

ABSTRACT
Mitochondrial oxidative phosphorylation (OXPHOS) generates ATP, but OXPHOS also supports biosynthesis during proliferation. In contrast, the role of OXPHOS during quiescence, beyond ATP production, is not well understood. Using mouse models of inducible OXPHOS deficiency in all cell types or specifically in the vascular endothelium that negligibly relies on OXPHOS-derived ATP, we show that selectively during quiescence OXPHOS provides oxidative stress resistance by supporting macroautophagy/autophagy. Mechanistically, OXPHOS constitutively generates low levels of endogenous ROS that induce autophagy via attenuation of ATG4B activity, which provides protection from ROS insult. Physiologically, the OXPHOS-autophagy system (i) protects healthy tissue from toxicity of ROS-based anticancer therapy, and (ii) provides ROS resistance in the endothelium, ameliorating systemic LPS-induced inflammation as well as inflammatory bowel disease. Hence, cells acquired mitochondria during evolution to profit from oxidative metabolism, but also built in an autophagy-based ROS-induced protective mechanism to guard against oxidative stress associated with OXPHOS function during quiescence.
Abbreviations: AMPK: AMP-activated protein kinase; AOX: alternative oxidase; Baf A: bafilomycin A1; CI, respiratory complexes I; DCF-DA: 2′,7′-dichlordihydrofluorescein diacetate; DHE: dihydroethidium; DSS: dextran sodium sulfate; ΔΨmi: mitochondrial inner membrane potential; EdU: 5-ethynyl-2’-deoxyuridine; ETC: electron transport chain; FA: formaldehyde; HUVEC; human umbilical cord endothelial cells; IBD: inflammatory bowel disease; LC3B: microtubule associated protein 1 light chain 3 beta; LPS: lipopolysaccharide; MEFs: mouse embryonic fibroblasts; MTORC1: mechanistic target of rapamycin kinase complex 1; mtDNA: mitochondrial DNA; NAC: N-acetyl cysteine; OXPHOS: oxidative phosphorylation; PCs: proliferating cells; PE: phosphatidylethanolamine; PEITC: phenethyl isothiocyanate; QCs: quiescent cells; ROS: reactive oxygen species; PLA2: phospholipase A2, WB: western blot.
Keywords: ATG4B, biosynthesis, cell death, electron transport chain, endothelial cells, mitochondria, oxidative phosphorylation, oxidative stress, reactive oxygen species
Introduction
Proliferation vs. quiescence is one of the biggest contrasts for a mammalian cell. Proliferating cells in developing organisms or in tumors engage in biosynthesis to build up biomass for rapid cell division and feature fast nucleic acid and protein turnover. In contrast, quiescent cells, which we define here as non-dividing metabolically active cells that represent most somatic cells in an adult organism, primarily need to maintain their long-lived components to support their extended life span [1]. For this reason, stress response pathways and quality control systems such as macroautophagy/autophagy and antioxidant defense are elevated during quiescence [2-6], and quiescent cells are better equipped to withstand oxidative insult [7-9]. Autophagy facilitates elimination of damaged organelles and proteins by segregation into specialized structures called autophagosomes for lysosomal delivery, cargo degradation and recycling [10]. Autophagy is induced by the AMP-activated protein kinase (AMPK) and inhibited by the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) via ULK1 (unc-51 like autophagy activating kinase 1) [11,12], a central switch in the canonical autophagic signaling. Autophagy can also respond to altered NADH and reactive oxygen species (ROS) levels [13-15]. However, these findings come predominantly from proliferating cells and the mechanisms that modulate stress resistance and autophagy during quiescence have not been fully resolved.
Mitochondrial oxidative phosphorylation (OXPHOS) is best known for its role in ATP generation. In this process, respiratory complexes I-IV of the electron transport chain (ETC) produce a proton gradient (ΔΨmi) across the inner mitochondrial membrane, which propels ATP synthase. ETC of OXPHOS also supports biosynthesis of essential metabolites, NADH/NAD+ redox-cycling by complex I (CI), and generation of ROS that arise when electrons transported through the ETC “leak” and react prematurely with oxygen [16]. ROS are potentially damaging but may also have physiological functions [17]. In proliferating cells ETC is required for biosynthesis, primarily for de novo synthesis of pyrimidines that enables DNA/RNA replication/transcription [18-22]. In contrast, ATP generation is considered the dominant function of OXPHOS in quiescent cells, as the limited need of quiescent cells for DNA/RNA synthesis predicts reduced dependence on de novo pyrimidine synthesis. Additional functions of OXPHOS during quiescence, including their contribution to physiology and disease, have not been explored.
Quiescent cells need protection from oxidative damage induced by ROS in pathological settings, such as during cancer therapy or inflammation. Indeed, resistance to oxidative stress maintains anti-inflammatory properties of quiescent vascular endothelium [6]. However, systematic examination of OXPHOS, a prominent intracellular source of damaging ROS, has never been performed in these settings. We hypothesized that OXPHOS could initiate mechanisms that regulate stress resistance of quiescent cells and tissues to counter oxidative damage inherent to its function. Using multiple in vitro and in vivo models of OXPHOS deficiency, we aimed to systematically explore possible mechanistic links between OXPHOS, quiescence, oxidative stress resistance and autophagy, as well as the role of OXPHOS-induced ROS-resistance in pathology.
Results
OXPHOS deficiency sensitizes to oxidative stress in vivo
To explore specific roles of OXPHOS in quiescent cells, we made use of the fact that several essential subunits of OXPHOS complexes (except for complex II) are encoded by mitochondrial (mt)DNA. Removal of mtDNA or a block in its transcription thus results in OXPHOS deficiency (Figure 1A), yielding mitochondria that lack the ETC and maintain ΔΨmi via ATP hydrolysis catalyzed by a subcomplex of ATP synthase (Figure 1A) [23,24]. We applied this approach in adult mice, as adult tissues contain mostly quiescent cells. We therefore generated a mouse model of conditional OXPHOS deficiency by systemic tamoxifen-induced deletion of TFAM (transcription factor A, mitochondrial) (Figure 1A and B), essential for mtDNA maintenance and transcription (tfam KO mice, see Materials and Methods, Figure 1B) [25]. Preliminary screening upon induction revealed that TFAM was most consistently deleted in the liver, kidney and pancreas. In these organs the TFAM protein and mtDNA-encoded subunits of OXPHOS complexes were markedly (albeit not fully) reduced in tfam KO animals on day 11 post induction (Fig. S1A and B), and in situ measurements of cytochrome c oxidase activity revealed that ETC was suppressed in most cells (Fig. S1C).
Figure 1.
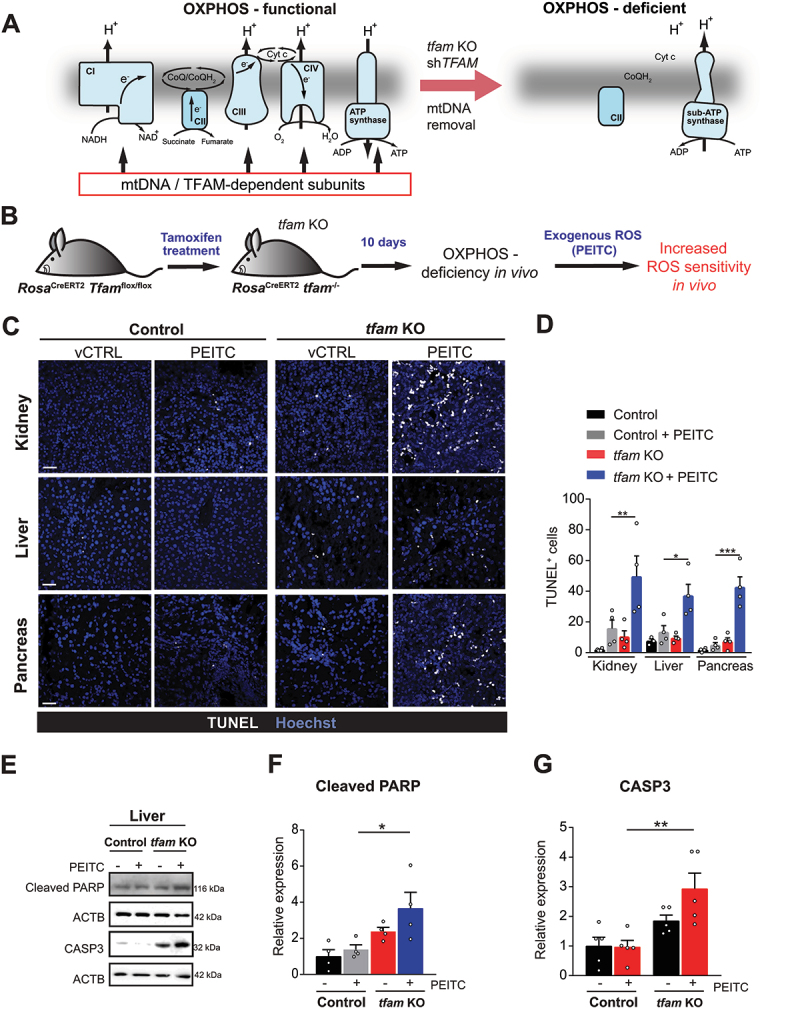
OXPHOS deficiency sensitizes to oxidative stress in vivo. (A) mtDNA removal, TFAM silencing or tfam deletion results in OXPHOS-deficient cells that lack ETC and contain a subcomplex of ATP synthase (sub-ATP synthase) that functions in reverse to hydrolyze ATP for ΔΨmi maintenance. (B) tfam deletion strategy in RosaCreERT2 Tfamflox/flox mice (referred to as tfam KO). (C) Representative images of cell death analyzed by TUNEL assay in kidney, liver and pancreas from vehicle-treated (vCTRL) and PEITC-treated control and tfam KO mice. Scale bar: 300 µm. (D) Quantification of TUNEL+ cells as shown in (C) (mean ± S.E.M., n = 4 mice per condition, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test). (E) Representative WB images of the cleaved PARP and cleaved CASP3 in liver tissue of control and tfam KO mice treated or not with PEITC. (F and G) Densitometric quantification of WB in E and 3-4 additional independent experiments (normalized to ACTB, mean ± S.E.M., n ≥ 3 mice per condition, *p < 0.05, **p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test).
We reasoned that if OXPHOS provides stress resistance during quiescence, then OXPHOS-deficient quiescent cells will be sensitized to cell death induced by exogenous administration of pro-oxidant agents. Hence, we challenged control and tfam KO mice with phenethylisocyanate (PEITC), a compound which induces ROS independent of the ETC by interfering with the glutathione system [26]. Strikingly, 16 h after a single dose of PEITC tfam KO mice displayed a large increase in the number of apoptotic (TUNEL-positive) cells in all tested organs (Figure 1C and D), and we detected increased apoptotic markers, such as CASP3 (caspase 3) activation and PARP1 (poly(ADP-ribose) polymerase 1) cleavage (Figure 1E-G). In contrast, little cell death was observed in untreated or PEITC-treated control mice.
OXPHOS-mediated ROS protection is specific to quiescent cells
To investigate whether OXPHOS deficiency selectively sensitizes quiescent cells to ROS-induced cell death (Figure 2A), we co-stained liver sections (that contain a minor fraction of proliferating cells) for TUNEL and 5-ethynyl-2’-deoxyuridine (EdU), a marker of proliferation. The increased TUNEL signal in PEITC-treated tfam KO animals was detected predominantly in EdU-negative cells (Figure 2B and C), suggesting that quiescent cells are indeed selectively sensitized to ROS by OXPHOS ablation in vivo.
Figure 2.
