

ABSTRACT
Rural environments and microbiota are linked to a reduction in the prevalence of allergies. However, the mechanism underlying the reduced allergies modulated by rural residency is unclear. Here, we assessed gut bacterial composition and metagenomics in urban and rural children in the EuroPrevall-INCO cohort. Airborne dusts, including mattress and rural henhouse dusts, were profiled for bacterial and fungal composition by amplicon sequencing. Mice were repeatedly exposed to intranasal dust extracts and evaluated for their effects on ovalbumin (OVA)-induced allergic airway inflammation, and gut microbiota restoration was validated by fecal microbiota transplant (FMT) from dust-exposed donor mice. We found that rural children had fewer allergies and unique gut microbiota with fewer Bacteroides and more Prevotella. Indoor dusts in rural environments harbored higher endotoxin level and diversity of bacteria and fungi, whereas indoor urban dusts were enriched with Aspergillus and contained elevated pathogenic bacteria. Intranasal administration of rural dusts before OVA sensitization reduced respiratory eosinophils and blood IgE level in mice and also led to a recovery of gut bacterial diversity and Ruminiclostridium in the mouse model. FMT restored the protective effect by reducing OVA-induced lung eosinophils in recipient mice. Together, these results support a cause-effect relationship between exposure to dust microbiota and allergy susceptibility in children and mice. Specifically, rural environmental exposure modulated the gut microbiota, which was essential in reducing allergy in children from Southern China. Our findings support the notion that the modulation of gut microbiota by exposure to rural indoor dust may improve allergy prevention.
KEYWORDS: Allergies, Aspergillus, gut microbiota, indoor microbiota, urban/rural environments, 16S rRNA-seq and metagenomics
Introduction
The prevalence of allergic diseases has increased dramatically in recent decades.1,2 Exposure to a diverse environmental microbiota is essential for the prevention of allergic diseases by shaping immune maturation in early life.3 Strong epidemiological evidence shows that school-aged children raised on traditional European or Amish farms are protected from asthma.4 We have previously reported that rural children (3.4%) have lower rates of asthma than their urban counterparts (6.9%) in South China (Guangzhou).5 A similar protective effect of rural residence has been reported in North China (Beijing).6
Children spend much time indoors. Indoor microbial communities are influenced by the outdoor environment (e.g., agricultural activities), indoor sources (e.g., building materials), and human and animal occupants.7,8 “House dust” is the main reservoir of bacteria and fungal spores in the domestic environment. The house dust microbiota reflects the indoor environment and is associated with the etiology of allergic diseases.9 The indoor microbiota studied during a child’s infancy, has been linked to asthmatic risk in children aged 10.5 years.10 In contrast, the indoor farm microbiota of non-farm households showed a protective effect against asthma in children.11 Farm dust and bacterial lipopolysaccharide are known to reduce allergic asthma induced by house dust mites through a mechanism of endotoxin tolerance mediated by ubiquitin-modifying enzyme A20, a negative feedback regulator of the NF-κB signaling pathway.12 Our recent study showed that environmental dusts from rural Conghua, China, induced a tolerance effect similar to that of European farms by inducing A20.13 However, the underlying mechanism of how exposure to rural environments modulates allergic susceptibility in humans is unknown.
Rapid urbanization in China has resulted in a more Westernized lifestyle, an overly hygienic indoor environment, and a shift in the gut microbial community toward a Westernized microbiota.14 There is increasing evidence that dysbiosis of the gut microbiome is associated with the development of allergic diseases.15 Recently, it has been hypothesized that the early life environment may shape the gut microbiota and influence the development of asthma and allergic diseases.16 One study demonstrated that environmental microbial exposures modulated the murine gut microbiome and had a beneficial effect on the mental health of mice.17
In this study, we aimed to investigate the effects of urban and rural environmental exposures on the gut microbiota concerning the outcome of allergic diseases both in humans and in mice using an in vivo mouse model.
Results
Rural children had a low prevalence of allergies in the EuroPrevall-INCO cohort and the case-control study
The overall study design is shown in Figure 1. The EuroPrevall-INCO cohort was established to evaluate the prevalence of allergy in children in China, India, and Russia using standardized methodology from the EU-funded project.18 The EuroPrevall-INCO cohort in China included 5,542 urban and 5,139 rural school-age children in Guangdong Province conducted by our laboratory. Screening survey revealed a significantly higher prevalence of diagnosed asthma, rhinitis, and eczema in urban children than in rural children (Table 1). The detailed questionnaire from the EuroPrevall-INCO cohort included 402 and 349 participants from urban and rural areas, respectively, with children from rural areas having a significantly lower percentage of food allergy (5.4% vs. 5.5%), asthma (2.9% vs. 10.7%), rhinitis (10.3% vs. 42.5%), and eczema (22.1% vs. 48.3%, Table 1).
Figure 1.
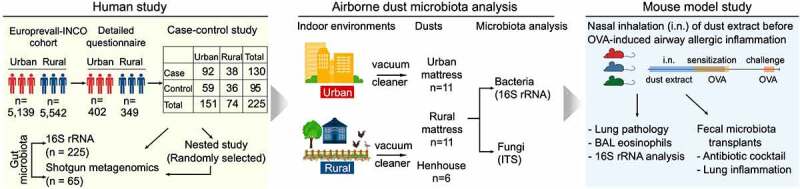
Overall study design. The study included (a) a human study, (b) an airborne dust microbiota analysis, and (c) a mouse model study. 16S rRNA and ITS, 16S rRNA and Internal Transcribed Spacer amplicon sequencing; BAL, bronchoalveolar lavage; EuroPrevall-INCO, a project to evaluate the prevalence of food allergy in China, India, and Russia using the standardized methodology of the EuroPrevall protocol;18 OVA, ovalbumin. Mouse color (panel c) indicates dust extracts from urban mattresses, rural mattresses, and henhouses. Individual dust from each environment was pooled and used in the mouse model study.
Table 1.
Characteristics of the EuroPrevall-INCO cohort and case-control study.
| Urban | Rural | p-value | |||||||
|---|---|---|---|---|---|---|---|---|---|
| EuroPrevall-INCO cohort (n = 10681) | |||||||||
| Response rate (%) | 94.3 | 91.8 | <0.001 | ||||||
| Participant number | 5542 | 5139 | |||||||
| Age (year, mean ± SD) | 8.9 ± 1.7 | <0.001 | |||||||
| Female (number, %) | 2704 (48.8) | 2523 (49.1) | 0.781 | ||||||
| Self-reported doctor-diagnosed allergic diseases (%) | |||||||||
| Food allergy | 4.0 | 3.5 | 0.154 | ||||||
| Asthma | 6.6 | 2.5 | <0.001 | ||||||
| Rhinitis | 23.2 | 5.3 | <0.001 | ||||||
| Eczema | 34.1 | 25.9 | <0.001 | ||||||
| Detailed questionnaire (n = 751) | p-value | ||||||||
| Response rate (%) | 76.9 | 70.5 | <0.001 | ||||||
| Participant number | 402 | 349 | |||||||
| Age (year, mean ± SD) | 10.3 ± 1.5 | 10.6 ± 1.5 | 0.168 | ||||||
| Female (number, %) | 192 (47.8) | 164 (47.0) | 0.123 | ||||||
| Self-reported doctor-diagnosed allergic diseases (%) | |||||||||
| Food allergy | 5.5 | 5.4 | <0.001 | ||||||
| Asthma | 10.7 | 2.9 | <0.001 | ||||||
| Rhinitis | 42.5 | 10.3 | <0.001 | ||||||
| Eczema |
48.3 |
|
|
|
22.1 |
|
|
<0.001 |
|
| |
Case |
Control |
p-value |
|
Case |
Control |
p-value |
p-value* |
p-value^ |
| Case-control study (n = 225) | |||||||||
| Participant number | 92 | 59 | 38 | 36 | |||||
| Race (Han Chinese, %) | 100 | 100 | 100 | 100 | |||||
| Age (year, mean ± SD) | 8.4 (1.5) | 8.6 (1.5) | 0.282 | 8.4 (1.3) | 8.3 (1.3) | 0.630 | 0.843 | 0.213 | |
| Female (number, %) | 39 (42.4) | 36 (61.0) | 0.039 | 14 (36.8) | 15 (41.7) | 0.852 | 0.697 | 0.105 | |
| BMI (kg/m2, mean ± SD) | 18.0 (4.0) | 17.5 (2.9) | 0.335 | 17.2 (2.2) | 17.1 (3.1) | 0.918 | 0.117 | 0.550 | |
| Exposure to rural environment (%) | |||||||||
| before 1 year old | 0 | 0 | - | 100 | 100 | - | - | - | |
| Blood eosinophils(×109/L, median, min, max) | 0.3 (0.2–0.5) | 0.1 (0.1–0.3) | <0.001 | 0.2 (0.1–0.3) | 0.1 (0.1–0.2) | <0.001 | <0.001 | 0.039 | |
| Sensitization (%)# | 100 | 0 | 100 | 0 | |||||
| house dust mite | 73.9 | 0 | 78.9 | 0 | |||||
| cockroach & | 4.3 | 0 | 39.5 | 0 | |||||
| cat dander | 15.2 | 0 | 0.0 | 0 | |||||
| shrimp | 6.5 | 0 | 57.9 | 0 | |||||
| egg | 17.4 | 0 | 10.5 | 0 | |||||
| cow milk | 12.0 | 0 | 5.3 | 0 | |||||
| Doctor-diagnosed allergic diseases (%) | |||||||||
| Food allergy alone | 17.4 | 0 | 0.002 | 44.7 | 0 | 0.002 | |||
| Food allergy with asthma | 28.3 | 0 | <0.001 | 0.0 | 0 | <0.001 | |||
| Food allergy with rhinitis | 51.1 | 0 | <0.001 | 10.5 | 0 | <0.001 | |||
| Food allergy with eczema | 67.4 | 0 | 0.097 | 50.0 | 0 | 0.097 | |||
| Food allergy with any of asthma/rhinitis/eczema | 82.6 | 0 | 0.002 | 55.3 | 0 | 0.002 | |||
*p-values were calculated between urban and rural cases. ^p-values were calculated between urban and rural controls. &Sensitization to cockroaches was determined by a skin prick test. Other allergens listed were determined by specific IgE using the ImmunoCAP reagent. #Only representative allergens in children from Southern China are listed. Refer to Fig. S1 for the complete list of sensitizations to 33 allergens by specific IgE and 23 allergens by skin prick test.
