

Abstract
Background
The interplay between cardiovascular disease (CVD) genetic risk indexed by a polygenic risk score (PRS) and diet quality still requires further investigation amongst older adults or those with established or treated CVD. The present study aimed to evaluate the relative contribution of diet quality, measured using the Australian Recommended Food Score (ARFS) and PRS, with respect to explaining variation in plasma lipids CVD outcomes in the Hunter Cohort.
Methods
The study comprised a secondary analysis of cross‐sectional data from the Hunter Cohort study. Single‐nucleotide polymorphisms from previously derived polygenic scores (PGSs) for three lipid classes were obtained: low‐density lipoprotein, high‐density lipoprotein and triglycerides, as well as PRS for coronary artery disease (CAD) from the PGS catalogue. Regression modelling and odds ratios were used to determine associations between PRS, ARFS and CVD risk.
Results
In total, 1703 participants were included: mean ± SD age 66 ± 7.4 years, 51% female, mean ± SD total ARFS 28.1 ± 8 (out of 74). Total diet quality and vegetable subscale were not significantly associated with measured lipids. By contrast, PGS for each lipid demonstrated a markedly strong, statistically significant correlation with its respective measured lipid. There was a significant association between CAD PRS and 5/6 CVD phenotypes (all except atrial fibrillation), with the largest effect size shown with coronary bypass. Adding dietary intake as a covariate did not change this relationship.
Conclusions
Lipid PGS explained more variance in measured lipids than diet quality. However, the poor diet quality observed in the current cohort may have limited the ability to observe any beneficial effects. Future research should investigate whether the diet quality of older adults can be improved and also the effect of these improvements on changes in polygenic risk.
Keywords: cardiovascular disease, cohort, diet quality, Hunter Community Study, polygenic risk
Variation in cardiovascular disease risk factors among older adults in the Hunter Community Study cohort: a comparison of diet quality versus polygenic risk score.

Key points
The Australian Recommended Food Score (ARFS) had little association with lipid and cardiovascular disease (CVD) endpoints.
Lipid polygenic score (PGS) explained more variance in measured lipids than diet quality.
The lipid PGS was associated with all three lipid parameters and some CVD endpoints, especially high cholesterol.
Coronary artery disease polygenic risk score was associated with CVD endpoints angina, coronary bypass, heart attack, high cholesterol and hypertension and some lipid parameters (high‐density lipoprotein cholesterol).
INTRODUCTION
Dietary intake is a modifiable, lifestyle factor contributing to the global burden of disease. 1 , 2 Higher diet quality, characterised by higher intakes of fruits, vegetables, wholegrains and lean sources of protein, as well as lower intakes of energy‐dense, nutrient‐poor foods including confectionery, biscuits, sugar sweetened beverages and take‐away foods (e.g., pizza, chips or deep‐fried foods) is associated with a lower risk of developing cardiovascular disease (CVD). 3 , 4 A recent systematic review of cohort studies identified that higher diet quality, measured by dietary indexes, was associated with significantly lower CVD incidence and mortality risk (relative risk = 0.80; 95% confidence interval [CI] = 0.78–0.82). 4
Emerging evidence shows that there is a relationship between genetic predisposition, dietary intake and CVD. 5 Although mRNA expression is partially controlled by genetic factors, it is also impacted directly by environmental effects such as dietary intake. 6 , 7 For example, associations between healthier dietary patterns such as the Prudent and Mediterranean diets and gene expression of inflammatory, immune response and cardiovascular pathways have been identified. 7 , 8 Other nutrient specific interactions include the ratio of omega‐6 to omega‐3 fatty acids consumed, which impacts on genes such as cytosolic PLA2 alpha or 5‐lipoxygenase, thus affecting inflammation and inflammatory related disease or conditions. 9 , 10 High‐density lipoprotein (HDL) and low‐density lipoprotein (LDL) concentrations can be influenced by variants in genes, such as vanin 1, as well as dietary intake. 11 Identification of individual variation in these pathways is termed nutrigenomics and these data can be used to inform personalised dietary advice targeting the biology of individuals. 9
Polygenic risk scores (PRSs) predict the likelihood of a specific outcome, such as CVD, by including known DNA variants into a statistical model. 12 In other words, the effect of variants on CVD are summed genome wide to derive an individual's genetic liability to the disorder. This allows development of models that stratify individuals by risk of developing specific diseases over time. PRSs have been used in observational studies to measure associations between lifestyle risk factors and genetic markers for disease. 13 , 14 , 15 , 16 These studies have shown promise, having identified that healthier lifestyles, including healthful dietary patterns, are associated with a lower risk of disease, including type 2 diabetes, CVD, CVD mortality and all‐cause mortality, even among those with a high PRS.
The majority of genome‐wide association studies (GWAS) evaluation to date have been in populations of European descent and, although studies in other ethnicities are underway, including Japanese and African‐American, 17 findings are not yet able to be used routinely in clinical settings. 12
The UK Biobank cohort has been a major resource for analysing the interplay between diet and lifestyle influences and genetics on CVD liability. A recent longitudinal analysis in middle aged (40–69 year old) adults with no history of CVD (n = 77,004) not only demonstrated a significant enrichment of coronary artery disease (CAD) PRS amongst participants who went on to develop CVD, but also identified that, among those with high PRS, a better diet quality was protective to some degree. 16 For example, for those with a high PRS, every one‐point higher Healthy Diet Indicator (higher diet quality) score was associated with a reduction in myocardial infarction risk (hazard ratio = 0.93, 95% CI = 0.88–0.99, p = 0.017). 16 Another study identified that participants without CVD, but with a familial predisposition to CVD and therefore higher PRS, had a lower risk of CVD among those with higher intakes of fish and a higher risk among those with higher processed‐meat intakes. 18 This study also identified a protective effect within those with a familial predisposition to CVD, and higher PRS showing a lower risk of CVD among those with higher intakes of cheese. 18 Although cheese consumption has historically been associated with an increased risk of CVD because of its high saturated fatty acid intake, 19 recent meta‐analyses of observational studies have identified an association between higher intakes of cheese and lower risk of CHD, 20 , 21 supporting the findings of a protective effect with higher cheese consumption. 18
The interplay between CVD genetic risk indexed by a PRS and diet quality still requires further investigation amongst older adults or those with established or treated CVD. Moreover, other dietary related CVD risk factors such as a measured lipids also have a strong genetic basis, although the extent to which diet modifies genetic predisposition to higher plasma lipids is also still relatively uncharacterised. The present study therefore aimed to evaluate the relative contribution of diet quality, measured using the Australian Recommended Food Score (ARFS) and PRS with respect to explaining variation in plasma lipids and liability to CVD phenotypes in the Hunter Community Study cohort.
