

Abstract
Four difluorenoheteroles having a central quinoidal core with the heteroring varying as furan, thiophene, its dioxide derivative and pyrrole have shown to be medium character diradicals. Solid‐state structures, optical, photophysical, magnetic, and electrochemical properties have been discussed in terms of diradical character, variation of aromatic character and captodative effects (electron affinity). Organic field‐effect transistors (OFETs) have been prepared, showing balanced hole and electron mobilities of the order of 10−3 cm2 V−1 s−1 or ambipolar charge transport which is first inferred from their redox amphoterism. Quantum chemical calculations show that the electrical behavior is originated from the medium diradical character which produces similar reorganization energies for hole and electron transports. The vision of a diradical as simultaneously bearing pseudo‐hole and pseudo‐electron defects might justify the reduced values of reorganization energies for both regimes. Structure‐function relationships between diradical and ambipolar electrical behavior are revealed.
Keywords: Ambipolar, Diradical Character, Electronic Couplings, Organic Field Effect Transistors, Reorganization Energies
Diradicals are electrically ambipolar owing to the semiaperture of the π‐electron shell which produces redox amphoteric behaviour and small reorganization energies for hole and electron transports.
Introduction
The use of organic π‐conjugated molecules as semiconducting materials in p–n ambipolar organic field effect transistors (OFETs) with balanced mobilities is a major target in organic electronics pursuing their implementation in complementary electrical circuits.[ 1 , 2 , 3 , 4 ] Closed‐shell small‐size organic molecules showing ambipolar charge transport encompass phthalocyanines, [5] indigo and diketopyrrolopyrrole derivatives, [6] antiaromatic [7] and fullerenes [8] compounds, acene [9] and cyano substituted oligothiophenes. [10] As for open‐shell diradical molecules with ambipolar transport is concerned, we have reported a diindenoanthracene diradical molecule [11] embedding a central quinoidal anthracenyl unit (DIAn, Figure 1) that displayed p–n balanced mobilities of ≈10−3 cm2 V−1 s−1 for the two regimes of transport. A similar hydrocarbon diradicaloid anti‐HDIP [12] in Figure 1 has also shown ambipolar transport, but with unbalanced behavior. Other examples of diradical OFETs based on benzoquinoidal cores are zethrene [13] (1) and diindenoperylene [14] in Figure 1 which only showed unipolar conduction. Non‐quinoidal polycyclic benzenoid diradicals with ambipolar behavior include, for instance, peri‐pentacenopentacene [15] (Figure 1); whereas quinoidal motifs based on thiophene, such as Q3 in Figure 1, have also enabled ambipolar transport with p–n mobilities of ≈10−4 cm2 V−1 s−1.[ 16 , 17 ] It turns out that DIAn, peri‐pentacenopentacene and anti‐HDIP share to have medium diradical character thus posing the question whether diradical character is mandatory to produce ambipolar and balanced transport in these systems?
Figure 1.
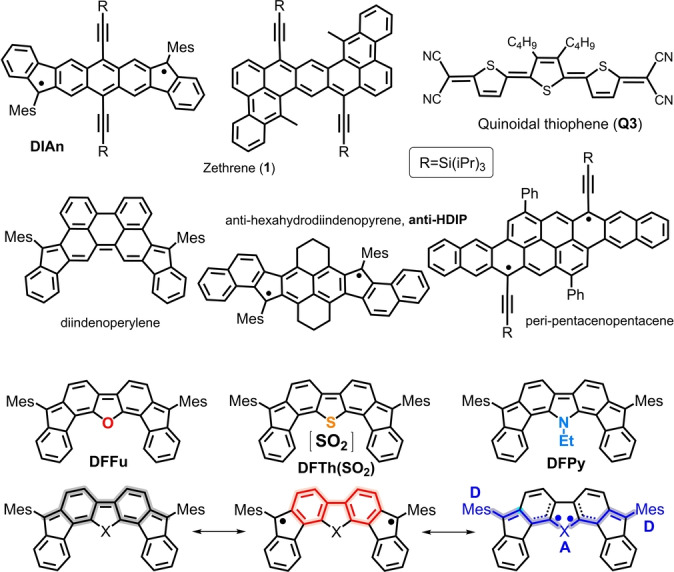
Quinoidal and diradical molecules implemented in OFETs and ambipolar OFETs together with the four difluorenoheterole compounds in this work. Bottom: resonant forms stabilizing the diradical structure: aromatic (red) and captodative (purple) forms.
Charge transport in organic semiconductors is a physical response to external electric fields that depends on intramolecular and intermolecular factors.[ 18 , 19 ] Intermolecular effects in the organic semiconducting layer (electronic couplings, V) and metal–organic (orbital energy alignment with the electrode Fermi energy level, E OM‐F) and semiconductor–dielectric interfaces are optimized by post‐preparation material processing, thus part of the focus of pre‐designing new molecular candidates is on the basis of enhancing intramolecular properties, such as intramolecular reorganization energy, λ. For instance, pentacene displays an outstanding semiconductor behavior given the fine balance between V, E OM‐F and λ, but it is the small reorganization energy for hole exchange, λ hole, that stands out in terms of tailoring intramolecular properties. [20] Pentacene bears mostly a closed‐shell structure with very incipient diradical character; indeed, longer acenes develop larger diradical character and chemical instability preventing further analysis. [21] Here, the hypothesis is that medium diradical character imparts a degree of electron confinement (i.e., electron unpairing) that gives rise to the simultaneous generation of similar pseudo‐hole and pseudo‐electron like‐particles on its frontier orbital electrons upon which further charge extraction/addition only produces small structural changes and reduced reorganization energies in similar degree for holes and electrons. In the two diradical extremes, less suitable conditions for ambipolar responses are attained given that large diradical character might restrict charge internal delocalization (strong confinement), whereas strong pairing (closed‐shell) generates large structural reorganization upon charging.