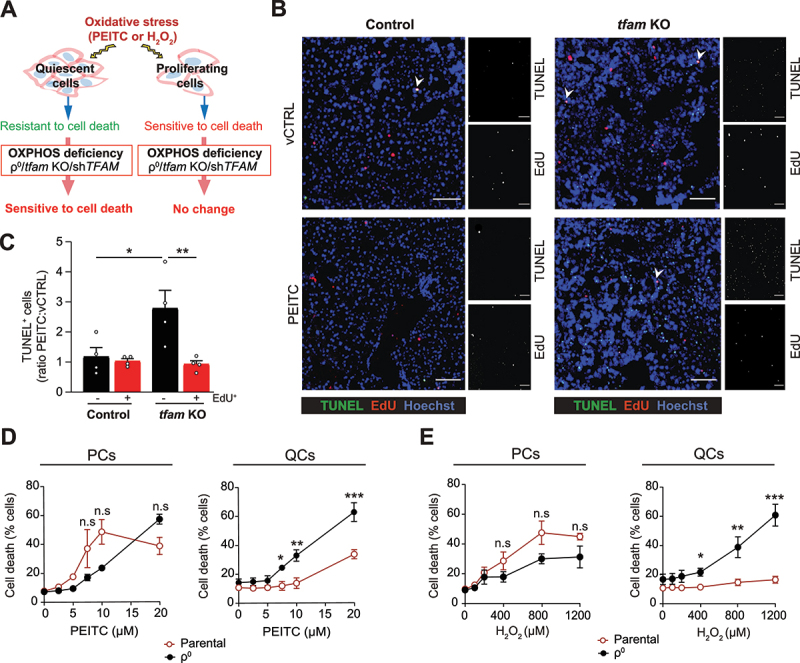
OXPHOS deficiency selectively sensitizes quiescent cells to oxidative stress. (A) OXPHOSdeficiency in quiescent cells leads to increased cell death upon oxidative damage. (B) Representative images of cell death and cell proliferation analyzed by TUNEL and EdU staining, respectively, in the liver of vCTRL and PEITC-treated mice. Scale bar: 500 µm. (C) Quantification of TUNEL positivity (ratio between PEITC and vCTRL) in EdU+ (proliferating, PCs) and EdU− (quiescent, QCs) cells (mean ± S.E.M., n = 4 mice per condition, *p < 0.05, **p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test). (D and E) Cell death in EA.hy926 PCs and QCs with (ρ0) or without OXPHOS deficiency and treated with (D) PEITC and (E) H2O2 for 22 h evaluated by ANXA5-FITC and PI using flow cytometry (mean ± S.E.M., n ≥ 4, n.s.p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 versus parental cells, two-way ANOVA with Tukey’s multiple comparisons test).
To confirm that OXPHOS provides oxidative stress protection selectively in quiescent cells, we used in vitro models where proliferation can be controlled by contact inhibition to directly compare proliferating (PCs) and contact-inhibited quiescent cells (QCs) under defined conditions. We reasoned that ideally the cellular model should not/minimally depend on OXPHOS for energy production. Endothelial cells generate most ATP via glycolysis [27,28], and are therefore expected to cope with the lack of OXPHOS-produced ATP. Indeed, OXPHOS-deficient immortalized EA.hy926 endothelial QCs (ρ0 cells, see Materials and methods, Fig. S1D) featured normal ATP levels (Fig. S1E), energy charge (Fig. S1F) and mitochondrial content (not shown). Furthermore, OXPHOSdeficiency did not compromise contact inhibition or entry into quiescence, as evidenced by correct adherence junction assembly (Fig. S1G), enrichment of cells in the G0 phase of the cell cycle (Fig. S1H), and arrest of proliferation (Fig. S1I) in the absence of senescence (Fig. S1J). Strikingly, however, OXPHOS-deficient EA.hy926 as well as primary human umbilical cord endothelial cells QCs (HUVEC, shTFAM, Fig. S2A) were markedly sensitized to cell death when exposed to pro-oxidants PEITC or hydrogen peroxide (H2O2) (Figure 2D and E, Fig. S2B), while no such effect was observed in corresponding PCs. Similarly, PEITC or H2O2 treatment induced more ROS in OXPHOS-deficient QCs (but not in PCs) than in parental cells (Fig. S2C-E). These results indicate that OXPHOS in endothelial QCs is required for oxidative stress resistance.
To evaluate whether OXPHOS provides ROS resistance also in QCs of non-endothelial origin, we eliminated OXPHOS by TFAM recombination/silencing in primary mouse embryonic fibroblasts (MEFs, isolated from RosaCreERT2 Tfamflox/flox embryos, see Materials and methods, Fig. S2F) and MCF10A (Fig. S2G) mammary epithelial immortalized cells, frequently used cellular models of quiescence [29]. Similarly to endothelial cells, we detected increased cell death (Fig. S2H and I) and exogenously induced ROS levels (Fig. S2J and K) upon PEITC treatment in QCs, whereas this was not observed in PCs.
Oxidative stress resistance during quiescence is provided by OXPHOS-stimulated autophagy
Next, we investigated if metabolic consequences of OXPHOS deficiency in QCs could underly the reduced oxidative stress resistance. While OXPHOS supports nucleotide synthesis in PCs [18,19,21,22,30,31], it was not required for this purpose in QCs (Figure 3A and B). In contrast, OXPHOS deficiency in QCs (but not in PCs) selectively reduced intracellular levels of free amino acids (Figure 3C and D).
Figure 3.
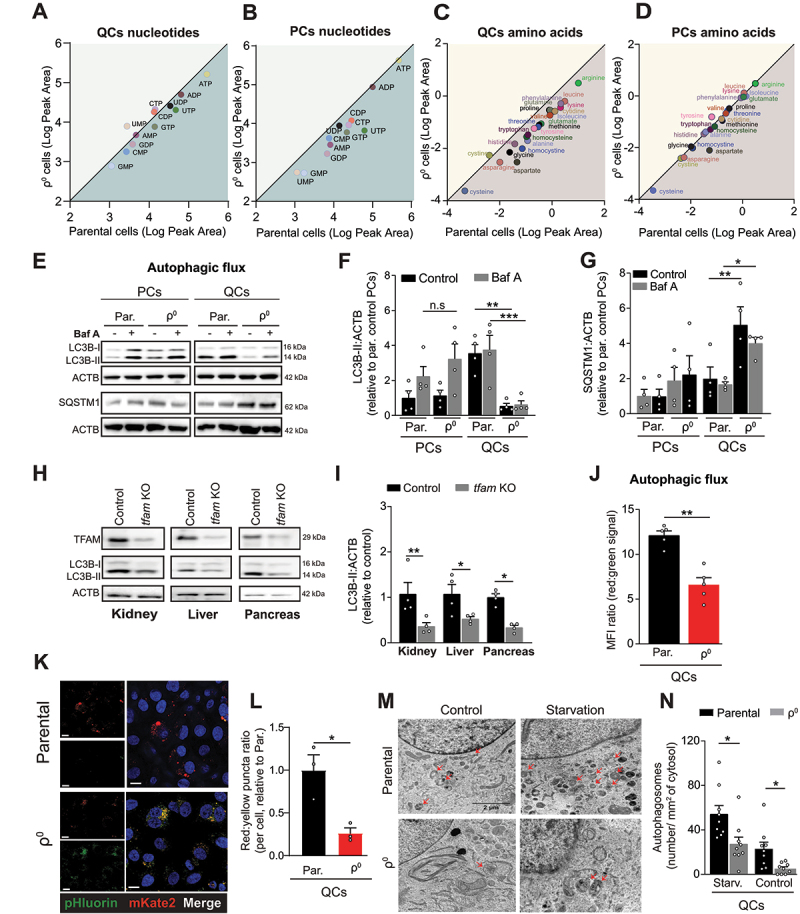
Autophagy is suppressed in OXPHOS-deficient quiescent cells and tissues. (A-D) Mass spectrometry quantification of intracellular levels of nucleotides and amino acids in parental and ρ0 EA.hy926 cells. Nucleotides QCs (A), nucleotides PCs (B), amino acids QCs (C), amino acids PCs (D). The plots show the logarithms of peak area for individual nucleotides and amino acids (mean of n > 3 independent experiments). The values below the diagonal are reduced in ρ0 compared to parental cells. (E) Representative WB images of activated LC3B (LC3B-II) and SQSTM1 in PCs and QCs in the presence or absence of 50 nM of Baf A for 4 h. (F and G) Densitometric quantification of LC3B-II (F) and SQSTM1 (G) (mean ± S.E.M., n = 4, n.s.p > 0.05, *p < 0.05, **p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test). (H) Representative WB images of TFAM protein expression and activated LC3B (LC3B-II) in kidney, liver and pancreas of control and tfam KO mice. (I) Densitometric quantification of activated LC3B (LC3B-II) as in H (mean ± S.E.M, n = 4 mice, *p < 0.05, **p < 0.01, unpaired two-tailed t test). (J) Fluorescent signal of the pHluorin-mKate2-LC3 autophagy flux reporter in parental and OXPHOS deficient EA.hy926 QCs assessed by flow cytometry (mean ± S.E.M., n = 5, **p < 0.01, unpaired two-tailed t test). Note: red:green fluorescence ratio is proportional to the native autophagic flux. (K and L) Representative images pHluorin-mKate2-LC3 flux reporter (K) and quantification of LC3 puncta (L) in parental and OXPHOS deficient EA.hy926 QCs assessed by confocal microscopy. Puncta counts were normalized to the number of cells (mean ± S.E.M., n = 3, **p < 0.01, unpaired two-tailed t test). Scale bar: 40 µm. (M) Representative transmission electron microscopy images and (N) autophagosome quantification of control and starved EA.hy926 parental and ρ0 QCs cells. Autophagosomes are indicated by red arrows (n = 9 cells per condition, *p < 0.05, unpaired two-tailed t test). Scale bar: 2 µm.
Decreased ROS resistance combined with reduced amino acid levels suggested a possible defect of autophagy, which targets (ROS-damaged) proteins and organelles into autophagosomes for lysosomal delivery and recycling and provides stress resistance as well as free amino acids [10,32,33]. Autophagy was selectively suppressed in OXPHOS deficient QCs, shown by the reduced level of activated microtubule associated protein 1 light chain 3 beta (LC3B-II) in ρ0 QCs (but not in PCs) (Figure 3E and F) and in tissues from tfam KO mice (Figure 3H and I). This was due to reduced autophagy flux, as revealed by negligible accumulation of LC3B-II in several types of OXPHOS-deficient QCs treated with bafilomycin A (Baf A), an inhibitor of lysosomal acidification (Figure 3E and F, Fig. S3A-C), and by elevated steady state levels of SQSTM1/p62, a canonical autophagy substrate (Figure 3E and G). A genetically encoded dual fluorescence reporter of autophagic flux pHluorin-mKate2-LC3 [34] (see Materials and methods for details), displayed a reduced red:green fluorescence ratio (Figure 3J, Fig. S3D) and a smaller fraction of red (lysosome-resident) LC3 puncta in OXPHOS-deficient QCs (Figure 3K and L), consistent with compromised delivery of LC3 into the lysosome. Similarly, transmission electron microcopy showed a lower number of autophagosomes in ρ0 QCs in both control conditions and upon starvation to induce autophagy (Figure 3M and N). These findings could not be explained by altered ATG7 (autophagy related 7) levels, which were similar in all tested conditions (Fig. S3E).
Next, we attenuated autophagy in parental QCs to ascertain its role in OXPHOS-mediated stress resistance (Figure 4A). Inhibition of autophagy by pre-treatment with Baf A or silencing of ATG5 (autophagy related 5), an essential autophagy enzyme (Figure 4B-D; Fig. S3F- H), sensitized parental (but not OXPHOS-deficient) QCs to PEITC (Figure 4E and F, Fig. S3I-K), without affecting respiration (Fig. S3L). OXPHOS-maintained autophagy thus provides oxidative stress resistance in QCs.
Figure 4.