METHODS
Study cohort
The present study comprises a secondary analysis of cross‐sectional baseline data from the Hunter Cohort study. The Hunter Community Study (HCS) cohort is a population‐based study comprised of men and women aged between 55 and 85 years at recruitment and who resided in the Newcastle region of New South Wales, Australia. Briefly, the participants were recruited by random selection from the electoral role between 2004 and 2007, with those agreeing providing their written informed consent for collection of a blood sample, attending a clinic visit, and answering a series of health‐related questionnaires on demographics, dietary intake, physical activity, morbidity, mental health and quality of life. Clinical measures were conducted by a trained nurse and included anthropometry, respiratory function, cardiovascular function, cognition, bone mineral density, functional capacity routine blood biomarkers, with DNA also extracted for genotyping. In total, 9784 individuals were sent invitation letters, 7575 responded (77.4%), 3877 agreed to participate via written informed consent and 3253 (response rate 44.5%) completed the study (47% men and 53% women). The full details of this cohort and the measures collected have been outlined elsewhere. 22 Although non‐response can affect bias, characteristics of the included population such as age and marital status, are reflective of the profiles of population of the Hunter, NSW and Australian population 22 ; however, the mean age is marginally younger.
The HCS protocol and all procedures involving study participants were approved by the University of Newcastle Human Research Ethics committee (H‐820–0504) with informed consent obtained from all participants. The current analysis was approved by the HCS data custodians and a ‘Memorandum of Understanding’ and ‘Confidentiality Statement’ signed by all authors.
Genotyping and imputation
Extracted DNA was genotyped using the Axiom Kaiser array (Affymetrix), with variants excluded with low call rate (<0.95), significant deviations from Hardy–Weinberg equilibrium (p < 1 × 10−6) and minor allele frequency less than 1%. Imputation and quality control of this cohort was described previously in Reay et al., 23 where imputation was performed using the Haplotype Reference Consortium panel via the Michigan Imputation Server, with high confidence (R 2 > 0.8) common variants retained post‐imputation that exhibited missingness <2%. 23 , 24 Unrelated individuals of European ancestry were selected for these analyses to account for the effect of population stratification and relatedness on the distribution of polygenic scores (PGSs). Physically genotyped common variants in relative linkage equilibrium, excluding known regions of long‐range linkage disequilibrium (LD), were the input for relatedness testing and principal component analysis (PCA) using PLINK, version 1.9 (https://www.cog-genomics.org/plink) to define population outliers via clustering the first two eigenvectors with that of each of the 1000 genomes phase 3 superpopulations via k‐means clustering. 25 , 26 PCA was then repeated using the same parameters in the defined European ancestry HCS cohort subset such that the principal components could be used as covariates in downstream analyses
PGSs
We obtained single nucleotide polymorphism (SNP) weights from previously derived PGS for three lipid classes: LDL, HDL and triglycerides (TGs), as well as PRSs for CAD from the PGS catalogue. 27 PGS is utilised hereafter to denote genetic scores related to the continuous lipid traits, whereas PRS relates to a disease phenotype, and thus, is more appropriate to designate the CAD scores. Two PGSs for each trait constructed using different methods were selected from the catalogue to profile in the HCS cohort. For the three lipid traits, two classes of PGS were considered: (i) clumping and thresholding (C + T) of established lipid loci in relative linkage equilibrium (GWAS, p < 5 × 10−8, r 2 < 0.1) trained using a GWAS meta‐analysis of up to 331,368 individuals (PGS catalogue accession IDs: PGS000064, PGS000065 and PGS000066) 28 and (ii) penalised regression (batch screening iterative LASSO–BASIL) for SNP selection trained in a subset of the UK Biobank (N = 255,256, PGS catalogue accession IDs: PGS000688, PGS000686, PGS000699). 29 The two CAD PRS selected were an LDPred derived score trained in the UK Biobank using an external CAD GWAS which is a Bayesian method that shrinks effect sizes to account for LD (PGS catalogue accession ID: PGS000013), with 0.001 selected as the optimal fraction of non‐zero effect sizes variants, and a ‘metaGRS’ approach that combined three pre‐existing CAD PRS (PGS catalogue accession ID: PGS000018). 30 , 31 In all instances, the PGS/PRS in the HCS were calculated by summing the j SNP weights () profiled in each individual i multiplied by their genotype dosage under an additive model (Equation 1).
| (1) |
We used the ‐‐score flag in PLINK 1.9 to calculate the scores in the HCS cohort, with each score then averaged by the number of non‐missing alleles in the score.
Dietary information
The ARFS was previously calculated in this cohort as a subscale of responses to a self‐administered 145‐item semiquantitative food frequency questionnaire (FFQ) completed by HCS participants. 32 The FFQ uses the NUTTAB90 nutrition database 33 ; however this nutrient database is not needed for the calculation of ARFS. The ARFS was scored directly from the frequency response data from the included questions on the FFQ. ARFS was previously been shown to be a valid measure of diet quality. 32 , 34 , 35 , 36 The ARFS sums seven categories: vegetables, fruit, protein foods, grains, dairy, fats and alcohol, with a maximum score of 74 points and higher ARFS indicative of a dietary pattern more closely aligned with the Australian Dietary Guidelines 37 and variety with food groups recommended in the Australian Guide to Health Eating. 38 One point was given for consumption of once or more per week for all foods, with upper limits on consumption of red meat and processed dairy foods such as flavoured milk and ice cream. Additional points were awarded for choosing whole grain bread, low fat milk and greater consumption of vegetables. ARFS scoring has been detailed further elsewhere. 39 We also considered the vegetable subscale of the ARFS, given the established association between vegetable intake, cardiovascular outcomes 40 and health usage. 41 , 42
CVD phenotypes
Total, HDL and TG were all measured from the fasting blood samples, whereas LDL was calculated using Friedewald's equation (LDL [mmol L–1] = total cholesterol − HDL − TG/2.2). Samples were obtained at baseline in the HCS, and thus were chosen as outcome phenotypes in assessing the relationship between diet quality and PGSs. Moreover, we investigated six CVD phenotypes that individuals self‐reported they had been previously diagnosed with at baseline: angina, atrial fibrillation, coronary bypass, heart attack, high cholesterol and hypertension.