Diradicals or diradicaloids are characterized by partially unpaired frontier electrons due to configurational mixing. One of the ways to attain diradicaloids is departing from quinoidal structures which might eventually undergo bond scission by aromatic stabilization such as shown in Figure 1. The diradical extent is measured by the so‐called diradical character, [22] y 0, a theoretical index where y 0=0 for closed‐shell molecules and y 0=1 for pure diradicals. Accordingly, we have: DIAn, y 0=0.62; peri‐pentacenopentace, y 0=0.64; anti‐HDIP, y 0=0.57; and zethrene 1, y 0=0.11. These values qualitatively pose connections between moderate‐medium y 0 and ambipolar electrical behavior. Nonetheless, the interest on diradical molecules is broader than that focused in electrical transport and other applications have been probed such as in non‐linear optics, [23] in singlet exciton fission [24] in photonics, in spintronics, [25] in thermopower devices, [26] etc.
We present here the study of benzoquinoidal polycyclic conjugated molecules with medium diradical character in which, additionally, systematic changes of their molecular structures are introduced with the finality of establishing patterns of structure–property relationships. These are four difluorenoheterole compounds (Figure 1) based on furan, pyrrole, thiophene and thiophene dioxide with identical chemical structures except for the mentioned heteroatom five‐membered rings which are embedded in a central fluorene with quinoidal structure. Such as in other medium diradicals, we found out ambipolar electrical behavior in all these molecules with different degrees of p–n balanced transport. The novelty of the present work is the elucidation of the connection between moderate/medium diradical character and ambipolar charge transport by means of the pivotal role of the small reorganization energies produced by such medium y0 ′s which, furthermore, are similar for holes and electrons thus fueling equivalent p‐ and n‐type transport. Difluorenoheteroles disclose medium diradical characters by an amalgam of electronic affinity, captodative effect and aromatic stabilization.
Results and Discussion
The synthesis of DFTh and DFPy is described in Scheme 1, while the synthesis of DFFu has been recently reported by us. [27]
Scheme 1.
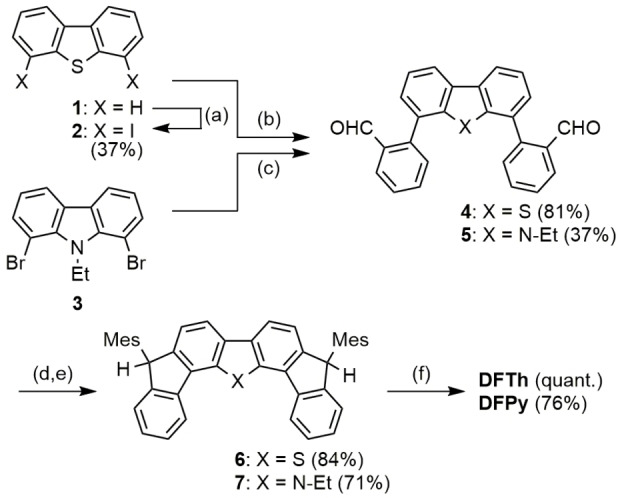
Synthesis of the studied compounds. Reagents and conditions: a) i) n‐BuLi, TMEDA, hexane, 60 °C. ii) CH2ICH2I, rt. b) 2‐formylphenylboronic acid, Pd2(dba)3⋅CHCl3, [(tBu)3PH][BF4], Cs2CO3, THF/H2O, 70 °C. c) 2‐formylphenylboronic acid, Pd(OAc)2, SPhos, K3PO4, toluene/H2O, 80 °C. d) MesMgBr, THF, rt. e) BF3⋅Et2O, CH2Cl2, rt. f) DDQ, toluene, 90 °C.
Direct lithiation of dibenzothiophene (1) at the 4‐ and 6‐positions using n‐BuLi in the presence of N,N,N′,N′‐tetramethylethylenediamine (TMEDA), followed by treatment with diiodoethane afforded iodide 2. A Pd‐catalyzed Suzuki–Miyaura coupling of 2 with 2‐formylphenylboronic acid provided aldehyde 4. The nucleophilic addition of mesitylmagnesium bromide (MesMgBr) to 4 gave the secondary alcohol, which was subjected to a Friedel–Crafts cyclization by BF3⋅Et2O to afford 6. Dehydrogenation of 6 with 2,3‐dichloro‐5,6‐dicyanobenzoquinone (DDQ) provided the desired DFTh. Oxidation of DFTh with MCPB acid produces DFThSO2 . In similar procedures, DFPy was synthesized from 1,8‐dibromocarbazole (3) in four steps. Additional synthetic details are in Scheme S1 together with detailed chemical characterization in Supporting Information.