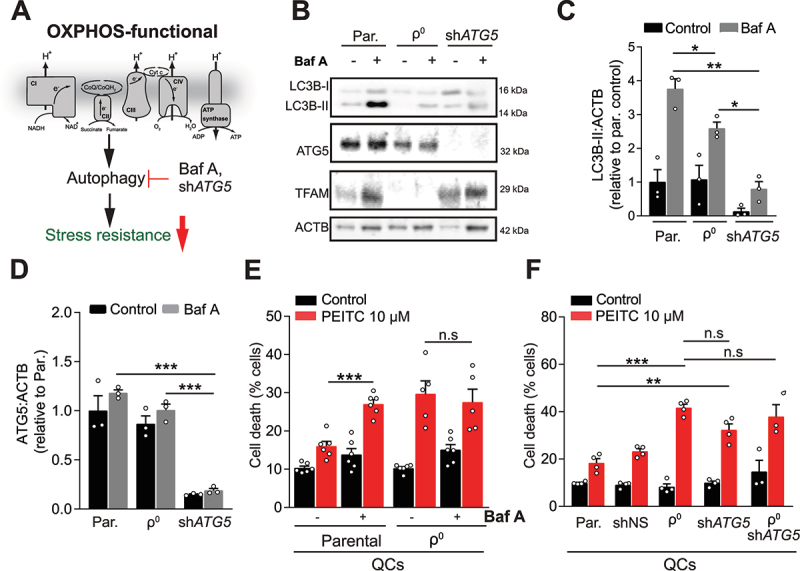
Suppression of autophagy in quiescent cells recapitulates the effects of OXPHOS deficiency on resistance to stress. (A) Pharmacological (Baf A) and genetic (shATG5) suppression of autophagy in parental QCs was used to explore its potential effects downstream of OXPHOS deficiency. (B, C, D) Representative WB images (B) and quantifications of LC3B-II (C) and ATG5 (D) in parental and ρ0 EA.hy926 QCs (mean ± S.E.M., n = 3, n.s.p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test). (E) Cell death in EA.hy926 QCs with or without OXPHOS deficiency, pre-incubated or not with 50 nM of Baf A for 2 h followed by 10 µM PEITC for 22 h, evaluated by ANXA5-FITC and PI (mean ± S.E.M., n ≥ 4, n.s.p > 0.05, ***p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test). (F) Cell death in indicated EA.hy926 QCs treated with 10 µM of PEITC for 22 h, evaluated by ANXA5-FITC and PI (mean ± S.E.M., n = 4, n.s.p > 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test).
Autophagy-mediated oxidative stress resistance during quiescence depends on ETC-derived ROS
We further investigated the mechanism of OXPHOS-maintained autophagy/stress resistance in QCs. We excluded a role for gene transcription/expression (Fig. S4A-D, Tables S3 and S4), canonical autophagy signaling via MTORC1, AMPK, or ULK1 [11,35] (Fig. S4E-K), and, in contrast to proliferating cells [14,15,36], the NADH:NAD ratio (Fig. S4L-S). However, the ETC is a major source of ROS and autophagy can respond to elevated intracellular ROS levels [13,37,38]. We hypothesized that continuous low-level endogenous ROS production from the ETC may be required to maintain basal autophagic flux (Figure 5A), paradoxically protecting the cell from exogenous ROS insult. Baseline ROS levels were reduced by OXPHOS deficiency both in mitochondria and in the cytoplasm (Figure 5B, Fig. S5A and B), showing that the ETC is crucial for endogenous ROS production in QCs. Furthermore, the elevation of antioxidant defense in QCs [2,6,7] was not compromised by OXPHOS deficiency (Fig. S5C-F), and likely further enhances depletion of ROS in OXPHOS-deficient QCs.
Figure 5.
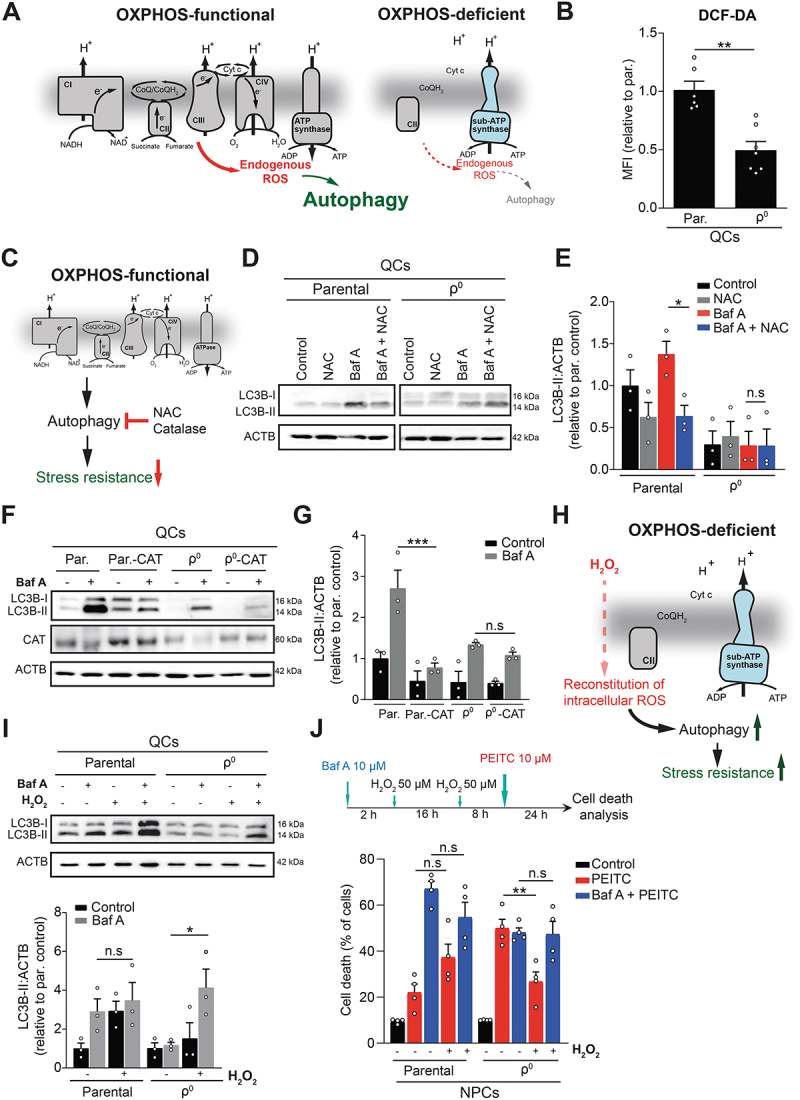
OXPHOS-derived ROS maintain autophagy and ROS resistance in quiescent cells. (A) OXPHOS-deficient QCs lack respiratory complexes and are therefore expected to produce less ROS from the ETC, possibly affecting autophagy. (B) Baseline ROS levels in EA.hy926 QCs assessed using DCF-DA fluorescent probe by flow cytometry (mean ± S.E.M., n ≥ 6, **p < 0.01, unpaired two-tailed t test). (C) Pharmacological (NAC) and genetic (catalase overexpression) reduction of endogenous ROS in parental QCs was used to recapitulated effects of OXPHOS deficiency on autophagy. (D and E) Representative WB image (D) and quantification (E) of activated LC3B (LC3B-II) in parental or ρ0 EA.hy926 QCs treated or not with NAC (1 mM) for 6 h and/or with Baf A (50 nM) for 4 h (mean ± S.E.M., n = 3, n.s.p > 0.05, *p < 0.05, unpaired two-tailed t test). (F and G) Representative WB image (F) and quantification (G) of activated LC3B (LC3B-II) protein levels in parental and ρ0 EA.hy926 QCs with or without overexpression of catalase in the presence or absence of Baf A (50 nM) (mean ± S.E.M., n = 3, n.s.p > 0.05, ***p < 0.001, unpaired two-tailed t test). (H) Treatment with low levels of exogenous ROS (H2O2) in OXPHOS deficient QCs was used to restore autophagy and cell death resistance. (I) Representative WB image (top) and quantification (bottom) of the activated LC3B (LC3B-II) protein expression in EA.hy926 parental and ρ0 QCs treated or not with H2O2 (1 mM) for 4 h in the presence or absence of Baf A (50 nM) (mean ± S.E.M., n = 3, n.s.p > 0.05, *p < 0.05, unpaired two-tailed t test). (J) PEITC-induced cell death in parental and ρ0 EA.hy926 QCs pre-treated or not with Baf A (10 µM), with or without pre-conditioning by H2O2 as shown in the scheme, measured by ANXA5 and PI (mean ± S.E.M, n = 4, n.s.p > 0.05, **p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test).
To functionally link ETC-produced ROS to autophagy, we manipulated basal ROS levels by (i) suppressing ROS in parental QCs and (ii) reconstituting ROS in OXPHOS-deficient QCs. First, we treated QCs with an antioxidant N-acetyl cysteine (NAC) or overexpressed catalase, an antioxidant enzyme (Figure 5C). Both manipulations reduced intracellular ROS levels and autophagic flux in parental QCs to the level found in ρ0 QCs (where they had no further effect) (Figure 5D-G, Fig. S5G and H). Vice versa, autophagic flux was rescued in ρ0 QCs by the administration of exogenous H2O2 (Figure 5H and I), and pre-conditioning with low concentrations of H2O2 restored ROS-resistance of ρ0 QCs to the parental cell levels (Figure 5J). The rescue effect of H2O2 was abrogated by the autophagy inhibitor Baf A (Figure 5J), indicating that the reactivation of autophagy is essential for the recovery of oxidative stress resistance. Overall, we show that endogenous ROS generated by the ETC maintain basal autophagy in quiescent cells, which is protective against exogenous ROS challenge.
ETC-derived ROS attenuate ATG4B activity to support autophagy and stress resistance during quiescence.
ROS can regulate autophagic flux transcriptionally via the ROS-responsive TFEB (transcription factor EB) [39,40], or post-translationally via phosphorylation of BECN1 (beclin 1) [41] or via ATG4B (autophagy related 4B cysteine peptidase) [38,42]. OXPHOS deficiency during quiescence neither suppressed TFEB-regulated transcripts (Fig. S6A) nor altered BECN1 phosphorylation in QCs (Fig. S6B and C). We thus focused on ATG4B, a protease sensitive to ROS-inhibition due to cysteine residues in its active site. Normally ATG4B promotes autophagy by converting pre-LC3B into LC3B-I, which, upon conjugation to phosphatidylethanolamine (PE), yields the active LC3B-II (Figure 6A). However, when ATG4B activity is too high, such as in the absence of ROS, it can also remove PE from LC3B-II and inhibit autophagy [38] (Figure 6A and B). Accordingly, optimal level of endogenous ROS, present in parental but not in OXPHOS-deficient QCs, may be needed to attenuate ATG4B activity and maintain basal autophagic flux (Figure 6B).
Figure 6.