Statistical analysis
All statistical analyses were conducted using R, version 3.6.0. 43 We derived four main subsets from the European, unrelated HCS cohort with genotype data available: (i) non‐missing cholesterol and ARFS; (ii) non‐missing cholesterol and ARFS, as well as non‐missing additional covariates of interest (ever smoked, educational attainment and statin usage); (iii) non‐missing self‐reported CVD diseases (the six described above) and ARFS; and (iv) non‐missing self‐reported CVD diseases and ARFS, as well as the covariates outlined in the second cohort. First, ARFS was regressed against each lipid class in the first cohort using linear regression covaried for age and sex, whereas each PGS/PRS was also regressed against the lipids, with the first‐five SNP derived principal components as additional covariates. A model with ARFS as a covariate was then utilised with the genetic scores. We calculated the variance explained in the lipids by the PGS for that same lipid using the (), where R 2 was the adjusted R 2 , the coefficient of determination adjusted for the number of predictors in the model. As result, the null model included age, sex, five principal components and the ARFS, whereas the full model included the PGS for each lipid trait. The PGS and ARFS were both scaled to have a mean of zero and unit variance. We also considered the impact of natural log transformation of the lipid outcomes on . An interaction term was the included between the PGS and ARFS in these models to test for a departure from additivity. The association of the PGS with each lipid was then also tested including the additional covariates of smoking status, educational attainment and self‐reported statin usage. Bonferroni correction was applied to the genetic results, with a significant association designated as p < 1.04 × 10−3 (0.05/[2x(3 × 8)]).
The CVD binary disease phenotypes were assessed for association with each PGS in a similar fashion, with these phenotypes the outcome variable in binomial logistic regression models. First, we tested the association of the ARFS and the eight PGS/PRS separately with each of the CVD phenotypes, using the entire rest of the cohort as controls, which would include other CVD cases. As a result, we then repeated these models by only retaining individuals who did not self‐report any of the six CVD phenotypes as controls for each model. Models with both ARFS and the genetic scores were also the constructed as in the lipid analyses, with the additional smoking, education and statin covariates included in additional models. The variance explained by the CAD PRS in each of these CVD binary phenotypes was estimated using Nagelkerke's R 2, which was the converted to the liability scale such that this metric was not biased by sample composition and population prevalence of the CVD trait, assuming the following population prevalences of 5%, 10%, 15% and 20%. 44 We used the most significantly associated CAD PRS from the two tested for each trait, with Nagelkerke's R 2 derived by subtracting the full model with the CAD PRS from the null covaried for age, sex, five principal components and the ARFS. In total, 192 genetic models were tested, and thus the threshold for significance was set as p < 2.60 × 10−4. Finally, as heart attack was the most severe CVD phenotype recorded, we tested whether people who had experienced this phenotype had an enrichment of CAD PRS relative to the remaining CVD traits.
RESULTS
Baseline characteristics of study population
Of the 3318 HCS cohort participants, there were 1703 unrelated, European ancestry individuals who survived genotyping quality control and had a non‐missing record of measured blood lipids (LDL, HDL and TG) and the ARFS diet quality score. The cohort mean ± SD age was 66.1 ± 7.4 years (51.32% female), whereas the mean ± SD total ARFS, vegetable subscale ARFS and lipids were: ARFS = 28.12 ± 8; ARFS vegetable subscale = 9.9 ± 3.6; LDL = 3.08 ± 0.91 mmol L–1; HDL = 1.35 ± 0.36 mmol L–1; and TG = 1.30 ± 0.70. A subset of these 1703 individuals (N = 1486) had smoking status, educational attainment and self‐reported statin usage recorded, and thus were retained for the sensitivity analyses that included those additional covariates. The sex and age profile of this subset was very similar (see Supporting information, Table S1). Furthermore, of the six binary self‐reported CVD phenotypes, there were 1678 individuals with non‐missing status for all of these traits. The number of cases ranged from 129 for heart attack to 758 for hypertension (see Supporting information, Table S2). There were 556 individuals who did not self‐report any of these conditions. Analogous to the lipid analyses, the majority of the cohort had non‐missing educational attainment, smoking status and statin usage (N = 1508) for those sensitivity analyses (see Supporting information, Table S3).
Lipid PGSs explain more variance in measured cholesterol than diet quality
We found that overall diet quality as measured by total ARFS score was not significantly associated with measured LDL or HDL, either in the baseline model adjusted for age and sex, or the full sensitivity model covaried for educational attainment, smoking and statin usage (see Supporting information, Table S4). However, there was a weak association between better diet quality and decreased plasma TG, with each SD increase (scaled to be one) in ARFS associated with a −0.035 mmol L–1 (95% CI = −0.001 to −0.069; p = 0.042) lower TG level (Figure 1a). This remained relatively consistent in the full sensitivity model. By contrast, the PGS for each lipid demonstrated a markedly stronger, statistically significant correlation with its respective measurement (Figure 1; see also Supporting information, Table S5). The The association between the best performing PGS for LDL, HDL, and triglycerides, respectively, with each measured lipid was as follows per standard deviation increase in PGS‐LDL: = 0.177 mmol L–1 (p = 9.35 × 10−17); HDL, = 0.134 mmol L–1 (p = 2.70 × 10−67); and TG, = 0.234 mmol L–1 (p = 7.89 × 10−47). Penalised regression (BASIL) scores performed best for HDL and TG, whereas the C + T approach was most significantly associated for LDL. Diet quality as measured by ARFS was then added as a covariate, with only a minute effect on estimated correlation between the score and measured lipids (Figure 1b). The variance explained () by each of the scores in the respective lipids relative to the null model covaried for age, sex, five SNP‐derived principal components, and the ARFS was 3.37%, 13.6% and 11.14% for LDL, HDL and TG, respectively. Natural log transformation of the outcome did not impact these estimates in any notable way (see Supporting information, Table S6). There was no significant interaction between ARFS and TG PGS on measured TG. The same sensitivity models applied to diet quality in this subset of the cohort did reduce the effect sizes for HDL and TG, however the PGS correlations with lipids all remained highly significant (p < 1 × 10−16) (Figure 1b). Interestingly, the addition of these other covariates resulted in an elevated correlation between the LDL PGS and measured LDL, likely because the large effect of statins was not accounted for in the baseline model (see Supporting information, Table S5). Genetic risk for CAD (CAD PRS) was associated with lower HDL but was not significantly correlated with measured TG or LDL (see Supporting information, Table S5). We next investigated whether the ARFS vegetable specific subscale would have differing effects than the total score. There were no statistically significant effects of vegetable subscale on plasma lipids levels (see Supporting information, Table S4). Moreover, adjusting PGS for ARFS vegetable subscale yielded very similar effects to that observed with the total ARFS (see Supporting information, Table S5).
Figure 1.
