerosis. Therefore, we hypothesized that CMA could be a very attractive target to alleviate cardiometabolic risk factors and prevent or ameliorate cardiovascular events.
We first established the cellular and temporal changes in CMA activity in the vasculature during atherogenesis using a fluorescent reporter (KFERQ-PS-Dendra2) mouse model that allows measuring CMA activity in vivo, together with analysis of the expression of LAMP2A (lysosomal-associated membrane protein 2A), the CMA receptor and rate-limiting component of this autophagic pathway. After an initial activation of CMA in early stages of disease, we found that CMA activity in plaque macrophages and smooth muscle cells gradually declines along plaque expansion.
To understand the consequences of reduced CMA activity in the vasculature and to determine if this autophagic pathway has a physiological protective effect on plaque development, we induced atherosclerosis in a mouse model with systemic blockage of CMA (whole body lamp2a−/−) through adeno-associated virus-mediated overexpression of PCSK9 (proprotein convertase subtilisin/kexin type 9; AAV-PCSK9) – which results in loss of LDLR (low density lipoprotein receptor) – in combination with a high-cholesterol diet. Plaque size and disease stage are considerably aggravated in the lamp2a−/− mice, coinciding with larger plaque necrotic cores and more pronounced local loss of contractile smooth muscle cells and macrophages (Figure 1) [1].
Figure 1.
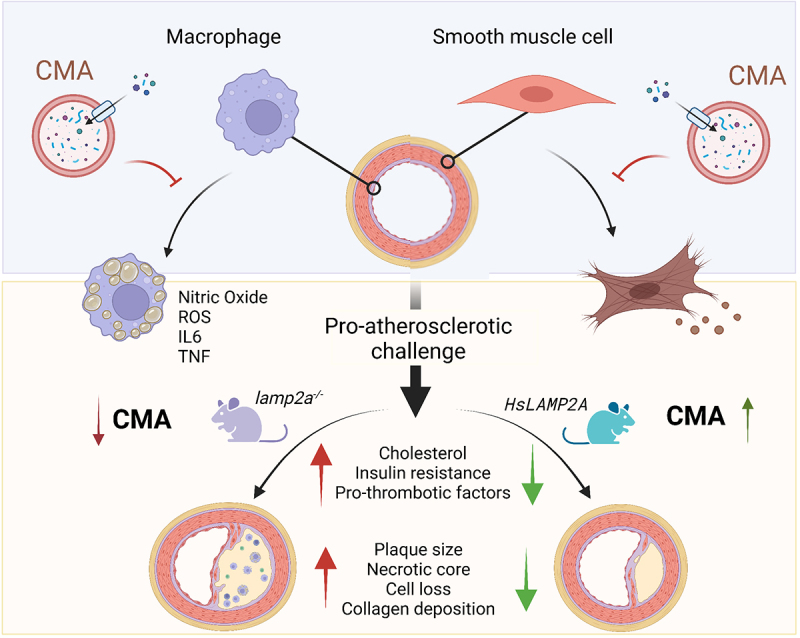
Protective role of CMA against atherosclerosis. Top: Active CMA in vascular cells is protective against lipotoxicity and prevents the dedifferentiation of vascular smooth muscle cells and the pro-inflammatory phenotype of macrophages associated with atherosclerosis. Bottom: Blockage of CMA in vivo increases vulnerability to pro-atherosclerotic challenges by accentuating systemic metabolic changes and worsening atherosclerotic plaque phenotype. Reverting the decrease in CMA activity observed during chronic exposure to pro-atherosclerotic challenges is effective in reducing vascular disease severity and slowing down disease progression.