Crystal structures of all the compounds were solved (X‐ray data in Figures S1–S4). [28] A common structural feature of all these difluorenoheteroles is the almost planar disposition of the fused rings with the mesityl rings pointing out perpendicularly to the main molecular plane. The CC bonds exocyclic to the innermost biphenyls (Figures S1–S4) that connect to the apical carbons are larger for DFTh (1.3940 Å) and DFFu (1.3938 Å) in agreement with their larger diradical characters (see discussion below). DFThSO2 conversely shows the shorter distance of this CC bond (1.3841–1.3756 Å) in Figure S4) and the largest diradical character in contraposition to DFTh and DFFu. DFTh forms dimers in the crystal in a head‐to‐tail manner with CC distances of 3.52 Å, further assembled in a zigzag array by CH–π interactions with distances of 2.86–2.99 Å. DFFu affords a π‐stacked column in a head‐to‐head and slipped arrangement with distances of 3.39–3.48 Å. All difluorenoheteroles display analogue intermolecular arrangements forming specific π‐dimer subunits that are very favourable for charge transport (see last section).
NICS scan values [29] have been calculated and represented in Figure 2, while the diradical character values, y 0, summarized for the four molecules are in Table 1. The central five‐member heterorings disclose the largest aromatic character for DFTh in agreement with the larger y0 compared to DFFu and DFPy revealing a dominant aromatic driving force for the formation of the open‐shell structures. DFThSO2 shows, however, a completely different trend in the central heteroring which is less aromatic than in any of the parent systems and, conversely, discloses the largest y 0. It can be argued that the internal benzenes gain larger aromaticity in DFThSO2 what would explain the largest y0 but this is insufficient to give account of the reduction of the distances in DFThSO2 for the biphenyl exocyclic C−C bonds.
Figure 2.
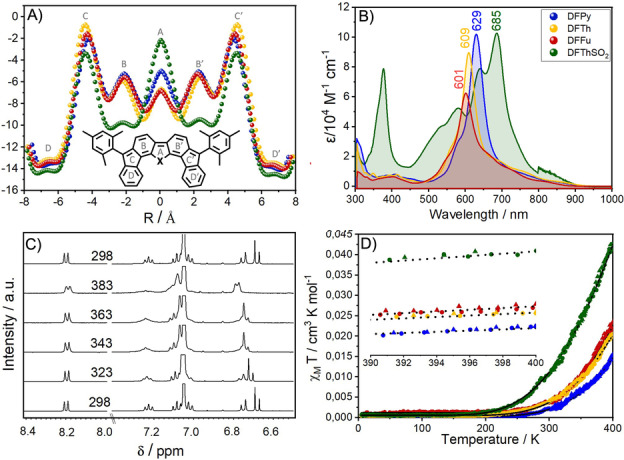
For the four compounds according to the following color codes: DFTh (red), DFFu (blue), DFPy (yellow) and DFThSO2 (green): A) NICS(1) scan values. B) Electronic absorption spectra in CH2Cl2 at room temperature. C) 1H NMR spectra in p‐xylene‐d 10 as a function of the temperature in K for DFTh. D) SQUID data in the heating and cooling processes together with the Bleany–Bowers adjusts. The insert is a zoom of the SQUID curve to distinguish between the adjustments (dotted black), heating (circles) and cooling (triangles) curves.
Table 1.
Redox potentials (in V vs. Fc/Fc+ from cyclic voltammetry), hole and electron mobilities from OFET measurements (in cm2 V−1 s−1) in vacuum, magnetic singlet–triplet gaps (ΔE ST) from SQUID (in kcal mol−1) and y 0 from PUHF calculations.
|
|
E ox |
E red |
E e‐gap |
μ h |
μ e |
ΔE ST |
y 0 |
|---|---|---|---|---|---|---|---|
|
DFPy |
0.17 |
−1.31 |
1.48 |
2×10−3 |
8×10−4 |
−5.0 |
0.541 |
|
DFFu |
0.32 |
−1.17 |
1.49 |
1×10−3 |
9×10−4 |
−4.9 |
0.555 |
|
DFTh |
0.31 |
−1.14 |
1.45 |
1×10−3 |
6×10−4 |
−4.3 |
0.570 |
|
DFThSO2 |
0.44 |
−0.99 |
1.43 |
2×10−4 |
3×10−4 |
−3.3 |
0.591 |
The diradical indexes, y 0, of these compounds in Table 1 have been estimated from quantum chemical calculations (Tables S1 and S2 computational details in Supporting Information) showing medium values varying as 0.541→0.555→0.570→0.591 on DFPy→DFFu→DFTh→DFThSO2 . Such as mentioned, only considering the aromatic character of the five‐membered heterocycles, which increases as thiophene‐SO2<furan<pyrrole<thiophene, the largest y0 would correspond to DFTh instead of to DFThSO2 . If electron affinity, which increases as S<N<O<SO2, of the three individual heteroatoms is considered together with the aromatic characters of the heterorings, their joint action qualitatively reproduces the changes of y 0 in the difluorenoheteroles. Thiophene with the largest aromatic character would display a large y 0 followed by furan with the largest captodative effect. The increase of the electron affinity of the sulfur in DFThSO2 produces a substantial increase of y 0 by captodative effect compensating the lack of aromaticity gaining in its five‐member ring. This connection of the stabilization of the diradical state with the electronic affinity might reveal an underlying mechanism of radical stabilization by captodative [30] effects in which a donor (D) and an acceptor (A) stabilize the radical placed in between. In Figure 1 we assign the role of the donor to the exocyclic vinylenes and the heteroatom plays as an acceptor, thus captodative effect would be more remarkable in DFThSO2 and DFFu. Interestingly, with the captodative effect taken into consideration, the observation that the biphenyl exocyclic CC bond lengths in the series is smaller in DFThSO2 nicely agrees with the donor–acceptor interaction in Figure 1.
