

Abstract
Special AT‐binding protein 1 (SATB1) is a chromatin‐binding protein that has been shown to be a key regulator of T‐cell development and CD4+ T‐cell fate decisions and function. The underlying function for SATB1 in peripheral CD8+ T‐cell differentiation processes is largely unknown. To address this, we examined SATB1‐binding patterns in naïve and effector CD8+ T cells demonstrating that SATB1 binds to noncoding regulatory elements linked to T‐cell lineage–specific gene programs, particularly in naïve CD8+ T cells. We then assessed SATB1 function using N‐ethyl‐N‐nitrosourea‐mutant mice that exhibit a point mutation in the SATB1 DNA‐binding domain (termed Satb1m1Anu/m1Anu). Satb1m1Anu/m1Anu mice exhibit diminished SATB1‐binding, naïve, Satb1m1Anu/m1Anu CD8+ T cells exhibiting transcriptional and phenotypic characteristics reminiscent of effector T cells. Upon activation, the transcriptional signatures of Satb1m1Anu/m1Anu and wild‐type effector CD8+ T cells converged. While there were no overt differences, primary respiratory infection of Satb1m1Anu/m1Anu mice with influenza A virus (IAV) resulted in a decreased proportion and number of IAV‐specific CD8+ effector T cells recruited to the infected lung when compared with wild‐type mice. Together, these data suggest that SATB1 has a major role in an appropriate transcriptional state within naïve CD8+ T cells and ensures appropriate CD8+ T‐cell effector gene expression upon activation.
Keywords: CD8+ T cell, chromatin, influenza A virus, memory T cell, naïve T cell
We examine the role of special AT‐binding protein 1 (SATB1), a chromatin‐binding protein, in the establishment of naïve and virus‐specific CD8+ T cells.
![]()
INTRODUCTION
Upon antigen recognition, naïve CD8+ T‐cell activation results in a program of proliferation and differentiation upon antigen recognition. This proliferative response correlates with acquisition of lineage‐specific effector function that includes expression of cytotoxic proteins such as granzymes (GZM) and inflammatory cytokines such as interferon‐gamma, all of which contribute to control and clearance of viral infection. Upon control of infection, the effector CD8+ T‐cell population contracts in number resulting in the establishment of a population of virus‐specific memory T cells that persist in the long term. In contrast to naïve CD8+ T cells, virus‐specific memory CD8+ T cell respond rapidly to secondary infection contributing to accelerated response and control of secondary infections.
Virus‐specific CD8+ T‐cell differentiation is associated with a stepwise progression of specific transcription factor expression that is critical for optimal CD8+ T‐cell responses. For example, upregulation of BATF, together with IRF4 and members of the JUN transcription factors family, drive immune, cell survival and metabolic gene transcription early after initial T‐cell activation. 1 , 2 Essential transcriptions for effective CD8+ T‐cell differentiation include RUNX3 and BLIMP1 (encoded by Prdm1). 1 , 2 , 3 , 4 , 5 T‐BET and RUNX3 function early upon T‐cell activation to drive acquisition of effector CD8+ T‐cell fucntion, 3 , 6 whereas terminal effector CD8+ T‐cell differentiation is dependent on BLIMP1. 4 By contrast, the naïve CD8+ T‐cell state is also associated with signature transcription factors such as TCF‐1, LEF‐1, FOXO1 and BACH2. More recently, FOXO1 was shown to repress effector CD8+ T‐cell differentiation by upregulating BACH2, which in turn occupies binding sites for AP1 family transcription factors that are required to drive effector CD8+ T‐cell differentiation. 7 , 8 Together, these data suggest that commitment of activated T cell to an effector differentiation state requires shutdown of a naïve CD8+ T‐cell transcriptional program.
Special AT‐binding protein 1 (SATB1) binds to nuclear matrix‐associated DNA regions and plays a key role in organizing higher‐order chromatin structures to regulate cell‐specific gene transcription. 9 SATB1 is highly expressed by immature T cells, with SATB1 deficiency resulting in dysregulation of T‐cell–specific genes and a block in T‐cell development. 10 It has multiple roles during T‐cell development including regulating appropriate transcription of the RAG locus within CD4+ and CD8+ T‐cell precursors ensuring appropriate T‐cell gene rearrangement, 11 binding to and regulating the interleukin (IL)‐2 receptor alpha (encoded by Il2ra) locus, 12 as well as playing a role in helping establish appropriate organization of the chromatin landscape at key T‐cell lineage–specifying genes prior to commitment. 13 Hence, SATB1 ensures appropriate temporal and cell‐specific regulation of T‐cell lineage developmental programs.
SATB1 is upregulated upon naïve CD4+ T‐cell activation under TH2 skewing conditions, where it plays a role in reorganizing the IL‐4/5/13 locus into a transcriptionally permissive chromatin landscape. 14 Deletion of SATB1 within mature TH17 CD4+ T cells results in a decrease in pathogenic effector function and protection from autoimmune disease in a murine model of experimental autoimmune encephalitis. In this case, SATB1 expression results in engagement of transcriptional programs that resulted in increased expression of pathogenic cytokines and decreased expression of checkpoint receptors such as PD‐1 (encoded by Pdcd1). 15 Similarly, overexpression of SATB1 in CD4+ T regulatory (Treg) cells overcame FOXP3‐mediated SATB1 repression and resulted in induction effector transcriptional programs. 16 Hence, SATB1 downregulation ensures appropriate lineage‐specific CD4+ Treg cell function.
The role of SATB1 in mature CD8+ T cells is less clear. We and others have recently showed that naïve CD8+ T‐cell activation results in initial SATB1 upregulation with SATB1 levels decreasing with extended T‐cell differentiation. 17 , 18 In a manner similar to TH17 cells, SATB1 was shown to repress PD‐1 expression in recently activated CD8+ T cells by directly targeting the Pdcd1 locus and recruiting the NuRD histone deacetylase complex. This results in histone deacetylation and repression of Pdcd1 transcription. 15 Despite these observations, the role of SATB1 in regulating virus‐specific CD8+ T‐cell responses has not been adequately addressed. Here we examine SATB1 expression and genome‐binding profiles within naïve and virus‐specific effector CD8+ T cells, as well as examining the virus‐specific CD8+ T‐cell response within mice that contain a point mutation in the SATB1 DNA‐binding domain (termed Satb1m1Anu/m1Anu mice). We demonstrate that diminished SATB1 binding within naïve CD8+ T cells resulted in a transcriptional landscape that was more similar to an effector‐like differentiation state. Interestingly, upon infection, the transcriptional profiles of effector wild‐type (WT) and Satb1m1Anu/m1Anu effector CD8+ T cells converged showing little difference. Satb1m1Anu/m1Anu mice showed delayed viral clearance, concurrent with reduced influenza A virus (IAV)–specific T cells numbers in the lungs. Generation of mixed bone marrow (BM) chimeras demonstrated this defect was immune cell specific rather a result of environmental effects. Overall, these data suggest that SATB1 expression in naïve CD8+ T cells is important for maintaining naïve CD8+ T cells and that disruption of SATB1 binding results in dysregulated transcriptional activation of key effector genes.
RESULTS
SATB1 binds to largely unique noncoding regulatory elements in naïve and IAV‐specific CD8+ T cells
We previously reported that SATB1 expression is dynamically regulated across human CD8+ T‐cell subsets, 17 and examination of our previously published RNA‐seq data from mice 18 demonstrated a consistent pattern, whereby naïve CD8+ T cells (CD44loCD62Lhi) have higher levels of SATB1 transcription, compared with effector (day 10 after infection, tetramer+CD8+) and memory (tetramer+CD44hi; > day 60) CD8+ T cells (Figure 1a). Naïve CD8+ T cells also exhibit higher SATB1 protein levels than effector CD8+ T cells (Figure 1b, c). Interestingly, central memory (CD44hiCD62Lhi) cells had higher SATB1 levels compared with effector memory T cells (TCD44hiCD62Llo; Figure 1b, c). Examination of SATB1 expression in human CD4+ and CD8+ T‐cell subsets also showed the same pattern of expression (Supplementary figure 1).
Figure 1.