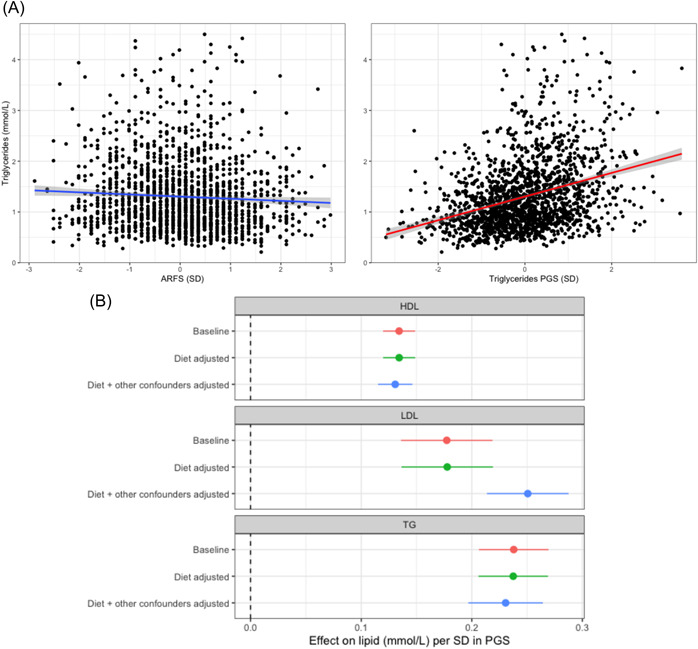
The relationship between diet, polygenic scores (PGSs) of lipids and measured lipids. (a) Scatterplots of distribution of measured triglycerides (TGs) relative to diet quality (Australian Recommended Food Score [ARFS]) and PGS of TGs. Grey shading around trend line denotes the 95% confidence interval (CI) from the linear model regressing either trait on the outcome of measured TGs. (b) Forest plot of the effect of a SD increase in the best performing polygenic score (PGS) for that lipid in three different models: (i) baseline = adjusted for age, sex, and five single nucleotide polymorphism derived principal components; (ii) diet adjusted = ARFS total score as an additional covariate to the first model; and (iii) diet + other confounders adjusted = smoking status, educational attainment, and statin usage in addition to covariates in the second model. Error bars represent 95% CIs of the beta coefficient
CAD PRS is associated with a broad range of CVD phenotypes independent of diet quality
We first tested the association of diet quality measured by total ARFS with six binary self‐reported CVD phenotypes and found that there were no strong relationships between diet and odds of any phenotypes (Table 1). These analyses were repeated after removing other CVD cases from the ‘control’ cohort. However, this did not reveal any diet related associations. The only exception was a potentially counterfactual positive relationship between ARFS and self‐reported atrial fibrillation, although this was very nominal (p = 0.03) and does not pass multiple‐testing correction. The ARFS vegetable subscale was then tested, with the same lack of association as with the total ARFS score. By contrast, genetic risk for CAD expressed as a PRS was strongly enriched amongst each class of CVD cases, with the exception that the CAD PRS was only associated with self‐reported atrial fibrillation in the reduced cohort where other CVD cases were removed as controls (Figure 2 and Table 1; see also Supporting information, Table S7).
Table 1.
Association between diet and polygenic risk for coronary artery disease and self‐reported cardiovascular disease phenotypes
| CVD phenotype | ARFS (log odds per SD)a | CAD PRS (log odds per SD)b |
|---|---|---|
| Unselected controls c | ||
| Angina | −0.01(0.01) | 0.37 (0.09)*** |
| Atrial fibrillation | 0.02 (0.01)* | 0.11 (0.08) |
| Coronary bypass | 0.01 (0.01) | 0.47 (0.09)*** |
| Heart attack | −0.01 (0.01) | 0.42 (0.10)*** |
| High cholesterol | −0.002 (0.01) | 0.25 (0.05)*** |
| Hypertension | −0.001 (0.01) | 0.19 (0.06)*** |
| Selected controls | ||
| Angina | −0.01 (0.11) | 0.60 (0.12)*** |
| Atrial fibrillation | 0.20 (0.09)* | 0.29 (0.09)** |
| Coronary bypass | 0.14 (0.10) | 0.69 (0.11)*** |
| Heart attack | 0.003 (0.11) | 0.51 (0.12)*** |
| High cholesterol | −0.001 (0.06) | 0.31 (0.06)*** |
| Hypertension | −0.01 (0.06) | 0.30 (0.06)*** |
Abbreviations: ARFS, Australian Recommended Food Score; CAD, coronary artery disease; CVD, cardiovascular disease; PRS, polygenic risk score.
Log odds (SE) of each CVD phenotype per SD increase in diet score (ARFS).
Log odds (SE) of each CVD phenotype per SD increase in best performing CAD PRS.
Cases who self‐reported the CVD phenotype in question were compared to all other participants (unselected controls), and participants who did not self‐report any CVD phenotypes (selected controls).
p < 0.05
p < 0.01
p < 0.001.
Figure 2.
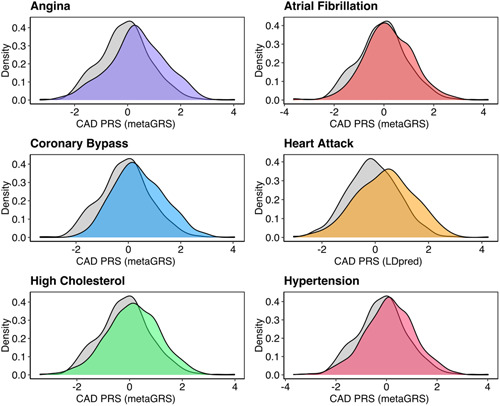
Distribution of coronary artery disease (CAD) polygenic risk score (PRS) in self‐reported cardiovascular disease cases and health controls. Kernel density estimation plot of the most significantly associated CAD PRS (metaGRS or LDpred, standardised to have SD units) for six cardiovascular disease phenotypes relative to participants who did not self‐report any of the phenotypes. For each plot, the cases are coloured, and the controls are grey
The largest effect size observed for a CAD PRS was between the ‘metaGRS’ CAD score and odds of a coronary bypass, odds ratio (OR) per SD = 2.01 (95% CI = 1.79–2.23; p = 3.74 × 10−10). Notably, the CAD PRS were still associated with the relevant CVD outcomes after the inclusion of total ARFS as a covariate, as well as the ARFS vegetable subscale score. The mean phenotypic variance explained by these CAD PRS on the liability scale when excluding other CVD cases as controls assuming a series of population prevalences was 9.5% for angina, 2.3% for atrial fibrillation, 11.2% for coronary bypass, 9% for heart attack, 2.6% for high cholesterol and 2.4% for hypertension (see Supporting information, Table S8). The lipid PGS were also associated with self‐reported high blood cholesterol, as expected, along with several other CVD phenotypes. Addition of smoking status, educational attainment and statin use as covariates did reduce the effect size of these associations, although most remained significant after multiple‐testing correction (see Supporting information, Table S9). For example, the association between the CAD PRS described above and coronary bypass was weakened upon adjustment for these additional factors but were still significant (OR per SD = 1.29; 95% CI = 1.16–1.43; p = 6.89 × 10−4). As a result, these genetic scores were independently associated with CVD phenotypes from diet quality, however, there was potentially some inflation of effect size because of factors such as smoking. We then tested whether heart attack would have a higher burden of CAD associated genetic risk relative to the five other CVD phenotypes considered, and we found some evidence that this was the case in this cohort. Specifically, each SD in CAD PRS was associated with a 42.70% (95% CI = 20.41%–63.93%) increase in the odds of a participant self‐reporting a heart attack relative to individuals who self‐reported at least one of the following: angina, atrial fibrillation, coronary bypass, high cholesterol or hypertension. We visualise the odds ratio of self‐reported attack for each quartile of CAD PRS and diet quality (total ARFS and vegetable subscale) in the three cohorts considered: (i) heart attack cases vs. participants without any other CVD phenotype (Figure 3a); (ii) heart cases vs. the remaining participants (Figure 3b); and (iii) heart attack cases vs. other CVD cases who did not self‐report a heart‐attack (Figure 3c). In all three instances, the odds of heart attack monotonically increased with each quartile of CAD PRS relative to the reference quartile, as expected, whereas there was no discernible relationship with diet quality.