The magnetic properties have been studied by using SQUID magnetometry in solid state together with variable temperature ESR and 1H NMR in solution in Figure 2 and Figures S5, S6 and S7). The χT–T data from SQUID have been adjusted to a Bleany–Bowers plot [31] using a dimer model providing the singlet‐triplet gaps, ΔE ST, shown in Table 1. The heating‐cooling SQUID experiment reveals no degradation of the samples in the temperature interval analyzed (i.e., thermogravimetric analysis in Figures S8, S9 and S10). Experimental and theoretical gaps correlated very well in Table 1. ΔE ST gaps evolve in line with the diradical indexes where the smallest diradical character for DFPy implies the larger magnetic ΔE ST gap terminating with DFThSO2 with the smaller gap and the larger y 0′s. [32] The solid samples also display a strong broad ESR signal centered at g=2.000 which emerges from thermally populated triplets at room temperature. The 1H NMR spectra in Figure 2 of DFFu, DFTh, DFPy and DFThSO2 at 298 K show well‐defined signals in the aromatic region that become broader upon increasing the temperature to 383 K due to the increasing population of thermally accessible triplets: signal broadening occurs in the order DFPy<DFFu<DFTh<DFThSO2 following the accessibility order of the triplets found by SQUID measurements.
The redox properties of the compounds from the cyclic voltammetries in Figure 3 display that the first reduction potential is only slightly different in DFFu than in DFTh but significantly smaller in DFThSO2 . The reduction stabilization by the larger aromaticity gaining in the anion of DFTh is compensated by the stabilization of the radical anion by the larger captodative effect by the oxygen of furan in DFFu (Figure 1). [32] On the other hand, the much larger electron accepting capacity of the dioxide sample produces the smallest reduction potential among the series. As for the first oxidation processes, the dioxide moiety makes its oxidation substantially less accessible than in DFFu and DFTh. The similarity of the first oxidations of DFFu and DFTh agrees with the similar aromatic character in the benzenes of the central biphenyl (Figure 2A). These results are in excellent agreement with predicted frontier orbital energies (Figure S11, Table S4).
Figure 3.
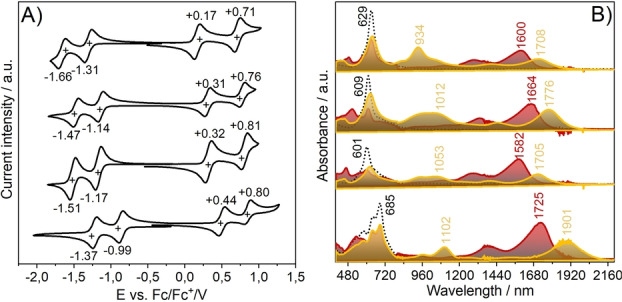
A) Cyclic voltammograms of the four compounds in CH2Cl2 containing 0.1 M [n‐Bu4N][PF6] as supporting electrolyte at a scan rate of 100 mV s−1. From the bottom: DFThSO2 , DFFu, DFTh and DFPy. B) Spectroelectrochemical UV/Vis‐NIR absorption spectra of the radical anions (red) and cations (yellow).
The absorption spectra of the four compounds in Figure 2 display the largest changes in DFThSO2 with a peak wavelength at 685 nm compared with DFTh at 609 nm. The UV/Vis‐NIR electronic absorption spectra of the radical anions and cations in Figure 3 and Figure S12 of the difluorenoheteroles have been obtained by spectroelectrochemical treatment. Quantum chemical calculations of the theoretical UV/Vis spectra of these charged species are in Figure S13 which reproduce well the experimental observations. We notice the appearance of well‐defined Vis‐NIR electronic absorption bands whose shapes are different for cations and anions, such as it has been recently reported for related diradicals. [32] Interestingly, the competition between captodative and aromatic effects in the radical anion of DFThSO2 provokes the reduction of the optical gap and the appearance of the lowest energy absorption bands at the longest wavelengths.
Transient absorption measurements in Figure 4 of the four compounds have been carried out (Figure S14). DFFu displays a positive band centered at 470 nm right upon excitation that disappears after 3 ps followed by a new band positioned at 630 nm that rises in 2 ps and decays upon 30 ps. These two bands are assigned to singlet excited states and given the similarity of the timescales for their rise/decay absorbances, a transformation of the first singlet species into a second one seems to take place. This is in agreement with the reported photophysics of known diradicals [33] that consists on ultrafast internal conversion from the pumped optical state to another dark singlet excited state, which in our case is responsible of the band at 630 nm and that clearly have double exciton character.[ 34 , 35 , 36 ] NEVPT2 quantum chemical calculations of the excited states of DFFu have been carried out in Table S5 and show the presence of the bright state at 566 nm and in the close vicinity, at 606 nm, a two‐photon excited state which corresponds to our dark state. The presence of this dark state justifies the absence of fluorescence in the studied compounds. The four compounds show this characteristic photophysical behavior consisting on S2→S1 internal conversion followed by S1→S0 relaxation all in pico‐second timescales such as shown in Figure S14, Figure S15 and Figure S16. A modulation of the relaxation kinetics is observed in the four compounds which systematically evolves as in 0.4/10 ps in DHPy, 0.6/14 ps in DFTh, 0.7/15 ps in DFFu and 1.3/14 ps in DFThSO2 .
Figure 4.