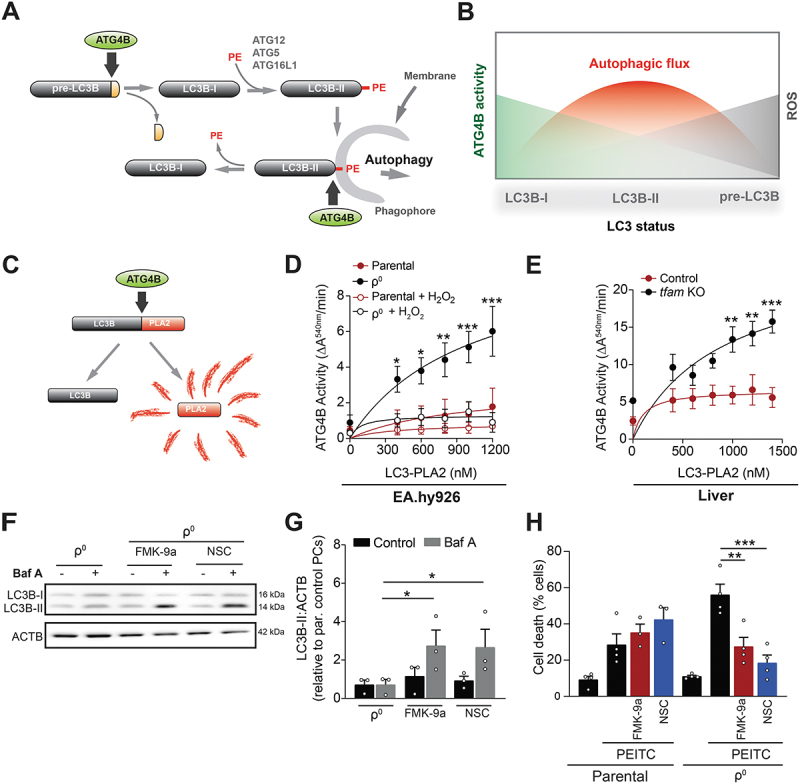
ETC-derived ROS regulate autophagy during quiescence via ATG4B. (A and B) ROS-sensitive ATG4B both activates (by cleaving pre-LC3) and inhibits (by removing PE) LC3B (A), leading to a complex relationship between ATG4B activity, ROS and autophagy (B). At optimal ROS levels ATG4B activity is partially attenuated, the activating pre-LC3 cleavage can proceed while the inhibitory removal of PE is suppressed, leading to the maximal autophagic flux. (C) The LC3B-PLA2 (phospholipase A2)-based reporter to assess ATG4B function. Active ATG4B liberates PLA2, producing signal in a fluorogenic assay. (D) ATG4B activity in cell lysates from EA.hy926 QCs treated or not with H2O2 (1 mM) for 4 h and incubated with increasing concentrations of LC3B-PLA2 fusion protein substrate (mean ± S.E.M., n ≥ 4, *p < 0.05, **p < 0.01, ***p < 0.001, versus parental cells, two-Way ANOVA with Tukey’s multiple comparisons test). (E) ATG4B activity in cell lysates from liver of control or tfam KO mice incubated with increasing concentrations of LC3B-PLA2 fusion protein substrate (mean ± S.E.M., n = 4 mice, **p < 0.01, ***p < 0.001, versus control cells, two-way ANOVA with Tukey’s multiple comparisons test). (F and G) Representative WB image (F) and quantification (G) of the activated LC3B (LC3B-II) protein in EA.hy926 ρ0 QCs treated with FMK-9a (1 µM) and NSC185058 (0.1 µM) for 4h (mean ± S.E.M., n = 3, *p < 0.05, unpaired two-tailed t test). (H) PEITC-induced cell death in parental and ρ0 EA.hy926 QCs pre-treated or not with FMK-9a (1 µM) and NSC185058 (0.1 µM) for 2 h, measured by ANXA5 and PI. (mean ± S.E.M, n = 4, n.s.p > 0.05, **p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test).
To assess the activity of ATG4B in OXPHOS-deficient QCs and test if it regulates autophagy/stress resistance, we used an ATG4B-specific phospholipase A2-based reporter (Figure 6C) [43,44]. ATG4B activity was high in OXPHOS-deficient and low in parental QCs (Figure 6D; Fig. S6D). Exposure of OXPHOS-deficient QCs to H2O2 reduced ATG4B activity to the level of parental cells (Figure 6D; Fig. S6D), consistent with the capacity of exogenous H2O2 to stimulate autophagy and restore stress resistance in the absence of functional OXPHOS (c.f. Figure 5I and J). Addition of recombinant human ATG4B further increased the signal of the ATG4B reporter in the lysates from OXPHOS-deficient but not from parental QCs (Fig. S6E and F), demonstrating that OXPHOS deficiency provides an environment permitting ATG4B overactivation. Consistently, ATG4B activity was elevated in liver and kidney tissue from tfam KO mice (Figure 6E, Fig. S6G). Next, we employed two distinct ATG4B inhibitors as well as RNA interference to normalize ATG4B activity in OXPHOS-deficient QCs to the level of parental cells (Figure S6H and I). Interestingly, both inhibitors, NSC185058 [45] and FMK-9a [46], upregulated LC3B-II in ρ0 QCs (Figure 6F and G) and restored resistance to PEITC (Figure 6H), while providing no additional protection for parental cells. Similarly, ATG4B silencing upregulated LC3B-II and recovered resistance to PEITC (Figure S6J and K). Accordingly, excessive ATG4B activity in OXPHOS-deficient QCs suppresses autophagy, leading to reduced oxidative stress resistance.
OXPHOS maintains specificity of ROS-based anticancer treatment by protecting normal tissue
Having shown that OXPHOS provides oxidative stress resistance in QCs, we next investigate if such protection has relevance in disease conditions. During cancer therapy, both tumor cells (mostly PCs) and normal somatic cells (mostly QCs) are exposed to ROS-inducing anticancer agents, yet cell death is selectively triggered in proliferating cancer cells [7,26]. We thus explored if OXPHOS-induced resistance to ROS in normal tissues underlies the therapeutic window, contributing to specificity. We grafted syngeneic B16 melanoma cells subcutaneously into tfam KO mice to generate respiration-competent tumors on an OXPHOS deficient background (Figure 7A). Tumor-bearing mice were treated with two ROS-inducing compounds: (i) PEITC, an experimental anticancer agent [26], or (ii) doxorubicin, a clinically used cancer therapeutic associated with ROS-linked toxicity in normal tissues (Fig. S7A) [47]. Subsequently, we assessed cell death in tissues and determined a specificity index (SI), defined as the ratio of cell death induction in the tumor versus non-malignant tissue (liver) (Figure 7A). In case the specificity of treatment is reduced, the SI should decrease. Indeed, the SI was reduced more than 2-fold in PEITC-treated tfam KO animals compared to PEITC-treated controls (Figure 7B, Fig. S7B), and a similar SI reduction was observed upon administration of doxorubicin (Figure 7B, Fig. S7B), mainly due to increased cell death in normal tissue (Fig. S7C). Inhibition of autophagy by chloroquine also lowered the specificity of PEITC treatment in tumor-bearing mice due to increased cell death in normal tissue (Figure 7C, Fig. S7D and E), similarly to OXPHOS deficiency. Hence, the selectivity of ROS-based anti-cancer therapy is, at least in part, determined by functional OXPHOS/autophagy that maintain oxidative stress resistance in non-malignant tissues.
Figure 7.
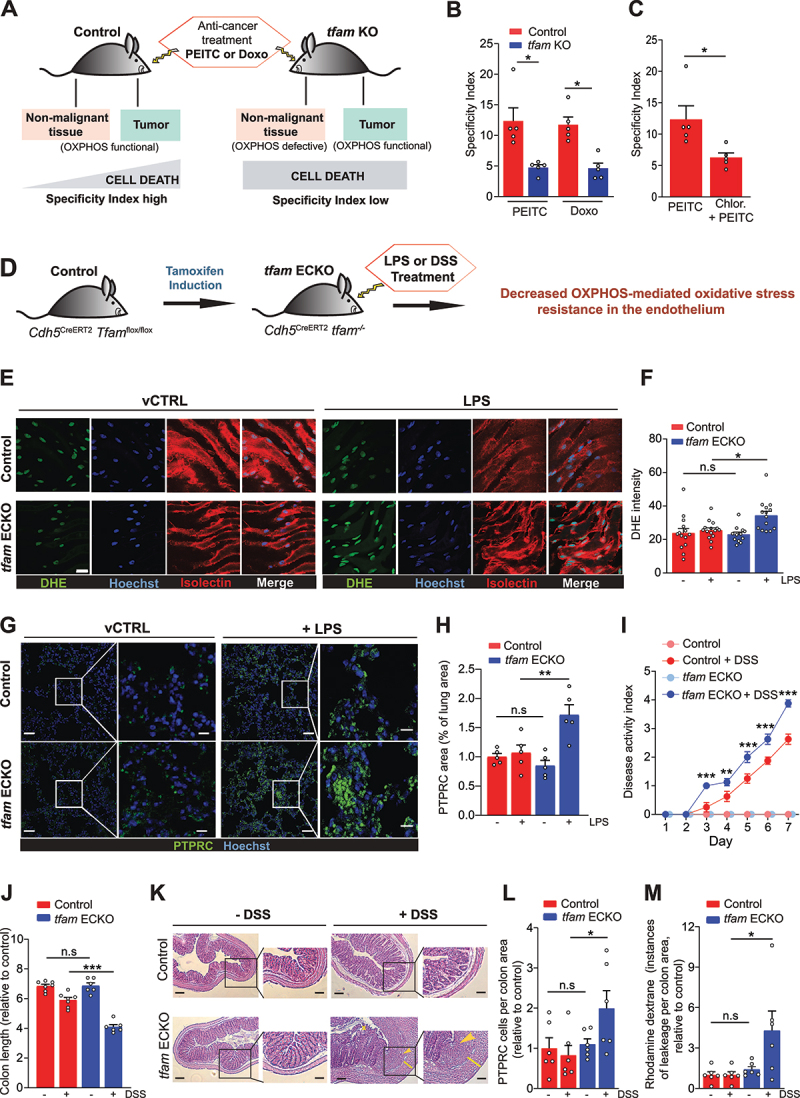
OXPHOS-mediated stress resistance during quiescence maintains therapeutic window in cancer and limits inflammation-linked pathology. (A) OXPHOS maintains therapeutic window by supporting resistance to ROS in non-malignant tissue during ROS-based cancer therapy. Treatment-induced cell death in non-malignant tissues (contain mostly QCs) is increased in tumor-bearing tfam KO mice, compromising specificity of treatment. (B) Specificity index (SI) assessed by quantification of the ratio of TUNEL+ cells in tumor and liver tissue from control and tfam KO mice after treatment with PEITC and doxorubicin (Doxo). (C) Specificity index (SI) assessed by quantification of the ratio of TUNEL+ cells in tumor and liver tissues from control mice treated with PEITC (1.25 mg/25 g mouse in corn oil) or PEITC + chloroquine (1 mg/20 g mouse in corn oil), (mean ± S.E.M., n = 5 mice per condition, *p < 0.05, **p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test). (D) Endothelium-specific tfam deletion strategy in Cdh5CreERT2 Tfamflox/flox mice (referred to as tfam ECKO). (E and F) Representative images (E) and quantification (F) of aorta open book preparations from control and tfam ECKO mice treated with LPS for 4 h, stained for DHE (green), the EC marker isolectin B4 (red) and nuclei (Hoechst-blue). Scale bar: 20 µm. (n ≥ 12 mice per group, n.s.p > 0.05, *p < 0.05, one-way ANOVA with Sidak’s multiple comparisons test). (G and H) Representative images (G) and quantification (H) of lungs from control and tfam ECKO mice 4 h after injection of LPS or vehicle solution stained with PTPRC/CD45 (green) and Hoechst (blue) (n = 5 mice per group, n.s.p > 0.05, **p < 0.01, one-way ANOVA with Sidak’s multiple comparisons test). Right panels in G are magnifications of the boxed areas on the left. Scale bar: 40 µm (left panels) and 20 µm (right panels). (I) Disease activity index of colitis in control and tfam ECKO mice during treatment with 2.5% DSS in the drinking water for 7 consecutive days (n = 6 mice, ***p < 0.001, two-way ANOVA Tukey’s multiple comparisons test). (J) Colon length in control and tfam ECKO mice after 2.5% DSS treatment (n = 6; 3 males, 3 females per group), n.s.p > 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with Sidak’s multiple comparisons test). (K) Representative images of hematoxylin and eosin-stained sections of colon from control and tfam ECKO mice treated as in I. Right panels are magnifications of the respective boxed areas. Asterisks denotes loss of crypts; arrowhead denotes more severe separation of the crypt base from the muscularis mucosa, infiltrated with inflammatory cells; yellow line visualizes the thickness of the colonic wall. Scale bar: 500 µm. (L) Quantification of colon PTPRC/CD45+ cells per total area of tissue from control and tfam ECKO mice after 2.5% DSS treatment (n = 6 mice, n.s.p > 0.05, *p < 0.05, one-way ANOVA with Sidak’s multiple comparisons test). (M) Quantification of vascular leakiness (dextran-rhodamine signal) in colon from control and tfam ECKO mice after 2.5% DSS treatment (n = 6 mice, n.s.p > 0.05, *p < 0.05, one-way ANOVA with Sidak’s multiple comparisons test).
OXPHOS controls ROS in quiescent endothelium in vivo and limits inflammation-linked pathology
To examine the role of OXPHOS-mediated oxidative stress resistance in pathologies other than cancer, we focused on inflammation, often accompanied by increased oxidative stress [48]. Reduced oxidative stress resistance of quiescent vascular endothelium exacerbates inflammation by decreasing endothelial barrier function, facilitating leukocyte extravasation, and increasing vessel permeability [6]. In addition, the endothelium does not rely on OXPHOS-derived ATP. We thus explored the role of OXPHOS in oxidative stress resistance specifically in the endothelium in the context of (i) systemic inflammation and (ii) inflammation of the colon linked to inflammatory bowel disease (IBD).