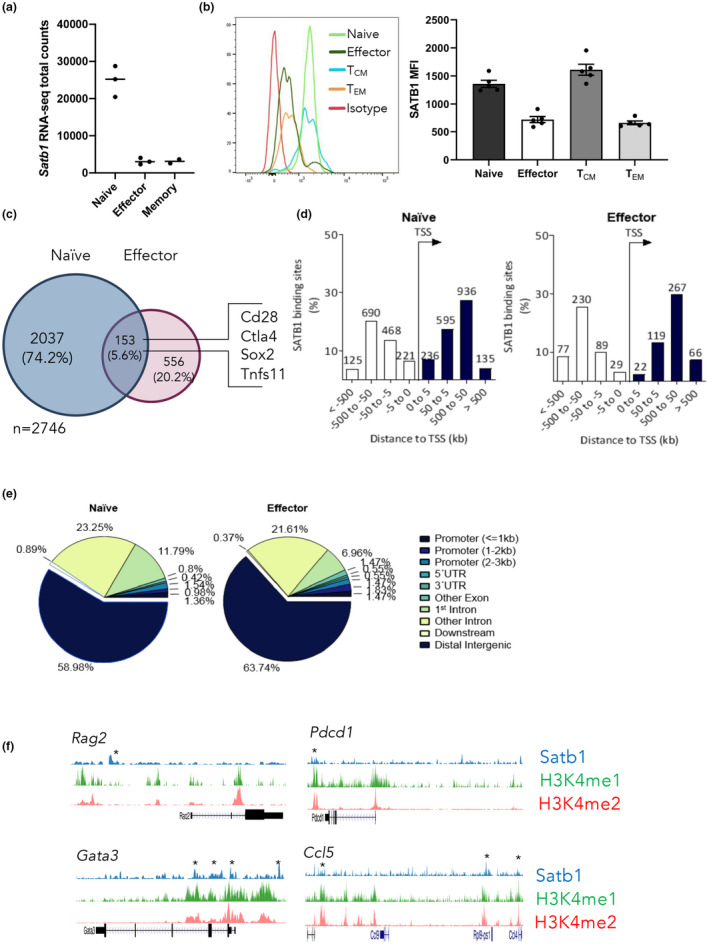
Special AT‐binding protein 1 (SATB1) is expressed in naïve CD8+ T cells and downregulated upon differentiation. (a) Analysis of RNA sequencing data 18 comparing SATB1 transcript levels in naïve (CD44loCD62Lhi), effector (day 10 after infection, tetramer+CD8+) and memory (tetramer+CD44hiCD62Llo; > day 60) CD8+ T cells before and after 5‐h peptide stimulation. (b) Protein expression of SATB1 in naïve, effector and memory including effector memory T cell or central memory T cell (tetramer+CD44hiCD62Lhi) CD8+ T cells. (c) SATB1 chromatin immunoprecipitation sequencing (ChIP‐seq) was carried out on pooled naïve (3) (CD44loCD62Lhi) or tetramer+ CD8+ influenza A virus–specific CD8+ T cells (10 mice/pool) in duplicate. Data were mapped back to the mouse genome (version mm10) and SATB1 peaks called. The number of peaks observed in naïve, effector or naïve and effector CD8+ T cells is shown. (d, e) SATB1 peaks identified in naïve or effector CD8+ T cells were mapped to the mouse genome and the distance (d) to the nearest neighboring transcriptional start site, and the type of genomic region (e) was determined. (f) SATB1‐binding tracks are shown overlaid with H3K4me1 and H3K4me2 ChIP‐seq data from naïve or effector CD8+ T cells. 21 Error bars show mean ± s.d. An unpaired t‐test was used. *P ≥ 0.05. [Colour figure can be viewed at wileyonlinelibrary.com]
The expression data suggest that SATB1 may contribute to maintenance of the naïve CD8+ T‐cell differentiation state. We therefore sought to identify genome‐wide targets of SATB1 binding within antigen‐specific, naïve and effector CD8+ T cells responding to an acute influenza A infection. To address this, we utilized our previously described adoptive transfer model where naïve (CD44lo, CD62Lhi) OT‐I T‐cell receptor (TCR) transgenic CD8+ T cells (specific for the ovalbumin peptide OVA257–264 presented by H2‐Kb) are adoptively transferred into congenic C57BL/6J (B6) hosts. This is followed by intranasal infection with the influenza A/HKx31‐OVA virus. 19 Naïve or effector (day 10 spleen, 4–8% of total CD8+ T cells; Supplementary figure 2a, b) were sort purified for subsequent SATB1 ChIP‐seq (Supplementary figure 2c, d). In total, 2190 SATB1‐binding sites were identified in naïve T cells, and 709 binding sites were identified in effector cells (Figure 1c, Supplementary figure 2c, d), with only a small proportion of SATB1‐binding peaks that overlapped between naïve and effector CD8+ T cells (about 6%; Figure 1c). This difference in binding sites is consistent with a downregulation of SATB1 expression during effector differentiation. To understand how SATB1 might influence T‐cell differentiation, unique binding sites observed within naïve or effector CD8+ T cell data sets were mapped relative to the transcriptional start site of nearest genes (Figure 1c). Interestingly, in both differentiation states, SATB1 bound most often at distal noncoding genomic regions (about 60% of sites) 50–500 kb from transcriptional start sites (Figure 1d, e). This supports earlier observations that SATB1 regulates transcription through interactions with noncoding regulatory elements. 12 , 15 Colocalization of H3K4me1 and H3K4me2 identifies putative transcriptional enhancers. 20 To explore whether SATB1 binding was enriched at transcriptional enhancers, we utilized our previously published H3K4me1 and H3K4me2 ChIP‐seq data 21 to assess the overlap of SATB1 binding with putative transcriptional enhancers. While SATB1 was observed to bind at enhancers of genes encoding important CD8+ T‐cell genes including Gata3 and Pdcd1 (Figure 1f), a broad analysis demonstrated that only a small proportion of SATB1 peaks overlapped with H3K4me1+me2+ transcriptional enhancers (about 14% of SATB1 peaks overlapped transcriptional enhancers; Supplementary figure 2e).
To assess whether SATB1 binding to noncoding genomic regions was linked to immune cell function, we utilized GREAT for gene ontology analysis 22 to gain a further understanding of the biological function of genes associated with SATB1 binding within the naïve T‐cell state (Supplementary figure 2f). SATB1 binding was evident at genomic regions associated with genes with both broad and T‐cell–specific immunological function (Supplementary figure 2f). Targets of SATB1 binding included genes such as immune receptors (Ccr7, Ccr5, Cxcr3, Cxcr5, Xcr1), costimulatory and inhibitory checkpoint molecules (Bcl2, Cd28, Cish, CD8 + T cella4, Havcr2, Icos, Pdcd1, Tigit), effector molecules (Gzmb, Gzmm, Ifng, Il7, Il12, Il15), cytokine receptors (Il2ra, Il2rb, Il4ra, Il7r, Il15ra, Il17ra) and key naïve (Bcl6, Bcl11b, Foxo1, Lef1, Tcf7) and effector (Bhlhe40, Gata3, Hic1, Irf4, Prdm1, Runx3, Stat5a, Stat5b, Tox, Zeb1, Zeb2) transcription factors. Together, these data show that SATB1 binds to genomic sites associated with key T‐cell biological processes and suggests SATB1 is key for regulation of CD8+ transcriptional programs.
Altered T‐cell selection and dysregulated naïve T‐cell generation in Satb1m1Anu/m1Anu mice
In a random N‐ethyl‐N‐nitrosourea mutagenesis screen aimed at identifying genes that regulate key aspects of adaptive immunity, we identified a pedigree in which some mice exhibited low CD44 expression on naïve CD4+ and CD8+ peripheral T cells (Supplementary figure 3a). The Satb1m1Anu mutation (NM_001163630.1:c.1179 T > A) was identified by exome sequencing of a mouse that exhibited low CD44 expression (Supplementary figure 3c). This mutation results in a position 393 Phe → Leu amino acid exchange within the N‐terminal DNA‐binding (CUT1) domain of SATB1. 23 Importantly, mice homozygous for the SATB1m1Anu mutation are viable, unlike global SATB1‐deficient mice that exhibit a shortened life expectancy. 24 The SATB1m1Anu mutation segregated with low CD44 expression in CD8+ T cells in a semidominant manner (Supplementary figure 3b). Immunoblot analysis demonstrated that Satb1m1Anu mutant mice expressed similar levels of SATB1 protein, indicating that this mutation did not impact protein translation or stability (Supplementary figure 3d). To determine whether the SATB1 m1Anu protein could bind DNA, we performed ChIP on double positive (CD4+CD8+) thymocytes from WT and Satb1m1Anu/m1Anu mice. While WT mice showed clear enrichment of the SATB1 binding at the Rag locus and SBS II upstream of Ccl5, there was a complete abolition of binding in Satb1m1Anu/m1Anu mice, suggesting that SATB1m1Anu‐mutant protein has altered DNA‐binding capacity (Supplementary figure 3d).
SATB1 has been previously demonstrated to play a role in the thymic development of conventional T cells, natural killer T cells and Foxp3+ Treg cells. 10 , 13 , 25 More recently, a preliminary examination of Satb1m1Anu/m1Anu mutant mice showed a defect in the thymic development of mucosal‐associated invariant T cells. 26 A key step in T‐cell selection is the deletion of self‐reactive thymocytes, where approximately half of all TCR‐signaled thymocytes undergo apoptosis prior to upregulating CCR7 and are marked by upregulation of the transcription factor HELIOS and PD‐1. 27 , 28 Compared with WT mice, Satb1m1Anu/m1Anu mice had a slightly higher frequency of HELIOS+ PD1+ thymocytes (Supplementary figure 4a). To examine this in more depth, we utilized BCL‐2–transgenic mice in which BCL‐2 expression rescues strongly TCR‐signaled thymocytes from BIM‐dependent apoptosis. 27 , 28 Interestingly, the extent of BCL‐2 rescue of HELIOS+ PD1+ thymocytes was greater in WT mice than in Satb1m1Anu/m1Anu mice (Supplementary figure 4b). Those findings suggest the Satb1m1Anu/m1Anu genotype confers a decrease in the frequency of CCR7− thymocytes that register a strong TCR signal as well as a decrease in the efficiency of apoptosis within these cells. At the subsequent CCR7+ CD4SP stage, the frequency of HELIOS+ FOXP3− cells was also increased in Satb1m1Anu/m1Anu mice in the absence of BCL‐2‐tg expression, consistent with a decrease in the efficiency of apoptosis (Supplementary figure 4c). We excluded the possibility that the increased frequency of HELIOS+ thymocytes in the Satb1m1Anu/m1Anu mice reflects an increase in the HELIOS+ thymocyte formation rate as a result of lowering of the TCR signaling threshold for HELIOS upregulation. Satb1m1Anu/m1Anu cells had lower expression of molecular markers of T‐cell activation, including BIM, CD5, CD69, ICOS and CD44, whereas TCRβ expression levels were similar (Supplementary figure 4d). In particular, the defect in BIM upregulation may explain the decreased efficiency of apoptosis in Satb1m1Anu/m1Anu thymocytes (Supplementary figure 4d). Altogether, these data indicate that the Satb1m1Anu/m1Anu mutation confers defects in TCR signaling and apoptosis in thymocytes.