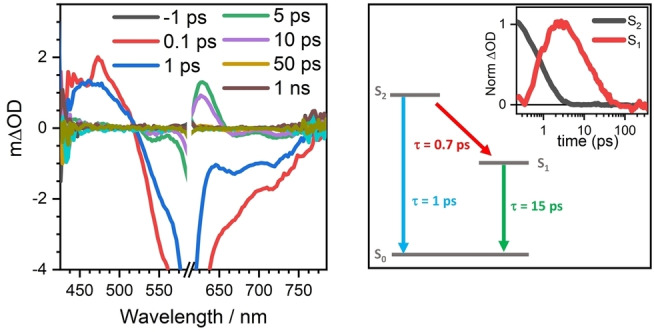
A) Femtosecond transient absorption spectra of a 10 μM DFFu solution in CH2Cl2 exciting at 600 nm with a power of 0.25 mW. B) Decays of the DFFu S2 and S1 species obtained by global analysis of the femtosecond transient absorption data.
Organic field effect transistors and electrical measurements were carried out in a top contact‐bottom gate OFET configuration for the four compounds. Different deposition conditions were tested, being the best performances achieved when the thin films were deposited by sublimation on HMDS‐treated substrates preheated at 80 °C. The devices were then finalized by gold electrodes deposition. Figure 5, Figure S17, S18 and Table S6 show the devices electrical features in which ambipolar charge transport is registered in all cases. Figures S19, S20 disclose the structural XRD and AFM characterization of the thin film substrates, while Figure S21 shows the stability of the OFET devices.
Figure 5.
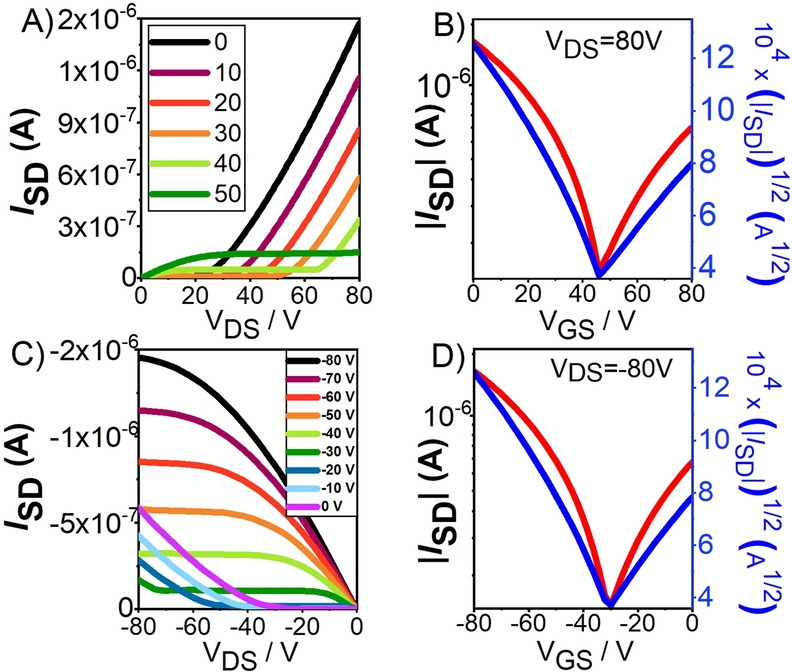
Electrical responses for: A), B) p‐transport, and C), D) n‐transport for the OFET devices of DFFu. Deposition of the semiconducting film was carried out on HMDS‐treated substrates preheated at 80 °C. A), C) Output and B), D) transfer characteristic curves, respectively.
The first noticeable aspect of the electrical behavior in Figure 5B–D concerns the typical “valley” structure in ambipolar semiconductors describing the transition between hole and electron transport regimes through the neutral charge point.[ 1 , 4 ] In our case, in the valley, it is verified, either for V DS=−80 V and +80 V, that V GS≈V DS/2 so that V T,hole≈V T,electron in line with the balanced p–n transport and that could be a unique feature of diradicals. [4]
Overall hole and electron mobilities, summarized in Table 1, are reasonably similar featuring balanced p–n charge transport for the four studied semiconductors. In particular, DFFu has well‐balanced mobilities of approx. 10−3 cm2 V−1 s−1 for hole and electron transport. Note, however, that the thiophene and pyrrole derivatives display more unbalanced charge conduction, with approximately twice p‐type mobility than n‐type mobility. In terms of thin film characterization, XRD data (Figure S19) shows partial crystallinity of the thin films which, in general, is slightly enhanced with deposition temperature. This crystallinity enhancement becomes also evident in the AFM images which show quite homogeneous films with small and compact grains in the case of films grown at room temperature. The grain sizes become slightly larger for films deposited on preheated substrates (80 °C), but maintaining the homogeneity. These results are in good agreement with the subtle field‐effect mobility enhancement with deposition temperature (Figure S20). Nonetheless the still low crystallinity degree of the films might explain the moderate recorded field‐effect mobilities.
