

Abstract
Diagnosing salivary gland tumors (SGTs) through fine‐needle aspiration (FNA) biopsies is challenging due to the overlapping cytomorphologic features between benign and malignant tumors. The authors developed an innovative, multiplexed cycling technology for the rapid analyses of single cells obtained from FNA that can facilitate the molecular analyses and diagnosis of SGTs. Antibodies against 29 protein markers associated with 7 SGT subtypes were validated and chemically modified via custom linker–bio‐orthogonal probes (FAST). Single‐cell homogenates and FNA samples were profiled by FAST cyclic imaging and computational analysis. A prediction model was generated using a training set of 151,926 cells from primary SGTs (N = 26) and validated on a separate cohort (N = 30). Companion biomarker testing, such as neurotrophic tyrosine receptor kinase (NTRK), was also assessed with the FAST technology. The FAST molecular diagnostic assay was able to distinguish between benign and malignant SGTs with an accuracy of 0.86 for single‐cell homogenate samples and 0.88 for FNA samples. Profiling of multiple markers as compared to a single marker increased the diagnostic accuracy (0.82 as compared to 0.65‐0.74, respectively), independent of the cell number sampled. NTRK expression was also assessed by the FAST assay, highlighting the potential therapeutic application of this technology. Application of the novel multiplexed single‐cell technology facilitates rapid biomarker testing from FNA samples at low cost. The customizable and modular FAST‐FNA approach has relevance to multiple pathologies and organ systems where cytologic samples are often scarce and/or indeterminate resulting in improved diagnostic workflows and timely therapeutic clinical decision‐making.
Keywords: biomarkers, fine‐needle biopsy, molecular diagnostic, salivary gland tumor, single‐cell analysis
Short abstract
This study reports a novel diagnostic assay based on a modular multiplexed cycling technology, which enables rapid molecular profiling of salivary gland tumors at single‐cell level.
INTRODUCTION
A fine‐needle aspiration (FNA) biopsy is often an integral first step in the workflow of any cancer workup. However, cytomorphology alone or in combination with a select number of immunocytology markers can often lead to an indeterminate result. Thus, one of the key clinical diagnostic challenges is the ability to perform deep cellular analyses when cytologic samples are limited, such as those with scant cellularity. Current single‐cell analysis techniques are not compatible with clinical workflows due to the prohibitive costs and the labor‐intensive infrastructure required, including bioinformatics support. To address this unmet clinical need, we have developed a same‐day, inexpensive, multiplex imaging approach for deep cellular phenotyping of single cells obtained from FNA biopsies by harnessing advances in chemistry, bioengineering, and computational analyses. Although this approach has been demonstrated to be successful for immune cell profiling of the tumor immune microenvironment, 1 we set out to expand its application to the molecular diagnosis of cancer cell subtypes with a proof‐of‐principle focus on salivary gland tumors (SGTs).
Primary tumors of the parotid and submandibular glands occur at a rate of approximately 1 per 100,000 adults each year resulting in 3200 incident cases per year in the United States. Approximately one‐third of these tumors are malignant whereas two‐thirds are benign. The management of these tumors can be associated with a high morbidity, given the location of the tumors within the parotid or submandibular glands, which places the facial nerve at high risk for tumor involvement and/or injury during surgery. Although tissue‐sparing procedures are favored for the treatment of benign tumors, more aggressive surgical resections, with wider margins and/or sacrifice of tumor‐infiltrated facial nerve branches, and possible neck dissection, are favored for malignancies. Thus, an accurate preoperative diagnosis of SGT is important in establishing both a surgical indication and for surgical planning.
Most SGTs are initially assessed by radiographic imaging followed by FNA for cytopathologic evaluation. Primary epithelial SGTs are classified into 31 tumor types. 2 Given the overlapping cytologic features among many benign and malignant SGTs (including those with low‐grade cytology using chromogenic stains), many FNA samples are indeterminate and, subsequently, limit preoperative surgical planning. 3 , 4 , 5 , 6 Subtype‐specific fusion genes have now been identified as oncogenic drivers in a majority of SGTs, 7 and the detection of the fusion proteins by immunohistochemistry (IHC) can help inform the classification of the SGT. Given the challenges with obtaining a cytopathologic diagnosis using FNA samples in combination with the presence of diagnostic fusion gene alterations, SGTs serve as a model system to evaluate the feasibility and accuracy of an innovative multiplexed molecular diagnostic imaging assay using pauci‐cellular cytologic samples.
We have recently shown that a bioorthogonal, multiplexed cycling technology (FAST) can be used to characterize the tumor immune microenvironment of pauci‐cellular FNA specimens (FAST‐FNA) 1 , 8 . The goal of the current study was to adapt the rapid chemical cycling technology to the deep cellular analysis of single tumor cells to establish the feasibility of obtaining an accurate diagnosis of complex cancer subtypes based on FNA sampling alone. To achieve this goal, we generated and validated over 30 FAST antibody conjugates tailored to the detection of SGT subtype‐specific molecular markers and performed clinical proof‐of‐principle analyses in patient samples. Each antibody was custom modified with a unique linker‐fluorochrome conjugate that allowed quenching after imaging and then re‐staining with new antibody sets. This workflow allowed for the quantitation of ~30 markers per single cell. In this study, we demonstrate that the minimally invasive molecular diagnostic FAST‐FNA assay enables the accurate diagnosis of primary SGTs from FNA sampling for the first time.
RESULTS
Rapid Cyclic and Quantitative Imaging of Multiple Biomarkers Is Feasible in Single‐Cell Samples of SGTs
Conventional processing of SGT FNA specimens involves staining with chromogens (Papanicolaou, Diff‐Quik, or hematoxylin‐eosin) and specialty histochemical stains (eg, mucicarmine). In some cases, when sufficient cellular material is present, a limited number of immunostains can be performed according to a list of markers associated with different SGT (Supporting Tables 1 and 2). The FAST‐FNA approach of SGT differs in that harvested single cells are repeatedly stained with fluorescently labeled antibodies, quenching the fluorescence after each image acquisition cycle. This is possible by designing custom linkers containing a trans‐cyclooctene (TCO) between an antibody of interest and a given fluorochrome (Fig. 1). The TCO acts as a specific site to which IF tetrazine (Tz)‐conjugated black hole quencher (BHQ) can be clicked during the quenching step. The click chemistry between the TCO and the Tz is exceptionally fast, namely with rate constants up to 30,000 M−1 s−1, which we discovered to be further accelerated (≥3300‐fold) by the FAST architecture, enabling >99% quenching in <10 seconds (Supporting Figure S2). 8 The FAST‐FNA workflow was devised through the careful design and validation of imaging conditions for maintaining the integrity of single cells. This approach enables the assessment of the binding of ~30 different antibodies against SGT biomarkers (Supporting Table 1) during 10 cycles of staining and fluorescent image capture, which can be completed in 1 day (Fig. 1C). The antibody panel was based on prior reports of molecular marker expression in different SGT histologies (Supporting Table 2). To validate each individual custom labeled antibody conjugate, we tested staining in cell lines (Supporting Table 2) and confirmed the positive correlation with flow cytometry analysis and immunohistochemistry. As in previous studies for immune cell profiling, 1 FAST‐antibodies for subsequent analyses were approved when i) these correlations were > 0.9, and ii) the cellular staining patterns matched with IHC or immunofluorescence on formalin‐fixed paraffin‐embedded (FFPE) tissues (Supporting Figures 3 and 4). In brief, protocols similar to those established for flow cytometry 9 or clinical IHC testing 10 were used.