To selectively disable OXPHOS in the endothelium in vivo, we used an established endothelium-specific tamoxifen-inducible Cdh5CreERT2 driver to ablate TFAM in the endothelium of adult mice (tfam ECKO mice, see Materials and methods) (Figure 7D). tfam deletion was confirmed by immunohistochemistry, which showed no colocalization of the TFAM signal with an endothelial marker ENG/CD105 in tfam ECKO animals (Fig. S7F). To induce systemic inflammation, control and tfam ECKO mice were challenged with LPS, a (patho)-physiological inducer of ROS in endothelial cells [49]. Interestingly, LPS-treated tfam ECKO mice featured elevated ROS levels in the endothelium in vivo, as demonstrated by dihydroethidium (DHE) staining of intact aortas (Figure 7E and F), and presented increased leukocyte infiltration in the lungs compared to LPS-treated controls (Figure 7G and H). In contrast, there was no apparent effect of tfam deletion in non-treated animals.
To assess the relevance of OXPHOS-linked oxidative stress resistance in IBD, we used a model of dextran sodium sulfate (DSS)-induced colitis. We previously established that increased susceptibility of the endothelium to oxidative stress promotes the disease by disrupting the endothelial barrier in the inflamed colon [6]. Indeed, whereas no adverse effects of TFAM deficiency were observed in non-treated animals, DSS-treated tfam ECKO mice had more severe disease as shown by a higher disease activity index (scoring body weight loss, stool consistency, and blood in the stool and anal region) (Figure 7I), shortened colon (Figure 7J, Fig. S7G), and showed more pronounced signs of colitis, including loss of crypts, separation of the crypt base from the muscularis mucosa and colonic wall thickening (Figure 7K). Furthermore, the DSS-treated tfam ECKO colons suffered from elevated leukocyte infiltration (Figure 7L, Fig. S7H) and increased vessel leakiness, demonstrated as rhodamine-dextran extravasation (Figure 7M, Fig. S7I). Co-treatment with tempol, an antioxidant [50], normalized the phenotype of DSS-treated tfam ECKO mice to the level of DSS-treated controls (Fig. S7J and K), confirming that ROS are responsible for the increased severity of colitis in tfam ECKO animals. To summarize, the selective ablation of endothelial OXPHOS exacerbates systemic inflammation as well as IBD, a frequent pathology in humans, demonstrating that oxidative stress resistance provided by OXPHOS in the endothelium is clinically relevant.
Discussion
Mitochondrial respiration sustained by the OXPHOS system is crucial for most living organisms. OXPHOS is a source of ATP and supports nucleotide biosynthesis in proliferating cells [18,19,21], yet whether OXPHOS has other essential roles specific to quiescent cells and tissues remained unclear. The key finding of our study therefore is that OXPHOS is necessary to combat oxidative stress in quiescent cells and tissues by supporting autophagy. Interestingly, a link between OXPHOS, autophagy and stress resistance was not found in proliferating cells, suggesting that regulation of autophagy by OXPHOS is proliferation-sensitive.
We provide evidence that OXPHOS/autophagy-mediated oxidative stress resistance during quiescence is relevant in several patho-physiological scenarios. First, we document that OXPHOS/autophagy ensure specificity of anti-cancer therapy by protecting normal non-proliferative tissues when tumors are treated with ROS-inducing anticancer agents, providing a therapeutic window. Hence, while many investigations focused on why tumors are sensitive to therapy, here we address an equally important, yet understudied question of why normal tissues are resistant. Our findings thus warrant caution when using autophagy inhibitors (chloroquine is in clinical trials in cancer) with ROS-inducing agents. Such a combination has the potential to compromise selectivity and thereby increase toxicity of cancer therapy.
Second, by selectively ablating OXPHOS in adult quiescent endothelium, which is relatively independent of ATP production via OXPHOS, we document increased susceptibility of mice to systemic as well as local inflammation, conditions associated with oxidative stress in endothelial cells. OXPHOS ablation exacerbated oxidative stress in endothelial cells during inflammation, leading to increased vessel permeability and leukocyte extravasation. A similar phenotype was observed when resistance of endothelial cells to oxidative stress was reduced by alternative manipulations [6]. This demonstrates that OXPHOS/autophagy protect endothelial cells during inflammatory conditions such as colitis associated with IBD, documenting a new role for OXPHOS as a guardian against oxidative stress in quiescent endothelium. Others have shown that OXPHOS-mediated biosynthesis is crucial for biomass buildup in proliferating endothelial cells during angiogenesis [51], demonstrating that distinct functions of OXPHOS during proliferation and quiescence (i.e., biomass buildup versus stress protection) are maintained in vivo.
Our findings point to stark differences in regulation of stress resistance/autophagy in proliferating and quiescent cells. While in quiescent cells OXPHOS deficiency abrogated autophagy and resistance to stress, it had no effect in proliferating cells. Furthermore, our results indicate that during quiescence ETC signals to autophagy via ROS, but not via NADH. In contrast, NADH has been found to regulate autophagy in proliferating cells [14,15], pointing to distinct autophagy regulation during proliferation and quiescence. Consequently, NADH may be the dominant regulator of autophagy in proliferating cells which require biosynthesis (NADH:NAD+ ratio is crucial for biosynthesis), while ROS become important in quiescent cells that accentuate stress resistance (ROS are important stressors). This is consistent with the dual role of OXPHOS which provides biosynthesis during proliferation and stress resistance during quiescence, reflecting the distinct needs of these two states (Figure 8).
Figure 8.
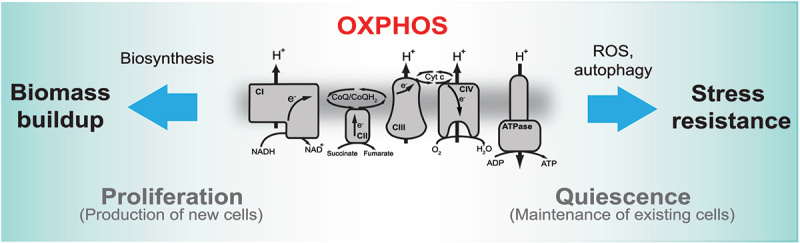
Distinct roles of OXPHOS during proliferation and quiescence. The proposed model depicts specific roles of OXPHOS in proliferating and quiescent cells. In proliferating cells OXPHOS supports biosynthesis, whereas in quiescent cells it provides stress resistance via ROS-stimulated autophagy.
How does OXPHOS, the main producer of intracellular ROS, induce protection against oxidative stress in quiescent cells? We propose that this is an active process where continuous low-level generation of endogenous ROS from the ETC attenuates the activity of ATG4B, a redox-regulated member of the autophagy cascade [38]. Such ROS-mediated ATG4B modulation sustains the basal, protective autophagy. The absence of ETC-produced ROS in OXPHOS-deficient cells is amplified by the quiescence-induced elevation of antioxidant defense, leading to unrestrained ATG4B activity that suppresses autophagy. Supplementation of OXPHOS-deficient quiescent cells with ROS normalized ATG4B activity and rescued autophagy as well as ROS resistance, which was phenocopied by direct attenuation of ATG4B activity by specific inhibitors or by RNA silencing. These findings favor a model where OXPHOS supports autophagy during quiescence by maintaining the optimal intracellular redox balance. In this way, OXPHOS/autophagy, along with the antioxidant defense, represent a basal, constitutive layer of ROS resistance in quiescent cells.
OXPHOS-mediated resistance to oxidative stress is conserved over multiple tissues and cell types, suggesting its ancient origin. This may have interesting evolutionary connotations. Eukaryotic cells acquired mitochondria to benefit from oxygen through oxidative metabolism for efficient ATP production and biosynthesis, making complex life possible. At the same time, they apparently built in an autophagy-based mechanism to protect against oxygen levels that rose throughout the evolution of multicellular life forms [52] and that predispose to oxidative stress. In addition, by coupling OXPHOS to autophagy evolution would devise a safety measure against ROS-induced damage inherent to energy generation by mitochondria. The OXPHOS system thus features unexpected plasticity and readily adapts to specific requirements of cellular proliferation and quiescence with prominent functional consequences for pathology.
Materials and methods
Mice
Whole-body model: C57BL/6J Tfamflox/flox mice (Jackson Laboratory, 026123)[53] were bred with C57BL/6J RosaCreERT2 mice (Jackson Laboratory, 008463) to produce C57BL/6J RosaCreERT2Tfamflox/flox mice. The mice were kept homozygous for both RosaCreERT2 and Tfamflox. TFAM deficiency was induced in 7-8-week-old mice by 3 doses of tamoxifen (Sigma-Aldrich, T5648), 0.25 mg/g body weight in corn oil (Sigma-Aldrich, C8267, concentration 9 mg/ml) every 12 h, intraperitoneally (i.p.). Tamoxifen-treated Tfamflox/flox mice served as controls. Experiments were performed 11 days post induction. At day 12-13 post induction, the mice started to show slowness of movement and apathy. Endothelial model: Cdh5CreERT2 Tfamflox/flox mice were produced by crossing C57BL/6J Cdh5CreERT2 mice [54] (kindly provided by Dr. R. Boon) with Tfamflox/flox animals. The Cdh5CreERT2 Tfamflox/flox mice were kept heterozygous for Cre. Endothelial specific TFAM deficiency was induced in 8-week-old-mice by 5 doses of tamoxifen, 0.10 mg/g body weight in corn oil every 24 h (i.p.). Tamoxifen-treated Tfamflox/flox mice were used as controls. Experiments were started 16 days post induction. Induced Cdh5CreERT2 Tfamflox/flox animals behaved normally and showed no adverse signs for 2 months post induction (we did not follow them longer). Animals were maintained under SPF conditions in individually ventilated cages with controlled temperature (22 ± 2°C) and humidity under a 12 h light/12 h dark cycle and with food and drink ad libitum. Animals were closely followed-up by the animal caretakers and the experimenters, with regular inspection by a veterinarian, as per the standard health and animal welfare procedures of the local animal facility. No statistical method was used to predetermine sample size. All animal experiments were approved by the Animal Ethics Committee of the Czech Academy of Sciences and were performed according to Czech guidelines for the Care and Use of Animals in Research and Teaching. None of the animals showed signs of distress during the experiments.
Cell lines
Primary cells: Mouse embryonic fibroblasts (MEFs) were isolated from non-induced 12 - 13 days embryos of RosaCreERT2 Tfamflox/flox mice using standard procedures and cultivated in high glucose (4.5 g/l) DMEM (Sigma-Aldrich, D6429) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, F7524), and 1% antibiotics (penicillin G, 100 U/ml; Sigma-Aldrich, P3032) and streptomycin (100 μg/ml; Sigma-Aldrich, S9137)). TFAM deficiency was induced by supplementation of 3 µM of 4-hydroxytamoxifen (Sigma-Aldrich, H6278) to the culture medium for 5 days. OXPHOS dysfunction was verified by respirometry and/or absence of TFAM. Human umbilical vein endothelial cells (HUVECs) were obtained from PromoCell (C-12208) and cultivated in endothelial cell basal medium (EGM2) (PromoCell, C22011) supplemented with endothelial cell growth medium supplement pack (PromoCell, C-30120) and were used before passage 5.