Quantum chemical calculations have been carried out to rationalize the connections between diradical character and ambipolar charge transport. Charge transport in organic semiconductors [18] has two relevant parameters for our discussion, the intramolecular reorganization energy, λ, related with the energetic cost resulting from the structural transformation from the neutral species to the charged ones: larger λs are detrimental for charge transport; and the intermolecular electronic coupling, V, related with the through space overlap between the electron wavefunctions within vicinal molecules: larger V′s favor large charge mobilities. [19] Transport through the yellow and green channels is favored by the largest calculated V values due to more efficient orbital overlap and shorter intermolecular distances (Figure 6). The electronic coupling values have been calculated for hole (radical cations) and electron (radical anions) transports which turn out to be rather similar, supporting similar transport mobilities for the two charge carriers types with slight preferences for hole conduction, such as observed experimentally. Intramolecular reorganization energies have been obtained for radical cations and radical anions in Table 2 (Table S8). These λ values are around 0.1 eV in magnitude which are rather small among organic semiconductors and comparable to the case of pentacene for hole transport. [20] Interestingly, the reorganization energies values are of the same magnitude for holes and electrons, with slight preference (i.e., smaller) again for holes. Taken together, these charge transport data are in line with the measurement of similar mobilities in both regimes of conduction and with the ambipolar character of the OFET devices. Ambipolar charge transport is fueled by the small reorganization energies combined with favorable level alignment and V values that, although smaller than those of pentacene, are in line with those of best p‐type/n‐type organic semiconductors and compatible with mobilities close to ca. 1 cm2 V−1 s−1 for perfect crystalline materials. [37] Compared to the ideal crystal expectations, the reported experimental mobilities are reduced by additional morphological issues such as grain size boundaries, defects, imperfect crystallinity etc.
Figure 6.
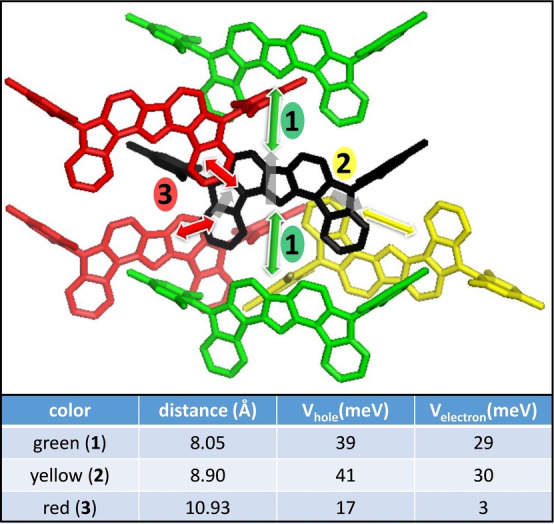
Selected cluster from the X‐ray structure of DFFu where electronic couplings, V, at the B3LYP/6‐31G* level are calculated. Dimers are formed between the black central molecule and the red, green and yellow neighbors, denoted by arrows.
Table 2.
Computed intramolecular reorganization energies (RB3LYP/6‐31G* and UB3LYP/6‐311G* level, in eV) using as reference the closed‐shell (CS) or the open‐shell (OS) structures of the neutral species.
|
Compound |
DFFu |
DFTh |
DFPy |
DFThSO2 |
|
|---|---|---|---|---|---|
|
CS |
hole |
0.210 |
0.212 |
0.190 |
0.258 |
|
electron |
0.216 |
0.222 |
0.237 |
0.209 |
|
|
OS |
hole |
0.104 |
0.101 |
0.093 |
0.123 |
|
electron |
0.114 |
0.113 |
0.136 |
0.102 |
|
Intramolecular reorganization energies have been also calculated for the closed‐shell structures (simulated situation with null diradical character, y 0=0) of the four derivatives and are gathered in Table 2. In comparison with the open‐shell diradical structure of the same molecules, a two‐fold increase of these reorganization energy values for the closed‐shell structures is predicted which is a key insight revealing the preferential conditions for charge transport in the diradical structure compared with the closed‐shell one. The diradical form produces a situation of non‐bonding frontier unpaired electrons in which the removal to produce holes does not largely affect the remaining molecular structure. On the other hand, the addition of an electron brings about a smaller amount of electronic repulsion compared with the situation of full pairing in the closed‐shell form.
Inspection of the computed bond lengths (Figures S23–S26) shows that the diradical open‐shell structure of neutral systems is more similar to the geometry of the charged species, which justifies the reduced reorganization energies. Such reduced geometry difference between neutral and charged species can be rationalized by simple electronic structure arguments, considering that the open‐shell structure results from the mixing of HOMO and LUMO closed‐shell orbitals. Thus, the open‐shell form of a diradical molecule, compared to the quinoidal closed‐shell, is expected to be somewhat similar to that of a HOMO→LUMO (or HOMO,HOMO→LUMO,LUMO) excitation (in which a hole is created in the HOMO and an electron is added to the LUMO). On the other hand, a similar geometry change is expected for the cation (in which a hole in the HOMO is created) or for the anion (where an electron is added to the LUMO). Such simple electronic structure considerations give the vision of the diradical as a structure in which coexist pseudo‐hole (partial de‐occupation of the HOMO) and pseudo‐electron (partial occupation of the LUMO) charge defects, a perspective that intuitively explains the reduced reorganization energies for diradicals either for the transport of holes and of electrons.