The case-control study included 225 children whose stool samples, clinical data and allergen sensitizations were collected, including specific IgE (sIgE) to 33 allergens and skin prick test (SPT) to 23 allergens (Figure 1a, Table 1). The cases had comparable age, gender, and body mass index (BMI) but higher level of blood eosinophils compared to controls in urban and rural areas. Rural cases had a significantly higher percentage of sensitization to cockroaches and shrimp but a lower percentage of sensitization to cat dander, egg, and cow’s milk than in urban cases (Table 1, Fig. S1). Both rural and urban cases had a higher percentage of sensitization to house dust mites. Consistent with the prevalence of allergic diseases found in the EuroPrevall-INCO cohort, we found that rural areas had significantly lower rates of asthma and rhinitis among participants diagnosed with allergic diseases (Table 1).
Urban or rural exposure dictated the gut microbiota in children
We used DADA2 workflow to generate a table of 10,677 amplicon sequence variants (ASVs) from 26,057,611 clean sequence reads (accounting for 43.9% of total reads) of 16S rRNA amplicon data after filtering, denoising and removing chimeras. Of them, 418 ASVs (3.9%) was assigned to 264 species by using exact string matching to the reference database (Table S1). At the genus level, the relative abundance of two bacterial taxa, Bacteroides and Prevotella, accounted for about 50.0% of the ASVs in both urban and rural participants (Figure 2a). However, while Prevotella_9 accounted for about 25.0% of the total ASVs in rural participants, it only accounted for ≤5.0% in urban participants, resulting in a higher Prevotella-to-Bacteroides ratio, a biomarker of rural lifestyle.19 The Prevotella-to-Bacteroides ratio was significantly higher in rural participants compared to urban after adjustment for age, gender, and BMI (p < 0.001) (Figure 2b). There was no significant difference in gut microbiota composition between the case and control in both urban and rural participants. Urban and rural participants did not differ in the Shannon index, whereas rural participants had a lower core abundance index, indicating a more diverse community with fewer shared taxa (adjusted p < 0.001, Figure 2b). In addition, rural environment had an increased Shannon index and Prevotella-to-Bacteroides ratio but a decreased core abundance index in cases when compared to those in urban environment (Fig. S2). Abundance-based Bray-Curtis matrices were generated for quantitative measures of compositional dissimilarity between urban and rural participants and between case and control participants using t-SNE ordination. Interestingly, urban-rural status (Adonis test, F = 6.98, p = 0.001), but not allergies (F = 1.06, p = 0.300), had a notable effect on the difference in community structure of the microbiota (Figure 2c). Two enterotypes (Figure 2d), dominated by Prevotella_9 or Bacteroides, were found in rural (43.2% and 6.0%, respectively) and urban participants (94.0% and 56.8%, respectively) (Adonis test, F = 6.98, p = 0.001) (Figure 2e). A composition biplot showed that Prevotellaceae was the dominant factor for variation in bacterial composition (Figure 2f). We found that one genus, Parasutterella, and two other species, Akkermansia muciniphila and Parabacteroides merdae, were less abundant among participants, while Bacteroidaceae, Bacteroidetes, Prevotellaceae, and Firmicutes, identified by MaAsLin2 analysis, showed notable differences between urban and rural status (Figure 2g-h, Table S2).
Figure 2.
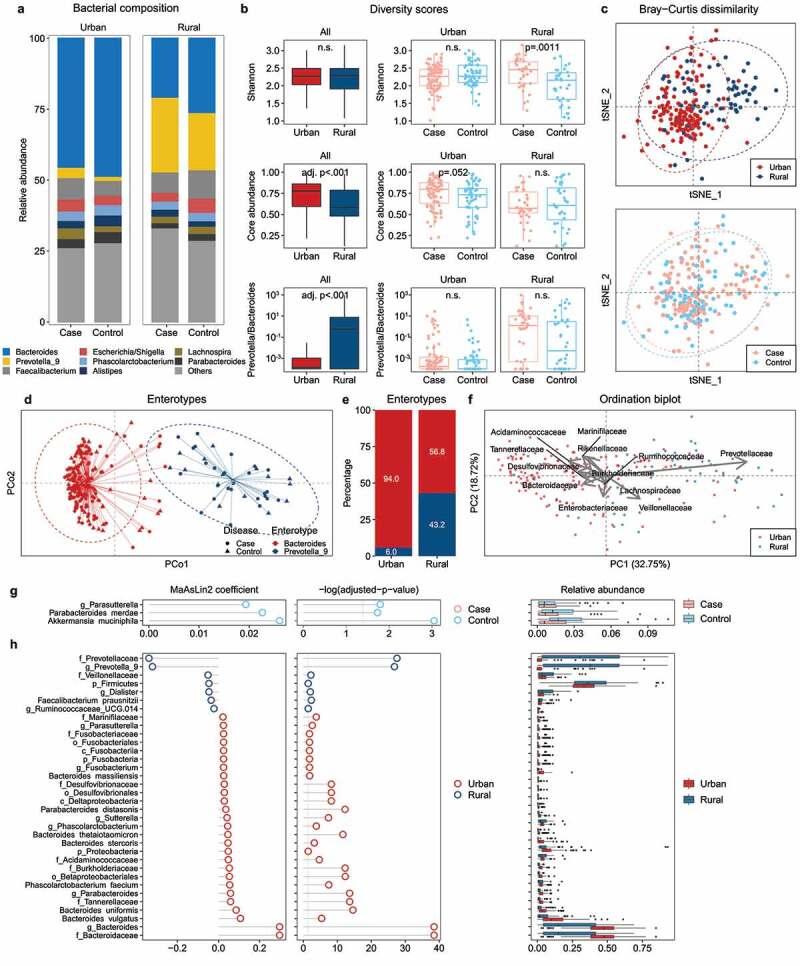
Gut microbiota profiles and differential bacterial taxa in children living in Southern China as determined by 16S rRNA sequencing. (a) Taxonomic profile of the bacterial community at the genus level. (b) Alpha diversity of species measured by three metrics, including Shannon diversity, core abundance (the relative proportion of the core species), and the Prevotella-to-Bacteroides ratio. (c) Beta diversity between urban and rural, case and control participants measured by the Bray-Curtis dissimilarity. (d, e) Weighted Principal Coordinate Analysis (PCoA) and percentage of enterotypes. (f) Biplot of sample dissimilarity and relative abundance of bacteria at the family level in the ordination space of Principal Component Analysis (PCA). (g, h) Association of differential gut bacterial taxa with allergy case vs. control (g), or urban vs rural status (h) as determined by the MaAsLin2 multivariate correlation after adjusting for age, gender, and BMI. Coefficients of linear models are shown. Benjamini–Hochberg adjusted p-values for multiple comparisons with a false discovery rate (FDR) < 0.25 are shown. Refer to Table S2 for MaAsLin2 associations. n.s., not significant.
Additionally, we randomly selected 65 participants with their stool DNA samples for shotgun metagenomic sequencing as part of the case-control study (Figure 1a). These 65 participants had clinical characteristics comparable to those of the case-control study participants (Table S3). Our MaAsLin2 analysis identified 14 microbial MetaCyc pathways associated with urban-rural status and nine MetaCyc pathways associated with allergy after adjustment for age, gender, and BMI (Figure 3). Metabolic pathways common in the urban group included GDP-mannose biosynthesis, a key substrate for glycoprotein formation, as well as tricarboxylic acid cycle and thiamine diphosphate (also known as vitamin B1) biosynthesis for energy generation and metabolism (Figure 3a-c). In addition, homolactic fermentation pathway, whose product is L-lactate, was positively associated with allergy; whereas the pathways of sugar acid degradation (PWY-6507, PWY-7242, GLUCUROCAT-PWY, and GALACT-GLUCUROCAT-PWY) and lipopolysaccharide component O-antigen biosynthesis (PWY-7323, PWY-7312, and PWY-5659, (Figure 3d-f), Table S4) were abundant in the microbiota of control participants.
Figure 3.
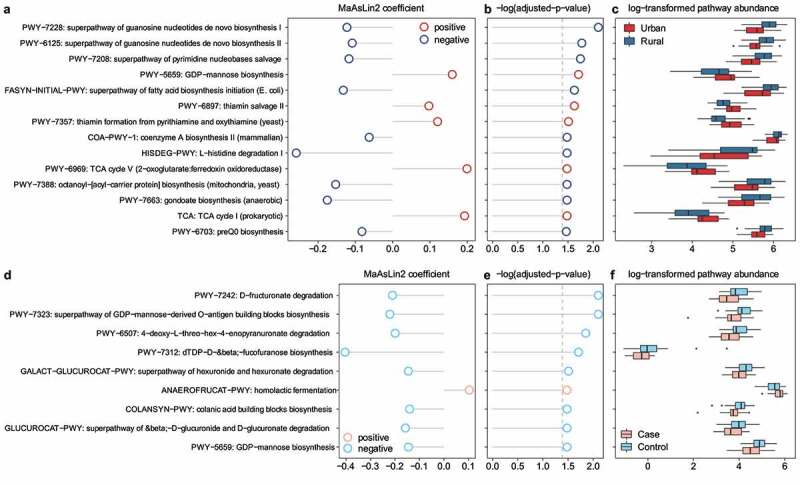
Significant enrichment of microbial MetaCyc pathways based on environments (urban vs. rural) (a–c) and allergies (d–f) in children’s gut microbiota. Associations with MetaCyc pathways are analyzed by MaAsLin2 and adjusted for age, gender, and BMI. (a, d) Positive and negative correlations of coefficients by MaAsLin2 as indicated by red and blue color, respectively. (b, e) P-values adjusted for multiple comparisons, and Benjamini–Hochberg adjusted p-values with an FDR < 0.25 shown. (c, f) Relative pathway abundance log-transformed and shown for urban vs. rural and case vs. control. Refer to Table S4 for the MaAsLin2 associations.