Figure 1.
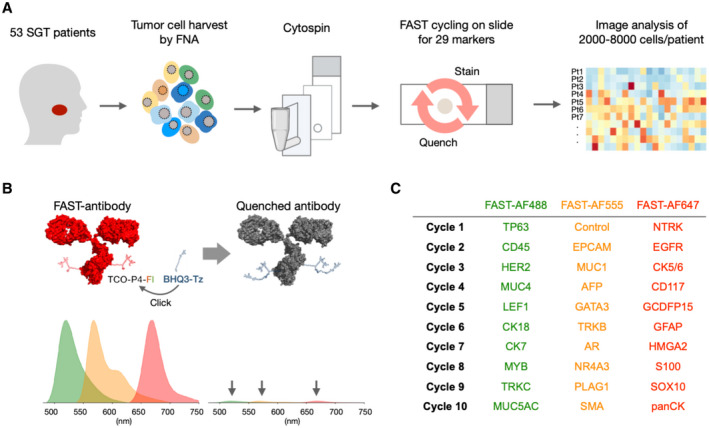
Overview of the FAST cyclic imaging method for rapid diagnostic analysis of salivary gland tumors (SGTs). (A) A total of 53 patients with salivary gland masses were enrolled. Cells harvested by ex vivo FNA were attached to glass slides by cytospin (see Supporting Fig. 1). Twenty‐nine antibodies validated against specific SGT markers were used to analyze the specimens at the single‐cell level. (B) FAST staining relies on a unique fluorochrome‐antibody linker containing a transcyclooctene (TCO), so that the fluorescence signal can be quenched within seconds after each staining cycle by the click reaction of TCO and tetrazine (Tz) attached to BHQ3 quencher. (C) Summary of biomarker assignment during the 10 cycles of FAST imaging.
Analysis of SGT Subtype‐Specific Biomarkers by FAST‐FNA
Figure 2A shows representative images of SGT specimens (pleomorphic adenoma [PA], mucoepidermoid carcinoma [MuEC], salivary duct carcinoma [SDC], and adenoid cystic carcinoma [AdCC]) stained with FAST‐labeled antibodies against subtype‐specific markers. We confirmed the expression of putative markers in different SGT subtypes: HMGA2 and GFAP in PA; p63, Muc1, Muc4, and Muc5ac in MuEC; AR, GATA3, and EpCAM in SDC; and CD117 and Myb in AdCC, for example. The fluorescence intensities measured in individual cells were used for determining the threshold for the positivity of each marker. After normalizing the peak intensity of negative populations of each biomarker, the threshold for calculating the positivity was set at the full width at half maximum (FWHM) away from the peak of the negative population in the aggregated intensity histogram of the first 15 samples (Supporting Fig. 5). Using these threshold values, the percentage of cells positive for each biomarker was then analyzed for the rest of samples. FAST‐FNA analysis showed high expression levels of putative biomarkers in the single cell samples (Fig. 2B). In MuECs, Muc1 was present in 100%, Muc5ac in 100%, Muc4 in 50%, and p63 in 50% of cases. In SDC, both GATA3 and AR were present in 100% of cases. We also stained all cell samples for other tumor markers. In malignant SGT cases, panCK and EPCAM were present in 93%, CK5/6 in 86%, CK7 in 79%, and CK18 in 71%, and the other markers at lower percentages. Notably, the same markers were much less common (ie, were not a “predominate stain”) in benign lesions. For example, in PA samples, panCK was present in 64%, EPCAM in 32%, CK5/6 in 31%, CK7 in 34%, and CK18 in 13% of cells on average.
Figure 2.
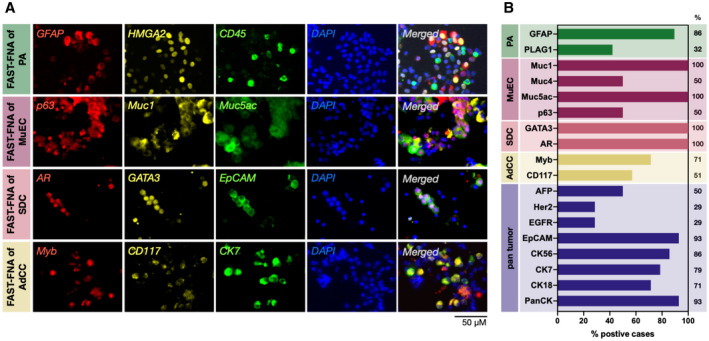
Representative examples of FAST‐fine‐needle aspiration (FNA) analysis. (A) Single‐cell staining with a panel of salivary gland tumor (SGT)‐relevant antibodies (pleomorphic adenoma [PA], mucoepidermoid carcinoma [MuEC], salivary duct carcinoma [SDC], adenoid cystic carcinoma [AdCC]). Antibodies were conjugated to FAST probes with one of the following fluorophores: Alexa fluor 488, Alexa fluor 555, or Alexa fluor 647. (B) FAST‐FNA analysis of SGT subtypes (PA, n = 19; MuEC, n = 3; SDC, n = 4; AdCC, n = 6; all malignant tumors n = 24) showing high expression levels of different biomarkers of each SGT subtype. Sample positivity was determined if more than 20% of tumor cells expressed the respective marker. The percentages refer to the number of patients in whom a given biomarker was positive.
Figure 5.