The underlying mechanism of the athero-protective effect of CMA is a combination of its systemic metabolic effects, as well as cell-autonomous regulatory functions of CMA in the vasculature. The lamp2a−/− mice present with a less favorable metabolic profile, i.e., higher body weight gain, reduced energy expenditure, hyperinsulinemia, and insulin resistance, all of which are well-known risk factors for CVD. Although cholesterol levels are increased in lamp2a−/− compared to WT mice, we found that the correlation between plaque size and cholesterol is lost in CMA-deficient mice, suggesting that local vascular effects are likely also contributing to the protective effect of CMA. Thus, we studied the effect of LAMP2A loss on vascular smooth muscle cells and macrophages using in vitro functional assays and comparative analysis of their transcriptome and of the cellular subproteome degraded by CMA in these cells (Figure 1). The lamp2a−/− vascular smooth muscle cells exhibit gene expression patterns indicative of dedifferentiation, inflammatory activation, aberrant cholesterol metabolism and cell death, mirrored by functional changes, such as intracellular lipid build up and subsequent cell death upon lipid challenges. In parallel, lamp2a−/− macrophages stimulated with pro-inflammatory triggers show a more pronounced pro-inflammatory protein expression pattern. Comparative proteomics of lysosomes isolated from WT and lamp2a−/− macrophages identified proteins involved in pro-inflammatory activation as CMA substrates and their inability to undergo degradation through this pathway as the cause for their elevated levels upon the pro-inflammatory stimuli. Notably, macroautophagy activity in lamp2a−/− cells in the vasculature is unchanged, pointing to an absence of autophagic counterbalance in this setting and that the observed effects are only mediated by CMA.
After establishing that CMA is protecting against atherosclerosis development, we studied if normalization of CMA activity after plaque development could be harnessed as a therapeutic approach. Atherosclerosis was induced, as described for the lamp2a−/− mice, in tamoxifen-inducible Lamp2a knockin (KI) mice and, importantly, expression of this CMA-limiting protein is activated after the observed decline in CMA activity following the initiation of plaques to closely mimic a therapeutic setting in humans. Reactivation of CMA, even when in presence of a pro-atherogenic diet for an additional 10-weeks, results in considerable halt of atherosclerotic plaque size and phenotype together with an improved metabolic profile, supporting the therapeutic potential of CMA activity (Figure 1).
The translational value of these findings was confirmed in four different cohorts of human carotid and coronary atherosclerosis using single-cell sequencing, microarray analysis combined with plaque phenotype analysis, and protein detection of LAMP2A in different disease stages and in relation to occurrence of future CV events. CMA activity in human plaque smooth muscle cells and macrophages declines with disease stage and this reduced CMA activity positively associates with an unfavorable plaque phenotype of inflammatory infiltration and intraplaque hemorrhage. Moreover, low plaque CMA activity at time of a first CV event is correlated with higher risk of a future secondary CV event. Because these data were obtained from cross-sectional studies and a prospective study with modest sample size, future human causality studies are warranted to provide full support of our observations.
In conclusion, we demonstrate a protective effect of CMA against atherosclerosis and prove the value of reactivation of CMA in slowing down disease progression and reducing clinical events. As CMA declines with age and upon sustained dietary lipid challenges, stimulation of CMA could have therapeutic value by decreasing the risk of vascular events and ameliorating disease severity and progression.
Funding Statement
This work was supported by the American Heart Association [17POST33650088]; Dutch Heart Foundation [Dr. Dekker fellowship 2016T060]; Fondation Leducq [15CVD04]; National Institutes of Health [AG021904, DK098408, AG031782]; Dutch Research council [VIDI 91718364, ASPASIA 015013064].
Disclosure statement
AMC is co-founder of the Selphagy program under Life Biosciences, and she consults for Generian Pharmaceuticals and Cognition Therapeutics. The other authors declare no competing interests.
Reference
- [1].Madrigal-Matute J, de Bruijn J, van Kuijk K, JC Sluimer and Cuervo AM et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc Natl Acad Sci U S A. 2022;119(14):e2121133119. [DOI] [PMC free article] [PubMed] [Google Scholar]