Urban indoor dusts had lower microbial diversity, but higher potential pathogenic bacteria and Aspergillus
To mimic the microbial exposure of children in urban and rural environments, we collected mattress dusts from ten urban and ten rural families and dusts from five rural families with rural henhouses (assuming that backyard poultries in rural families might contribute to the indoor microbial community) (Figure 1b). A total of 62,479 and 7,326 ASVs, belonging to bacteria and fungi, respectively, were identified in the dust samples. There are 3835 ASVs (6.1%) and 2024 fungi ASVs (27.6%) assigned to 1134 bacterial species and 669 fungal species, respectively (Table S1).
In terms of bacterial composition, Enterobacteriaceae and Rhizobiaceae were predominant in the urban dust microbiota but not in the microbiota from rural indoor or henhouse dusts (Figure 4a). The microbiota from rural dusts had significantly higher α-diversity and endotoxin content than the microbiota from urban/rural henhouse dusts (Figure 4b-c). Although no difference in these metrics was observed between the microbiota from urban and rural henhouses, their overall composition profiles differed in beta diversity (Figure 4d). Interestingly, the urban dusts microbiota had a significantly higher proportion of potentially pathogenic bacteria predicted by BugBase (Figure 4f).
Figure 4.
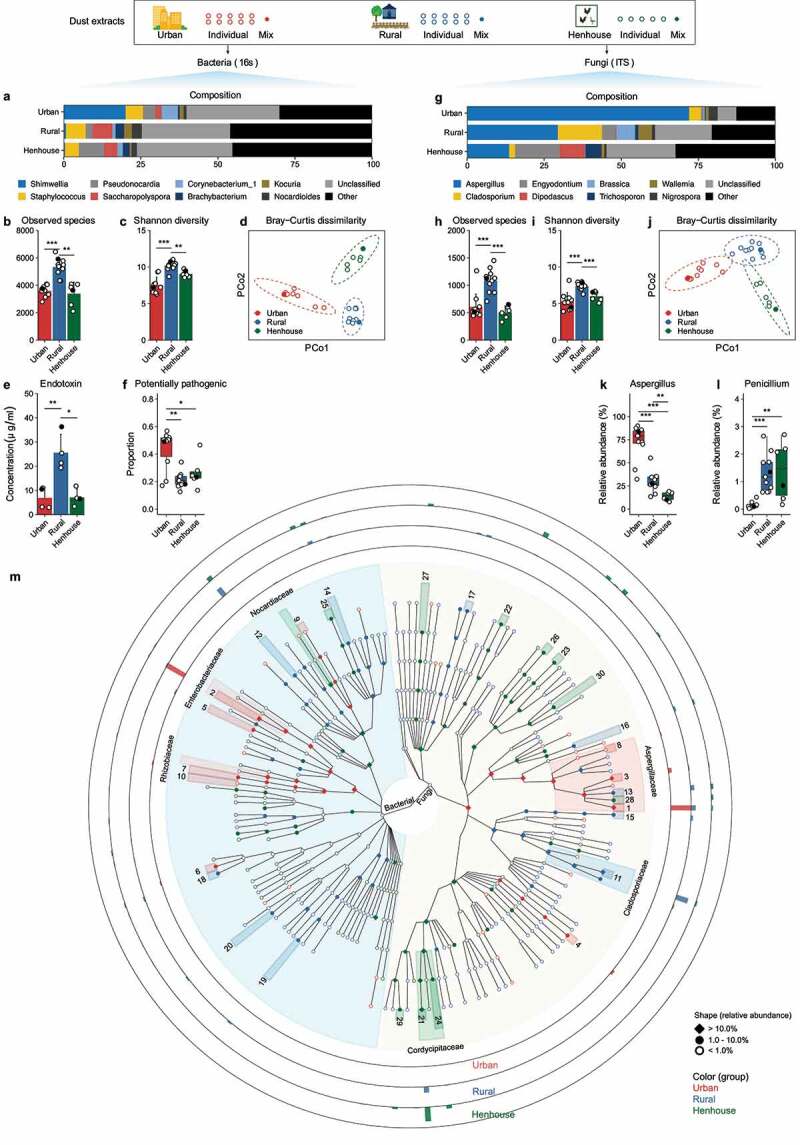
Characterization of urban/rural indoor environmental and rural henhouse dust microbiota. Individual dust extracts (open circle) or a mixture of each group (solid dot, mixture used for the mouse study in Figure 4) were determined for both 16S rRNA and ITS gene amplicons. (a–d, g–i) Genus level microbial composition and diversity metrics of bacterial and fungal species. (e–f) Endotoxin concentration, proportion of potentially pathogenic bacteria, and relative abundance of selected bacterial taxa. (k–l) Relative abundance of selected fungal taxa. (m) GraPhlAn plot of LEfSe2 linear discriminant analysis of bacterial and fungal microbial markers (linear discriminant analysis (LDA) score > 3.0). The relative abundance of microbial markers is indicated as Rhombus for >10.0%, a solid dot represents 1.0–10.0%, and an open circle represents less than 1.0%. The color of the microbial markers represents the corresponding enriched group. The microbial marker with >10.0% abundance at the family level is indicated. The height of the outer rings, normalized to the most abundant taxa (Aspergillus penicillioides, 20.8%) indicates the geometric mean of the relative abundance at the genus or species level in urban, rural, and rural henhouses. Refer to Table S2 for labels of microbial markers with the indicated number. * p < 0.05, ** p < 0.01, and *** p < 0.001 indicate different levels of significant differences calculated by a t test or Wilcoxon test. Refer to Table S5 for differentially abundant bacteria and fungi identified by LEfSe2.
In terms of fungal composition, Aspergillaceae dominated the urban dust microbiota, whereas Trichocomaceae (i.e., the genus Penicillium) was more prevalent in the rural indoor and henhouse dust microbiota (Figure 4g). It is worth noting that Aspergillus, a well-known genus of allergenic fungi, accounted for more than 75% of the relative abundance at the genus level in the urban dust microbiota. In contrast, Penicillium, another allergenic fungus, was less abundant in the urban dust microbiota but more abundant in the rural and henhouse dust microbiota, despite its overall low abundance (< 2.0%, Figure 4l) in both environments. The rural dust microbiota had consistently higher fungal diversity in both observed species and Shannon index (Figure 4h-j).
Furthermore, a total of 166 bacterial and 206 fungal markers were differentially enriched by LEfSe2 (Linear discriminant analysis Effect Size) analysis (Figure 4m). The top 10 most enriched microbial markers are reported in Table S5. Of these ten microbial markers, two fungal species, Aspergillus penicillioides (20.8%) and Aspergillus conicus (3.3%), and one bacterial genus, Shimwellia (20.3%), were highly enriched in the urban dust microbiota. The microbiota in rural indoor dusts was dominated by two fungal species (Cladosporium halotolerans, 12.5% and Aspergillus sydowii, 3.8%) and one bacterial genus (Saccharopolyspora, 9.4%). Nine of the top 10 microbial markers in the microbiota of rural henhouses were fungi, with Engyodontium album (14.5%) being the most abundant species (Figure 4m, Table S5).
Repeated intranasal exposure to rural indoor dusts protected mice against OVA-induced airway allergic inflammation
Next, we tested the hypothesis that exposure to environmental dusts may modulate allergic disease by altering the gut microbiota using a mouse model with repeated intranasal exposure (Figure 1c, Figure 5a). In mice exposed to dust alone, no significant changes were observed in the number of inflammatory cells infiltrating in the bronchoalveolar lavage (BAL) in general and in lung pathology (Fig. S3).
Figure 5.
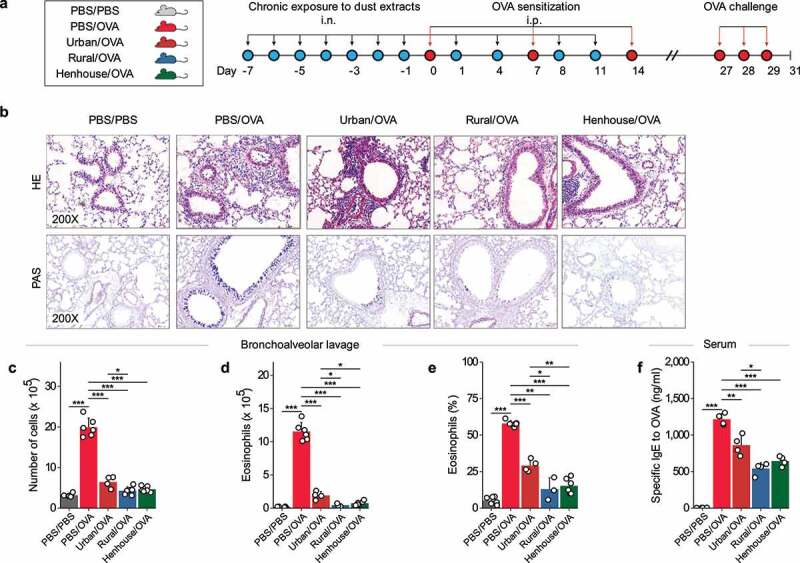
Repeated nasal inhalation (n = 11) of dust extracts regulated OVA-induced allergic airway inflammation in mice. (a) Study protocol illustrating the dust extracts, intranasal exposure, and sensitization, as well as challenge by OVA. The dust extract corresponds to the mixture of extracts from each group in Figure 3 (solid dot). (b) Histopathology of lung tissue (Hematoxylin and Eosin (H&E) and Periodic Acid-Schiff (PAS) staining) in each group as indicated above. (c–f) Inflammatory cell counts, number and percentage of eosinophils in bronchoalveolar lavage fluid, and serum-specific IgE level against OVA in the serum. * p < 0.05, ** p < 0.01, and *** p < 0.001 indicate the levels of different significant differences calculated by a t test or Wilcoxon test.