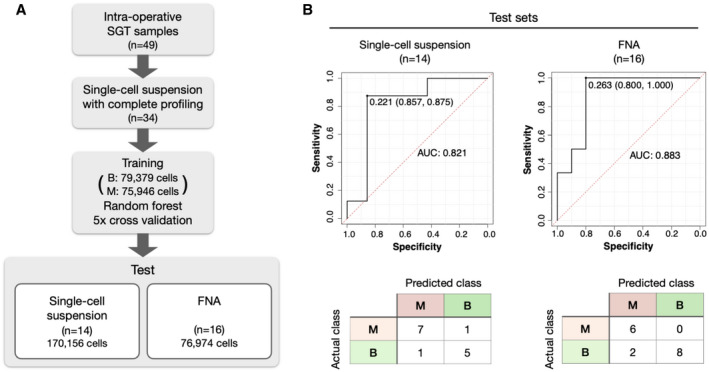
Prediction model for identifying malignant salivary gland tumor (SGT) cases based on the FAST‐fine‐needle aspiration (FNA) analysis. (A) A random forest classifier was trained on over 150,000 cells to predict the malignancy of SGT cases. Out of 34 samples whose expression level of all 29 markers were analyzed by FAST imaging, 20 single‐cell suspension samples, including 79,379 cells from 13 benign cases (B; all pleomorphic adenoma [PA]), and 75,946 cells from 7 malignant cases (M; 2 adenoid cystic carcinoma [AdCC], 1 salivary duct carcinoma, 1 mucoepidermoid carcinoma [MuEC], 1 myoepithelial carcinoma, 1 lympho‐epithelial carcinoma, and 1 acinic cell carcinoma) were used for the training. The remaining 14 samples were then used as a test cohort. The test cohort included 6 PA, 5 AdCC, 2 SDC, and 1 MuEC. The single‐cell suspension samples in the test cohort included 81,404 cells from benign cases and 88,752 cells from malignant cases, 170,156 cells in total. FNA samples (n = 16) were used as another test cohort, with 46,568 cells from benign cases and 30,406 cells from malignancy cases, 76,974 cells in total. (B) Receiver operating characteristic (ROC) curves and confusion matrix show the diagnostic accuracy of the prediction model. In each ROC curve, the cutoff value for determining a sample’s malignancy (0.221 for single‐cell suspension, 0.263 for FNA samples) is shown under the curve, with corresponding specificity and sensitivity in parentheses. The accuracy was 0.86 for single‐cell suspension samples (n = 14) and 0.88 for FNA samples (n = 16). AUC indicates area under curve.
Quantitative 30‐Plex Biomarker Analysis of SGT
Routine clinical FNA obtained by manual palpation or image guidance are often indeterminate for SGTs because of i) insufficient material for ancillary studies, ii) overlapping cytologic features between benign and low‐grade malignancies assessed by Papanicolaou or Diff‐Quik staining, 3 , 4 , 5 , 6 and iii) tumor heterogeneity. A hypothesis in our study is that multiplexed single‐cell profiling of FNA samples can improve the diagnostic accuracy of FNA sampling of SGTs. To test this hypothesis, we set out to map the comprehensive biomarker expression across the remaining cohort of SGT samples. Out of 53 SGT cases profiled, 52 cases had sufficient numbers of cells for analysis (ranging from 527 to 45,905 cells per patient specimen). All samples were stained with the same panel of diagnostic or cell–type‐specific protein antibodies, and the positive signals in individual cells were captured and quantified through rapid cycling of immunofluorescence. 4′,6‐Diamidino‐2‐phenylindole (DAPI) was used for alignment of fields of view obtained during the repeated cycles. Manual or automated image analysis was then used to quantify typical cell characteristics such as cell size, nuclear size, and fluorescence intensity in individual cells.
To assess whether the analyzed cell number per sample influenced diagnostic accuracy, we compared the positive fraction of putative markers in 19 cases of PA, which had cell counts ranging from 527 to 10,712. Figure 3A summarizes the fraction of HMGA2, GFAP, and PLAG1‐positive cells per specimen and the total cell counts of each specimen. Regardless of the number of cells stained and analyzed, the expression of one or multiple PA markers was detected in the PA specimens. There was no correlation or association between biomarker positivity and cell numbers in a given sample (Fig. 3A; HMGA2: R2 = 0.0004, P = .79; GFAP: R2 = 0.0003, P = .84; PLAG1: R2 = 0.003, P = .82), suggesting that an accurate diagnosis is independent of the cell number sampled and is possible with even a pauci‐cellular sample.
Figure 3.
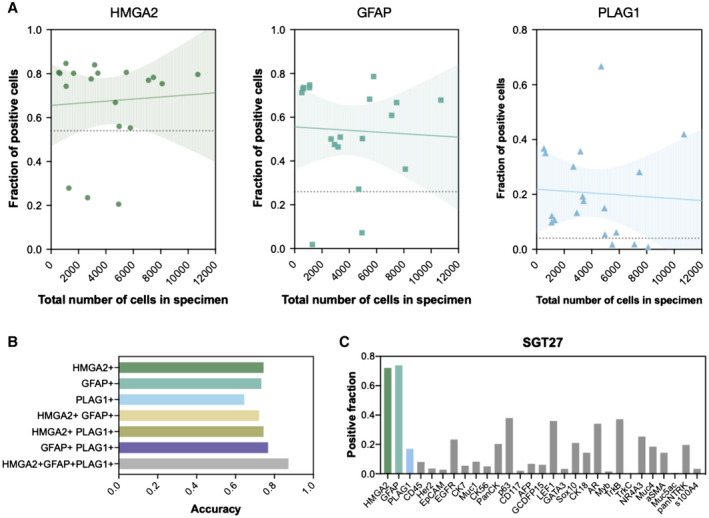
Biomarkers for identification of pleomorphic adenoma. (A) The correlation between biomarker positivity and harvested cell number revealed that the 3 biomarkers for pleomorphic adenoma (PA) (HMGA2, GFAP, and PLAG1) were detected regardless of the total cell count, indicating that the sample size of a sample does not affect the diagnosis of PA within the tested range of 527 to 10,712 cells. Solid lines show fitted linear model with 95% confidence interval in the shaded area (HMGA2: R2 = 0.0004, P = .79; GFAP: R2 = 0.0003, P = .84; PLAG1: R2 = 0.003, P = .82). Dashed lines indicate the cutoff level determined as the fraction of positive cells that maximizes the sum of sensitivity and specificity of diagnoses based on the expression level of each biomarker. (B) Accuracy of PA diagnosis using single markers or combination of markers was compared. Expression levels of each of the 3 markers were quantified on both PA samples and non‐PA samples for calculation of the diagnostic accuracy (see Supporting Fig. 6 for supporting information). Diagnosis based on all 3 markers (HMGA2, GFAP, or PLAG1) showed the highest accuracy (0.82), as compared to those based on single markers (HMGA2: 0.74, GFAP: 0.73, PLAG1: 0.65) or different combinations (HMGA2/GFAP: 0.72, HMGA2/PLAG1: 0.74, GFAP/PLAG1: 0.77). (C) Multiplexed FAST‐fine‐needle aspiration analysis of a PA specimen shows the expression profile of 29 biomarkers including HMGA2, GFAP, and PLAG1. Interestingly, a fraction of cells were also positive for AR, TrkB, NR4A3, and panNTRK antibodies, arguing that no single marker is exclusively specific for a tumor type. The fraction of cells with fluorescent intensity above the intensity threshold was calculated as described in Supporting Figure 6.
Next, the accuracy of PA diagnosis using single markers or combination of PA‐specific markers was compared based on the positive fractions. The cutoff value was determined based on the receiver operating characteristic (ROC) such that the sum of sensitivity and specificity is maximized for a PA diagnosis (Supporting Fig. 6). The diagnostic accuracy was calculated as the ratio of correctly identified specimen to total PA specimens. Diagnosis based on all 3 markers (HMGA2, GFAP, and PLAG1) showed the highest accuracy (0.82), as compared to those based on single markers (HMGA2: 0.74, GFAP: 0.73, PLAG1: 0.65) or different marker combinations (HMGA2/GFAP: 0.72, HMGA2, PLAG1: 0.74, GFAP, PLAG1: 0.77) (Fig. 3B).