The impact of Satb1m1Anu mutation on the peripheral CD8+ T‐cell naïve repertoire
Given Satb1m1Anu/m1Anu mice exhibited dysregulated thymic development, we next sought to characterize the peripheral T‐cell compartment. In comparison to WT mice, Satb1m1Anu/m1Anu mice had reduced proportions of CD3+CD8+ T cells, CD4+ T cells and γδ T cells (Figure 2a). While there was no difference in the total number of lymphocytes between WT and Satb1m1Anu/m1Anu mice (Figure 2b), there was a decrease in absolute numbers of conventional CD3+CD8+ T and γδ T cells in Satb1m1Anu/m1Anu mice (Figure 2b).
Figure 2.
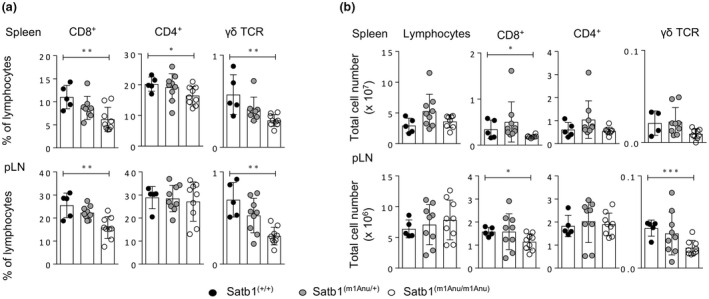
Assessment of peripheral T‐cell subsets in wild‐type (WT) and Satb1m1Anu/m1Anu mice. (a) The proportion of CD8+, CD4+ and γδ T cells was determined for lymphocytes in the spleen and peripheral lymph nodes from WT or Satb1m1Anu/m1Anu mice. (b) Quantitation of total numbers of CD8+, CD4+ and γδ T cells from WT or Satb1m1Anu/m1Anu mice. Error bars show mean ± s.d. of five mice for three independent experiments. Unpaired Student's t‐tests were used. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. pLN, popliteal lymph node; SATB1, special AT‐binding protein 1; TCR, T‐cell receptor.
To further characterize the peripheral CD4+ and CD8+ T‐cell repertoire, we assessed the expression of the surface markers CD44, CD25, the glucocorticoid‐induced tumor necrosis factor–related (GITR) protein and the GATA3 transcription factor (both identified as a SATB1 target; Supplementary table 1). As observed for developing thymocytes, both naïve CD4+ and CD8+ T cells expressed lower levels of CD44 (Figure 3a–c), and increased levels of GITR (Figure 3a–c); however, there was no difference in CD25 expression. Further, GATA3 was upregulated in both the spleen and popliteal LN‐derived CD8+ T cells (Figure 3d, e). Importantly, no difference was observed in SATB1 expression between Satb1m1Anu/m1Anu and WT CD8+ T cells (Figure 3d, e). These data suggest that dysregulated SATB1 binding in naïve CD8+ T cells results in the altered naïve T‐cell expression profiles.
Figure 3.
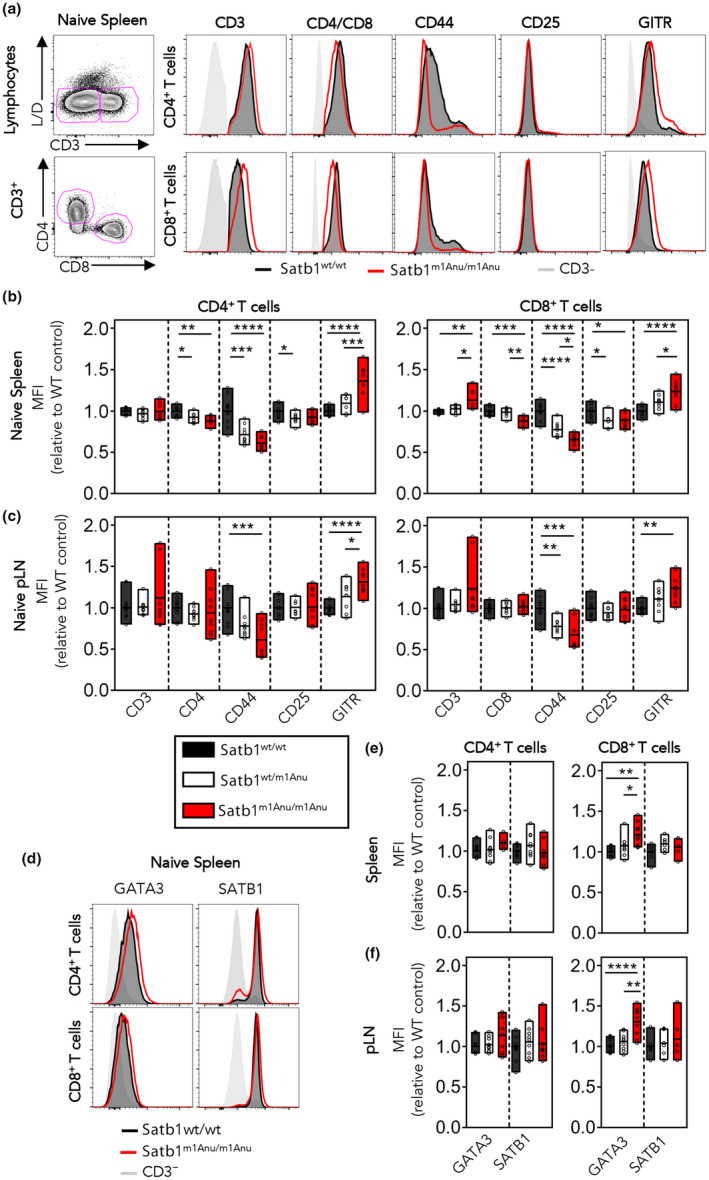
Dysregulated expression of cell surface proteins within Satb1m1Anu/m1Anu T cells. (a) Representative flow cytometry plots and histograms for CD3, CD4 or CD8 with CD44, CD25 and GITR on lymphocytes from the spleen of naïve SATB1+/+, Satb1m1Anu/+ and Satb1m1Anu/m1Anu1 mice. (b, c) Fold change in mean fluorescence intensity (MFI) of cell surface markers on CD4+ or CD8+ T cells from the spleen (b) or lymph node (c). (d) Representative histograms of GATA3 and SATB1 expression within naïve CD4+ or CD8+ T cells, or CD3− lymphocytes. (e, f) Fold change in GATA3 and SATB1 MFI within CD4+ or CD8+ T cells from the spleen (e) or lymph node (f) from wild‐type (WT), Satb1m1Anu/+ or Satb1m1Anu/m1Anu1 mice. Error bars show mean ± s.d. of three or five mice from two independent experiments. Unpaired Student's t‐tests were used. *P < 0.05, **P < 0.01, ***P < 0.001, ****P ≤ 0.0001. pLN, popliteal lymph node; SATB1, special AT‐binding protein 1. [Colour figure can be viewed at wileyonlinelibrary.com]
Virtual memory T cells (TVMs) are a subset of semidifferentiated T cells that are antigen naïve, but exhibit heightened responsiveness to TCR stimulation and common γ‐chain cytokines IL‐7 and IL‐15. 29 , 30 They can be identified via high expression of CD44, CD62L but low‐level CD49d expression. 31 Hence, the observed alterations in CD44 expression within the naïve Satb1m1Anu/m1Anu CD8+ T‐cell compartment may reflect alterations in the proportion of TVM. Evaluation of CD44hiCD49dlo TVM within the naïve CD8+ T‐cell compartment (Figure 4a) demonstrated that Satb1m1Anu/m1Anu mice did in fact exhibit both a lower proportion and fewer total TVMs compared with WT mice (Figure 4b). While there was no difference in the total number of naïve (CD44loCD49dlo) CD8+ T cells, the decrease in TVM coincided with an increase in the proportion of memory phenotype CD8+ T cells, but not necessarily total number (Figure 4b). Together, these data suggest that SATB1 plays a role in regulating TVM within the naïve CD8+ T‐cell compartment.
Figure 4.