Mice treated with PBS/OVA exhibited severe infiltration of inflammatory cells, mucus secretion in lung tissue, eosinophilic infiltration of airways in BAL, and increased sIgE level for OVA (Figure 5b-f). In mice pre-exposed to one of the three dust extracts, allergic inflammation was relieved to varying degrees after OVA induction (Figure 5c). However, preexposure to urban indoor dusts followed by OVA induction (urban/OVA group) did not improve infiltration of inflammatory cells, as seen in hematoxylin and eosin-stained tissues (Figure 5b). Compared to control mice (PBS only), mice exposed to rural house dusts (rural/OVA) showed the strongest suppression of allergic inflammation in terms of reduction in the number of BAL eosinophils (96.1% vs. 83.5%, p = 0.011), BAL eosinophil abundance (77.6% vs. 49.8%, p = 0.041), and the level of sIgE to OVA (55.3% vs. 28.9%, p = 0.032). Mice exposed to urban indoor dusts (urban/OVA) showed the least suppression in each of the categories tested above, and mice exposed to rural henhouse dusts (henhouse/OVA) exhibited a modest suppression of allergic inflammation (Figure 5d-f, Fig. S4).
Pre-exposed to dust extracts restored gut microbiota and alleviated lung eosinophils
The composition of the microbiota of mice exposed to urban indoor dusts (urban/OVA) differed from that of microbiota of mice exposed to rural indoor dusts (rural/OVA and henhouse/OVA), as determined by α-diversity metrics and interindividual dissimilarities (Figure 6a-d). We identified 12,970 unique ASVs in mouse gut microbiota, 560 ASVs (4.3%) were assigned to 62 species by DADA2 (Table S1). Specifically, PBS/OVA and urban/OVA treatment significantly reduced the number of species within the microbiota and increased compositional contrasts between the microbiota of mice in these two groups and other groups. However, preexposure to rural indoor dusts or henhouse dusts maintained the diversity indices in both groups at a similar level to the control group. In the Bray-Curtis dissimilarity test, clear separation was observed in all samples (Figure 6d). The PBS/OVA and urban/ OVA samples were clearly separated from the other groups after the first Principal Coordinate Analysis (PCoA) component (17.0% explained variance), whereas all samples showed less separation after the second PCoA component, except for some PBS/OVA samples. In addition, mice exposed to rural/OVA showed the lowest increase in the proportion of potentially pathogenic bacteria, as predicted by BugBase. Analysis of microbial markers using LEfSe2 showed that the relative abundance of Bacteroidales increased and the abundance of Clostridiales, including both Lachnospiraceae and Ruminococcaceae families, decreased in mice treated with PBS/OVA and urban/OVA. No significant difference was observed between mice treated with PBS/PBS and rural/OVA (Figure 6e-h, Figs. S5, S6). Mice treated with henhouse/OVA showed an increased abundance of Ruminococcaceae compared to that in the PBS/OVA group (Figure 6h). Interestingly, we found some moderate but statistically significant correlations between the relative abundances of BAL eosinophils and Bacteroides (r=0.59, p=0.001), Ruminiclostridium (r=-0.45, p=0.05), respectively, in the gut (Figure 6j). This suggests that preexposure to rural indoor dust decreased the abundance of Bacteroides in the gut, but increased the abundance of Ruminiclostridium, which in turn, may suppress BAL eosinophils, resulting in less allergic inflammation. To further validate the effect of alteration of the gut microbiota on allergic airway inflammation, we exposed mice to PBS or dust extracts from rural henhouses (based on the requirement for dust and its availability) and prepared suspensions containing the fecal microbiota altered by dust exposure (Figure 6k). Pretreatment with the antibiotic cocktail significantly reduced bacterial load, Shannon diversity, and the proportion of common bacteria colonizing the intestine of the mice (i.e., Lachnospiraceae, data not shown) by 16S rRNA analysis. The proportion of BAL eosinophils in the recipient mice that received transplants from dust-exposed mice decreased significantly (Figure 6l, Fig. S7).
Figure 6.
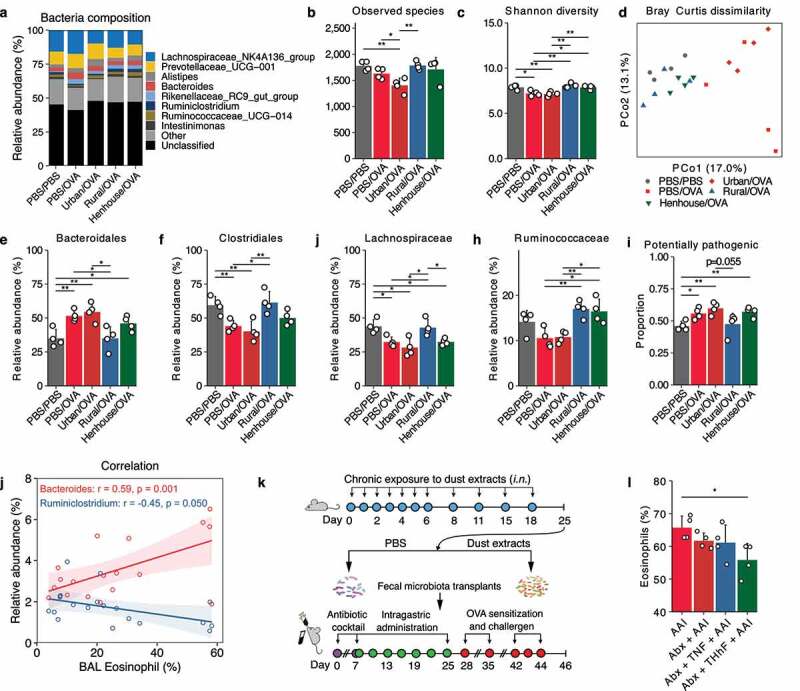
Repeated nasal inhalation of dust extracts modulated the gut microbiota in a mouse model. (a–d) Genus level bacterial composition and diversity metrics of the gut microbiota. (e–h) The relative abundance of selected microbial markers that were highly abundant in the gut microbiota as identified by LEfSe2 analysis (refer to Fig. S6, S7 for LEfSe2 results). (i) Proportion of potentially pathogenic bacteria predicted by BugBase. (j) Correlations between the percentage of BAL eosinophils and the relative proportion of Bacteroides, Ruminiclostridium, respectively. (k) Scheme of fecal microbiota transplants with PBS or henhouse dust extracts. (l) Effect of fecal microbiota transplants on the percentage of eosinophils in the BAL fluid. AAI, PBS/OVA group; Abx, pretreatment with antibiotic cocktail; TNF, transplant with feces from PBS exposed mice; THhF, transplant with feces from henhouse dust extract exposed mice. * p < 0.05, ** p < 0.01, and *** p < 0.001 indicate the levels of significant differences calculated by a t test or Wilcoxon test.
Discussion
In this study, we demonstrated that the diverse microbiota of rural environments reduced the risk of allergic diseases by modulating the gut microbiota in children and mice compared to the microbiota of urban indoor environments. We reported that the microbiota in urban indoor environments in Southern China was typically characterized by higher levels of Aspergillus and potentially pathogenic bacterial species, in addition to a low α-diversity for both bacterial and fungal communities, in contrast to the microbiota in rural indoor environments. We presented evidence that the indoor microbiota modulates the gut microbiota of children as well as their immune response to allergic diseases. A change in the gut microbiota toward Bacteroides was evident in urban children and mice exposed to urban dust extracts. In contrast, the gut microbiota of rural children and mice exposed to rural dust extracts were rich in Prevotella and Clostridiales, respectively. We also showed that eosinophils in mouse’s lungs were correlated with the relative abundance of Bacteroides and Ruminiclostridium in the gut after exposure to indoor dust, and eosinophils in mouse lungs were attenuated by FMT of mice exposed to rural dusts (Figure 7).
Figure 7.
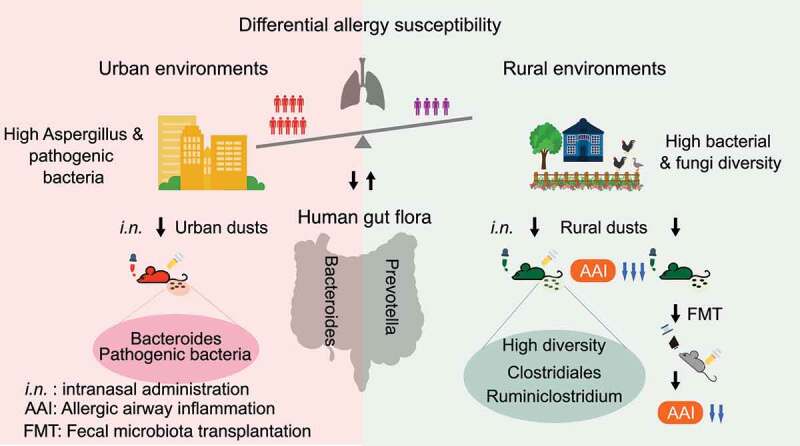
Schematic summary of the relationship between exposure to environmental microbiota and allergy susceptibility in children and mice.
Exposure to environmental microorganisms is an important factor in allergic diseases.20 However, it has been reported that microbial communities in urban air have a lower abundance and diversity of fungal and bacterial communities due to urbanization.21 In this study, we found that increased bacterial diversity and endotoxin content in the environmental microbiota were associated with a lower risk of allergy, which is consistent with findings of the earlier studies.20 Some bacterial-derived products, such as gram-negative bacterial endotoxins and extracellular polysaccharides from gram-positive bacteria, have been reported to play a protective role in asthma or atopy.22,23 More recently, bacterial richness, bacterial load, and the FaRMI index (representing the ‘farm-like’ nature of an indoor environment) are thought to play an important role in protecting children from asthma.11 The FaRMI index suggests that the abundance of the Streptococcaceae family, which includes common opportunistic respiratory pathogens, contributes to the protective effect. Although we did not observe an enrichment of Streptococcaceae in urban indoor dusts in our study, this assumption was supported by the finding that urban indoor dusts contained more potentially pathogenic bacteria predicted by BugBase.