Figure 6.
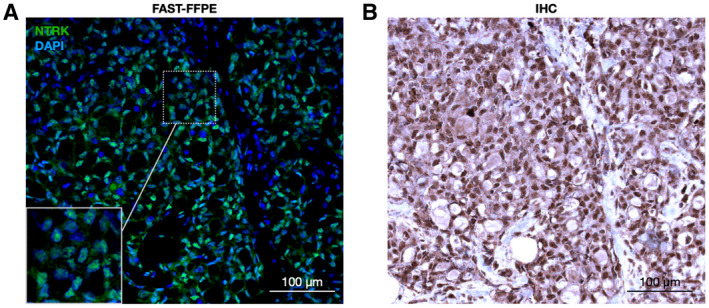
Neurotrophic tyrosine receptor kinase (NTRK) detection in secretory carcinoma by FAST‐formalin‐fixed paraffin‐embedded (FFPE). In this example of an NTRK fusion‐positive case, a FAST antibody against NTRK shows strong nuclear signal (green, NTRK; blue, 4′,6‐diamidino‐2‐phenylindole; A) similar to immunohistochemistry (brown, NTRK; blue, hematoxylin; B) in adjacent FFPE tissue sections of a secretory carcinoma case. Scale bar = 100 μm.
Prediction Model for Diagnosing Tumor Malignancy
One of the key clinical interests is to be able to differentiate benign and malignant SGT based on FNA. To assess this, we first applied uniform manifold approximation and projection (UMAP) to visualize marker expression levels in a given sample. Figure 4A shows a UMAP of 34 different patient samples that cluster into benign and malignant categories as annotated in green (benign) and magenta (malignant). Figure 4B shows a number of specific samples superimposed onto the entire UMAP collection. Interestingly, the malignant cases of the same subtypes clustered closely (eg, SDC and AdCC), whereas those of different subtypes were distant from each other. This indicates that not only benign and malignant cases but also different subtypes of SGT can be distinguished based on their protein expression profile.
Figure 4.
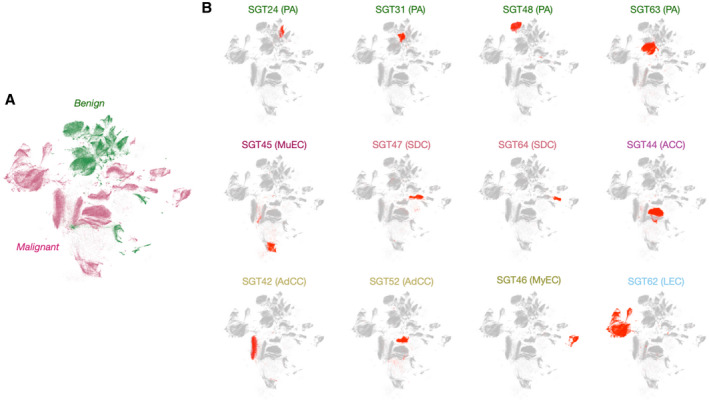
Uniform manifold approximation and projection (UMAP) clustering of salivary gland tumor (SGT) specimens. (A) UMAP analysis revealed that the benign SGT specimens cluster tightly among themselves. Clusters in green represent benign populations and clusters in pink represent malignant populations. (B) Twelve representative cases including pleomorphic adenoma (PA), mucoepidermoid carcinoma (MuEC), acinic cell carcinoma (ACC), adenoid cystic carcinoma (AdCC), salivary duct carcinoma (SDC), myoepithelial carcinoma (MyEC), and lympho‐epithelial carcinoma (LEC) were superimposed on the UMAP of 34 primary SGT samples whose expression level of all 29 markers were analyzed (gray).
In an effort to enable rapid malignancy prediction of SGT samples, computational models were then developed and tested (Fig. 5). We obtained single‐cell suspensions from 26 intraoperative SGT samples and trained a random forest classifier on 151,926 of these cells. The trained classifier was then tested on a separate set of single‐cell suspensions (n = 14) or FNA (n = 16). ROC analyses were then performed on both test sets. Our results show that the classifier performed well in both the FNA and single‐cell analysis cohorts, with accuracy of 0.86 for single‐cell suspension samples and 0.88 for FNA samples. In the FNA samples, the algorithm correctly classified all malignant cases. Notably, the method was able to correctly diagnose malignancy in 7 of 8 SGT cases that had been nondiagnostic using conventional processing.
Selective Biomarker Analysis Informs the Presence of Drug Targets
Tropomyosin receptor kinases (TRK) kinases play a critical role in cell proliferation, survival, and differentiation through the Ras/mitogen‐activated protein kinase (MAPK), the phosphatidylinositol 3‐kinase (PI3K) and phospholipase C (PLC‐γ) pathways. 11 The National Comprehensive Cancer Network recommends the use of TRK inhibitors (larotrectinib, entrectinib, and repotrectinib) 12 in patients with neurotrophic tyrosine receptor kinase (NTRK) fusion‐positive tumors who fulfill certain clinical criteria. Although next‐generation sequencing (NGS) is the diagnostic test of choice to define the specific type of fusions, the test has a long turnaround time and high cost, both of which can delay treatment. Conversely, pan‐TRK protein expression analysis is widely available in clinical laboratories for FFPE. To adapt this to FAST analysis in cells or FFPE tissue sections, we conjugated the TCO linker to a pan‐TRK monoclonal antibody for analysis. 13 Figure 6 shows a prototypical example of a proven NTRK‐fusion sample from a patient with secretory carcinoma (SC). The pan‐TRK staining was predominantly nuclear as would be expected and the results were available the same day. NTRK presence in FNA or tissue sections can guide therapeutic decision‐making to give TRK inhibitors (larotrectinib and entrectinib), which have been Food and Drug Administration (FDA)‐approved for the treatment of patients with SC with NTRK fusion.
DISCUSSION
Routine FNA sampling of solid tumors from any anatomic site can allow for the broad categorization of benign and malignant categories but the cellular samples are often not sufficient for additional contextual biomarker assessment. When sufficient cellular material is present, cell blocks can be prepared for a limited number of single channel IHC stains. These challenges can limit the current utility of FNA sampling in establishing a surgical indication as well as preoperative surgical planning. To overcome the limitations of capturing a limited number of cells in FNA, the field has explored the use of gene expression analysis from FNA, such as the Afirma test for thyroid nodules, to classify cytologically indeterminate nodules as either benign or suspicious to assist with clinical decision‐making. However, gene expression profiling in FNA is limited in distinguishing whether the gene expression profile is derived from the tumor or immune cells that restrains its use in settings of high immune cell infiltration. 14 Analogous gene expression profiling tests for many tumor types, including SGTs, are currently not available.