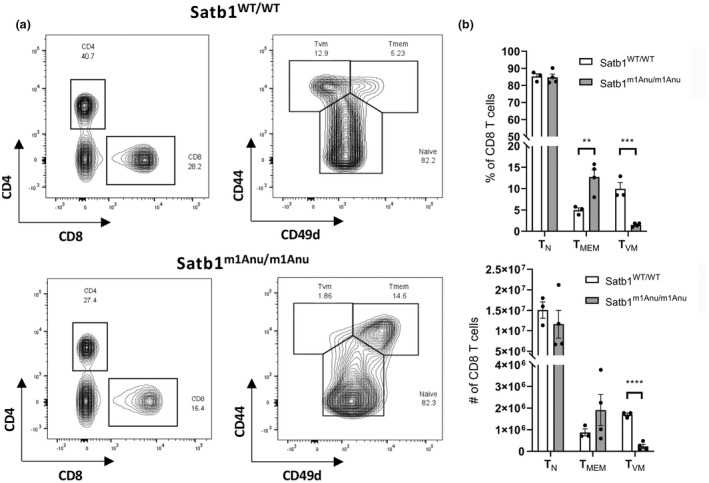
Loss of the virtual memory (TVM) CD8+ T cells in Satb1m1Anu/m1Anu mice. (a) Representative fluorescence‐activated cell sorting plot gating of wild type (WT) and Satb1m1Anu/m1Anu to determine proportion of naïve (TN), memory (TMEM) and TVM CD8 T cells. (b) Proportion and number of TN, TMEM and TVM CD8 T cells. (c) Proportion of CD44lo and CD44int CD8 TN in WT versus Satb1m1Anu/m1Anu in the spleen. WT n = 3; Satb1m1Anu/m1Anu n = 4; representative of two independent experiments. Unpaired Student's t‐tests were used. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). SATB1, special AT‐binding protein 1.
The Satb1m1Anu mutation dysregulates transcription primarily in naïve CD8+ T cells
Given the observation that the Satb1m1Anu/m1Anu mutation altered DNA binding to target gene loci, and the observed perturbation of expression of some cell surface markers and GATA3, RNA sequencing was performed on naïve (CD62Lhi) and IAV‐specific effector CD8+ T cells from WT and Satb1m1Anu/m1Anu mice. For IAV infection, mice were challenged with 104 plaque‐forming units A/HKx31 (H3N2) virus with tetramer+ (DbPA224 and DbNP366)‐specific CD8+ T cells isolated 10 days after infection.
To determine global differences in data sets and to assess data quality, multidimensional scaling was performed. This analysis showed a tight clustering of biological replicates, with the largest difference observed between the naïve splenic CD8+ T cells of WT and Satb1m1Anu/m1Anu mice, with the transcriptional profiles converging following IAV‐specific effector (tetramer+ CD44hi; day 10 spleen) differentiation (Figure 5a, b). A larger number of genes were upregulated in naïve Satb1m1Anu/m1Anu CD8+ T cells relative to the WT, including immune checkpoint–encoding genes Pdcd1, Lag3 and CTLA‐4; transcription factors such as Foxp3 and Zbtb32 as well as chemokines such as Ccl5 (Figure 5c; fold change of > 1.5; false discovery rate < 0.05). Interestingly, there was also increased transcription of TCR gamma constant chains (Tcrg‐C2, Tcrg‐C4), which may indicate altered TCR gene regulation (Figure 5c). Despite convergence of differential gene expressions between effector Satb1m1Anu/m1Anu and WT IAV‐specific CD8+ T cells (Figure 5a, d), effector molecules such as Gzma and Gzmb, and cell surface markers such as Tigit and Itgae were upregulated in Satb1m1Anu/m1Anu IAV‐specific CD8+ T cells (Figure 5d). Unexpectedly, TCR‐gamma chain–encoding genes such as Tcrg‐c2, Tcrg‐v2 and Tcrg‐v4 were also upregulated Satb1m1Anu/m1Anu IAV–specific CD8+ T cells (Figure 5d).
Figure 5.
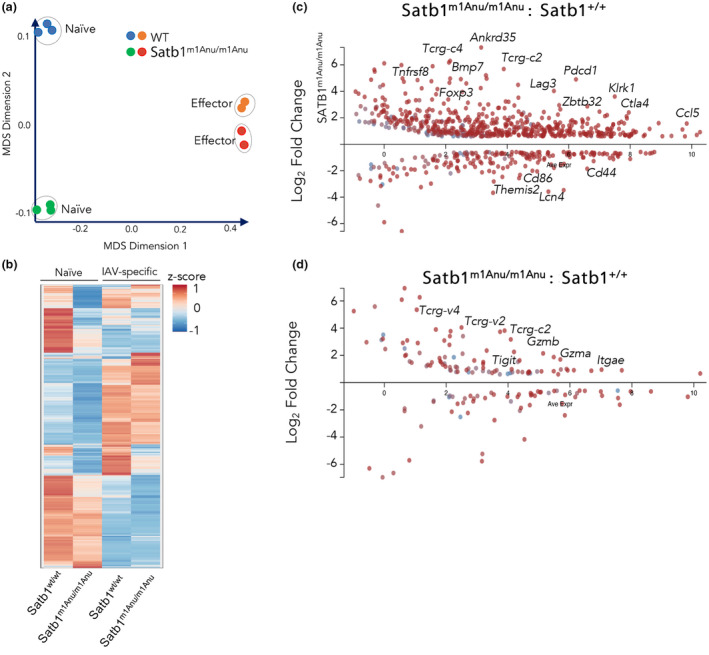
Differential transcriptional profiles between naïve CD8+ T cells from wild‐type (WT) and Satb1m1Anu/m1Anu mice. RNA sequencing was performed on either naïve (CD44loCD62Lhi) CD8+ T cells from uninfected WT or Satb1m1Anu/m1Anu mice, or on purified DbNP366/DbPA224 influenza A virus–specific CD8+ T cells 10 days after primary infection with A/HKx31. (a) Multidimensional scaling (MDS), (b) hierarchical clustering based on Z‐score and volcano plots of (c) naïve or (d) effector transcriptional profiles with highlighted genes are shown. RNA‐seq libraries were generated in triplicate from pooled samples of three to five mice per replicate. SATB1, special AT‐binding protein 1. [Colour figure can be viewed at wileyonlinelibrary.com]
Given the higher levels of effector mRNA for PD‐1 and GZM within naïve Satb1m1Anu/m1Anu CD8+ T cells in the steady state, we examined whether the Satb1m1Anu/m1Anu mutation also resulted in greater PD‐1 and granzyme protein expression upon activation (Supplementary figure 5). Naïve WT or Satb1m1Anu/m1Anu CD8+ T cells were activated in vitro with α‐CD3/α‐CD28 and exhibited similar levels of cell division as measured by Cell Trace Violet dilution (Supplementary figure 5a). Satb1m1Anu/m1Anu CD8+ T cells expressed higher levels of PD‐1 across all time points assayed (Supplementary figure 5b). A greater proportion of activated Satb1m1Anu/m1Anu CD8+ T cells expressed GZMB and GZMA at days 3 and 5, but not at day 7 after activation compared with WT CD8+ T cells (Supplementary figure 5c, d). Satb1m1Anu/m1Anu CD8+ T cells also expressed higher levels of GZMB (Supplementary figure 5c), but not GZMA on a per‐cell basis (Supplementary figure 5d) at days 3 and 5 after activation compared with WT CD8+ T cells. Overall, these data support the notion that Satb1m1Anu/m1Anu CD8+ T cells exhibit a heightened effector state reflected by early upregulation of effector markers upon activation (Figure 6).
Figure 6.
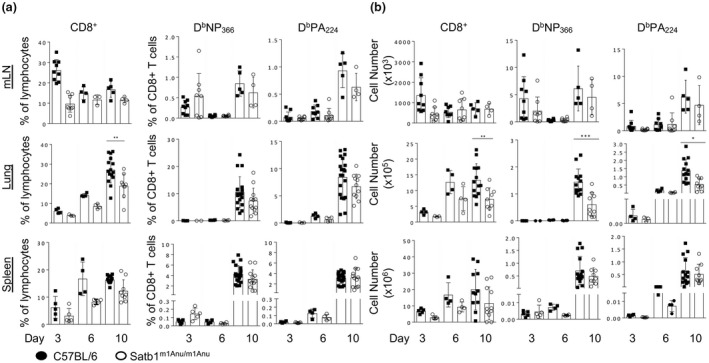
Analysis of primary influenza A virus–specific CD8+ T‐cell responses in wild‐type (WT) and Satb1m1Anu/m1Anu mice. WT and Satb1m1Anu/m1Anu mice were infected intranasally with A/HKx31 and lymphocytes from the mediastinal lymph node (mLN), lung tissue and spleen were analyzed. The (a) proportion and (b) total number of CD8+, DbNP366‐ and DbPA224‐specific CD8+ T cells in mLN, lung and spleen 3, 6 and 10 days after primary infection. Error bars show mean ± s.d. of five mice from two independent experiments. Unpaired Student's t‐tests were used. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. SATB1, special AT‐binding protein 1.