Figure 3.
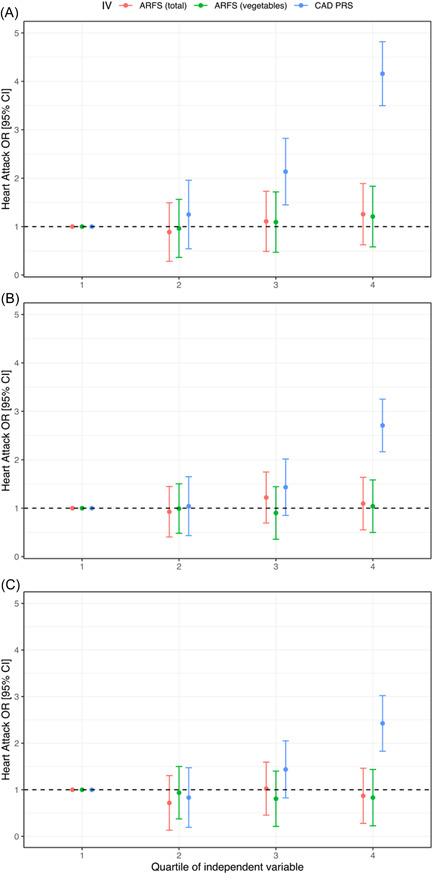
Effect size of diet and coronary artery disease polygenic risk score (PRS) per quartile of variable. Points denote the odds ratio (OR) estimate of that quartile relative to the lowest quartile (1, reference category), error bars denote 95% confidence intervals (CIs). Heart attack cases were compared to the following three cohorts: (a) participants with no self‐reported binary cardiovascular disease (CVD) phenotypes; (b) all other participants with relevant dietary data available; and (c) participants who self‐reported one or more other CVD phenotypes but not heart attack. ARFS, Australian Recommended Food Score, with the total and vegetable specific subscale tested
The key findings are summarised in Figure 4.
Figure 4.
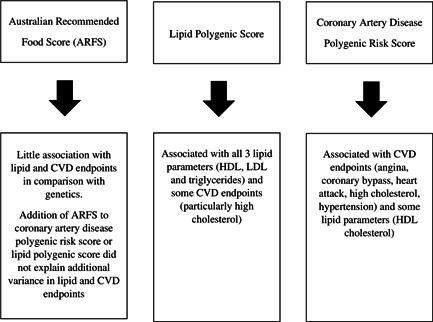
Summary of findings. ARFS, Australian Recommended Food Score; CVD, cardiovascular disease; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein
Investigation of the effect of energy intake
Given that total energy intake is known to be related to diet quality, 45 the influence of total energy intake in this cohort was evaluated in a sensitivity analysis. Total energy intake was positively correlated with total ARFS in this sample (R 2 = 0.186), with each SD increase in ARFS associated with a 0.432 (95% CI = 0.389–0.475) SD elevation in total energy intake (scaled such that SD = 1 in both instances). The association between energy intake and vegetable ARFS subscale was comparatively smaller (R 2 = 0.107) but still highly statistically significant. Covariation for total energy intake did not change the association between ARFS and lipids, with TGs still the only nominally significant association with ARFS. Similarly, adding total energy intake to the model along with ARFS did not impact the association between each lipid PGS and its respective lipid in any notable capacity (see Supporting information, Table S10). Total energy intake also did not ablate the association between CAD PRS and any of the CVD phenotypes (see Supporting information, Table S11). In summary, the additional covariate of total energy intake does not alter any of the conclusions drawn from these analyses.
DISCUSSION
We found that overall diet quality as measured by total ARFS score was not significantly associated with measured LDL or HDL, either in the baseline model adjusted for age and sex, or the full sensitivity model covaried for educational attainment, smoking and statin usage. There were also no strong relationships between diet and the odds of any of the six phenotypes. Of note is that the mean diet quality at 28 points was very low in this population, and considered poor in comparison to a sample of over 93,000 in a cross‐sectional population sample where diet quality was calculated using the ARFS via the publicly available website, the Healthy Eating Quiz 46 where the mean was 34, with the subset of 2111 adults aged 65–74 years scoring an average of 37. 47 A SD of eight within this study also highlights the narrow range in diet quality with relatively few considered to have dietary patterns reflecting that which would reduce disease risk. Previous research has shown that ARFS scores higher than those scores seen in the present study are associated with a lower risk of hypertension and non‐fatal CVD, 48 and ARFS‐vegetable sub‐scores were associated with higher HDL cholesterol. 49 The overall poor diet quality observed in the current cohort and the small SD in ARFS scores, may have limited the ability to observe any beneficial effects of diet quality in those with higher PRS.
Currently, over 27,500 Australians die each year from preventable deaths secondary to poor diet quality, 50 whereas 7 million live with at least one diet‐related chronic disease. 51 Surprisingly, the current study found that lipid PGSs explained more variance in measured lipids and CAD phenotypes than did diet quality. Considering the current cohort were older adults who, in some cases, had existing CVD and, furthermore, that the mean diet quality scores were relatively low, this may have reduced the ability to detect relationships between diet quality and PRS. Additionally, although genetics did explain more of the variance in the lipid levels, genetics are not amendable to change, whereas diet is. It is possible that diet interventions need to occur earlier, before CVD progression, and this is where Accredited Practising Dietitians (APDs) may make an important contribution in delivery of medical nutrition targeting diet related risk factors. Evidence‐based health guidelines recommend medical nutrition therapy (MNT) interventions as first‐line treatment for CVD. 52 , 53 Yet, in 2018–2019, only 1% of eligible Australians had a Medicare (the Australian public health care insurance scheme) funded MNT consult from an APD. 54 Receiving MNT interventions not only improves individual health outcomes, but also counselling by APDs confers annual healthcare savings of $830 to $1893 per patient, with patients taking fewer medications and hospital admissions. 55 This suggests that personalised dietary interventions, targeted to specific lifestage and diet related risk factors, commencing earlier in life and/or triggered by lifestage or risk factor screening programs, are required to cost‐effectively reduce diet‐related CVD risk.