Cell lines: Human endothelial EA.hy926, epithelial MCF10A and B16 melanoma cell lines were obtained from the ATCC (CRL-2922, CRL-10317, CRL-6323, respectively). The Ea.hy926 cells were cultured in high glucose (4.5 g/l) DMEM media supplemented with 10% FBS, 2% hypoxanthine aminopterin thymidine (HAT) supplement (Thermo Fisher Scientific, 21060017) and 1% antibiotics (penicillin G 100 U/ml and streptomycin 100 μg/ml). MCF10A cells were cultured in DMEM:F12 (Lonza, 12-719F) supplemented with 5% horse serum (Sigma-Aldrich, H1138), 0.1% human insulin, 20 ng/ml (Sigma-Aldrich, I9278) epidermal growth factor (Thermo Fisher Scientific, PHG0311), 0.5 mg/ml hydrocortisone (Sigma-Aldrich, H0888), 100 ng/ml cholera toxin (Sigma-Aldrich, C8052) and 1% antibiotics (penicillin G, 100 U/ml and streptomycin, 100 μg/ml). B16 melanoma cells were maintained in DMEM supplemented with 10% FBS and 1% antibiotics (penicillin G 100 U/ml and streptomycin 100 μg/ml). For quiescent cultures, the cells were seeded into 24-well plates at 80% density and cultured for 4 days post-confluence. For proliferating cultures, cells were seeded into 6-well plates at 2 × 105 cells per well 2 days before experiment. Ea.hy926 cells deficient in mtDNA (ρ0 phenotype) were prepared by long-term culture with 0.2 μg/ml of ethidium bromide as described (Tan et al., 2015). OXPHOS-deficient MCF10A cells are described below. OXPHOS deficient cells were cultured in the media corresponding to the given cell type supplemented with 1 mM sodium pyruvate (Sigma-Aldrich, P5280) and 50 µg/ml uridine (Sigma-Aldrich, U3003) unless indicated otherwise. Under these culture conditions, all OXPHOS deficient cell showed normal viability. All cultures were maintained in a 5% CO2, 37°C incubator.
Gene silencing and overexpression
HUVEC, EA.hy926 and MCF10A cells were silenced for ATG5, NDUFV1 and TFAM genes by lentiviral transduction using the following predesigned shRNAs in pLKO1 vector: ATG5-TRCN0000150940; NDUFV1-TRCN0000025872; TFAM-TRCN0000329819; non-silencing control-SHC002 (Sigma-Aldrich). Silencing efficacy was more than 85% in all cases. Expression of CAT (catalase) from the pLenti6/V5-MCS plasmid, a gift of S. Lortz (Hannover Medical School, Hannover, Germany) [55] and pHluorin-mKate2-hLC3 reporter from the FUGW lentiviral plasmid (a gift of I. Tanida; Addgene, 61460) [34] was achieved by lentiviral transduction. Cells were used for experiments 2-4 weeks posttransduction.
Western blotting (WB)
Cells were lysed in RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40 [Fluka, 74385], 0.5% sodium deoxycholate [Sigma-Aldrich, D6750], 0.1% SDS) supplemented with 1% of protease (SERVA, 3910201) and 1% of phosphatase (Sigma-Aldrich, P5726) inhibitor cocktail. The lysate was cleared by centrifugation and total protein determined by BCA assay (Thermo Fisher Scientific, 23227). Murine tissues were homogenized 3 x 20s, 5000 rpm, with an interval of 2 min on ice, using a Precellys 24 tissue homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France) in prefilled lysis tubes (Bertin, KT03961-1-203.05) in RIPA buffer containing 1% protease and 1% phosphatase inhibitors. 30 mg of total protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes, and probed with the following primary antibodies: TFAM (Abcam, ab131607); ATG5 (Cell Signaling Technology, cs12994); LC3B (Cell Signaling Technology, cs2775); NDUFV1 (Abcam, ab55535); MTOR (Cell Signaling Technology, cs2972); phospho-MTOR (Cell Signaling Technology, cs2971); PRKAA/AMPKα (Cell Signaling Technology, cs2532); phospho-PRKAA/AMPKα (Cell Signaling Technology, cs2531); ULK1 (D8H5; Cell Signaling Technology, cs8054); phospho-ULK1 (Ser757; Cell Signaling Technology, cs6888); phospho-ULK1 (Ser555; Cell Signaling Technology, cs5869); RPS6KB/p70S6 kinase (Santa Cruz Biotechnology, sc-8418); phospho-RPS6KB/p70S6 kinase (Santa Cruz Biotechnology, sc-8416); cleaved CASP3 (Asp175, 5A1E; Cell Signaling Technology, 9664); cleaved CASP3 (ENZO, 31A1067); cleaved-PARP (Cell Signaling Technology, cs9541); MT-ND5 (Abcam, ab92624) MT-CO1 (Abcam, ab110258); SOD2 (Acris, AP03024-PU-N); GPX1 (Abcam, ab108427, BECN1 (Santa Cruz Biotechnology, sc-11427), phospho-BECN1 (Ser93; Cell Signaling Technology, 14717); SQSTM1/p62 (Santa Cruz Biotechnology, sc-2557); ATG7 (Cell Signaling Technology, d12b11); PTPRC/CD45 (Thermo Fisher Scientific, 14-0451-85); ENG/CD105 (Bio-Techne R&D Systems, AF1320) and anti-ACTB/β-Actin HRP conjugate (Cell Signaling Technology, cs5125).
Autophagic flux measurements
Cells were treated or not with 50 nM of bafilomycin A1 (Santa Cruz Biotechnology, sc-201550) for 4 h and collected for WB analysis to detect non-activated LC3B-I and activated, phosphatidylethanolamine (PE)-conjugated, LC3B-II protein, and SQSTM1. Where indicated, cells were pre-incubated with the antioxidant N-acetylcysteine 1 mM (NAC; Sigma-Aldrich, A7250-10G) for 2 h or with MTOR inhibitor everolimus 20 µM (Invivogen, tlrl-eve) for 4 h.
Autophagy flux was also assessed using a genetically encoded fluorescent reporter, pHluorin-mKate2-LC3. This reporter contains pHluorin (green) and mKate2 (red) fluorescent proteins fused to LC3B for autophagosome targeting. The green fluorescence of pHluorin is pH-sensitive and is quenched when the autophagosome enters the low pH of the lysosome, while the red fluorescence of mKate2 is pH-independent and is retained. Therefore, red:green (or red:yellow) ratio is proportional to the native autophagic flux. Cells expressing pHluorin-mKate2-LC3 were washed with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) trypsinized, resuspended in PBS and analyzed by flow cytometry (LSRFortessaTM SORP, Becton Dickinson, New Jersey, USA). To detect pHluorin (green) and mKate2 (red) fluorescence, 488 nm and 561 nm lasers were used for excitation. Emission was measured at 530±15 nm for pHluorin and 610±10 nm for mKate2. For microscopy experiments cells expressing pHluorin-mKate2-LC3 were washed, fixed with 4% of formaldehyde (FA; VWR, 9713.1) and nuclei were counterstained with Hoechst 33342 (Sigma-Aldrich, B2261). Imaging was performed using the Carl Zeiss LSM880 NLO inverted point scanning microscope (Carl Zeiss, Jena, Germany) using a 63x objective. Puncta of mKate2 and pHluorin were quantified using “Particle analyzer” in ImageJ and the ratio of mKate2-positive (red) to mKate2+pHluorin-positive (yellow) puncta per cell was calculated.
Transmission electron microscopy
Cells were seeded on a glass coverslip and grown in culture medium (as described above) or kept in HBSS medium (Sigma-Aldrich, H6648) for 24 h to induce starvation. After the incubation, cells were quickly washed with Sörensen buffer (SB; 0.1 M sodium/potassium phosphate buffer, pH 7.3) at 37°C, fixed with 2.5% glutaraldehyde (Sigma-Aldrich, G5882) in SB for 2 h (1 h at room temperature and 1 h in 4°C), washed with SB, and post-fixed with 1% OsO4 (Sigma-Aldrich, 75632) solution in SB for 2 h at room temperature. Cells were dehydrated in series of acetone (Penta, 10060-11000) with increasing concentration and embedded in Epon-Durcupan resin (Sigma-Aldrich, 45345, 44611-13) and left to polymerize for 72 h at 60°C. Polymerized blocks were cut into 80-nm ultrathin sections and collected on 200-mesh size copper grids (Agar Scientific, AGG2200C). The sections were examined at 80 kV in FEI Morgagni 268 transmission electron microscope (FEI, Netherlands) using Mega View III CCD camera (Olympus Soft Imaging Solutions, Germany), in JEOL JEM-1011 (Jeol, Japan) equipped with Veleta CCD camera (Olympus Soft Imaging Solution, Germany) and in Jeol JEM-1400 FLASH equipped with Matataki camera (Jeol, Japan). Multiple images at 25,000x and 30,000x were taken from each analyzed cell to cover the whole area of the cell profile in ultrathin section. Quantification was performed using Ellipse image analysis program (ViDiTo, Slovakia).
Cloning, expression, and purification of LC3B-PLA2
The LC3B-PLA2 fusion was generated using custom-made codon-optimized DNA template gene-string synthesis protocol (Thermo Fisher Scientific, see Table S1 for the corresponding DNA and protein sequences). The coding sequence was PCR amplified with a pair of gene-specific primers (Table S2) and cloned into the pMT/BiP/SF-TEV vector in frame with the BiP secretion signal and the N-terminal Twin-Strep purification tag [56]. The identity of the expression plasmid, designated pMT/SLIN-LC3B-PLA2, was verified by Sanger sequencing.
The expression and purification of the LC3B-PLA2 fusion protein was carried out using protocols described previously [56]. Briefly, the pMT/SLIN-LC3B-PLA2 plasmid, together with pCoBLAST which confers resistance to Blasticidin (InvivoGen, ant-bl-1), were transfected into Schneider’s S2 cells using Effectene (Qiagen, 301425) according to the manufacturer’s protocol. Transfectants were selected in the Insect-Xpress medium (Lonza, BELN12-730Q) supplemented with 10% v:v FBS and 40 µg/ml Blasticidin. Four weeks post transfection, the Blasticidin-resistant S2 cells were transferred into the SFX medium, expanded and expression of SF-LC3B-PLA2 induced by the addition of 0.7 mM CuSO4 (Sigma-Aldrich, C8027) at a cell density of 1 x 106/ml. Seven days post induction, cells were harvested by centrifugation, the conditioned medium supplemented with recombinant Streptavidin (10 μg/ml, produced in-house) and loaded on a StrepTactin XT affinity column (IBA, 2-4010-025). Following extensive washing with 100 mM Tris-HCl, 300 mM NaCl, pH 8.0, StrepTactin-bound fusion protein was eluted with 5 mM D-Biotin (IBA, 2-1016-005) in 100 mM Tris-HCl, 300 mM NaCl, pH 8.0. Pooled elution fractions containing pure SF-LC3B-PLA2 were concentrated to 1 mg/ml, snap-frozen in liquid nitrogen and stored at -80°C until further use. The typical yield of SF-LC3B-PLA2 was approximately 15 mg per liter of S2 culture with purity > 95%.
ATG4 activity assay
The assay was performed as described [43]. Cells and murine tissues were harvested in lysis buffer (25 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM CaCl2, 5 mM MgCl2, 5% glycerol, 0.1% NP-40). Cell lysates were sonicated on ice (2 x 2 s) and tissues were homogenized 3 x 20s, 5000 rpm, with an interval of 2 min on ice, in prefilled lysing kits using a Precellys 24 tissue homogenizer and clarified using centrifugation. Cellular ATG4B activity was assessed by incubation of equal amounts of lysate with various concentrations of LC3B-PLA2 fusion protein substrate (200-1400 nM) or ATG4B (200, 400 and 600 nM) in reaction buffer (20 mM Tris-HCl, pH 8.0, 2 mM CaCl2) in the presence of 20 mM Red/Green BODIPY PC-A2 (Thermo Fisher Scientific, A10072). If required, cells were pretreated with 1 mM of H2O2 (Sigma-Aldrich, 516813) for 4 h, or with 1 µM FMK-9a (MedChemExpress, HY-100522) or 0.1 µM NSC185058 (Cayman Chemical, 23957) for 2 h prior to harvesting. Fluorescence intensity was measured for 1 h at 2-min intervals (60 measurements per data point) using a Tecan Infinite M200 Microplate Reader (TECAN, Mannedorf, Switzerland) at 30°C with excitation and emission wavelength of 485 nm and 530 nm, respectively. Signal was normalized to the protein content determined by the BCA assay. To assess catalytic efficiency, the Km of ATG4B on LC3B-PLA2 was determined using Michaelis Menten nonlinear regression fitting of a series of progress curves.