Satb1m1Anu/m1Anu mice exhibit impaired IAV‐specific CD8+ T cells recruitment to the lung
To examine whether there was a potential impact of the SATB1m1Anu mutation on virus immunity, WT and Satb1m1Anu/m1Anu mice were infected with the A/HKx31 influenza virus and lungs harvested on different days after infection to study the kinetics of viral clearance (Supplementary figure 6). While no significant differences in body weight loss were observed between WT and Satb1m1Anu/m1Anu mice (Supplementary figure 6a), Satb1m1Anu/m1Anu mice showed a slight delay in viral clearance on day 7 after infection (Supplementary figure 6b). The delay in viral clearance could be indicative of a diminished CD8+ T‐cell response. To assess this, WT and Satb1m1Anu/m1Anu mice were infected with A/HKx31 and lymphocytes sampled from the spleen, lung and draining (mediastinal) lymph node (mesenteric LN) at days 3, 5 (early expansion) or day 10 (peak expansion) after infection. IAV‐specific CD8+ T cells were assessed using DbPA224 and DbNP366 tetramers (Supplementary figure 6c). Despite Satb1m1Anu/m1Anu mice exhibiting a greater proportion of CD8+ T cells in the mesenteric LN at day 3 after infection, the number of tetramer‐positive CD8+ T cells at days 3 and 5 was at the limit of detection, with tetramers in the LN and lung making observations inconclusive. A greater sampling size within spleen enabled analysis demonstrating no difference in T‐cell numbers during the early phases of the T‐cell responses to IAV infection (Figure 6a, b). At the peak of the response (day 10), Satb1m1Anu/m1Anu mice demonstrated reduced proportions and absolute numbers of tetramer+ CD8+ T cells in the lung (Figure 6a, b). The decrease in tetramer+ CD8+ T cells was also reflected in total CD8+ T‐cell number recruited to the lungs of Satb1m1Anu/m1Anu mice (Figure 6a, b). There was no difference in the functional capacity of WT and Satb1m1Anu/m1Anu mice with a similar proportion of interferon‐gamma or tumor necrosis factor production observed after 5 h of peptide stimulation (Supplementary figure 6d). Thus, taken together, these data indicate that there is a reduced number of virus‐specific effector CD8+ T cells in the infected lung in Satb1m1Anu/m1Anu mice, consistent with the delayed viral clearance observed in these mice.
We next examined expression of PD‐1 on naïve CD4+ and CD8+ T cells (Supplementary figure 7a, b). In the CD4+ T‐cell compartment, naïve Satb1wt/wt T cells exhibited low levels of PD‐1 with increased levels of PD‐1 in CD44int and CD44hi subsets (Supplementary figure 7a, c; day 0). In line with our RNA‐seq data (Figure 4b) and in vitro activation data (Supplementary figure 5b), higher levels of PD‐1 expression were observed on naïve Satb1m1Anu/m1Anu CD4+ T cells, particularly those found within the CD44int and CD44hi subset (Supplementary figure 7a, c; day 0). In the CD8+ T‐cell compartment, naïve CD8+ T cells of WT mice expressed low levels of PD‐1, regardless of CD44 status (Supplementary figure 7b, d), while CD44int and CD44hi CD8+ T cells of Satb1m1Anu/m1Anu mice had increased PD‐1 expression (Supplementary figure 7b, d), consistent our RNA‐seq data. After IAV infection, PD‐1 expression was upregulated on days 10, 14 and 30 on splenic CD4+ (Supplementary figure 7c) and CD8+ T cells (Supplementary figure 7d). In line with PD‐1 expression in the steady state, Satb1m1Anu/m1Anu CD4+ and CD8+ T cells exhibited higher levels of PD‐1 across all time points, compared with WT. These results suggest that the reduced IAV‐specific CD8+ T‐cell numbers observed in the lung after A/HKx31 infection within Satb1m1Anu/m1Anu mice could be because of higher levels of PD‐1 expression in the naïve state and over the course of infection.
Satb1m1Anu/m1Anu mice exhibit altered peripheral Foxp3 T regulatory T‐cell numbers
Treg cells express the lineage‐specific transcription factor, FOXP3, and have been shown to limit expansion of virus‐specific CD8+ T cells. 32 , 33 Our RNA‐seq data showed that Satb1m1Anu/m1Anu CD8+ T cells had higher levels of Foxp3 transcript compared with Satb1wt/wt CD8+ T cells. Moreover, given the role of Treg cells in limiting virus‐specific CD8+ T‐cell responses, 32 , 33 , 34 and SATB1 regulation of FOXP3, 16 , 25 we investigated whether the Satb1m1Anu mutation might impact peripheral Treg cell numbers in uninfected and infected mice. Naïve Satb1m1Anu/m1Anu mice exhibited an increased proportion and number of conventional CD4+ Treg cells (Foxp3hiGITR+) as well as an unconventional Foxp3 intermediate (Foxp3int) CD4+ population (Figure 7a, c). As overall CD4+ T‐cell numbers were similar between WT and Satb1m1Anu/m1Anu mice, the increase in Foxp3+ CD4+ T cells is consistent with the observed decrease in the virus‐specific CD8+ T‐cell response in the infected lungs. Despite the overall reduction in CD8+ T cells, both CD8+ Treg and CD8+ FOXP3int Treg cells were increased proportionally in naïve and effector Satb1m1Anu/m1Anu CD8+ T cells compared with WT mice (Figure 7b, d). Taken together, these data suggest the Satb1m1Anu mutation results in a perturbation of both CD4+ and CD8+ Treg cell populations, which in turn results in the compromised virus‐specific CD8+ T‐cell responses we observed following influenza challenge of these mice.
Figure 7.
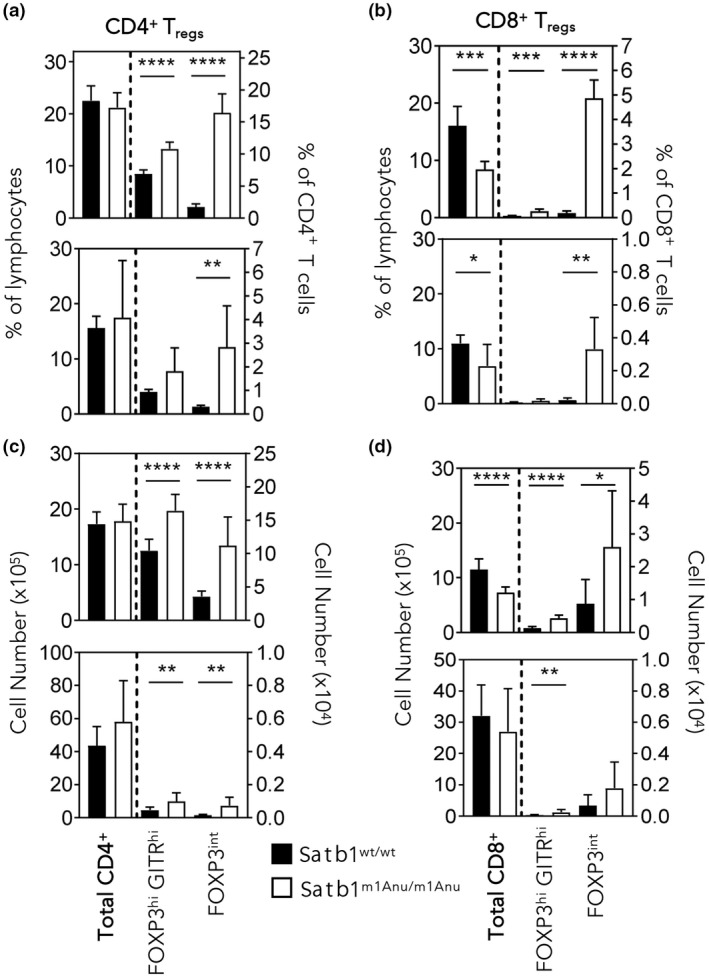
Satb1m1Anu/m1Anu mice exhibit increased frequencies of CD4+ and CD8+ FOXP3+ T cells. The (a, b) proportion and (c, d) total numbers of CD4+ and CD8+ cells with T regulatory (Treg) cells phenotypes (see Supplementary figure 7 for gating of FOXP3hiGITRhi and FOXP3int subsets) from the spleens of uninfected wild‐type (WT) and Satb1m1Anu/m1Anu mice (a–d, top panels) or from mice 14 days after primary A/HKx31 infection (a–d, bottom panels). Error bars show mean ± s.d. of three or five mice per group from three independent experiments. Unpaired Student's t‐tests were used. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. SATB1, special AT‐binding protein 1.
Reduced IAV‐specific CD8+ T‐cell responses in Satb1m1Anu/m1Anu mixed BM chimeras
To investigate whether the altered virus‐specific CD8+ T‐cell phenotypes could also be explained by T‐cell–intrinsic defects, BM chimeras were generated whereby irradiated WT (CD45.1+) or Satb1m1Anu/m1Anu (CD45.2+) recipients received either WT (CD45.1+, CD45.2+) or Satb1m1Anu/m1Anu (CD45.2+) BM, or an equal mix of both (WT CD45.1+/2+, Satb1m1Anu/m1Anu CD45.2+; Supplementary figure 8a, b).