Frequent exposure to fungal products, such as beta-(1,3)-glucans and extracellular polysaccharides, was associated with a lower risk of atopic wheezing.22 In contrast, high indoor concentrations of allergenic fungi, e.g., Cladosporium, Alternaria, and Aspergillus, increase the risk of developing asthma and exacerbate asthma symptoms.24 Consistently, we found an unexpectedly high prevalence of the fungus Aspergillus, more specifically Aspergillus penicillioides (with a relative frequency of over 20%), in urban indoor environments in Southern China. Aspergillus is commonly found in house dusts,25 while A. penicillioides isassociated with house dust mite allergenicity26 and exhibits serological reactivity and IgE-binding activity.27 This finding is of clinical significance as it suggests that the indoor microbiota, rich in potentially pathogenic bacteria and/or with an altered abundance of fungi, particularly Aspergillus, may contribute to an increased risk of allergy when living in urban environments.
Cowsheds of Western farms have been shown to host a high bacterial load, high microbial diversity, and more protective bacteria species, like Acinetobacter lwoffii F78 and Lactococcus lactis G121.28 Because cowsheds are uncommon in Chinese rural backyards, we utilized henhouses as the counterpart of a cowshed in our epidemiological study.8 In our study, henhouse dusts had, to a certain extent, similarity with rural indoor dusts in terms of bacterial composition and pathogen load, but significantly reduced diversity. LEfSe2 analysis identified more differential fungal species in rural henhouse dusts compared to those in urban and rural indoor dusts, which could be due to the damp and moldy poultry housing. In addition, participants in our study living in urban areas had a higher rate of sensitization to cat dander, suggesting a potential role of domestic pets (e.g., cats) on dust microbiota in urban families. Considering the cohabitants in urban and rural household, backyard animals or domestic pet-derived microorganisms may contribute to the indoor dust microbiota; however, further investigations are needed.
The gut composition of microbiota and microbial products have been associated with allergic diseases in many studies.29,30 Thus, children with allergic diseases harbor fewer anti-inflammatory bacterial species, such as Akkermansia muciniphila,31 Parabacteroides merdae,32 and Parasutterella.33 Using functional enrichment analysis of shotgun sequencing data of the gut microbiota, we found that products of the gut microbiota, such as L-lactate and lipopolysaccharides, may be new triggers for allergic diseases. Additionally, we found that Clostridiales, Lachnospiraceae, and Ruminococcaceae were abundant in the gut microbiota of mice exposed to rural indoor dusts. Interestingly, these taxa are bacteria that produce short-chain fatty acids,34 suggesting that the microbiota in rural indoor environments may be beneficial in suppressing allergic lung inflammation.35 Our findings support the notion that the composition of the gut microbiota and its metabolites contribute to the etiology of allergic diseases.
The alteration of the gut microbiota by environmental factors is a possible cause of allergic diseases, including food allergies.36 Our study showed that changes in Prevotella_9 and the ratio of Prevotella-to-Bacteroides were primarily responsible for the differences in microbial composition and diversity in the gut microbiota between urban and rural children. A Prevotella-dominant enterotype was generally associated with a high-fiber diet, which is more common in non-Western participants, whereas a Bacteroides-dominant enterotype is common in Western participants on a high-fat diet.37 Since gut microbial enterotypes have been associated with long-term dietary patterns, and our previous study reported the difference between urban and rural diets,38 the role of diets in modulating gut microbiota could not be excluded. Nevertheless, we have shown that dust exposure modulates the composition and diversity of the gut microbiota in a mouse model with allergic inflammation. Our findings that OVA induction reduced gut bacterial diversity and abundance of Lachnospiraceae were consistent with a previous study,39 which showed similar protective effects when mice were treated with commercial soil prior to exposure. The effect of reported commercial soil is comparable to that of urban dusts in our study, as no difference in Bacteroidale abundance was observed with or without treatments with urban dusts or soil. In contrast, rural dusts reduce Bacteroidales in the gut after OVA induction. The reverse correlation between gut Bacteroidales and BAL eosinophils hints that rural dusts suppress allergic inflammatory response. This finding is consistent with a recent study which demonstrated that Bacteroidales in the gut promoted sensitization to allergens.40 Our study also shows that rural dust exposure shifts the gut microbiota of mice toward a healthier gut microbiota and that FMT from dust-exposed donor mice reduces eosinophils in the lungs of recipient mice, suggesting the significant therapeutic and interventional role of modulating gut microbiota in preventing allergic inflammation. Consistent with our findings, infants in an Amish community had a lower risk of asthma, and transplantation of the fecal microbiota from Amish infants provides the protective gut microbiota to germ-free piglets.41 Another study indicated that 3-month exposure to a swine farm environment could alter the gut microbiota of children.42 In conclusion, our observations show that natural microbial interventions, i.e., exposure to a protective farm or rural environment, can mitigate allergic diseases.3
How could the dusts affect the gut microbiota? Liddicoat et al. provided evidence that trace-level of dusts from a high biodiversity soil can change mouse gut microbiota and result in a beneficial effect on the mental health, representing an important supplementary source of the mammalian gut microbiota.17 Similarly, other studies showed that air pollutants have the potential to alter the gut microbiota.43 We speculate that dusts modulate the gut microbiota through the following mechanisms:43 1) environmental microorganisms and metabolites in the respiratory tract can arrive at the intestinal tract through blood circulation and thus affect the gut microbiota; 2) respiratory exposure induces a local immune response involving immune cells and cytokines that can reach to the intestine through lymphatic circulation, which would then affect the intestinal immunity; 3) airborne particles in the airway are transported to the oropharynx via mucociliary clearance and are subsequently ingested and reach to the gastrointestinal tract. In addition, it is possible that a portion of intranasally administrated dusts might pass to the colon through swallow, which mirrors natural exposures in human. However, how the exposure to dusts contributes to the alteration to the airway microbiota remains to be further elucidated.
The major strength of this study is the multidimensional analysis of the environmental and gut microbiota from a pediatric cohort with varying susceptibility to allergy and the validation study using an in vivo mouse model. One important finding is that the exposure to an abundance of Aspergillus in indoor microbiota can contribute to the increased risk of allergies in urban children. A limitation of this study is that the influence of diets38 cannot be excluded when interpreting the difference in gut microbiota between urban and rural children, although we adjusted for several confounders. Furthermore, we employed a strategy based on short-read sequencing of 16S rRNA variable regions and DADA2 workflow, which limited our analysis in providing taxonomic resolution at species level. Further studies targeting important species-level taxa are suggested to take full advantage of a full-length 16S gene sequencing or single-molecule long-read sequencing strategy.44
In conclusion, our study indicates that a diverse environmental microbiota in rural China reduces allergies by modulating the gut microbiota. Our findings provide insight into exposure to a protective environment as a novel strategy in preventing allergic diseases.
Patients and Methods
Study design
The EuroPrevall-INCO study was a multi-staged, cross-sectional survey on the prevalence of allergic diseases in random samples of 35,549 children from the general population, with a nested case-control design. The study was conducted in China, India, and Russia. The study methodology, analysis of sensitization, and prevalence of allergic diseases have been described previously.18,38,45,46 In China, for the screening stage, 5,542 urban in Guangzhou city and 5,139 rural Shaoguan school-age children (7 to 10 years) participated by answering a standardized, one-page questionnaire. Those who had recorded both food-adverse reactions (FAR) and doctor-diagnosed food allergy (FA) in the screening questionnaire (223 urban and 181 rural cases) and age-matched children without allergies and other diseases (300 urban and 300 rural) were invited for the case-control study to complete the detailed questionnaire. A total of 190 urban and 116 rural participants with both FAR and FA, as well as 212 urban and 233 rural children without allergies and other diseases responded, with a response rate of 85.2%, 64.1%, 70.7%, and 77.7%, respectively (Table 1). Skin prick tests (SPTs) and specific IgE determinations for common food and airborne allergens were conducted (see below). We defined ‘cases’ as participants with FAR and FA and any positive results of allergen sensitizations, while those without any documentation of allergies and other diseases, as well as allergen sensitizations were defined as ‘controls’ (Figure 1). Finally, a total of 225 participants who met the above criterion and whose stool for DNA was available were included (Table 1). Total DNA were extracted from stool samples from these participants in batches using the QIAamp Stool DNA kit (Qiagen, Hilden, Germany), following the manufacturer’s protocol and stored at – 80°C. In the case-control study, the effect of the gut microbiota on allergy in children exposed a rural or urban environment was investigated. In addition, we randomly selected stool DNA of age- and gender-matched participants (n = 65) of cases and controls from urban and rural areas for shotgun metagenomic analysis (Table S3).
A serum specific IgE (sIgE) test was performed by drawing a blood sample from a vein. For determination of allergen sensitizations, sIgE against five groups of food mixtures (epcx1, epcx2, epcx3, fx5, and fx6; Thermo Fisher Scientific, Waltham, MA USA) was measured, followed by assessment of 27 individual food allergens and 6 airborne allergens. Food allergens included egg, milk, fish, wheat, maize, rice, sesame, buckwheat, peanut, soy, hazelnut, shrimp, crab, tomato, carrot, apple, kiwi, celery, melon, mustard, mango, banana, peach, poppy, lentil, walnut, and sunflower. Airborne allergens included house dust mites (Dermatophagoides pteronyssinus), cockroaches (Blatella germanica), cat dander, birch (Betula verrucosa), timothy grass (Phleum pratense), mugwort (Artemisia vulgaris), and Parietaria pollen. A panel of 18 food allergens (mango, crab, egg, date, mussel, orange, milk, soybean, hazelnut, shrimp, wheat, peanut, fish, peach, beef, apple, lemon, and tomato) and 5 airborne allergens (house dust mite, cockroach, cat dander, timothy grass, mugwort and cat dander) were used for the SPT (ALK, Horsholm, Denmark). A positive skin reaction was defined as a wheal size ≥ 3 mm after elimination of the negative control. The determination of sIgE was considered positive if the sIgE level was greater than 0.35 kU/L.