In the present study, we found that a penalised regression lipid PGS, explained more phenotypic variance in measured HDL and TG levels, whereas a more restricted clumping and thresholding (C + T) approach was optimal for measured LDL. The C + T approach only retains the most confidently associated genetic variants with each lipid biomarker and filters out variants that are statistically likely to be inherited together, such that each variant is independent. By contrast, a penalised regression approach includes more variants from throughout the genome and models, rather than explicitly excluding, co‐inherited variants. Both approaches were significantly associated with all three lipid subsets, demonstrating that different methodological approaches for polygenic scoring are useful to investigate, and further work is required to define the most effective types of scores.
Interestingly, the addition of smoking status, educational attainment and statin usage as covariates in addition to age, sex, five SNP derived principal components and ARFS, resulted in an elevated correlation between the LDL PGS and measured LDL, likely because the large effect of statins was not accounted for in the baseline model.
Genetic risk for CAD (CAD PRS) was associated with lower HDL but was not significantly correlated with measured TG or LDL. This further highlights that there is genetic overlap between CAD and plasma lipids. Previous evidence obtained using summary‐based data from GWAS supports this relationship, as represented by a genetic correlation between the effect of variants on lipids and on CAD. 56 Further work is required to refine all potential mechanisms that could contribute to this overlap. However, it has been shown in CAD genetic studies that the genetic signal associated with CAD is disproportionately enriched amongst pathways related to lipid transport, metabolism and signalling. 57 Genetic risk for CAD expressed as a PRS was strongly enriched amongst each class of CVD cases, with the exception of atrial fibrillation, indicating that genetic risk for CAD was not as strongly associated as for atrial fibrillation, which may reflect the differing pathophysiology in terms of dietary risk factors. CAD PRS demonstrated the strongest statistical association with coronary bypass, which is not unexpected because it is a more severe CVD phenotype. Additionally, adjusting for diet did not alter the strong enrichment of CAD PRS amongst those with self‐reported CAD. The effect size observed when comparing the different CVD phenotypes is partially related to statistical power. Hence, bypass having the largest effect size is a function of that.
Each SD in CAD PRS was associated with a 42.7% increase in the odds of a participant self‐reporting a heart attack relative to individuals who self‐reported either angina, atrial fibrillation, coronary bypass, high cholesterol or hypertension. This highlights that individuals who self‐reported a heart attack appear to carry a higher burden of CAD associated genetic risk, as indexed by a PRS, compared to other participants with a CVD phenotype without heart attack. This supports the idea that a higher genetic risk may also be a marker of severity. Therefore, further clinical research investigating the feasibility of CAD PRS together with traditional risk factors to stratify risk of heart attack is warranted.
Strengths, limitations and future directions
This cohort study included over 3000 individuals, providing a strong sample size to investigate the relative contribution of diet quality and PRS with respect to explaining variation in plasma lipids and CAD outcomes. Future research may determine whether providing feedback on genetic predisposition, measured by PRS, leads to improved diet quality, potentially by increasing an individual's motivation to make healthful dietary changes, or not, and, additionally, evaluate whether any strategies to improve diet quality have greater effectiveness if utilised as a preventative strategy for those at highest genetic risk. There are a number of limitations that should be acknowledged. Despite the ARFS previously having a association with reduced medicare costs, and fewer medications and hospital admissions, the diet quality score was not designed to detect negative nutrients, namely nutrients that likely increase CAD risk. A final limitation is the small variability in diet quality score and therefore few individuals at the highest and lowest ends of the score. This may limit our ability to detect a relationship between diet quality and measured lipids or CAD phenotypes.
CONCLUSIONS
Lipid PGSs explained more variance in measured lipids and CAD phenotypes than diet quality. However, the less than average diet quality observed in the current cohort, along with small variance, may have limited the ability to observe any beneficial effects. Future research should investigate whether the diet quality of older adults can be improved, as well as the effect of these improvements in relation to polygenic risk.
AUTHOR CONTRIBUTIONS
Clare Elizabeth Collins, Murray J. Cairns, William R. Reay, John Attia and Rebecca Haslam contributed to project conception. William R. Reay and Murray J. Cairns analysed the data. All authors contributed to data interpretation. William R. Reay and Clare Elizabeth Collins drafted the initial paper. All authors revised and approved the final version of the manuscript submitted for publication.
ETHICAL STATEMENT
University of Newcastle Human Research Ethics committee (H‐820–0504) approved the present study.
TRANSPARENT PEER REVIEW
This paper has undergone a Transparent Peer review.
Supporting information
Supporting information.
ACKNOWLEDGEMENTS
This publication is based on data that was collected as part of the Hunter Community Study (HCS). We thank the men and women who participated in the HCS and the staff, investigators and collaborators who have supported or been involved in HCS to date. The current analysis did not receive specific funding. William R. Reay is supported by a National Health and Medical Research Council (NHMRC) grant (1147644). Murray J. Cairns is supported by an NHMRC Senior Research Fellowship (1121474) and a University of Newcastle College of Health Medicine and Wellbeing, Gladys M. Brawn Senior Fellowship. Open access publishing facilitated by The University of Newcastle, as part of the Wiley‐The University of Newcastle agreement via the Council of Australian University Librarians.
Biographies
William R. Reay is a career researcher who recently completed his PhD candidature at the University of Newcastle and the Hunter Medical Research Institute. He is passionate about the potential of genetics to rapidly improve patient outcomes for variety of disorders. His research seeks to combine statistical approaches with large genetic datasets to identify new uses for existing drugs and targets for drug development. William completed a Bachelor of Biomedical Science with first class honours at the University of Newcastle and first joined the laboratory during undergraduate research placement. He specialises in a number of important techniques in this field, including genome‐wide association studies, polygenic risk scoring, causal inference strategies such as Mendelian randomisation, and integration of genetics with functional genomics data such as RNA sequencing. In his PhD candidature, William has already been an author on in excess of 10 publications, with first author papers published in journals including Nature Reviews Genetics, Molecular Psychiatry, Neuropsychopharmacology and eLife.
Rebecca Haslam, PhD, is an early career researcher in Nutrition and Dietetics at the University of Newcastle and an Accredited Practising Dietitian. Her research focuses on investigating optimal dietary interventions for the prevention and treatment of chronic disease and how telehealth interventions can increase access to personalised nutrition support. Rebecca has published over 30 peer‐reviewed journal articles and been awarded close to $200k in research funding.
Murray J. Cairns, Professor, received his PhD in Biochemistry and Molecular Genetics from the University of New South Wales, Sydney, NSW, Australia. He then worked as an industry postdoctoral scientist at Johnson and Johnson Research and the biotech startup Nucleics Pty Ltd in Sydney before moving to Newcastle University to take an academic research position supported by the Schizophrenia Research institute. Since 2017, Professor Cairns has been supported by an NHMRC Senior Research fellowship to pursue research focused on the application of systems biology in complex psychiatric disorders, both to improve our understanding of the pathogenesis of these conditions and identify novel opportunities for treatment. This research is using both population‐based cohort studies and individualised analysis of both genomic and transcriptomic variation to identify opportunities for drug repositioning and precision intervention. Professor Cairns also has a long‐standing interest in functional genomics and the application of model systems to determine the biological significance of disease‐associated genomic variation.