Prior to infection, examination of BM engraftment based on congenic marker expression demonstrated that about 80% of circulating lymphocytes from mice receiving donor cells from a single source were of donor origin, while those receiving an equal mixture of WT and Satb1m1Anu/m1Anu BM showed approximately equal engraftment for each donor source (Supplementary figure 8c, d). Interestingly, surface staining of mixed chimeras showed that CD44 expression was reduced on CD8+ T cells derived from Satb1m1Anu/m1Anu (CD45.2+), but not WT donors (CD45.1/2+; Supplementary figure 8e), indicating that the reduced CD44 expression observed for Satb1m1Anu/m1Anu mice is T‐cell intrinsic. Mice that received Satb1m1Anu/m1Anu BM, either alone or mixed, exhibited a decrease in the proportions of CD4+ and CD8+ T cells (Supplementary figure 8f). Taken together, these data indicate that there is an intrinsic defect in the naïve T‐cell population of Satb1m1Anu/m1Anu mice.
To determine whether the altered immune phenotypes of virus‐specific CD8+ T cells observed after IAV infection of Satb1m1Anu/m1Anu mice was also a result of a T‐cell intrinsic defect, BM chimeras established above were infected with A/HKx31 IAV and tissue‐specific DbNP366 and DbPA224‐specific responses examined at the peak of the primary response (Figure 8). While similar numbers of lymphocytes were observed within all BM chimeras, mice that received mixed WT (CD45.1/2) and Satb1m1Anu/m1Anu (CD45.2) BM showed a decreased frequency of CD45.2+ lymphocytes in the bronchoalveolar lavage and lung (Figure 8a, b). We enumerated IAV‐specific CD8+ T cells in the bronchoalveolar lavage, lung, mLN and spleen, and noted that recipients of Satb1m1Anu/m1Anu BM alone or mixed (WT and Satb1m1Anu/m1Anu) exhibited a lower proportion of CD8+ T cells across multiple organs (Figure 8c, d). Satb1m1Anu/m1Anu derived IAV–specific CD8+ T cells were found at lower proportions in lungs in both single and mixed BM recipients (Figure 8c, d). Consistent with findings in global Satb1m1Anu/m1Anu mice, Satb1m1Anu/m1Anu‐derived CD8+ T cells in the mixed BM chimeras showed a reduction in the proportion of CD44int and CD44hi cells and increased expression of PD‐1 and CD8+ CTLA‐4 (Figure 8e). Taken together, the data show that the diminished IAV‐specific CD8+ T‐cell immunity observed for Satb1m1Anu/m1Anu mice largely results from a CD8+ T‐cell–intrinsic defect.
Figure 8.
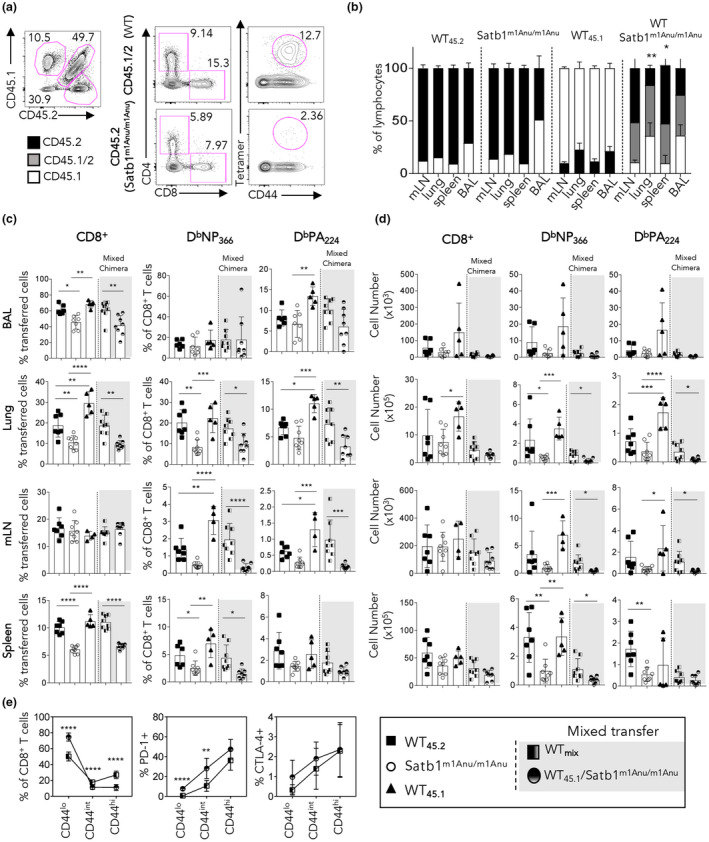
Satb1m1Anu/m1Anu influenza A virus–specific CD8+ T cells exhibit an intrinsic defect in localizing to the infected lung. Bone marrow chimeras were generated as per the “Methods” section and Supplementary figure 8. (a) Representative flow cytometry plots to determine the extent of chimerism. (b) Determination of the extent of chimerism in mixed bone marrow chimeras. (c, d) Mice that received mixed bone marrow from wild type (WT) (CD45.1+/45.2+) and/or Satb1m1Anu/m1Anu (CD45.2+) were infected with A/HKx31. Lymphocytes from the draining lymph node (mLN), lung, spleen and bronchoalveolar lavage (BAL) were isolated 10 days after infection and stained with DbNP366‐ or DbPA224‐specific tetramers. Shown are the (c) proportions and (d) absolute number of CD8+tetramer+ T cells. (e) The proportion of CD8+CD44lo, CD44int and CD44hi subsets from mixed bone marrow chimeras expressing PD‐1 and CTLA‐4 on WT or Satb1m1Anu/m1Anu CD8+ T cells within mixed bone marrow chimeras. Error bars show mean ± s.d. of five or eight mice per bone marrow group from three independent experiments. Unpaired Student's t‐tests were used. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. SATB1, special AT‐binding protein 1. [Colour figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
SATB1 is a chromatin‐binding protein that has been described as a key regulator of T‐cell lineage commitment and fate determination. 10 , 11 , 14 , 16 , 24 , 35 , 36 Here we demonstrate that SATB1 is highly expressed in naïve CD8+ T cells and is downregulated upon effector CD8+ T‐cell differentiation, consistent with earlier observations made in polyclonal human T‐cell subsets. 17 Within naïve CD8+ T cells, SATB1 binding was enriched at genomic regions related to immune lineage function, with binding being decreased following effector CD8+ T‐cell differentiation. We also describe the Satb1m1Anu/m1Anu mouse line, identified by N‐ethyl‐N‐nitrosourea mutagenesis, where a point mutation in the CUT1 domain diminishes SATB1 DNA binding. Satb1m1Anu/m1Anu mice exhibited dysregulated thymic development, a reduction in peripheral CD8+ T cells, alterations in the phenotype of naïve CD8+ T cells and diminished IAV‐specific CD8+ T‐cell responses.
SATB1 contains multiple DNA‐binding domains, including two CUT domains that act to increase the affinity of chromatin binding. 37 We report the generation of Satb1m1Anu/m1Anu‐mutant mice that have a point mutation within the SATB1 CUT1 domain, which does not impact on overall protein expression. SATB1 is highly expressed at the CD4+CD8+ double positive (CD4+CD8+) thymocytes stage of immature T‐cell development, with lower levels of SATB1 expression upon T‐cell commitment to single‐positive thymocytes. 10 , 35 While conditional SATB1 deletion results in accumulation of double positive (CD4+CD8+) thymocytes and fewer CD4SP and CD8SP thymocytes, 10 , 35 we observed a significant impact on thymic selection events in Satb1m1Anu/m1Anu mice. The thymic phenotype of Satb1m1Anu/m1Anu mice suggests that the Satb1m1Anu mutation diminishes TCR signaling and impedes apoptosis of those thymocytes that do perceive strong TCR signaling. This may reflect the role of SATB1 in licensing noncoding regulatory elements prior to cellular commitment, as has been observed for licensing transcriptional super‐enhancers that ensure appropriate FOXP3 expression within CD4+ Treg cells. 25
Compared with effector CD8+ T cells, more extensive SATB1 binding was observed in naïve CD8+ T cells and was associated with genomic regions linked to immune T‐cell activation, differentiation and function. While the Satb1m1Anu mutation within naïve CD8+ T cells did not alter protein expression, it did result in a decrease of chromatin binding at target genomic loci and a distinct transcriptional signature that included dysregulated CD44 expression, and upregulation of checkpoint molecules such as PD‐1, CD8+ CTLA‐4 and LAG‐3. SATB1 has been reported to transcriptionally repress the Pdcd1 gene, with downregulation of SATB1 correlating with increased PD‐1 expression. 15 This is also consistent with the observation that SATB1 expression is repressed in lymphocytic choriomeningitis virus–mediated CD8+ T‐cell exhaustion, where PD‐1 expression is stably and highly expressed. 38 SATB1 has roles in promoting and repressing CD4+ T‐cell fates and function. For example, SATB1 upregulation serves to promote CD4+ TH2 cytokine expression, 39 and pathogenic CD4+ TH17 lineage commitment. 40 Conversely, downregulation of SATB1 is required for ensuring CD4+ Treg cell suppressive function. 16 Given we and others 17 , 38 have observed that SATB1 is downregulated upon CD8+ T‐cell differentiation, it was therefore of interest that the transcriptional profiles of WT and Satb1m1Anu/m1Anu virus–specific effector CD8+ T cells converged. It appears likely that the role of SATB1 in naïve CD8+ T cells is to repress inappropriate activation rather than to actively instruct differentiation outcomes, as in TH2 cells. Further, SATB1 has been reported to be key for ensuring self‐renewal and limiting cell fate commitment of hematopoietic stem cells. It is therefore tempting to speculate that SATB1 serves to maintain the naïve T‐cell state by preventing inappropriate transcriptional activation of effector lineage–specific genes.