Here, we demonstrate that an innovative FAST‐FNA technology allows for high dimensional cellular profiling of tumor cells sampled from FNA that can result in an accurate tumor diagnosis within a few hours of cellular sampling. These diagnostic capabilities are comparable to those of histopathologic and IHC analysis of surgically removed whole tissue but resulted in a shorter turn around and with lower morbidity given its minimally invasive approach. The multiplex single‐cell assessment is based on interdisciplinary methodologies involving new bioorthogonal chemistry, cyclic imaging, and data analysis.
Cycling Methods Allow Deep Cellular Profiling
Most antibody‐based cycling methods 15 , 16 , 17 were originally developed for paraffin‐embedded tissue sections that have the architectural resilience to withstand harsh destaining conditions. Unfortunately, these harsh conditions use oxidants for bleaching and extremes of pH and are not compatible with cellular, scant samples such as FNA. We have developed gentler DNA barcoded antibody technologies for cellular profiling such as ABCD 18 , 19 and SCANT. 20 Although technically feasible, these early methods suffered from moderate signal‐to‐noise ratio (SNR), complex blocking regimens to reduce background signal, and long destaining times comparable to other cycling techniques, bringing challenges when translating the technology into the clinical setting. A more recent complementary technology is the FAST approach 8 that bypasses the above shortcomings (cost, limited SNR, and sensitivity) and allows extremely fast cycling in the seconds time frame while still being gentle on cells. Chemically, the FAST method takes advantage of a hyper‐accelerated click reaction between black hole quencher Tz and TCO‐linked fluorophores, achieving >99% quenching in <10 seconds. This eliminates the requirement to destroy, rinse, or remove the previous cycle stain. Here, we show that the unique FAST linkers can be successfully attached to diagnostic antibodies relevant for SGT subtype analyses and that the resulting reagents perform exceptionally well. Specifically, we show that it is possible to perform 10 cycles of rapid antibody labeling and image capture and analyses without cellular loss and in a reasonable amount of time.
FAST‐FNA of SGT
The majority of SGT express unique protein biomarkers that can complement histological staining to obtain a definitive pathologic diagnosis. In the clinical arena, this is typically done after surgical resection of the SGT in the form of immunohistochemical staining of FFPE whole tissue sections and less commonly in FNA samples due to scant cellularity. This clinical workflow limits surgical preoperative planning. Therefore, we assessed both the feasibility and accuracy of diagnosing SGT subtypes through the FAST‐FNA technology. First, we identified and attributed various cellular markers to each histologic SGT subtype and validated the specificity of the combinatorial cellular markers via conventional IHC staining on whole tissue sections. We determined there was a good correlation between the immunostaining and histopathologic diagnosis (Figs. 2 and 3), resulting in a defined panel of SGT‐specific markers (Supporting Table 1). One of the important findings was the fact that molecular profiling of SGTs enabled the correct diagnosis even in samples with scant cellularity, such as those with as few as hundreds of cells that is a typical yield from FNA sampling (Fig. 3). Furthermore, because this assay is able to assess for the presence of fusion proteins, there is a natural extension of the utility of the FAST‐FNA SGT assay to guide patient eligibility for targeted therapies. NTRK expression was of particular interest not only because of its diagnostic but also therapeutic implications. To explore this possibility, we evaluated NTRK expression in SC of the salivary gland using the FAST‐FNA antibody probes. NTRK activation can result from various genomic NTRK alterations, including mutations, splice variants, copy number variations, and fusions, the latter being the most common mechanisms for TRK activation. 21 NTRK activation is most commonly diagnosed by IHC when ≥1% tumor cells show either membranous, cytoplasmic, or nuclear staining above background. 13 Diagnosis of NTRK activation has emerged as clinically significant because of the recent FDA approval of NTRK inhibitors. As shown in Figure 6, we were able to identify nuclear NTRK in a SC of the salivary gland. Although not performed in this study, it may be possible to even monitor drug response to NTRK inhibitors (eg, entrectinib and larotrectinib) by serial FNA sampling, similarly as has been done for other kinase inhibitors. 20
Computational Methods Enable Prediction of the Malignancy of SGT
The ultimate clinical goal of multiplexed FAST‐FNA analysis is to provide rapid and accurate answers that can inform clinical decision‐making. To achieve this goal, computational approaches are necessary to streamline data analysis and decision‐making. We obtained comprehensive SGT marker expression profile from single‐cell suspensions from 34 primary SGTs of varying subtypes and trained a machine learning classifier on over 150,000 cells from 20 training cohort. The classifier was then tested on separate cohorts, showing good accuracy. Although these studies were performed as proof‐of principle, we anticipate that training on larger subsets of diverse tumor subtypes will further improve diagnostic accuracy. These computational analyses are readily amenable to automation and broadly applicable to a number of different clinical contexts. 1 , 22
Opportunities for Future Improvement
The goal of the current study was to show proof‐of‐principle of being able to profile the tumor phenotype through FNA analysis. To accomplish this goal we initially focused on the synthesis of new antibody conjugates, validation against accepted gold standards, automating image analysis and developing diagnostic algorithms. Cytology and expert morphometric analyses will likely continue to play an important role in future analyses. In practice, we would envision using the FAST‐FNA approach as a pre‐screen. Our original goal was to use biomarker positivity to see if automated analyses could be performed as a prescreen. It is interesting that correct diagnoses were ultimately established in >90% cases, presumably because of the deeper profiling of harvested cells. Future clinical studies will have to be designed to compare diagnostic accuracies across methods either alone or combined.
There remain several opportunities for future improvements. First, one could extend the molecular profiling with an even higher number of cycles without significant loss of cellular integrity or increase the number of nonoverlapping fluorochromes imaged to obtain comprehensive tumor and/or immune cell phenotypes and functional states. Using both approaches, we estimate that up to 60 to 100 markers could potentially be identified within single cells. Future research is required to determine the optimal number of biomarkers needed for routine clinical diagnostics. Expanded biomarker panels will have advantages in that one could perform more detailed differential diagnosis, assess the tumor immune cell environment (that is relevant for the increasing application of immuno‐oncology), 1 and stain for therapeutically important pathway markers, 20 all from a simple FNA procedure. Second, sample processing could be automated to minimize manual workloads and preserve valuable reagents. This could be achieved with microfluidics and other point‐of‐care approaches. Third, as discussed above, improvements in the artificial intelligence algorithm and training of larger data sets could be performed to improve the accuracy of testing. Fourth, the diagnostic capabilities and algorithm can be further validated with future prospective clinical trials with primary FNA samples obtained by image (eg, ultrasound) or manual guidance. Fifth, FAST‐FNA has the potential to be cost‐effective and inexpensive. The amount of antibodies required is substantially less than for tissue staining. We estimate that a 30‐biomarker panel currently costs ~$30 per patient. This could be further reduced by further optimizing reagent production and use of bulk supplies. This cost compares favorably to the Afirma test that costs approximately $3000 to $3600 per FNA sample.