George Moschonis, PhD, is a Professor of Dietetics and Human Nutrition at La Trobe University in Melbourne, Australia. During the last 17 years, he has structured a strong research profile that is recognised at an international level, mainly because of his role as chief investigator in several interdisciplinary multicentre studies funded by the European Commission. His main research interests are the assessment of nutritional status and the design, implementation, and evaluation of the effectiveness of personalised nutrition and lifestyle optimisation intervention programs, which aim to correct nutrient insufficiencies and improve health and performance throughout the lifespan. Dr George Moschonis is a highly‐cited researcher who (up to date) has authored 214 research articles published in international peer‐reviewed scientific journals with 14,121 citations and has an h‐index of 39 (Scopus).
Erin Clarke, PhD, is an Accredited Practising Dietitian and early career researcher at The University of Newcastle, Australia. She completed her Bachelor of Nutrition and Dietetics in 2016 and a PhD in Nutrition and Dietetics in 2021, both at the University of Newcastle. She works both clinically as a practising dietitian and as a researcher. Her research interests include dietary assessment, dietary biomarkers and diet quality.
John Attia is a clinical epidemiologist and general physician with a diploma in Palliative Care. He trained at McMaster University (Canada) in general internal medicine (MD) and obtained his fellowship with the Royal College of Physicians of Canada and the Royal Australasian College of Physicians. He also obtained a BSc in Physiology (Faculty scholar at McGill University), an MSc in Epidemiology (McMaster University) and a PhD in Molecular Genetics (University of Toronto). From 2008 to 2018, he was founding academic director of general medicine at John Hunter Hospital responsible for the advanced training program, as well as the founding director of the Clinical Research Design, IT, and Statistical Support (CReDITSS) Unit, a unit consisting of two epidemiologists, 10 statisticians, two bioinformaticians and a clinical informatics officer, which provides epidemiological and statistical methodological advice to clinical researchers across the university and health district. He has straddled the university and the health system for his entire academic life; he took up the role of Assistant Dean Research for the Faculty of Health in 2019, and is still active in clinical medicine providing a consult service in Palliative Medicine. He has been awarded > $36 million in competitive research grants in his career to date, and has published > 670 scientific papers. He brings expertise in clinical, molecular and genetic epidemiology with a strong methodological bent, particularly around observational epidemiology and the use of directed acyclic graphs.
Clare Elizabeth Collins, Laureate Professor, is a Fellow of the Australian Academy of Health and Medical Sciences, Nutrition Society of Australia and Dietitians Australia. L/Prof Collins research focusses on personalised nutrition technologies and programs evaluating impact on diet‐related health across life stages and chronic disease.
Reay WR, Haslam R, Cairns MJ, Moschonis G, Clarke E, Attia J, et al. Variation in cardiovascular disease risk factors among older adults in the Hunter Community Study cohort: A comparison of diet quality versus polygenic risk score. J Hum Nutr Diet. 2022;35:675–688. 10.1111/jhn.13031
REFERENCES
- 1. Vos T, Lim S, Abbafati C, Abbas KM, Abbasi M, Abbasifard M, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990‐2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang YB, Pan XF, Chen J, Cao A, Xia L, Zhang Y, et al. Combined lifestyle factors, all‐cause mortality and cardiovascular disease: a systematic review and meta‐analysis of prospective cohort studies. J Epidemiol Community Health. 2021;75:92–9. [DOI] [PubMed] [Google Scholar]
- 3. Schwingshackl L, Bogensberger B, Hoffmann G. Diet quality as assessed by the healthy eating index, alternate healthy eating index, dietary approaches to stop hypertension score, and health outcomes: an updated systematic review and meta‐analysis of cohort studies. J Acad Nutr Diet. 2018;118:74–100.e111. [DOI] [PubMed] [Google Scholar]
- 4. Morze J, Danielewicz A, Hoffmann G, Schwingshackl L. Diet quality as assessed by the healthy eating index, alternate healthy eating index, dietary approaches to stop hypertension score, and health outcomes: a second update of a systematic review and meta‐analysis of cohort studies. J Acad Nutr Diet. 2020;120:1998–2031.e1915. [DOI] [PubMed] [Google Scholar]
- 5. Voruganti VS. Nutritional genomics of cardiovascular disease. Curr Genet Med Rep. 2018;6:98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barnes S. Nutritional genomics, polyphenols, diets, and their impact on dietetics. J Am Diet Assoc. 2008;108:1888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouchard‐Mercier A, Paradis AM, Rudkowska I, Lemieux S, Couture P, Vohl MC. Associations between dietary patterns and gene expression profiles of healthy men and women: a cross‐sectional study. Nutr J. 2013;12:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Camargo A, Delgado‐Lista J, Garcia‐Rios A, Cruz‐Teno C, Yubero‐Serrano EM, Perez‐Martinez P, et al. Expression of proinflammatory, proatherogenic genes is reduced by the Mediterranean diet in elderly people. Br J Nutr. 2012;108:500–8. [DOI] [PubMed] [Google Scholar]
- 9. Mullins VA, Bresette W, Johnstone L, Hallmark B, Chilton FH. Genomics in personalized nutrition: can you “eat for your genes”? Nutrients. 2020;12:3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chilton FH, Murphy RC, Wilson BA, Sergeant S, Ainsworth H, Seeds MC, et al. Diet‐gene interactions and PUFA metabolism: a potential contributor to health disparities and human diseases. Nutrients. 2014;6:1993–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hegele R. Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet. 2009;10:109–21. [DOI] [PubMed] [Google Scholar]
- 12. Sugrue LP, Desikan RS. What are polygenic scores and why are they important? JAMA. 2019;321:1820–1. [DOI] [PubMed] [Google Scholar]
- 13. Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ye Y, Chen X, Han J, Jiang W, Natarajan P, Zhao H. Interactions between enhanced polygenic risk scores and lifestyle for cardiovascular disease, diabetes, and lipid levels. Circ Genom Precis Med. 2021;14:e003128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rutten‐Jacobs LC, Larsson SC, Malik R, Rannikmäe K, Sudlow CL, Dichgans M, Markus HS, et al. Genetic risk, incident stroke, and the benefits of adhering to a healthy lifestyle: cohort study of 306 473 UK Biobank participants. BMJ. 2018;363:k4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Livingstone KM, Abbott G, Bowe SJ, Ward J, Milte C, McNaughton SA. Diet quality indices, genetic risk and risk of cardiovascular disease and mortality: a longitudinal analysis of 77 004 UK Biobank participants. BMJ Open. 2021;11:e045362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roselli C, Chaffin MD, Weng LC, Aeschbacher S, Ahlberg G, Albert CM, et al. Multi‐ethnic genome‐wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang H, Zeng Y, Yang H, Hu Y, Hu Y, Chen W, et al. Familial factors, diet, and risk of cardiovascular disease: a cohort analysis of the UK Biobank. Am J Clin Nutr. 2021;114:1837–46. [DOI] [PubMed] [Google Scholar]