Naïve Satb1m1Anu/m1Anu CD8+ T cells proliferated normally following in vitro activation and exhibited robust effector function. While there was an increase in the total CD8+ T‐cell numbers within the draining mesenteric LN 3 days after infection, we were unable to convincingly demonstrate any difference at this early time point in virus‐specific CD8+ T cells. More importantly, we did observe fewer virus‐specific CD8+ T cells in lungs of Satb1m1Anu/m1Anu mice at the peak of infection. Activation of CD4+ Treg cell is a key factor in limiting tissue damage to respiratory virus infection. 32 , 33 , 34 Moreover, naïve Satb1m1Anu/m1Anu CD8+ T cells exhibited an increase in Foxp3 transcription, and FOXP3+ CD8+ cells were observed following infection. Hence, it remains possible that the increase in CD4+ Treg cells, and/or Treg cells function in FOXP3+ CD8+ T cells in Satb1m1Anu/m1Anu mice may play a role in limiting lung CD8+ T‐cell responses.
Another, but not mutually exclusive, explanation could be perturbation of cytokine signaling required to recruit activated CD8+ T cells from the periphery into the infected lung. CXCR3 and production of CCL5 on virus‐specific CD8+ T cells are key for optimal recruitment to the lung tissue after respiratory infection. 41 While little difference in dysregulated chemokine receptor expression was observed in naïve or effector Satb1m1Anu/m1Anu IAV–specific CD8+ T cells, there may be dysregulated production of chemokines by lung stromal cells in Satb1m1Anu/m1Anu mice. This may result in an altered cytokine microenvironment that may not support proper T‐cell proliferation and/or recruitment. Examination of cytokine and chemokine microenvironment in the infected lungs of Satb1m1Anu/m1Anu mice would provide more insights into this.
While extrinsic factors may be a formal possibility in the diminished IAV‐specific CD8+ T‐cell responses in Satb1m1Anu/m1Anu mice, mixed bone chimera experiments demonstrated that it was Satb1m1Anu/m1Anu IAV‐specific CD8+ that were specifically impacted, pointing to an intrinsic defect. An element of the dysregulated effector transcriptional program observed in Satb1m1Anu/m1Anu naïve CD8+ T cells included upregulation of immune checkpoint molecules such as PD‐1, LAG‐3 and CTLA‐4. Hence, the dysregulated expression of these checkpoint molecules on recently activated CD8+ T cells may impact the ability to fully activate and recruit Satb1m1Anu/m1Anu IAV‐specific CD8+ T cells to the infected lung. It would be of interest to see if blocking antibodies to PD‐1, LAG‐3 or CTLA‐4 restore CD8+ T‐cell numbers in the lung.
SATB1 deficiency is associated with increased autoimmune disease prevalence such as Sjögren's syndrome and systemic lupus erythematosus, 10 , 42 while conditional deletion of SATB1 in T cells results in an increased resistance to experimental autoimmune encephalomyelitis. 43 Thus, taken together, these studies indicate that pharmacological interventions that lead to ectopic expression of SATB1 in T cells may have utility in a number of disease states. As a potential key regulator that may be involved in both ‐cell exhaustion and tolerance, SATB1 could be a promising novel immunotherapy target for a multitude of chronic conditions caused or exacerbated by T‐cell–mediated exhaustion or tolerance.
METHODS
Mice, viruses and infections
C57BL/6J (WT and B6) mice with congenic markers (CD45.1+, CD45.1.2+ or CD45.2+) were bred in the Department of Microbiology and Immunity, The Peter Doherty Institute for Infection and Immunity, or at the Monash Animal Research Platform, Monash University. Satb1m1Anu/m1Anu mice were generated by intraperitoneal injection of male B6 mice with N‐ethyl‐N‐nitrosoiurea (Sigma Aldrich, Melbourne, Australia) (100 mg per kg body weight) once a week for 3 weeks. Over 300 pedigrees were screened by flow cytometry and Satb1m1Anu/m1Anu mice were identified by decreased CD44 expression that segregated in an incompletely dominant manner. All experiments were approved by institutional ethics committees (AEC ID 1614025, University of Melbourne; AEC ID 24568, Monash University). For primary IAV infection, mice were anesthetized and infected intranasally with 104 plaque‐forming units of A/HKx31 virus. For secondary infection, mice were primed intraperitoneal with 107 plaque‐forming units of recombinant A/PR8 virus followed by infection with 104 plaque‐forming units of HKx31‐OVA intranasally 4–6 weeks later.
Generation of congenic bone marrow chimeras
BM chimeras were generated by irradiating B6 (CD45.1) recipient mice twice with 550 Rads, 3 h apart. BM from hind legs of donor mice was flushed and T cells depleted with anti‐CD4 (clone RL172), anti‐CD8 (clone 3.168), anti‐Thy1 (clone Jlj) and rabbit complement. Incubation of BM with antibodies was performed for 30 min on ice followed by cell resuspension in 1 mL of complement for 20 min at 37°C. About 5 × 106 T‐cell–depleted BM cells were injected intravenously into recipient mice; 24 h after irradiation, recipient mice were injected intraperitoneally with 100 μL of α‐CD4 and α‐CD8 T‐cell monoclonal antibodies (clone RL172 and clone 3.168, respectively) to eliminate radioresistant T cells. Submandibular bleeds were performed > 8 weeks after irradiation to determine BM reconstitution by staining blood with a cocktail of α‐CD45.1 (eBioscience, Fischer Scientific, Waltham, MA, USA), α‐CD45.2 (BioLegend, San Diego, CA, USA), anti‐CD4 (BioLegend), α‐CD8 (BioLegend) and α‐CD62L (BioLegend). Live cells were discriminated with a fixable LIVE/DEAD stain (Life Technologies, Carlsbad, CA, USA).
Flow cytometry
Tissue samples taken at various time points after infection were prepared as previously described 18 and expression of phenotypic markers was determined with a BD Canto or BD Fortessa (BD Biosciences, Nth Ryde, Australia) and analyzed with FlowJo software (TreeStar, Ashland, OR, USA). For intracellular staining of cytokines we restimulated cells for 5 h with 1 μm of the OVA257–264 peptide in the presence of GolgiPlug (BD Biosciences, San Jose, CA, USA) and 10 U mL−1 rhIL‐2 (Roche, Diagnostics, Mannheim, Germany). Cells were then permeabilized and stained using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's instructions. For intranuclear staining of transcription factors we permeabilized and stained cells using the FoxP3/Transcription Factor Staining kit (eBioscience, San Diego, CA, USA) according to the manufacturer's instructions. Live cells were discriminated with a fixable LIVE/DEAD stain (Life Technologies).
RNA sequencing, chromatin immunoprecipitation (ChIP) and ChIP‐seq
Total RNA sequencing, ChIP and ChIP‐seq were carried out according to Russ et al.18,21 and Li et al. 44 Sequencing was carried out on a Hiseq2000 instrument at the Australian Genome Research Facility, the Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia. The Degust (Monash Bioinformatics Platform) package was used to determine differential gene expression with a false discovery rate of < 0.05 and log2 fold change > 1.2.
AUTHOR CONTRIBUTIONS
Simone Nüssing: Conceptualization; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Lisa Miosge: Formal analysis; investigation; resources; visualization. Kah Lee: Formal analysis; investigation; visualization; writing – review and editing. Moshe Olshansky: Data curation; formal analysis; methodology; software; visualization; writing – review and editing. Adele Barugahare: Data curation; formal analysis; methodology; software; visualization; writing – review and editing. Carla Roots: Investigation; methodology; resources; visualization. E Bridie Clemens: Investigation; supervision; writing – review and editing. Marios Koutsakos: Investigation; methodology. Katherine Kedzierska: Funding acquisition; resources; supervision; writing – review and editing. Christopher Goodnow: Funding acquisition; resources; writing – review and editing. Brendan Russ: Conceptualization; formal analysis; investigation; methodology; supervision; visualization; writing – original draft; writing – review and editing. Stephen Daley: Conceptualization; funding acquisition; investigation; methodology; resources; visualization; writing – review and editing. Stephen Turner: Conceptualization; funding acquisition; project administration; supervision; writing – original draft; writing – review and editing.
CONFLICT OF INTEREST
Authors have no conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported by grants from the National Health and Medical Research Council of Australia (program grant number 5671222 awarded to SJT and KK; project grant number APP1003131 awarded to SJT) and an Australian Research Council Discovery Grant (DP170102020 awarded to SJT) and National Institute of Health, USA grants AI52127, AI054523, AI100627 (awarded to CCG). SJT is supported by an NHMRC Principal Research Fellowship; KK is supported by an NHMRC Leadership Investigator Grant (number 1173871). We thank the Monash Genomics Platform (Micromon) for high‐throughput sequencing and the Monash Bioinformatics Platform for data analysis. Open access publishing facilitated by Monash University, as part of the Wiley ‐ Monash University agreement via the Council of Australian University Librarians.