In summary, the results of our study show that an accurate diagnosis of SGT subtype can be obtained on FNA‐based molecular diagnostics alone, using a panel of ~30 biomarkers assessed on a few thousand single cells. The FAST‐FNA SGT panel can not only differentiate SGTs into benign and malignant tumors but also can provide the histologic subtype diagnoses. Furthermore, given the modular platform of the FAST‐FNA antibody conjugates, unique companion biomarkers can be rapidly incorporated into any FAST‐FNA panel to inform eligibility for targeted therapies. Finally, the current approach could be extended to other pathologies and organ systems where cytologic samples are often scarce and/or indeterminate to improve diagnostic workflows with the application of multiplex single‐cell technologies tailored for minimally invasive sampling approaches.
MATERIALS AND METHODS
Overall Design
The overall goal of the study was to determine whether deep multiplexing of cellular biomarkers could allow the correct diagnosis of different salivary gland tumors in short periods of time. Secondary goals were to determine whether the fraction of “nondiagnostic” samples could be reduced and whether biomarker analysis could be used for therapeutic decision‐making (eg, NTRK inhibitors). To achieve the above, we first identified the cellular markers of various SGT subtypes and find appropriate antibodies. We decided to focus on the most common SGT types (Table 1) and identify markers that have been previously reported to be expressed in specific SGT subtypes (Supporting Table 2). We then obtained multiple antibody clones from vendors in carrier‐free solution so that the antibodies could be modified with FAST probes by amine‐reactive chemical groups (Supporting Figure 2). The FAST‐conjugated antibodies were then tested in cell lines (Supporting Table 2). Immunoglobulin G (IgG) isotypes were used for negative controls. Once we had identified the ideal FAST‐antibody conjugate for a given molecular target, we performed additional immunohistochemistry to validate that the FAST‐labeled antibodies could appropriately detect molecular markers compared to gold standard histology (Supporting Figs. 3, 4, and 8 and Fig. 6 for NTRK). Once appropriately validated, we used the FAST‐labeled antibody panel to develop a single‐cell analysis pipeline that could ultimately be used for FNA. This proof‐of‐principle study, however, was done with surgical specimen, so that we would have a gold standard for direct comparison. With these tools in hand, we proceeded to analyze multiple SGT subtypes (n = 53) (Figs. 2, 4, and 5 and Table 1). The overall study design is summarized in Supporting Figure 9. The sections below detail the different steps.
TABLE 1.
Clinical Demographic Characteristics of SGT Cases
| Tumor Type | Malignancy | Patients | M/F | Mean Age (Range) |
|---|---|---|---|---|
| Pleomorphic adenoma | Benign | 29 | 11/17 | 55 (22‐79) |
| Adenoid cystic carcinoma | Malignant | 7 | 3/4 | 64 (24‐77) |
| Salivary duct carcinoma | Malignant | 4 | 3/1 | 54 (24‐64) |
| Secretory carcinoma | Malignant | 3 | 2/1 | 61 (49‐72) |
| Mucoepidermoid carcinoma | Malignant | 3 | 1/2 | 40 (27‐54) |
| Myoepithelial carcinoma | Malignant | 3 | 1/1 | 62 (55‐69) |
| Lymphoepithelial carcinoma | Malignant | 2 | 0/2 | 75 (66‐83) |
| Acinic cell carcinoma | Malignant | 1 | 1/0 | 76 |
| Intraductal carcinoma | Malignant | 1 | 0/1 | 63 |
| Total | 53 | 23/30 |
Abbreviations: F, female; M, male; SGT, salivary gland tumor.
SGT Tissue Specimens
Tissue collection from head and neck cancer patients was performed under an institutional review board–approved protocol at the Massachusetts General Hospital (IRB 2014P000559). A total of 53 patients were enrolled into the study, including a range of SGT pathologies (Table 1). When the size of surgical removed tissue allowed for FNAs, 5 to 10 passes of minimally invasive FNAs were obtained ex vivo. FNA samples with an insufficient number of cells for immune profiling (<200 cells total) were excluded from the analysis. In total, there was 1 such case out of 53 (1.89%). Nondiagnostic samples in conventional cytopathology often range between 15% and 30%. 23 , 24 Tumor tissues were homogenized into 1‐mm or smaller pieces with clean scissors in RPMI 1640 media and ground against a cell strainer (50 μm) using the back end of 1 mL syringe plunger for single‐cell isolation. The FNA and single‐cell suspension samples were fixed immediately in 4% paraformaldehyde (PFA), washed in phosphate‐buffered saline (PBS), and then stored in Cyto‐Last buffer (Biolegend) with 0.1% sodium azide until imaging. All samples were de‐identified and blinded for the clinical information for imaging and image processing.
Synthesis of Fluorochrome/Quencher Pair
FAST probes were constructed as a modular linker that connects fluorochromes and antibodies with an embedded TCO for clicking with a Tz‐quencher. FAST probes were custom synthesized as described in detail in our previous study. 8 FAST probes were stored as the carboxylic acids and activated for antibody labeling with our in situ NHS/TFP activation chemistry. The dTCO‐PEG6‐CO2H blocking reagent was synthesized from dTCO‐PNP and amino‐dPEG6‐CO2H and characterized by liquid chromatography‐mass spectrometry. All reagents were obtained from commercial sources at the highest grade available. Fluorophores were purchased from Click Chemistry Tools or Fluoroprobes; BHQ‐3 amine from LGC Biosearch Technologies (5 or 25 mg aliquots); N‐α‐Boc‐N‐ε‐Fmoc‐lysine from Chem‐Impex; and amino‐dPEGn‐carboxylic acids (n = 4,6) from Quanta BioDesign. Dry solvents and coupling reagents were obtained from Sigma Aldrich.
Antibody Modifications
Antibodies without carrier were purchased (Supporting Table 1) to be labeled with FAST probes as previously described. 1 A Zeba column (Thermo Fisher) was used to buffer‐exchange the antibodies into bicarbonate buffer (pH 8.4). Antibodies (1‐3 mg/mL) were incubated with a 5‐ to 10‐fold molar excess of the FAST probe with 10% DMSO for 25 minutes at room temperature in dark. For desalting and removal of unreacted dye molecules after the conjugation reaction, FAST‐antibody conjugate was loaded onto another 40K Zeba column equilibrated with PBS. The absorbance spectrum of the FAST‐labeled antibody was measured using a Nanodrop 1000 (Thermo Scientific) to determine the degree of labeling (DOL). The known extinction coefficients of the dye (AF488, AF555, and AF647), IgG antibody, and correction factor for the dye absorbance at 280 nm were used for calculation of DOL. The FAST‐labeled antibodies were stored protected from light at 4°C in PBS. Information of the antibodies used for the single‐cell profiling of clinical samples are summarized in Supporting Table 1. Antibodies were tested and validated on positive cell lines or peripheral blood mononuclear cells (Innovative Research Inc) before usage.