Dust samples
Environmental dust samples were collected from families in the EuroPrevall-INCO cohort in urban Guangzhou and rural Shaoguan. Dusts were collected and extracted following our previously published methods with minor modification.5,18,47 Briefly, participants had been living in their homes for at least 1 year before dust collection. Participants were asked not to replace their bedclothes for 4 weeks prior to dust collection. Dusts were collected from the bedsheet, pillowcase, pillow, quilt, and mattress of each child’s bedding, whereas henhouse dusts were collected from the housing facilities for poultry in rural areas close to their residence building by trained technicians using a hand-held 1200 W vacuum cleaner equipped with a filter trap (ALK-Abello, Hørsholm, Denmark). Dusts weighted less than 0.1 gram were excluded. Ten dust samples from representative urban and rural families, regardless of their case or control status, were randomly selected. In order to identify the potential source of microbial communities in rural indoor environments, we sampled dusts from the backyard poultry housing facilities from five rural families. For the dusts from each group (urban n = 10, rural n = 10, and henhouse dust n = 5), we pooled together 200 mg from individual dust in that group to generate one mixed pool of dusts. Pooled (n = 3) and individual (n = 25) dust samples were weighed, filtered through a fine dust sifter to remove large particles, extracted by using low-endotoxin phosphate-buffered saline (PBS) on a rotor at 25°C for 18 hours at a concentration of 100 mg/mL, centrifuged, and then the supernatant was filtered through a 0.22-μm membrane filter for sterility. The filtered extract solution was stored at −20°C until analysis. The concentration of the dust extract was defined as the weight of dry dust per milliliter of PBS suspension before filtration. The level of endotoxin in the dust extracts was quantified using the QCL-1000 Endpoint Chromogenic LAL Assay (Lonza, Visp, Swiss), following the manufacturer’s protocol. Genomic DNA (gDNA) was extracted from all dust samples using the PowerSoil DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA, USA), according to the manufacturer’s protocol.
Mouse model
Female C57BL/6 mice, 6–8 weeks old, were purchased from GemPharmatech Co., Ltd (Guangzhou, China) and housed in a specific pathogen-free facility. Animals were housed at least for 1 week prior to the initiation of the experiment to allow animals to habituate to the new facilitiy and surroundings. Mice were randomized into different groups prior to the start of the experiment to limit cage effects. A inflammation was induced following our previous protocol.48 Briefly, mice were sensitized on day 0, 7 and 14 by intraperitoneal (i.p.) injections of 25 μg ovalbumin (OVA, Sigma-Aldrich, Darmstadt, Germany) in conjunction with 2 mg of the adjuvant aluminum hydroxide, and then challenged by aerosolized OVA at days 27, 28, and 29 and euthanized on day 31. In order to investigate the diverse effects of exposure to urban and rural environments, we pre-exposed mice to PBS or dust extracts from pooled dust samples from urban mattresses, rural mattresses, and rural henhouses at one week (day −7) before the first OVA-sensitization. The mice were subject to 11 intranasal (i.n.) administrations with 50 μL of 100 mg/mL dust extract or PBS (control group) at the indicated time points (every day from −7 to −1 day and day 1, 4, 8, 11, Figure 4a), as previously described.49,50 Forty-eight hours after the final challenge (day 31), the mice were anesthetized. Bronchoalveolar lavage (BAL), lung tissue, and serum were collected as previously described.48 The cells from BAL fluids were counted and stained with anti-CD11c (eBioscience, Waltham, MA USA) and anti-Siglec-F (BD Biosciences, San Jose, CA, USA) monoclonal antibodies for eosinophils (SiglecF+CD11c−) using a fluorescence activator cell sorting FACSCalibur™ system (BD Biosciences, San Jose, CA, USA). The lungs were fixed in a 4% formaldehyde solution for 48 h and then embedded in paraffin. Tissue sections (3-μm thick) were prepared and stained with hematoxylin and eosin (H&E) (Sigma-Aldrich, Darmstadt, Germany) or Periodic acid-Schiff-alcian blue (PAS-AB) following standard procedures. The level of OVA-specific IgE was determined by enzyme-linked immunosorbent assay (ELISA, R&D Systems, Minneapolis, MN, USA). Luminal contents of the small intestines of mice were collected immediately after being euthanized into a sterile tube, were snap-frozen in liquid nitrogen and stored at −80°C until further analysis. Genomic DNA was then isolated from the luminal contents using the PowerSoil DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA, USA).
Fecal microbiota transplantation
To validate the effect of dust exposure altered gut microbiota on allergic airway inflammation, fresh fecal pellets were collected from specific pathogen-free (SPF)-mice that were intranasally administrated 11 times with PBS or henhouse dust extracts. Stools from donor mice were pooled; every 400 mg of stool was mixed and resuspended in 1 mL of sterile PBS, and the suspension was stored after centrifugation. Recipient mice were re-treated in every 3 days after 1 week by oral gavage with 200 μL of the suspension containing fecal microbiota pre-treated with a cocktail of broad-spectrum antibiotics, including 0.4 mg/mL kanamycin sulfate, 0.035 mg/mL gentamicin sulfate, 850 U/mL colistin sulfate salt, 0.215 mg/mL metronidazole, 0.045 mg/mL vancomycin hydrochloride.
Amplicon sequencing (16S rRNA and internal transcribed spacer)
The quality of gDNA was verified by TapeStation 2200 (Genomic DNA Screen Tape; Agilent Technologies, Santa Clara, CA, USA). The 16S rRNA V3-V4 region were amplified from 12.5 ng of gDNA and sequenced on an Illumina MiSeqDX instrument (Illumina, San Diego, CA, USA). Two sets of primers were used to amplify the V3-V4 region of 16S rRNA. For human samples, the 341 F primer (5’- CCTACGGGNGGCWGCAG-3’) and the 805 R primer (5’-GACTACHVGGGTATCTAATCC-3’) were used. For gDNA samples extracted from dusts and intestinal luminal contents, the 341 F primer and 806 R primer (5’-GGACTACHVGGGTATCTAAT-3’) were used. Illumina sequencing adapters and dual‐index barcodes were added to the gene-specific primers following Illumina’s 16S Metagenomics Sequencing Library Preparation Guide. Primers ITS3_KYO2 (5’-GATGAAGAACGYAGYRAA-3’) and ITS4 (5’-TCCTCCGCTTATTGATATGC-3’) were used to amplify the fungal ITS region 2 from gDNA of dust extracts.
The concentrations of the amplicon libraries were determined by Qubit 3.0 assay (Thermo Fisher Scientific, Waltham, MA USA). After dilution, about 10 ng of DNA library was run on 2200 TapeStation with D1000 Screentape to validate the library and determine the library size. The library concentration (in nM) was calculated based on the size of amplicons determined above, using the following formula: (concentration in ng/μl)/ (660 g/mol x average library size) x 106. Sequencing was performed on an Illumina MiSeqDX instrument using pair-end 300 cycles (2 x 150 reads, human samples) or 250 cycles (2 x 125 reads, dust extract and mice samples).
Shotgun metagenomic sequencing
TruSeq DNA PCR-free libraries were prepared manually from 400 ng fecal gDNA following the manufacturer’s protocol. Library concentrations were measured by a qPCR assay on a QuantStudio 7 Flex Real-Time PCR system (Applied Biosystems, Foster, CA, USA) using a Kapa library quantification kit (Roche, Basel, Switzerland). Sequencing clusters were generated on cBot using a HiSeq4000 PE cluster kit. Sequencing was performed on an Illumin HiSeq 4000 using HiSeq SBS reagents with 150 × 2 paired-end reads (Illumina, San Diego, CA, USA).
Amplicon data analysis
The amplicon libraries of each participant were sequenced in one to three MiSeqDX runs. Reads from different runs were combined using the cat command line and processed in R version 3.5.1 (R Foundation, Vienna, Austria) with DADA2, a high-resolution amplicon sequence processing pipeline that corrects for errors introduced in PCR, which generate amplicon sequence variants as compared to the traditional operational taxonomic units.50 The higher resolution afforded by ASV provided better ability to distinguish species from others. Pair-end reads were trimmed for PCR primers (341 F/805 R, 341 F/806 R, ITS3_KYO2/ITS4) with cutadapt v1.18 and truncated for enough overlap and then dereplicated. Built-in training models were utilized to learn error rates and denoise. After removing chimeras, taxonomic classification was assigned using the DECIPHER and SILVA_SSU_r132_March2018 database.51 The assignSpecies method in DADA2 returns species-level assignments if there is no ambiguity by using exact string matching. The generated count table of amplicon sequence variants (ASVs) and metadata were then loaded into phyloseq52 for downstream analysis.
Sequencing analysis
For taxonomic composition analysis, we agglomerated taxa at the family or genus level using the aggregate_taxa function using R package ‘microbiome’ (http://microbiome.github.io/microbiome/, v1.5.28) and the “compositional” abundances were returned as relative abundances. The ratio of two dominate genera, Bacteroides and Prevotella, were calculated as described by Gorvitovskaia et al.19 For diversity measurement, taxa were agglomerated at the species level, and the Shannon index was estimated with the function estimate_richness. ‘Core microbiota’ was defined as community of microbes and their functions that are shared in majority of hosts. An ‘Abundance-Occurrence’ approach was proposed to quantify the core microbiota in a conservative way.53 Follow this approach, the core taxa in our study were defined as those that exceed the given population prevalence threshold (prevalence > 50.0%) at given detection level (abundance > 0.1%). The core_abundance method, implemented in R package ‘microbiome’, then calculated community core abundance index, which gave the relative proportion of the core species. The Bray-Curtis dissimilarity, representing beta-diversity, was calculated and visualized by dimension reduction technology t-SNE with perplexity 50 (https://github.com/opisthokonta/tsnemicrobiota). PERMANOVA test measured by “bray” distance was performed with 999 permutations using Adonis test using R package vegan v2.5.4. To characterize bacterial contributors for enterotypes, we used R package biotyper v0.1.3 as described elsewhere (http://enterotypes.org). To visualize this contribution, count data were centered, log-transformed, and PCA compositional biplots were generated.