Conclusion
We have studied four different difluorenoheterole compounds by changing the heteroring from furan to thiophene (and dioxide thiophene) and pyrrole which are diradicals owing to the gaining of aromatic character in the central molecular part at expenses of the quinoidal closed‐shell structure. The diradical character and magnetic properties finely vary as a function of the heteroatom which also modulates the electrochemical redox and photophysical properties by the combined action of aromaticity and captodative effects. The electrical properties of the four molecules have been analyzed in OFET configurations showing ambipolar balanced hole and electron mobilities of the order of 10−3 cm2 V−1 s−1. We have addressed the presence of ambipolar electrical transport as a unique feature of the diradicaloid state in these fused planar molecules. The partial aperture of the closed‐shell configuration generates a pseudo‐hole and pseudo‐electron defect in the neutral state that, upon charging, provide similar conditions for stabilization and transport of holes and electrons resulting in amphoteric redox behavior first and then ambipolar electrical behavior. This is substantiated in the rather small and reduced reorganization energies found theoretically either for hole and for electron exchanges. In contrast, the closed‐shell structures unbalance reorganization energies in favor of holes or of electrons, destroying the best scenario for ambipolar response. In comparison to the benchmark pentacene, our difluorenoheteroles display similar reorganization energies around 0.1 eV, with the outstanding distinction that these are for holes and electrons. Electronic couplings which are also similar for holes and electrons in the most efficient intermolecular conduction channels (also in support of ambipolarity), are however smaller than in pentacene but compatible with relevant mobilities that might be reached in single crystal measurements. Grain boundaries and imperfect crystallinity thus explains the still reduced observed holes and electron mobilities. The connection between diradical character and ambipolar electrical behavior shown here might have general validity for the class of oligoacene‐like planar fused diradicals which represents a very important and new structure‐function connection that will guide future molecular design in the field.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors thank the Spanish Ministry of Science and Innovation (projects MINECO/FEDER PGC2018‐098533‐B‐100, and PID2019‐110305GB‐I00), the Junta de Andalucía and Generalidad Valenciana, Spain (UMA18FEDERJA057, P18‐FR‐4549 and Prometeo/2019/076) and JSPS KAKENHI grant (JP21K05042 for S.‐i.K., JP21K04995 and JP21H05489 for R.K., JP21H01887 and JP20K21173 for M.N.). S.‐i.K. gratefully acknowledges the Asahi Glass Foundation for financial support. We also thank the Research Central Services (SCAI) of the University of Málaga, Unidad de Espectroscopía Vibracional (Dra. Capel y Dr. Zafra) and Unidad de Optica No‐Lineal y Espectroscopía Ultrarápida (Dr. Román). This work was partially supported by the Cooperative Research Program “Network Joint Research Center for Materials and Devices” (Kyushu University). We thank Prof. Shuhei Higashibayashi (Keio University) for assistance with synthesis. Mass spectrometric data were collected at Hiroshima University (N‐BARD: Ms. Tomoko Amimoto). Theoretical calculations were partly performed using Research Center for Computational Science (R‐CCS), Okazaki, Japan. F.N and Y.D. acknowledge support from “Valutazione della Ricerca di Ateneo” (VRA)—University of Bologna. Y.D. acknowledges Ministero dell'Università e della Ricerca (MUR) for her Ph.D. fellowship.
S. Mori, S. Moles Quintero, N. Tabaka, R. Kishi, R. González Núñez, A. Harbuzaru, R. Ponce Ortiz, J. Marín-Beloqui, S. Suzuki, C. Kitamura, C. J. Gómez-García, Y. Dai, F. Negri, M. Nakano, S.-i. Kato, J. Casado, Angew. Chem. Int. Ed. 2022, 61, e202206680; Angew. Chem. 2022, 134, e202206680.
Contributor Information
Prof. Fabrizia Negri, Email: fabrizia.negri@unibo.it.
Prof. Shin‐ichiro Kato, Email: kato.s@mat.usp.ac.jp.
Prof. Juan Casado, Email: casado@uma.es.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Sirringhaus H., Adv. Mater. 2014, 26, 1319; < [DOI] [PMC free article] [PubMed] [Google Scholar]; Zaumseil J., Sirringhaus H., Chem. Rev. 2007, 107, 1296–1323. [DOI] [PubMed] [Google Scholar]
- 2. Ren Y., Yang X., Zhou L., Mao J.-Y., Han S.-T., Zhou Y., Adv. Funct. Mater. 2019, 29, 1902105. [Google Scholar]
- 3. Kim J., Baeg K.-J., Khim D., James D. T., Kim J.-S., Lim B., Yun J.-M., Jeong H.-G., Amegadze P. S. K., Noh Y.-Y., Kim D.-Y., Chem. Mater. 2013, 25, 1572. [Google Scholar]
- 4. Higashino T., Mori T., Phys. Chem. Chem. Phys. 2022, 24, 9770. [DOI] [PubMed] [Google Scholar]
- 5. Tada H., Touda H., Takada M., Matsushige K., Appl. Phys. Lett. 2000, 76, 873. [Google Scholar]