DATA AVAILABILITY STATEMENT
The accession number for ChiP‐seq and RNA‐seq data reported in this paper is SEO: SRP049743 (Russ et al. 2014) 14 ; SATB1 ChIP‐seq and WT and Satb1m1Anu/m1Anu RNA‐seq data are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Kurachi M, Barnitz RA, Yosef N, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 2014; 15: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xin A, Masson F, Liao Y, et al. A molecular threshold for effector CD8+ T cell differentiation controlled by transcription factors Blimp‐1 and T‐bet. Nat Immunol 2016; 17: 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cruz‐Guilloty F, Pipkin ME, Djuretic IM, et al. Runx3 and T‐box proteins cooperate to establish the transcriptional program of effector Ctls. J Exp Med 2009; 206: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kallies A, Xin A, Belz GT, Nutt SL. Blimp‐1 transcription factor is required for the differentiation of effector CD8+ T cells and memory responses. Immunity 2009; 31: 283–295. [DOI] [PubMed] [Google Scholar]
- 5. Wang D, Diao H, Getzler AJ, et al. The transcription factor Runx3 establishes chromatin accessibility of cis‐regulatory landscapes that drive memory cytotoxic T lymphocyte formation. Immunity 2018; 48: 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Intlekofer AM, Takemoto N, Wherry EJ, et al. Effector and memory CD8+ T cell fate coupled by T‐bet and Eomesodermin. Nat Immunol 2005; 6: 1236–1244. [DOI] [PubMed] [Google Scholar]
- 7. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 2012; 12: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Russ BE, Denton AE, Hatton L, Croom H, Olson MR, Turner SJ. Defining the molecular blueprint that drives CD8+ T cell differentiation in response to infection. Front Immunol 2012; 3: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dickinson LA, Joh T, Kohwi Y, Kohwi‐Shigematsu T. A tissue‐specific MAR/SAR DNA‐binding protein with unusual binding site recognition. Cell 1992; 70: 631–645. [DOI] [PubMed] [Google Scholar]
- 10. Kondo M, Tanaka Y, Kuwabara T, Naito T, Kohwi‐Shigematsu T, Watanabe A. SATB1 plays a critical role in establishment of immune tolerance. J Immunol 2016; 196: 563–572. [DOI] [PubMed] [Google Scholar]
- 11. Hao B, Naik AK, Watanabe A, et al. An anti‐silencer‐ and SATB1‐dependent chromatin hub regulates Rag1 and Rag2 gene expression during thymocyte development. J Exp Med 2015; 212: 809–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yasui D, Miyano M, Cai S, Varga‐Weisz P, Kohwi‐Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 2002; 419: 641–645. [DOI] [PubMed] [Google Scholar]
- 13. Kakugawa K, Kojo S, Tanaka H, et al. Essential roles of SATB1 in specifying T lymphocyte subsets. Cell Rep 2017; 19: 1176–1188. [DOI] [PubMed] [Google Scholar]
- 14. Cai S, Lee CC, Kohwi‐Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet 2006; 38: 1278–1288. [DOI] [PubMed] [Google Scholar]
- 15. Stephen TL, Payne KK, Chaurio RA, et al. SATB1 expression governs epigenetic repression of PD‐1 in tumor‐reactive T cells. Immunity 2017; 46: 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beyer M, Thabet Y, Muller RU, et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol 2011; 12: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nussing S, Koay HF, Sant S, et al. Divergent SATB1 expression across human life span and tissue compartments. Immunol Cell Biol 2019; 97: 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russ BE, Olshanksy M, Smallwood HS, et al. Distinct epigenetic signatures delineate transcriptional programs during virus‐specific CD8+ T cell differentiation. Immunity 2014; 41: 853–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jenkins MR, Webby R, Doherty PC, Turner SJ. Addition of a Prominent epitope affects influenza a virus‐specific CD8+ T cell Immunodominance hierarchies when antigen is limiting. J Immunol 2006; 177: 2917–2925. [DOI] [PubMed] [Google Scholar]
- 20. Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39: 311–318. [DOI] [PubMed] [Google Scholar]
- 21. Russ BE, Olshansky M, Li J, et al. Regulation of H3k4me3 at transcriptional enhancers characterizes acquisition of virus‐specific CD8+ T cell‐lineage‐specific function. Cell Rep 2017; 21: 3624–3636. [DOI] [PubMed] [Google Scholar]
- 22. Mclean CY, Bristor D, Hiller M, et al. GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 2010; 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamasaki K, Akiba T, Yamasaki T, Harata K. Structural basis for recognition of the matrix attachment region of DNA by transcription factor SATB1. Nucleic Acids Res 2007; 35: 5073–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alvarez JD, Yasui DH, Niida H, Joh T, Loh DY, Kohwi‐Shigematsu T. The MAR‐binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T‐cell development. Genes Dev 2000; 14: 521–535. [PMC free article] [PubMed] [Google Scholar]
- 25. Kitagawa Y, Ohkura N, Kidani Y, et al. Guidance of regulatory T cell development by Satb1‐dependent super‐enhancer establishment. Nat Immunol 2017; 18: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koay HF, Su S, Amann‐Zalcenstein D, et al. A divergent transcriptional landscape underpins the development and functional branching of MAIT cells. Sci Immunol 2019; 4: eaay6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daley SR, Hu DY, Goodnow CC. Helios Marks strongly autoreactive CD4+ T cells in two major waves of Thymic deletion distinguished by induction of PD‐1 or NF‐κB. J Exp Med 2013; 210: 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Randall KL, Law HD, Ziolkowski AF, Wirasinha RC, Goodnow CC, Daley SR. DOCK8 deficiency diminishes Thymic T‐regulatory cell development but not Thymic deletion. Clin Transl Immunology 2021; 10: e1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haluszczak C, Akue AD, Hamilton SE, et al. The antigen‐specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med 2009; 206: 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee JY, Hamilton SE, Akue AD, Hogquist KA, Jameson SC. Virtual memory CD8 T cells display unique functional properties. Proc Natl Acad Sci USA 2013; 110: 13498–13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sosinowski T, White JT, Cross EW, et al. CD8α+ dendritic cell trans presentation of IL‐15 to naive CD8+ T cells produces antigen‐inexperienced T cells in the periphery with memory phenotype and function. J Immunol 2013; 190: 1936–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ballesteros‐Tato A, Leon B, Lund FE, Randall TD. CD4+ T helper cells use CD154‐CD40 interactions to counteract T Reg cell‐mediated suppression of CD8+ T cell responses to influenza. J Exp Med 2013; 210: 1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fulton RB, Meyerholz DK, Varga SM. Foxp3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8+ T cell response during respiratory syncytial virus infection. J Immunol 2010; 185: 2382–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu C, Chen W. Influenza virus infection selectively triggers the accumulation and persistence of more potent Helios‐expressing Foxp3+ regulatory T cells in the lungs. Immunol Cell Biol 2021; 99: 1011–1025. [DOI] [PubMed] [Google Scholar]
- 35. Gottimukkala KP, Jangid R, Patta I, et al. Regulation of SATB1 during Thymocyte development by TCR Signaling. Mol Immunol 2016; 77: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Notani D, Gottimukkala KP, Jayani RS, et al. Global regulator SATB1 recruits Beta‐catenin and regulates TH2 differentiation in Wnt‐dependent manner. PLoS Biol 2010; 8: e1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghosh RP, Shi Q, Yang L, et al. Satb1 integrates DNA binding site geometry and torsional stress to differentially target nucleosome‐dense regions. Nat Commun 2019; 10: 3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wherry EJ, Ha SJ, Kaech SM, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007; 27: 670–684. [DOI] [PubMed] [Google Scholar]
- 39. Cai S, Han HJ, Kohwi‐Shigematsu T. Tissue‐specific nuclear architecture and gene expression regulated by SATB1. Nat Genet 2003; 34: 42–51. [DOI] [PubMed] [Google Scholar]
- 40. Yasuda K, Kitagawa Y, Kawakami R, et al. Satb1 regulates the effector program of Encephalitogenic tissue Th17 cells in chronic inflammation. Nat Commun 2019; 10: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kohlmeier JE, Reiley WW, Perona‐Wright G, et al. Inflammatory chemokine receptors regulate CD8+ T cell contraction and memory generation following infection. J Exp Med 2011; 208: 1621–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tanaka Y, Sotome T, Inoue A, et al. SATB1 conditional knockout results in Sjogren's syndrome in mice. J Immunol 2017; 199: 4016–4022. [DOI] [PubMed] [Google Scholar]
- 43. Akiba Y, Kuwabara T, Mukozu T, Mikami T, Kondo M. Special AT‐rich sequence binding protein 1 is required for maintenance of T cell receptor responsiveness and development of experimental autoimmune encephalomyelitis. Microbiol Immunol 2018; 62: 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li J, Hardy K, Olshansky M, et al. KDM6B‐dependent chromatin Remodeling underpins effective virus‐specific CD8+ T cell differentiation. Cell Rep 2021; 34: 108839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for ChiP‐seq and RNA‐seq data reported in this paper is SEO: SRP049743 (Russ et al. 2014) 14 ; SATB1 ChIP‐seq and WT and Satb1m1Anu/m1Anu RNA‐seq data are available from the corresponding author upon reasonable request.