Shotgun metagenomics reads were quality filtered with KneadData (version 0.7.2) under default parameters and further analyzed for biochemical pathways using HUMAnN2ʹs (v0.11.2, http://huttenhower.sph.harvard.edu/humann2) tiered search workflow.54,55 Briefly, putative taxonomic abundances were calculated with MetaPhlAn2 v2.7.7, which generated a ‘bug list’ for the most abundant organisms (> 0.01% abundance). Nucleotide-level mapping was performed with a custom Bowtie2 database from a subset of the ChocoPhlAn v0.1.1 pangenome database corresponding to the organisms in the bug list. Unmapped reads were further aligned to either the UniRef90 or UniRef50 database56 (uniref50_annotated.1.1.dmnd) using the Diamond v0.9.21 translated search. Further analysis was based on the UniRef50 profiling output because of the high mapping rate and consistency. The HUMAnN2 output table (genefamilies) of each participant was merged using humann2_join_tables and normalized to count per million (CPM) and relative abundance using humann2_renorm_table helper scripts. The genefamilies table was also regrouped to MetaCyc metabolic pathways.57
In order to identify associations between variables of clinical interest and the abundance of individual microbial taxa or MetaCyc pathway after adjusting for potential confounders, we tested general linear models with R package MaAsLin2 (http://huttenhower.sph.harvard.edu/maaslin2, v0.2.3).58 The following covariates were forced into the MaAsLin2 model: urban-rural status, case-control category, age, gender, and body mass index (BMI). The minimum abundance for each feature was set to 0.01.
In order to identify microbial markers of the environment and gut microbiota, we used LEfSe2 in a docker environment (biobakery/lefse:1.0.0_dev_9adc3a62460e).59 The cutoff of the Benjamini–Hochberg multiple testing corrected p-value was set at 0.25. The LDA score was set at above 3.0 because the default setting with LDA score > 2.0 might be prone to a higher false discovery rate.60 LEfSe2 (LDA>3.0), used in our analysis, achieved similar results compared to a consensus approach suggested by Nearing et al.’s benchmark study60 regarding differential abundance analysis (refer to Suppl file 1 in our analysis report). Other parameters were set as the default. Differential associated taxa were visualized using Graphlan (http://huttenhower.sph.harvard.edu/graphlan) in a docker environment (biocontainers/graphlan:v1.1.3-1-deb_cv1).61
Supplementary Material
Acknowledgments
We thank the Biobank for Respiratory Diseases in the National Clinical Research Center for Respiratory Disease (BRD-NCRCRD, Guangzhou, Southern China).
Funding Statement
This study was supported by the National Natural Science Foundation of China under Grant 82161138020, the Guangdong Basic and Applied Basic Research Foundation under Grant 2021A1515011037 and 2017B020226006, the Open Project of State Key Laboratory of Respiratory Disease under Grant SKLRD-Z-202112 and SKLRD-OP-202004, the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program under Grant 2017BT01S155, the Science and Technology Program of Guangzhou under Grant 202102010011, and the Zhongnanshan Medical Foundation of Guangdong Province under Grant ZNSA-2020003 and ZNSA-2020013, Hong Kong Research Grant Council Award CUHK 14119320, and TS Lo Foundation.
Authors’ contributions
JL, CW, ZS conceived and designed the study. ZY drafted the manuscript and annotated all the results together with ZC with input from all authors. ZS guided mouse model experiments. ZY carried out the bioinformatics data analysis. ZC, MX, MJ, XX, and WL contributed to data analysis. XL, SY, XN, WY, and XR performed mouse and environmental experiments. MX, NL, MF, WF, ZL, ZZ, JZ, and NW performed the analysis of clinical specimens. GW, MR, and BS initialized the EuroPrevall-INCO surveys and contributed to the manuscript. JL and CW revised and finalized the manuscript.
Disclosure statement
The authors report there are no competing interests to declare.
Ethics approval and consent to participate:
The study was approved by the Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University (Guangzhou, China). Written informed consent was obtained from parents at the time of enrollment. The mice were housed in a specific pathogen-free animal facility at the Guangzhou Institutes of Biomedicine and Health (Guangzhou, China). All experiments were carried out in accordance with the National Animal Protection Guidelines of China.
Data availability statement
The dataset supporting the conclusions of this article is available in the Sequence Read Archive (SRA) with the accession code PRJNA786015. Contaminant sequence reads from human in metagenomic libraries were identified and filtered using KneadData (v0.10.0) before deposition.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2125733
References
- 1.Eder W, Ege MJ, von Mutius E.. The asthma epidemic. N Engl J Med. 2006;355(21):2226–20. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 2.Leung ASY, Tham EH, Li J, Pacharn P, Takizawa T, Lee E, Xing Y, Leung T-F, Hong S-J, Wong GWK. The role of the environment in shaping the trends of childhood asthma - An Asian perspective. Pediatr Allergy Immunol Off Publ Eur Soc Pediatr Allergy Immunol. 2021;32(6):1152–1164. doi: 10.1111/pai.13508. [DOI] [PubMed] [Google Scholar]
- 3.von Mutius E, Smits HH. Primary prevention of asthma: from risk and protective factors to targeted strategies for prevention. Lancet Lond Engl. 2020;396(10254):854–866. doi: 10.1016/S0140-6736(20)31861-4. [DOI] [PubMed] [Google Scholar]
- 4.von Mutius E. The microbial environment and its influence on asthma prevention in early life. J Allergy Clin Immunol. 2016;137(3):680–689. doi: 10.1016/j.jaci.2015.12.1301. [DOI] [PubMed] [Google Scholar]
- 5.Feng M, Yang Z, Pan L, Lai X, Xian M, Huang X, Chen Y, Schröder PC, Roponen M, Schaub B, et al. Associations of early life exposures and environmental factors with asthma among children in rural and urban areas of Guangdong, China. Chest. 2016;149(4):1030–1041. doi: 10.1016/j.chest.2015.12.028. [DOI] [PubMed] [Google Scholar]
- 6.Huang S, Garshick E, Weschler LB, Hong C, Li J, Li L, Qu F, Gao D, Zhou Y, Sundell J, et al. Home environmental and lifestyle factors associated with asthma, rhinitis and wheeze in children in Beijing, China. Environ Pollut. 2020;256:113426. doi: 10.1016/j.envpol.2019.113426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilbert JA, Stephens B. Microbiology of the built environment. Nat Rev Microbiol. 2018;16(11):661–670. doi: 10.1038/s41579-018-0065-5. [DOI] [PubMed] [Google Scholar]
- 8.Xing Y, Wang MH, Leung T-F, Wong C-K, Roponen M, Schaub B, Li J, Wong GWK. Poultry exposure and environmental protection against asthma in rural children. Allergy. 2022. doi: 10.1111/all.15365. [DOI] [PubMed] [Google Scholar]
- 9.Lee MK, Wyss AB, Carnes MU, Richards M, Parks CG, Beane Freeman LE, Thorne PS, Umbach DM, Azcarate-Peril MA, Peddada SD, et al. House dust microbiota in relation to adult asthma and atopy in a US farming population. J Allergy Clin Immunol. 2021;147(3):910–920. doi: 10.1016/j.jaci.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karvonen AM, Kirjavainen PV, Täubel M, Jayaprakash B, Adams RI, Sordillo JE, Gold DR, Hyvärinen A, Remes S, von Mutius E, et al. Indoor bacterial microbiota and development of asthma by 10.5 years of age. J Allergy Clin Immunol. 2019;144(5):1402–1410. doi: 10.1016/j.jaci.2019.07.035. [DOI] [PubMed] [Google Scholar]
- 11.Kirjavainen PV, Karvonen AM, Adams RI, Täubel M, Roponen M, Tuoresmäki P, Loss G, Jayaprakash B, Depner M, Ege MJ, et al. Farm-like indoor microbiota in non-farm homes protects children from asthma development. Nat Med. 2019;25(7):1089–1095. doi: 10.1038/s41591-019-0469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, Madeira FB, Beyaert R, van Loo G, Bracher F, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science. 2015;349(6252):1106–1110. doi: 10.1126/science.aac6623. [DOI] [PubMed] [Google Scholar]
- 13.Krusche J, Twardziok M, Rehbach K, Böck A, Tsang MS, Schröder PC, Kumbrink J, Kirchner T, Xing Y, Riedler J, et al. TNF-α-induced protein 3 is a key player in childhood asthma development and environment-mediated protection. J Allergy Clin Immunol. 2019;144(6):1684–1696.e12. doi: 10.1016/j.jaci.2019.07.029. [DOI] [PubMed] [Google Scholar]
- 14.Winglee K, Howard AG, Sha W, Gharaibeh RZ, Liu J, Jin D, Fodor AA, Gordon-Larsen P. Recent urbanization in China is correlated with a Westernized microbiome encoding increased virulence and antibiotic resistance genes. Microbiome. 2017;5(1):121. doi: 10.1186/s40168-017-0338-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hufnagl K, Pali-Schöll I, Roth-Walter F, Jensen-Jarolim E. Dysbiosis of the gut and lung microbiome has a role in asthma. Semin Immunopathol. 2020;42(1):75–93. doi: 10.1007/s00281-019-00775-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sbihi H, Boutin RC, Cutler C, Suen M, Finlay BB, Turvey SE. Thinking bigger: how early-life environmental exposures shape the gut microbiome and influence the development of asthma and allergic disease. Allergy. 2019;74(11):2103–2115. doi: 10.1111/all.13812. [DOI] [PubMed] [Google Scholar]
- 17.Liddicoat C, Sydnor H, Cando-Dumancela C, Dresken R, Liu J, Gellie NJC, Mills JG, Young JM, Weyrich LS, Hutchinson MR, et al. Naturally-diverse airborne environmental microbial exposures modulate the gut microbiome and may provide anxiolytic benefits in mice. Sci Total Environ. 2020;701:134684. doi: 10.1016/j.scitotenv.2019.134684. [DOI] [PubMed] [Google Scholar]
- 18.Wong GWK, Mahesh PA, Ogorodova L, Leung TF, Fedorova O, Holla AD, Fernandez-Rivas M, Clare Mills EN, Kummeling I, van Ree R, et al. The EuroPrevall-INCO surveys on the prevalence of food allergies in children from China, India and Russia: the study methodology. Allergy. 2010;65(3):385–390. doi: 10.1111/j.1398-9995.2009.02214.x. [DOI] [PubMed] [Google Scholar]