Assessment of cell death and ROS production
Cells were seeded as above, harvested by trypsin, washed by PBS and cell death was quantified using ANXA5/Annexin V-FITC (Exbio, P030-F500) and propidium iodide (PI) (Sigma-Aldrich, P4170) by flow cytometry. If required, the cells were pre-incubated with H2O2 or PEITC (Sigma-Aldrich, 253731) for 22 h. In some cases, 10 - 50 nM bafilomycin A1 for 2 h, or 1 µM FMK-9a or 0.1 µM NSC185058 for 2 h, were applied prior to ROS inducers. Cells positive for ANXA5, PI, or ANXA5 and PI were considered dead. For experiments including Ndi1 (rotenone-insensitive NADH-ubiquinone oxidoreductase) and AOX expressing cells, ANXA5-Dy647 (Exbio, EXB0023) and Hoechst33258 (Sigma-Aldrich, 861405) was used instead (these cells express also GFP and RFP).
For ROS measurements, cells in the basal state or exposed to H2O2 or PEITC for 1 h were incubated for 20 min with 5 μM 2′,7′-dichlordihydrofluorescein diacetate (DCF-DA) (Sigma-Aldrich, D6883) and the levels of ROS were evaluated by flow cytometry. Data are expressed as geometric mean normalized to the control.
Targeted metabolomic analysis
For measurement of intracellular amino acids, cells were rinsed twice with ice-cold 0.9% NaCl (Lach-Ner, 30093-APO), scraped, washed at 15,000 g, 10 min at 4°C and stored at -80°C. Metabolites were extracted with ice-cold 75% acetonitrile (Fluka, 34967-1L) containing internal standards (Chromsystems, 55000) sonicated (10 times for 0.5 s), spun (12,000 g, 10 min at 4°C) and supernatant was filtered using Multiscreen filter plate (Merck, MSRLN0410). The levels of metabolites were estimated by hydrophilic interaction liquid chromatography coupled to Qtrap 5500 mass spectrometer (Sciex, MA, USA) as described [57]. The data were analyzed using Skyline software[58] and processed according to Dunn et al [57,59].
For measurement of intracellular nucleotides and energy charge, cells grown in a 6-well plate for 24 h without uridine and pyruvate supplementation were washed with ice cold 0.9% NaCl solution, scraped into cold extraction buffer (80% LC-MS grade methanol in water) and stored at -80°C. Next, proteins were pelleted by centrifugation at 20,000 g for 15 min at 4°C. Part of the supernatant was transferred to LC-MS vials. The cell pellet was lysed in 200 mM NaOH for 20 min at 95°C. After centrifugation at 1,000 g for 10 min, protein levels were measured by BCA for normalization purposes. Target measurements of nucleotides and energy balance ([ATP + ½ ADP]/[ATP+ADP+AMP]) were performed using a Dionex UltiMate 3000 LC System (Thermo Fisher Scientific, Massachusetts, USA) in-line connected to a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Massachusetts, USA). Ten microliters of the sample extracts (collected in 80% LC-MS grade methanol in water as described above) were separated on an Acquity HSS T3 UPLC column (Waters Corp, 2.1 mm x 150 mm, 1.8 µm particle size) using an Ultimate 3000 HPLC (Dionex, Thermo Fisher Scientific, MA, USA). Column temperature was held at 40°C throughout the analysis. Elution of metabolites was performed using a quaternary solvent system consisting of solvent A (10 mM Tributylamine, 15 mM acetic acid 5% methanol in water) and solvent B (100% methanol). The flow rate is kept constant at 250 µl/min and the following linear gradient is applied: at 0 min 0% B, from 2 to 7 min an increase to 37% B is accomplished; from 7 min to 14 min an increase to 41% B is carried out, from 14 to 26 min the gradient increases to 100% B and is maintained until 30 min. At 31 min the gradient progressively decreases to 0% B and lasts until 40 min. The data was then collected in “centroid data acquisition” mode, with negative electron spray ionization over a mass range of 70 – 1050 m/z and an extra filter of 300-850m850 m/z starting from the 8th min, both at a mass resolution of 70,000 (at m/z 200). The detector was an Orbitrap Qexactive (Thermo Fisher Scientific, MA, USA). We used following source settings: sheath gas flow rate at 50, aux gas flow rate at 10, spray voltage at 4 kV, capillary temperature at 350°C and the S-lens RF level was set at 60. Data processing: mass spectrometry files were converted to the mzXML format using msConvert available from the proteowizard toolkit [60]. Analysis was performed using the Quan software (Thermo Fisher Scientific, Xcalibur version 4.0) and manually verified.
Determination of cell death sensitivity in vivo - TUNEL assay
Tamoxifen-induced mice were treated with a single dose of PEITC (1.25 mg/25 g mouse in corn oil) or vehicle-control on day 10 postinduction. Where indicated, mice were grafted with B16 melanoma cells (1 x 106 in 100 ml PBS) 24 h after the last tamoxifen injection and treated with a single dose of PEITC (1.25 mg/25 g mouse in corn oil) or doxorubicin (0.2 mg/20 g mouse in 0.9% NaCl; Sigma-Aldrich, D1515) on day 10. Where indicated, mice were treated with chloroquine (Sigma-Aldrich, 50-63-5; 1 mg/20 g mouse in corn oil) (on 3 consecutive days, the last dose being co-administered with PEITC (as above). Mice were sacrificed 16 h postapplication and non-malignant as well as tumor tissues were dissected, fixed in 4% FA overnight, dehydrated, embedded in paraffin or in cryo embedding matrix (OCT) (Leica, 14020108926) and sectioned. Paraffin sections (8 μm) were deparaffinized using xylene and decreasing concentration of ethanol (100% - 70%), rehydrated in PBS followed by permeabilization using 20 µg/ml of proteinase K (Sigma-Aldrich, P5568) for 1 h. Fixed frozen sections (5 μm) were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, T8787) 5 min at room temperature. Cell death was detected using the In Situ Cell Death Detection Kit, Fluorescein (Roche, 11684795910) according to the manufacturer’s instructions. The positive and negative control tissue slides were incubated with DNAase (Sigma-Aldrich, D4527-10KU) (for 12 min) and/or TUNEL-label solution (1 h), respectively. Where indicated, TUNEL assay was combined with EdU staining for detection of proliferation. Mice were injected with 50 µg of EdU (Cayman Chemical, 20518) in PBS 4 h prior to the end of the experiment. EdU was detected by a Click reaction using 100 mM copper sulfate (Thermo Fisher Scientific, C10337) 200 mg/ml sodium ascorbate (Sigma-Aldrich, A4034) and 10 µM Alexa Fluor™ 647 Azide, (Invitrogen, A10277) for 45 min at room temperature. Hoechst 33342 (10 µg/ml) was used to visualize cell nuclei. Imaging was performed using the Carl Zeiss LSM880 NLO inverted point scanning microscope. Cell death was quantified as number of TUNEL-positive cells per area of the tissue.
In situ detection of ROS in the endothelium
Tamoxifen induced control and tfam ECKO mice were treated for 4 h with LPS (10 mg/kg in PBS, i.p.) (Sigma-Aldrich, L2630) or vehicle control. Aortas were harvested, opened longitudinally and stained with 20 µM of dihydroethidium (DHE) (Sigma-Aldrich, 37291) in PBS for 30 min in the dark at 37°C, followed by staining with Hoechst 33342 for 15 min and Isolectin B4 (Thermo Fisher Scientific, 132450) at 10 µg/ml for 1 h as previously described [6]. Imaging was performed using a Leica TCS SP8 WLL SMD-FLIM microscope (Leica Microsystems, Wetzlar, Germany) and DHE fluorescence intensity was quantified using Fiji/ImageJ software. Seven images per aorta were analyzed. The images were segmented based on the Hoechst signal and the average intensity of the DHE signal in the nuclear region was calculated using Fiji/ImageJ for each image. Results for each animal were averaged.
Leukocyte infiltration into the lungs
Lungs from LPS-treated mice were dissected and fixed in 4% FA overnight, dehydrated, embedded in paraffin, and sectioned. Immune cells were stained using rat anti-mouse PTPRC/CD45 antibody (Invitrogen, 14-0451-85) followed by an appropriate secondary antibody. Nuclei were counterstained with Hoechst 33342. Imaging was performed using a Carl Zeiss LSM880 NLO inverted point scanning microscope and the PTPRC/CD45-positive area was quantified using Fiji/ImageJ software. The infiltration of leukocytes is expressed as a percentage of PTPRC/CD45-positive region per lung section area. Results for each animal were averaged.
DSS-induced acute colitis model
Tamoxifen induced control and tfam ECKO mice were exposed to 2.5% dextran sulfate sodium (DSS), 36,000-50,000 M. Wt. (MP Biomedicals, ICNA0216011080) in the drinking water for 7 consecutive days. Where indicated mice were co-treated with 15 mg/kg/day of 4-Hydroxy-TEMPO (Sigma-Aldrich, 176141) in PBS or with solvent control administered every 12 h by subcutaneous injections. The disease activity index, which scores body weight loss, stool consistency, and blood in the stool and the anal region, indicates the severity of colitis under DSS treatment (score 0: no weight loss, normal stool consistency, no bleeding; score 1: 1-5% weight loss, normal stool consistency, occult blood; score 2: 5-10% weight loss, loose stool, occult blood; score 3: 10-15% weight loss, loose stool, occult blood; score 4: > 15% weight loss, diarrhea, gross bleeding). On day 7, mice were injected via the tail vein with 0.25 mg dextran-rhodamine, 70,000 kDA (Thermo Fisher Scientific, D1818) and 10 min after injection, mice were perfused and fixed using 1% FA. Colon was harvested and fixed in 4% FA overnight, dehydrated, embedded in paraffin, and sectioned. Colon sections were stained with Hematoxylin/Eosin for morphological analyses, with anti-ENG/CD105 antibody (RD Systems, AF1320) to identify blood vessels and with an anti-PTPRC/CD45 antibody to analyze immune cell infiltration, followed by incubation with an appropriate secondary antibody. Nuclei were counterstained with Hoechst 33342. Imaging was performed using a Carl Zeiss LSM880 NLO inverted point scanning microscope. Fiji/ImageJ software was used to quantify the number of PTPRC/CD45-positive cells per colon area as well as the instances of the extravascular dextran-rhodamine leakages per colon area. Results were averaged per mouse.
Quantification and statistical analysis
Data were analyzed in GraphPad Prism 5.04 software (GraphPad Software) using unpaired Student’s t-test analysis, one-way ANOVA or two-way ANOVA for comparisons of more than two parameters. Data shown are mean values ± S.E.M. of at least 3 independent experiments. A statistical difference of p < 0.05 was considered significant.
Supplementary Material
Acknowledgment
We acknowledge Imaging Methods Core Facility of Faculty of Science, Charles University at BIOCEV and the Electron Microscopy Core Facility of Institute of Molecular Genetics, Czech Academy of Sciences, for assistance with light and electron imaging. We thank the Laboratory of Electron Microscopy, Faculty of Science, Charles University, for the possibility to use the transmission electron microscope JEOL JEM-1011. Nucleotide analysis was performed at the Metabolomics Expertise Center, VIB, Leuven, Belgium. Proteomics LC/MS analysis was done by K. Harant and P. Talacko from Laboratory of Mass Spectrometry, Biocev, Faculty of Science, Charles University. We thank B. Havlinova (Institute of Biotechnology, Czech Academy of Sciences) for help with LC3B-PLA2 fusion protein construction, T. Michalcikova and I. Bukova (Czech Center for Phenogenomics/Institute of Molecular Genetics, Czech Academy of Sciences) for help and advice on MEF isolation and to R. Boon (Goethe University, Frankfurt am Mein, Germany) for providing the Cdh5CreERT2 mice.