- 6. Ashizawa A., Masuda N., Higashino T., Kadoya T., Kawamoto T., Matsumoto H., Mori T., Org. Electron. 2016, 35, 95. [Google Scholar]
- 7. Yamaguchi Y., Ogawa K., Nakayama K.-i., Koganezawa T., Katagiri H., J. Am. Chem. Soc. 2013, 135, 19095. [DOI] [PubMed] [Google Scholar]
- 8. Anthopoulos T. D., Tanase C., Setayesh S., Meijer E. J., Hummelen J. C., Blom P. W. M., De Leeuw D. M., Adv. Mater. 2004, 16, 2174. [Google Scholar]
- 9. Katsuta S., Miyagi D., Yamada H., Okujima T., Mori S., Nakayama K.-i., Uno H., Org. Lett. 2011, 13, 1454. [DOI] [PubMed] [Google Scholar]
- 10. Ponce Ortiz R., Facchetti A., Marks T. J., Casado J., Zgierski M. Z., Kozaki M., Hernandez V., López Navarrete J. T., Adv. Funct. Mater. 2009, 19, 386. [Google Scholar]
- 11. Rudebusch G. E., Zafra J. L., Jorner K., Fukuda K., Marshall J. L., Arrechea-Marcos I., Espejo G. L., Ponce Ortiz R., Gómez-García C. J., Zakharov L. N., Nakano M., Ottosson H., Casado J., Haley M. M., Nat. Chem. 2016, 8, 753. [DOI] [PubMed] [Google Scholar]
- 12. Jousselin-Oba T., Mamada M., Okazawa A., Marrot J., Ishida T., Adachi C., Yassar A., Frigoli M., Chem. Sci. 2020, 11, 12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zong C., Zhu X., Xu Z., Zhang L., Xu J., Guo J., Xiang Q., Zeng Z., Hu W., Wu J., Li R., Sun Z., Angew. Chem. Int. Ed. 2021, 60, 16230; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 16366. [Google Scholar]
- 14. Sbargoud K., Mamada M., Marrot J., Tokito S., Yassar A., Frigoli M., Chem. Sci. 2015, 6, 3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jousselin-Oba T., Mamada M., Marrot J., Maignan A., Adachi C., Yassar A., Frigoli M., J. Am. Chem. Soc. 2019, 141, 9373. [DOI] [PubMed] [Google Scholar]
- 16. Chesterfield R. J., Newman C. R., Pappenfus T. M., Ewbank P. C., Haukaas M. H., Mann K. R., Miller L. L., Frisbie C. D., Adv. Mater. 2003, 15, 1278. [Google Scholar]
- 17. Ribierre J. C., Fujihara T., Watanabe S., Matsumoto M., Muto T., Nakao A., Aoyama T., Adv. Mater. 2010, 22, 1722. [DOI] [PubMed] [Google Scholar]
- 18. Yavuz I., Martin B. N., Park J., Houk K. N., J. Am. Chem. Soc. 2015, 137, 2856. [DOI] [PubMed] [Google Scholar]
- 19. Coropceanu V., Cornil J., Da Silva Filho D. A., Olvier Y., Silbey R., Brédas J. L., Chem. Rev. 2007, 107, 926. [DOI] [PubMed] [Google Scholar]
- 20. Gruhn N. E., da Silva Filho D. A., Bill T. G., Malagoli M., Coropceanu V., Kahn A., Brédas J.-L., J. Am. Chem. Soc. 2002, 124, 7918. [DOI] [PubMed] [Google Scholar]
- 21. Tönshoff C., Bettinger H. F., Angew. Chem. Int. Ed. 2010, 49, 4125; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4219. [Google Scholar]
- 22. Nakano M., Champagne B., J. Phys. Chem. Lett. 2015, 6, 3236. [Google Scholar]
- 23. Lukman S., Richter J. M., Yang L., Hu P., Wu J., Greenham N. C., Musser A. J., J. Am. Chem. Soc. 2017, 139, 18376. [DOI] [PubMed] [Google Scholar]
- 24. Hu X., Wang W., Wang D., Zheng Y., J. Mater. Chem. C 2018, 6, 11232. [Google Scholar]
- 25. Yuan D., Huang D., Medina Rivero S., Carreras A., Zhang C., Zou Y., Jiao X., McNeill C. R., Zhu X., Di C., Zhu D., Casanova D., Casado J., Chem 2019, 5, 964. [Google Scholar]
- 26.
- 26a. Doehnert D., Koutecky J., J. Am. Chem. Soc. 1980, 102, 1789; [Google Scholar]
- 26b. Salem L., Rowland C., Angew. Chem. Int. Ed. Engl. 1972, 11, 92; [Google Scholar]; Angew. Chem. 1972, 84, 86. [Google Scholar]
- 27. Mori S., Akita M., Suzuki S., Asano M. S., Murata M., Akiyama T., Matsumoto T., Kitamura C., Kato S.-i., Chem. Commun. 2020, 56, 5881. [DOI] [PubMed] [Google Scholar]
- 28.Deposition Numbers 1974502 (for DFFu), 2117204 (for DFTh), 2117205 (for DFPy) and 2165958 (for DFThO 2 ) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 29. Gershoni-Poranne R., Stanger A., Chem. Eur. J. 2014, 20, 5673–5688. [DOI] [PubMed] [Google Scholar]
- 30. Viehe H. G., Janousek Z., Merényi R., Stella L., Acc. Chem. Res. 1985, 18, 148. [Google Scholar]
- 31. Bleaney B., Bowers K. D., Proc. R. Soc. London Ser. A 1952, 214, 451. [Google Scholar]
- 32. Hayashi H., Barker J. E., Cárdenas Valdivia A., Kishi R., MacMillan S. N., Gómez-García C. J., Miyauchi H., Nakamura Y., Nakano M., Kato S.-i., Haley M. M., Casado J., J. Am. Chem. Soc. 2020, 142, 20444. [DOI] [PubMed] [Google Scholar]
- 33. Rose B. D., Shoer L. E., Wasielewski M. R., Haley M. M., Chem. Phys. Lett. 2014, 616, 137. [Google Scholar]
- 34. Di Motta S., Negri F., Fazzi D., Castiglioni C., Canesi E. V., J. Phys. Chem. Lett. 2010, 1, 3334. [Google Scholar]
- 35. Varnavski O., Abeyasinghe N., Aragó J., Serrano-Pérez J. J., Ortí E., López Navarrete J. T., Takimiya K., Casanova D., Casado J., Goodson T., J. Phys. Chem. Lett. 2015, 6, 1375. [DOI] [PubMed] [Google Scholar]
- 36. Canola S., Casado J., Negri F., Phys. Chem. Chem. Phys. 2018, 20, 24227. [DOI] [PubMed] [Google Scholar]
- 37. Canola S., Negri F., Phys. Chem. Chem. Phys. 2014, 16, 21550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.