- 19.Gorvitovskaia A, Holmes SP, Huse SM. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome. 2016;4(1):15. doi: 10.1186/s40168-016-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ege MJ, Mayer M, Normand A-C, Genuneit J, Wocm C, Braun-Fahrländer C, Heederik D, Piarroux R, von Mutius E; GABRIELA Transregio 22 Study Group . Exposure to environmental microorganisms and childhood asthma. N Engl J Med. 2011;364(8):701–709. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- 21.Flies EJ, Clarke LJ, Brook BW, Jones P. Urbanisation reduces the abundance and diversity of airborne microbes - but what does that mean for our health? A systematic review. Sci Total Environ. 2020;738:140337. doi: 10.1016/j.scitotenv.2020.140337. [DOI] [PubMed] [Google Scholar]
- 22.Schram-Bijkerk D, Doekes G, Douwes J, Boeve M, Riedler J, Ublagger E, von Mutius E, Benz MR, Pershagen G, van Hage M, et al. Bacterial and fungal agents in house dust and wheeze in children: the PARSIFAL study. Clin Exp Allergy J Br Soc Allergy Clin Immunol. 2005;35(10):1272–1278. doi: 10.1111/j.1365-2222.2005.02339.x. [DOI] [PubMed] [Google Scholar]
- 23.Heederik D, von Mutius E. Does diversity of environmental microbial exposure matter for the occurrence of allergy and asthma? J Allergy Clin Immunol. 2012;130(1):44–50. doi: 10.1016/j.jaci.2012.01.067. [DOI] [PubMed] [Google Scholar]
- 24.Sharpe RA, Bearman N, Thornton CR, Husk K, Osborne NJ. Indoor fungal diversity and asthma: a meta-analysis and systematic review of risk factors. J Allergy Clin Immunol. 2015;135(1):110–122. doi: 10.1016/j.jaci.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Visagie CM, Hirooka Y, Tanney JB, Whitfield E, Mwange K, Meijer M, Amend AS, Seifert KA, Samson RA. Aspergillus, Penicillium and Talaromyces isolated from house dust samples collected around the world. Stud Mycol. 2014;78(1):63–139. doi: 10.1016/j.simyco.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hay DB, Hart BJ, Douglas AE. Effects of the fungus Aspergillus penicillioides on the house dust mite Dermatophagoides pteronyssinus: an experimental re-evaluation. Med Vet Entomol. 1993;7(3):271–274. doi: 10.1111/j.1365-2915.1993.tb00687.x. [DOI] [PubMed] [Google Scholar]
- 27.De León J G, González Méndez R, Cadilla CL, Rivera-Mariani FE, Bolaños-Rosero B. Identification of Immunoglobulin E-Binding Proteins of the Xerophilic Fungus Aspergillus penicillioides Crude Mycelial Mat Extract and Serological Reactivity Assessment in Subjects with Different Allergen Reactivity Profiles. Int Arch Allergy Immunol. 2018;175(3):147–159. doi: 10.1159/000484898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Debarry J, Garn H, Hanuszkiewicz A, Dickgreber N, Blümer N, von Mutius E, Bufe A, Gatermann S, Renz H, Holst O, et al. Acinetobacter lwoffii and Lactococcus lactis strains isolated from farm cowsheds possess strong allergy-protective properties. J Allergy Clin Immunol. 2007;119(6):1514–1521. doi: 10.1016/j.jaci.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 29.Lee-Sarwar KA, Lasky-Su J, Kelly RS, Litonjua AA, Weiss ST. Gut microbial-derived metabolomics of asthma. Metabolites. 2020;10(3):E97. doi: 10.3390/metabo10030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu C, van Meel ER, Medina-Gomez C, Kraaij R, Barroso M, Kiefte-de Jong J, Radjabzadeh D, Pasmans SGMA, de Jong NW, de Jongste JC, et al. A population-based study on associations of stool microbiota with atopic diseases in school-age children. J Allergy Clin Immunol. 2021;148(2):612–620. doi: 10.1016/j.jaci.2021.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog. 2017;106:171–181. doi: 10.1016/j.micpath.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Maltz RM, Keirsey J, Kim SC, Mackos AR, Gharaibeh RZ, Moore CC, Xu J, Somogyi A, Bailey MT. Social stress affects colonic inflammation, the gut microbiome, and short-chain fatty acid levels and receptors. J Pediatr Gastroenterol Nutr. 2019;68(4):533–540. doi: 10.1097/MPG.0000000000002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ju T, Kong JY, Stothard P, Willing BP. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019;13(6):1520–1534. doi: 10.1038/s41396-019-0364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin-Gallausiaux C, Marinelli L, Blottière HM, Larraufie P, Lapaque N. SCFA: mechanisms and functional importance in the gut. Proc Nutr Soc. 2021;80(1):37–49. doi: 10.1017/S0029665120006916. [DOI] [PubMed] [Google Scholar]
- 35.Cait A, Hughes MR, Antignano F, Cait J, Dimitriu PA, Maas KR, Reynolds LA, Hacker L, Mohr J, Finlay BB, et al. Microbiome-driven allergic lung inflammation is ameliorated by short-chain fatty acids. Mucosal Immunol. 2018;11(3):785–795. doi: 10.1038/mi.2017.75. [DOI] [PubMed] [Google Scholar]
- 36.Rachid R, Stephen-Victor E, Chatila TA. The microbial origins of food allergy. J Allergy Clin Immunol. 2021;147(3):808–813. doi: 10.1016/j.jaci.2020.12.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen -Y-Y, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Zheng W, Yung E, Zhong N, Wong GWK, Li J. Frequency of food group consumption and risk of allergic disease and sensitization in schoolchildren in urban and rural China. Clin Exp Allergy. 2015;45(12):1823–1832. doi: 10.1111/cea.12532. [DOI] [PubMed] [Google Scholar]
- 39.Ottman N, Ruokolainen L, Suomalainen A, Sinkko H, Karisola P, Lehtimäki J, Lehto M, Hanski I, Alenius H, Fyhrquist N. Soil exposure modifies the gut microbiota and supports immune tolerance in a mouse model. J Allergy Clin Immunol. 2019;143(3):1198–1206.e12. doi: 10.1016/j.jaci.2018.06.024. [DOI] [PubMed] [Google Scholar]
- 40.Nomura A, Matsubara A, Goto S, Takahata J, Sawada K, Ihara K, Nakaji S. Relationship between gut microbiota composition and sensitization to inhaled alergens. Allergol Int Off J Jpn Soc Allergol. 2020;69(3):437–442. doi: 10.1016/j.alit.2019.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Dhakal S, Wang L, Antony L, Rank J, Bernardo P, Ghimire S, Bondra K, Siems C, Lakshmanappa YS, Renu S, et al. Amish (rural) vs. non-Amish (urban) infant fecal microbiotas are highly diverse and their transplantation lead to differences in mucosal immune maturation in a humanized germfree piglet model. Front Immunol. 2019;10:1509. doi: 10.3389/fimmu.2019.01509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun J, Liao X-P, D’Souza AW, Boolchandani M, S-H L, Cheng K, Luis Martínez J, Li L, Feng Y-J, Fang L-X, et al. Environmental remodeling of human gut microbiota and antibiotic resistome in livestock farms. Nat Commun. 2020;11(1):1427. doi: 10.1038/s41467-020-15222-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey MJ, Naik NN, Wild LE, Patterson WB, Alderete TL. Exposure to air pollutants and the gut microbiota: a potential link between exposure, obesity, and type 2 diabetes. Gut Microbes. 2020;11(5):1188–1202. doi: 10.1080/19490976.2020.1749754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson JS, Spakowicz DJ, Hong B-Y, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):5029. doi: 10.1038/s41467-019-13036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Ogorodova LM, Mahesh PA, Wang MH, Fedorova OS, Leung TF, Fernandez-Rivas M, Mills ENC, Potts J, Kummeling I, et al. Comparative study of food allergies in children from China, India, and Russia: the EuroPrevall-INCO surveys. J Allergy Clin Immunol Pract. 2020;8(4):1349–1358.e16. doi: 10.1016/j.jaip.2019.11.042. [DOI] [PubMed] [Google Scholar]
- 46.Yang Z, Zhao J, Wei N, Feng M, Xian M, Shi X, Zheng Z, Su Q, Wong GWK, Li J. Cockroach is a major cross-reactive allergen source in shrimp-sensitized rural children in southern China. Allergy. 2018;73(3):585–592. doi: 10.1111/all.13341. [DOI] [PubMed] [Google Scholar]
- 47.Zhang C, Gjesing B, Lai X, Li J, Spangfort MD, Zhong N. Indoor allergen levels in Guangzhou city, southern China. Allergy. 2011;66(2):186–191. doi: 10.1111/j.1398-9995.2010.02465.x. [DOI] [PubMed] [Google Scholar]
- 48.Gao X, Ren X, Wang Q, Yang Z, Li Y, Su Z, Li J. Critical roles of regulatory B and T cells in helminth parasite-induced protection against allergic airway inflammation. Clin Exp Immunol. 2019;198(3):390–402. doi: 10.1111/cei.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ng N, Lam D, Paulus P, Batzer G, Horner AA. House dust extracts have both TH2 adjuvant and tolerogenic activities. J Allergy Clin Immunol. 2006;117(5):1074–1081. doi: 10.1016/j.jaci.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 50.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. DECIPHER: database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84(4):524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neu AT, Allen EE, Roy K. Defining and quantifying the core microbiome: challenges and prospects. Proc Natl Acad Sci U S A. 2021;118(51):e2104429118. doi: 10.1073/pnas.2104429118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15(11):962–968. doi: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 56.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinforma Oxf Engl. 2007;23(10):1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 57.Caspi R, Billington R, Keseler IM, Kothari A, Krummenacker M, Midford PE, Ong WK, Paley S, Subhraveti P, Karp PD. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 2020;48(D1):D445–53. doi: 10.1093/nar/gkz862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol. 2021;17(11):e1009442. doi: 10.1371/journal.pcbi.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nearing JT, Douglas GM, Hayes MG, MacDonald J, Desai DK, Allward N, Jones CMA, Wright RJ, Dhanani AS, Comeau AM, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022;13(1):342. doi: 10.1038/s41467-022-28034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ. 2015;3:e1029. doi: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset supporting the conclusions of this article is available in the Sequence Read Archive (SRA) with the accession code PRJNA786015. Contaminant sequence reads from human in metagenomic libraries were identified and filtered using KneadData (v0.10.0) before deposition.