Funding Statement
This work was supported by Czech Science Foundation (20-18513S, 17-24441S, 20-05942S, 18-02550S, 19-10354S, 21-04607X, 22-34507S, 18-24753Y), Czech Health Research Council (17-30138A, 17-32727A, NU21-03-00545, NU20J-02-00035), Charles University Grant Agency (1552218, 1506318, 1435320). J.K. is supported by AIAS-CO-FUND II: GA: MSCA: 754513, Lundbeckfonden: R307-2018-3667, Carlsberg Fonden: CF19-0687, Kræftens Bekæmpelse: R302-A17296, A.P. Møller Fonden: 20-L-0317, Riisfort Fonden and Steno Diabetes Center Aarhus (SDCA). P.C. is supported by long-term structural Methusalem funding by the Flemish Government, FWO-Vlaanderen, a Novo Nordisk Foundation Laureate grant, and an ERC Advanced Research Grant (EU-ERC743074). K.R is supported by Marie Skłodowska-Curie Individual Fellowship (101027977) and by EMBO Installation Grant (IG 5068-2022). Infrastructure support was from ERDF and MEYS CR (CZ.02.1.01/0.0/0.0/16_013/0001775, CZ.02.1.01/0.0/0.0/18_046/0016045), ESIF and MEYS CR (CZ.02.1.01/0.0/0.0/18_046/0015861), MEYS CR (LM2018129, LM2015040, LM2018126) and from Czech Academy of Sciences (RVO:86652036, RVO:68378050, RVO:67985823).
Disclosure statement
The authors declare no competing interests.
Data availability
All raw sequencing data are available in Gene Expression Omnibus under accession number GSE146025. Raw proteomics data are available in the Pride database under accession number PXD021376.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- 1.Arrojo EDR, Lev-Ram V, Tyagi S. et al. Age Mosaicism across Multiple Scales in Adult Tissues. Cell Metab. 2019;30(2):343–351 e343. PubMed PMID: 31178361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kops GJ, Dansen TB, Polderman PE. et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–321. PubMed PMID: 12239572 [DOI] [PubMed] [Google Scholar]
- 3.Naderi J, Hung M, Pandey S.. Oxidative stress-induced apoptosis in dividing fibroblasts involves activation of p38 MAP kinase and over-expression of Bax: resistance of quiescent cells to oxidative stress. Apoptosis. 2003;8(1):91–100. PubMed PMID: 12510156 [DOI] [PubMed] [Google Scholar]
- 4.Legesse-Miller A, Raitman I, Haley EM. et al. Quiescent fibroblasts are protected from proteasome inhibition-mediated toxicity. Mol Biol Cell. 2012;23(18):3566–3581. PubMed PMID: 22875985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valentin M, Yang E.. Autophagy is activated, but is not required for the G0 function of BCL-2 or BCL-xL. Cell Cycle. 2008;7(17):2762–2768. PubMed PMID: 18758240 [DOI] [PubMed] [Google Scholar]
- 6.Kalucka J, Bierhansl L, Conchinha NV. et al. Quiescent Endothelial Cells Upregulate Fatty Acid beta-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018;28(6):881–894 e813. PubMed PMID: 30146488 [DOI] [PubMed] [Google Scholar]
- 7.Blecha J, Novais SM, Rohlenova K. et al. Antioxidant defense in quiescent cells determines selectivity of electron transport chain inhibition-induced cell death. Free Radic Biol Med. 2017;112:253–266. PubMed PMID: 28774815 [DOI] [PubMed] [Google Scholar]
- 8.Rohlena J, Dong LF, Kluckova K. et al. Mitochondrially targeted alpha-tocopheryl succinate is antiangiogenic: potential benefit against tumor angiogenesis but caution against wound healing. Antioxid Redox Signal. 2011;15(12):2923–2935. PubMed PMID: 21902599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong LF, Swettenham E, Eliasson J. et al. Vitamin E analogues inhibit angiogenesis by selective induction of apoptosis in proliferating endothelial cells: the role of oxidative stress. Cancer Res. 2007;67(24):11906–11913. PubMed PMID: 18089821 [DOI] [PubMed] [Google Scholar]
- 10.Dikic I, Elazar Z.. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. PubMed PMID: 29618831 [DOI] [PubMed] [Google Scholar]
- 11.Egan DF, Chun MG, Vamos M. et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59(2):285–297. PubMed PMID: 26118643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Kundu M, and Viollet B. et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132. PubMed PMID: 21258367: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scherz-Shouval R, and Elazar Z.. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17(9):422–427. PubMed PMID: 17804237 [DOI] [PubMed] [Google Scholar]
- 14.Santidrian AF, Matsuno-Yagi A, Ritland M. et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123(3):1068–1081. PubMed PMID: 23426180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas HE, Zhang Y, Stefely JA. et al. Mitochondrial Complex I Activity Is Required for Maximal Autophagy. Cell Rep. 2018;24(9):2404–2417 e2408. PubMed PMID: 30157433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. PubMed PMID: 19061483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills EL, Kelly B, Logan A. et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167(2):457–470 e413. PubMed PMID: 27667687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birsoy K, Wang T, Chen WW. et al. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162(3):540–551. PubMed PMID: 26232224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullivan LB, Gui DY, Hosios AM. et al. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015;162(3):552–563. PubMed PMID: 26232225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Titov DV, Cracan V, Goodman RP. et al. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science. 2016;352(6282):231–235. PubMed PMID: 27124460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bajzikova M, Kovarova J, Coelho AR. et al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019;29(2):399–416 e310. PubMed PMID: 30449682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Reyes I, Cardona LR, Kong H. et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature. 2020;585(7824):288–292. PubMed PMID: 32641834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Appleby RD, Porteous WK, Hughes G. et al. Quantitation and origin of the mitochondrial membrane potential in human cells lacking mitochondrial DNA. Eur J Biochem. 1999;262(1):108–116. PubMed PMID: 10231371 [DOI] [PubMed] [Google Scholar]
- 24.Buchet K, Godinot C.. Functional F1-ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA-depleted rho degrees cells. J Biol Chem. 1998;273(36):22983–22989. PubMed PMID: 9722521 [DOI] [PubMed] [Google Scholar]
- 25.Larsson NG, Wang J, Wilhelmsson H. et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18(3):231–236. PubMed PMID: 9500544 [DOI] [PubMed] [Google Scholar]
- 26.Trachootham D, Zhou Y, Zhang H. et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10(3):241–252. PubMed PMID: 16959615 [DOI] [PubMed] [Google Scholar]
- 27.De Bock K, Georgiadou M, Schoors S. et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–663. PubMed PMID: 23911327 [DOI] [PubMed] [Google Scholar]
- 28.Falkenberg KD, Rohlenova K, Luo Y. et al. The metabolic engine of endothelial cells. Nat Metab. 2019;1(10):937–946. PubMed PMID: 32694836 [DOI] [PubMed] [Google Scholar]
- 29.Aragona M, Panciera T, Manfrin A. et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154(5):1047–1059. PubMed PMID: 23954413 [DOI] [PubMed] [Google Scholar]
- 30.Tan AS, Baty JW, Dong LF. et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21(1):81–94. PubMed PMID: 25565207 [DOI] [PubMed] [Google Scholar]
- 31.King MP, Attardi G.. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246(4929):500–503. PubMed PMID: 2814477 [DOI] [PubMed] [Google Scholar]
- 32.Kuma A, Hatano M, Matsui M. et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. PubMed PMID: 15525940 [DOI] [PubMed] [Google Scholar]
- 33.Carroll B, Korolchuk VI, Sarkar S.. Amino acids and autophagy: cross-talk and co-operation to control cellular homeostasis. Amino Acids. 2015;47(10):2065–2088. PubMed PMID: 24965527 [DOI] [PubMed] [Google Scholar]
- 34.Tanida I, Ueno T, Uchiyama Y.. A super-ecliptic, pHluorin-mKate2, tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. PLoS One. 2014;9(10):e110600. PubMed PMID: 25340751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Kundu M, Viollet B. et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. PubMed PMID: 21258367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cardenas C, Lovy A, Silva-Pavez E. et al. Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum-to-mitochondria Ca(2+) transfer for survival. Sci Signal. 2020;13(640):eaay1212. PubMed PMID: 32665411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filomeni G, De Zio D, Cecconi F.. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388. PubMed PMID: 25257172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scherz-Shouval R, Shvets E, Fass E. et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. PubMed PMID: 17347651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Cheng X, Yu L. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109. PubMed PMID: 27357649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Settembre C, Di Malta C, Polito VA. et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. PubMed PMID: 21617040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo QQ, Wang SS, Zhang SS. et al. ATM-CHK2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020;39(10):e103111. PubMed PMID: 32187724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng X, Yang Z, Gu Q. et al. The protease activity of human ATG4B is regulated by reversible oxidative modification. Autophagy. 2020;16(10):1838–1850. PubMed PMID: 31880198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shu CW, Drag M, Bekes M. et al. Synthetic substrates for measuring activity of autophagy proteases: autophagins (Atg4). Autophagy. 2010;6(7):936–947. PubMed PMID: 20818167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qiao S, Dennis M, Song X. et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014. PubMed PMID: 25916556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akin D, Wang SK, Habibzadegah-Tari P. et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10(11):2021–2035. PubMed PMID: 25483883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiu Z, Kuhn B, Aebi J. et al. Discovery of Fluoromethylketone-Based Peptidomimetics as Covalent ATG4B (Autophagin-1) Inhibitors. ACS Med Chem Lett. 2016;7(8):802–806. PubMed PMID: 27563406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Konorev EA, Kotamraju S. et al. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. J Biol Chem. 2004;279(24):25535–25543. PubMed PMID: 15054096 [DOI] [PubMed] [Google Scholar]
- 48.Incalza MA, D’Oria R, Natalicchio A. et al. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. PubMed PMID: 28579545 [DOI] [PubMed] [Google Scholar]
- 49.Kratzer E, Tian Y, Sarich N. et al. Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am J Respir Cell Mol Biol. 2012;47(5):688–697. PubMed PMID: 22842495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Araki Y, Sugihara H, Hattori T.. The free radical scavengers edaravone and tempol suppress experimental dextran sulfate sodium-induced colitis in mice. Int J Mol Med. 2006;17(2):331–334. PubMed PMID: 16391834 [PubMed] [Google Scholar]
- 51.Diebold LP, Gil HJ, Gao P. et al. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab. 2019;1(1):158–171. PubMed PMID: 31106291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Planavsky NJ, Reinhard CT, Wang X. et al. Earth history. Low mid-Proterozoic atmospheric oxygen levels and the delayed rise of animals. Science. 2014;346(6209):635–638. PubMed PMID: 25359975 [DOI] [PubMed] [Google Scholar]
- 53.Hamanaka RB, Glasauer A, Hoover P. et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci Signal. 2013;6(261):ra8. PubMed PMID: 23386745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pitulescu ME, Schmidt I, Benedito R. et al. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010;5(9):1518–1534. PubMed PMID: 20725067 [DOI] [PubMed] [Google Scholar]
- 55.Lortz S, Gurgul-Convey E, Naujok O. et al. Overexpression of the antioxidant enzyme catalase does not interfere with the glucose responsiveness of insulin-secreting INS-1E cells and rat islets. Diabetologia. 2013;56(4):774–782. PubMed PMID: 23306382 [DOI] [PubMed] [Google Scholar]
- 56.Barinka C, Ptacek J, Richter A. et al. Selection and characterization of Anticalins targeting human prostate-specific membrane antigen (PSMA). Protein Eng Des Sel. 2016;29(3):105–115. PubMed PMID: 26802163 [DOI] [PubMed] [Google Scholar]
- 57.Yuan M, Breitkopf SB, Yang X. et al. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc. 2012;7(5):872–881. PubMed PMID: 22498707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MacLean B, Tomazela DM, Shulman N. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–968. PubMed PMID: 20147306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dunn WB, Broadhurst D, Begley P. et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6(7):1060–1083. PubMed PMID: 21720319 [DOI] [PubMed] [Google Scholar]
- 60.Chambers MC, Maclean B, Burke R. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30(10):918–920. PubMed PMID: 23051804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data are available in Gene Expression Omnibus under accession number GSE146025. Raw proteomics data are available in the Pride database under accession number PXD021376.