Sample Processing, Immunostaining, and Quenching
Cells were fixed for 10 minutes in 4% PFA and attached to glass slide by cytocentrifugation (Cytospin4 Thermo Scientific). In brief, fixed cells were loaded onto Octospot well strips or Cytofunnel, held in place to the glass slide by Cytoclip for centrifugation (Thermo Scientific). Cells were permeabilized for 25 minutes with 0.5% Triton‐X100 before staining. Immunostaining for FAST imaging was performed in accordance with typical immunofluorescence protocols. After blocking with Intercept Blocking buffer (LI‐COR Biosciences) for 30 minutes, cells were stained with FAST‐conjugated antibodies. Antibodies were diluted to 2 to 5 μg/mL in Intercept Blocking buffer before staining. Cells were labeled with antibodies for 30 minutes for surface markers, or 1 hour for intracellular/nuclear markers. Stained cells were washed 3 times for 5 minutes each with PBS before imaging. Following image acquisition, cells were briefly incubated with 10 μM Tz‐BHQ (<10 seconds) in PBS‐bicarbonate buffer (pH 9) to quench imaging signal before the round of staining. Tz‐BHQ solution was removed by 3 washes with PBS‐bicarbonate buffer, and the cells were imaged again in the same fields of view to record the quenched signal. Before antibody staining of the subsequent cycle, cells were incubated in a solution of 20 μM dTCO‐PEG6‐CO2H for 1 minute to block any residual Tz‐BHQ3 from reacting with FAST antibodies of the next cycle. The same staining, imaging, and quenching cycle was repeated until all of the target proteins were imaged. Each cycle took 60 to 90 minutes including antibody staining, washing, and imaging before and after quenching. On average, it thus took ~12 hours to image all 29 markers on a sample. A fraction of the sample was set aside and incubated with isotype antibodies as a negative control. The controls were imaged every cycle following the same protocol as described above.
Fluorescence Microscopy
An Olympus BX‐63 upright automated epifluorescence microscope was used to acquire fluorescent images. DAPI, FITC, Cy3, and Cy5 filter cubes were used to image DAPI nuclear stains, AF488, AF555, and AF647 fluorophores, respectively. Depending on the cellularity of specimen and the density of cells on the glass slides, 15 to 30 fields of view were imaged to capture a total cell population sufficient for analysis. The same cells in the 15 to 30 fields of view were imaged each stain‐image‐quench cycle. X‐Y coordinates for each field of view were saved to enable automatic imaging of the same set of cells in every cycle using Multi Dimensional Acquisition in Metamorph software.
Analysis of Surgical Tissue Sections
We performed a number of different studies in FFPE tissue sections from surgical resections to i) test and optimize the FAST cycling methods in tissues (Supporting Fig. 3), ii) directly compare FAST‐FFPE against immunohistochemistry (Supporting Fig. 8), iii) analyze the spatial distribution of biomarker expression (Supporting Fig. 4), iv) compare FNA to tissue based analyses, and v) determine whether the FAST method could be used to detect therapeutically actionable biomarkers such as NTRK fusions. FFPE tissue sections were cut to 5 μm and then processed for immunohistochemistry or FAST cycling. FFPE tissue sections were deparaffinized, rehydrated, and antigen‐retrieved in pH 9 antigen retrieval buffer. Then sections were blocked with Intercept Blocking buffer (LI‐COR) for 30 minutes before antibody staining for FAST‐FFPE. For IHC, sections were blocked with Dako Antibody Diluent (Agilent Technologies) for 30 minutes followed by blocking with avidin and biotin for 30 minutes each (Vector Laboratories). The same antibodies for FAST‐FFPE were used as primary antibodies for IHC, and the development of DAB staining was done using Dako EnVision FLEX system (Agilent Technoliges). NTRK fusions were identified by screening for nuclear NTRK expression after staining with an anti‐panTRK antibody (Abcam, clone EPR17341).
Image Analysis
CellProfiler 25 was used for image registration, cell segmentation, and measurement of the fluorescent intensities in individual cells. Acquired images were corrected for an illumination function and aligned using normalized cross correlation to compensate for minor pixel shifts that occur during cyclic imaging. For background correction, fluorescence signals measured in quenched images were subtracted pixel‐by‐pixel from the immunostained images of the following cycle. Cells were identified using the DAPI signal of the last image cycle as input, and the cell boundaries were segmented using maximum projection of intensity measured in all channels. A small fraction of cells (typically well below <10%) that were lost during repeat imaging cycles were excluded from the image analysis. In every identified cell, an areal mean fluorescence intensity was used for subsequent computational analyses.
Dimensional Reduction and Predictive Modeling
Intensity normalization, dimensional reduction, and predictive modeling were performed using a custom R code. To correct for the multiplicative batch effect that results from variations in the illumination power and the degree of antibody labeling, we divided the intensity values of each marker in each sample by the 10% percentile such that the rising edge of the intensity distribution of the negative population is located at 1. We confirmed that the end results were not sensitive to the choice of the percentile value between 5 and 10 because every sample included a number of negative cells for each marker. For dimensionality reduction, we used the UMAP algorithm with its R implementation in the uwot package. 26 UMAP was used with the following parameter setting: n_neighbors = 10, min_dist = 0.3, metric = “euclidean,” and n_epochs = 500. Specimens with measurement of all 29 biomarkers were included in the UMAP analysis. A random forest model was used for classification of cells derived from benign or malignant tumor. The model was trained using a data set of 79,379 cells from 13 benign tumor digests and 75,946 cells from 7 malignant tumor digests, totaling 155,325 cells from 20 tumor digests. Twenty‐seven markers (excluding CD45 and aSMA from the full list of markers as they target nontumor cells) and the nucleus area were used as predictors. A 5‐fold cross validation was performed to determine the optimal hyper‐parameters of the random forest model. The trained model was tested on 14,390 other cells (7219 benign and 7171 malignant) that were not included in the training. The probability of each cell belonging to a malignant tumor was predicted using the model, and a ROC curve was drawn to evaluate its performance as a binary classifier and determine the optimal probability threshold. For each new tumor sample, the fraction of cells classified as being from malignant tumors was calculated to predict its malignancy.
Statistics
GraphPad Prism was used for statistical analysis. Results were expressed as mean ± SEM. Statistical tests included one‐way ANOVA followed by Tukey’s or Dunnett’s multiple comparison test. When applicable, the unpaired one‐tailed and two‐tailed Student’s t tests using Welch’s correction for unequal variances were used. The threshold for a given biomarker was determined based on the normalized intensity distribution of the aggregated data (Supporting Figure 4) using a custom R code. The location and FWHM of the negative population peak were determined by applying a kernel density estimation to the normalized intensity histogram followed by peaks function in IDPmisc package. The threshold for determining whether a cell is positive for a marker was set FWHM away from the negative peak intensity. We confirmed that the end results were not sensitive to a small variation in the threshold setting.
FUNDING SUPPORT
Certain aspects of this work were supported by CSB (R01CA257623, R01CA206890, P01CA069246, and P01CA240239). Juhyun Oh was supported in part by a Tosteson MGH Research Institute Fellow award.
CONFLICT OF INTEREST DISCLOSURES
The authors declare the filing of a patent that was assigned to Massachusetts General Hospital. Sara I. Pai reports grants from AbbVie, AstraZeneca/MedImmune, Cue Biopharma, Merck, and Tesaro; and consulting fees from AbbVie, AstraZeneca/MedImmune, Cue Biopharma, Fusion Pharmaceuticals, MSD/Merck, Newlink Genetics, Oncolys Biopharma, Replimmune, Scopus Biopharma, and Sensei Bio. Ralph Weissleder is a consultant to ModeRNA, Tarveda Pharmaceuticals, Lumicell, Seer, Earli, Alivio Therapeutics, Aikili Biosystems, and Accure Health. The other authors made no disclosures.
AUTHOR CONTRIBUTIONS
Juhyun Oh: Conceptualization, experiments, data analysis, and writing. Tae Yeon Yoo: Data analysis and writing. Talia M. Saal: Experiments and writing. Lisa Tsay: Experiments and writing. William C. Faquin: Clinical samples and writing. Jonathan C.T. Carlson: Synthesis of imaging probes and writing. Daniel G. Deschler: Clinical samples and writing. Sara I. Pai: Conceptualization, clinical samples, data analysis, and writing. Ralph Weissleder: Conceptualization, clinical samples, data analysis, and writing.
Supporting information
Appendix S1 Supporting Information
We are grateful to the patients who participated in this study, and we acknowledge the help and many discussions with our clinical colleagues from the Department of Otolaryngology‐Head and Neck Surgery at Massachusetts Eye and Ear and Harvard Medical School, Boston, Massachusetts.
Contributor Information
Sara I. Pai, Email: Sara.Pai@mgh.harvard.edu.
Ralph Weissleder, Email: rweissleder@mgh.harvard.edu.
REFERENCES
- 1. Oh J, Carlson JCT, Landeros C, et al. Rapid serial immunoprofiling of the tumor immune microenvironment by fine needle sampling. Clin Cancer Res. 2021;27:4781‐4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . WHO Classification of Head and Neck Tumors. Naggar AK, Chan JKC, Grandis JR, et al, eds. IARC; 2017. [Google Scholar]
- 3. Griffith CC, Pai RK, Schneider F, et al. Salivary gland tumor fine‐needle aspiration cytology: a proposal for a risk stratification classification. Am J Clin Pathol. 2015;143:839‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Griffith CC, Schmitt AC, Pantanowitz L, Monaco SE. A pattern‐based risk‐stratification scheme for salivary gland cytology: A multi‐institutional, interobserver variability study to determine applicability. Cancer Cytopathol. 2017;125:776‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hughes JH, Volk EE, Wilbur DC; Cytopathology Resource Committee, College of American Pathologists. Pitfalls in salivary gland fine‐needle aspiration cytology: lessons from the College of American Pathologists Interlaboratory Comparison Program in Nongynecologic Cytology. Arch Pathol Lab Med. 2005;129:26‐31. [DOI] [PubMed] [Google Scholar]
- 6. Colella G, Cannavale R, Flamminio F, Foschini MP. Fine‐needle aspiration cytology of salivary gland lesions: a systematic review. J Oral Maxillofac Surg. 2010;68:2146‐2153. [DOI] [PubMed] [Google Scholar]
- 7. Di Villeneuve L, Souza IL, Tolentino FDS, Ferrarotto R, Schvartsman G. Salivary gland carcinoma: novel targets to overcome treatment resistance in advanced disease. Front Oncol. 2020;10:580141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ko J, Oh J, Ahmed MS, Carlson JCT, Weissleder R. Ultra‐fast cycling for multiplexed cellular fluorescence imaging. Angew Chem Int Ed Engl. 2020;59:6839‐6846. [DOI] [PubMed] [Google Scholar]
- 9. Kalina T, Lundsten K, Engel P. Relevance of antibody validation for flow cytometry. Cytometry A. 2020;97:126‐136. [DOI] [PubMed] [Google Scholar]
- 10. MacNeil T, Vathiotis IA, Martinez‐Morilla S, et al. Antibody validation for protein expression on tissue slides: a protocol for immunohistochemistry. Biotechniques. 2020;69:460‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cocco E, Scaltriti M, Drilon A. NTRK fusion‐positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15:731‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murray BW, Rogers E, Zhai D, et al. Molecular characteristics of repotrectinib that enable potent inhibition of TRK fusion proteins and resistant mutations. Mol Cancer Ther. 2021;20:2446‐2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Solomon JP, Linkov I, Rosado A, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33:38‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papoian V, Rosen JE, Lee W, Wartofsky L, Felger EA. Differentiated thyroid cancer and Hashimoto thyroiditis: utility of the Afirma gene expression classifier. J Surg Oncol. 2020;121:1053‐1057. [DOI] [PubMed] [Google Scholar]
- 15. Lin JR, Fallahi‐Sichani M, Sorger PK. Highly multiplexed imaging of single cells using a high‐throughput cyclic immunofluorescence method. Nat Commun. 2015;6:8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gerdes MJ, Sevinsky CJ, Sood A, et al. Highly multiplexed single‐cell analysis of formalin‐fixed, paraffin‐embedded cancer tissue. Proc Natl Acad Sci U S A. 2013;110:11982‐11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schubert W, Bonnekoh B, Pommer AJ, et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 2006;24:1270‐1278. [DOI] [PubMed] [Google Scholar]
- 18. Ullal AV, Peterson V, Agasti SS, et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine‐needle aspirates. Sci Transl Med. 2014;6:219ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Agasti SS, Liong M, Peterson VM, Lee H, Weissleder R. Photocleavable DNA barcode‐antibody conjugates allow sensitive and multiplexed protein analysis in single cells. J Am Chem Soc. 2012;134:18499‐18502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giedt RJ, Pathania D, Carlson JCT, et al. Single‐cell barcode analysis provides a rapid readout of cellular signaling pathways in clinical specimens. Nat Commun. 2018;9:4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruiz‐Cordero R, Ng DL. Neurotrophic receptor tyrosine kinase (NTRK) fusions and their role in cancer. Cancer Cytopathol. 2020;128:775‐779. [DOI] [PubMed] [Google Scholar]
- 22. Weissleder R, Lee H. Automated molecular‐image cytometry and analysis in modern oncology. Nat Rev Mater. 2020;5:409‐422. [Google Scholar]
- 23. Park JH, Cha YJ, Seo JY, Lim JY, Hong SW. A retrospective cytohistological correlation of fine‐needle aspiration cytology with classification by the Milan System for Reporting Salivary Gland Cytopathology. J Pathol Transl Med. 2020;54:419‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saraph S, Cohen H, Ronen O. Effect of needle gauge on thyroid FNA diagnostic rate. Endocrine. 2021;74:625‐631. [DOI] [PubMed] [Google Scholar]
- 25. McQuin C, Goodman A, Chernyshev V, et al. CellProfiler 3.0: next‐generation image processing for biology. PLoS Biol. 2018;16:e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single‐cell data using UMAP. Nat Biotechnol. 2019;37:38‐44 2018. doi: 10.1038/nbt.4314 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information