- 19. Geiker NRW, Mølgaard C, Iuliano S, Rizzoli R, Manios Y, van Loon L, et al. Impact of whole dairy matrix on musculoskeletal health and aging‐current knowledge and research gaps. Osteoporos Int. 2020;31:601–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexander DD, Bylsma LC, Vargas AJ, Cohen SS, Doucette A, Mohamed M, et al. Dairy consumption and CVD: a systematic review and meta‐analysis. Br J Nutr. 2016;115:737–50. [DOI] [PubMed] [Google Scholar]
- 21. Jakobsen MU, Trolle E, Outzen M, Mejborn H, Grønberg MG, Lyndgaard CB, et al. Intake of dairy products and associations with major atherosclerotic cardiovascular diseases: a systematic review and meta‐analysis of cohort studies. Sci Rep. 2021;11:1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McEvoy M, Smith W, D'Este C, Duke J, Peel R, Schofield P, et al. Cohort profile: the Hunter Community Study. Int J Epidemiol. 2010;39:1452–63. [DOI] [PubMed] [Google Scholar]
- 23. Reay WR, El Shair SI, Geaghan MP, Riveros C, Holliday EG, McEvoy MA, et al. Genetic association and causal inference converge on hyperglycaemia as a modifiable factor to improve lung function. eLife. 2021;10:e63115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loh PR, Danecek P, Palamara PF, Fuchsberger C, A Reshef Y, K Finucane H, et al. Reference‐based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Price AL, Weale ME, Patterson N, Myers SR, Need AC, Shianna KV, et al. Long‐range LD can confound genome scans in admixed populations. Am J Hum Genet. 2008;83:132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lambert SA, Gil L, Jupp S, Ritchie SC, Xu Y, Buniello A, et al. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat Genet. 2021;53:420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuchenbaecker K, Telkar N, Reiker T, Walters RG, Lin K, Eriksson A, et al. The transferability of lipid loci across African, Asian and European cohorts. Nat Commun. 2019;10:4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sinnott‐Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53:185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F, et al. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol. 2018;72:1883–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith W, Mitchell P, Reay EM, Webb K, Harvey PW. Validity and reproducibility of a self‐administered food frequency questionnaire in older people. Aust N Z J Public Health. 1998;22:456–63. [DOI] [PubMed] [Google Scholar]
- 33. Department of Community Services and Health . NUTTAB90 nutrient data table for use in Australia. Canberra. 1990.
- 34. Li PF, McEvoy MA, McKiernan S, Schofield PW, MacDonald‐Wicks LK, Patterson AJ. Diet quality and cognitive performance in australian adults aged 55–85 years: a cross‐sectional analysis of the Hunter Community Study cohort. Nutrients. 2021;13:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collins CE, Young AF, Hodge A. Diet quality is associated with higher nutrient intake and self‐rated health in mid‐aged women. J Am Coll Nutr. 2008;27:146–57. [DOI] [PubMed] [Google Scholar]
- 36. Collins CE, Burrows TL, Rollo ME, Boggess MM, Watson JF, Guest M, et al. The comparative validity and reproducibility of a diet quality index for adults: the Australian Recommended Food Score. Nutrients. 2015;7:785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. National Health and Medical Research Council . Australian dietary guidelines Canberra National Health and Medical Research Council. 2013.
- 38. National Health and Medical Research Council . Australian guide to healthy eating. https://www.eatforhealth.gov.au/guidelines/australian-guide-healthy-eating (2013). Accessed 10 August 2021.
- 39. Ashton L, Williams R, Wood L, Schumacher T, Burrows T, Rollo M, et al. Comparison of Australian Recommended Food Score (ARFS) and plasma carotenoid concentrations: a validation study in adults. Nutrients. 2017;17:888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhan J, Liu YJ, Cai LB, Xu FR, Xie T, He QQ. Fruit and vegetable consumption and risk of cardiovascular disease: a meta‐analysis of prospective cohort studies. Crit Rev Food Sci Nutr. 2017;57:1650–63. [DOI] [PubMed] [Google Scholar]
- 41. Baldwin JN, Ashton LM, Forder PM, Haslam RL, Hure AJ, Loxton DJ, et al. Increasing fruit and vegetable variety over time is associated with lower 15‐year healthcare costs: results from the Australian longitudinal study on women's health. Nutrients. 2021;13:2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baldwin JN, Forder PM, Haslam R, Hure A, Loxton D, Patterson AJ, et al. Lower vegetable variety and worsening diet quality over time are associated with higher 15‐year health care claims and costs among Australian women. J Acad Nutr Diet. 2021;121:655–68. [DOI] [PubMed] [Google Scholar]
- 43. R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2020. [Google Scholar]
- 44. Lee SH, Goddard ME, Wray NR, Visscher PM. A better coefficient of determination for genetic profile analysis. Genet Epidemiol. 2012;36:214–24. [DOI] [PubMed] [Google Scholar]
- 45. Collins CE, Burrows TL, Rollo ME, Boggess MM, Watson JF, Guest M, et al. The comparative validity and reproducibility of a diet quality index for adults: the Australian Recommended Food Score. Nutrients. 2015;7:785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Collins C. Healthy eating quiz. https://healthyeatingquiz.com.au/ (2013). Accessed 24 Aug 2021.
- 47. Williams RL, Rollo ME, Schumacher T, Collins CE. Diet quality scores of Australian adults who have completed the healthy eating quiz. Nutrients. 2017;9:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jackson JK, MacDonald‐Wicks LK, McEvoy MA, Forder PM, Holder C, Oldmeadow C, et al. Better diet quality scores are associated with a lower risk of hypertension and non‐fatal CVD in middle‐aged Australian women over 15 years of follow‐up. Public Health Nutr. 2020;23:882–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Owen AJ, Abramson MJ, Ikin JF, McCaffrey TA, Pomeroy S, Borg BM, et al. Recommended intake of key food groups and cardiovascular risk factors in australian older, rural‐dwelling adults. Nutrients. 2020;12:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Crosland P, Ananthapavan J, Davison J, Lambert M, Carter R. The health burden of preventable disease in Australia: a systematic review. Aust N Z J Public Health. 2019;43:163–70. [DOI] [PubMed] [Google Scholar]
- 51. Australian Institute of Health Welfare . Australia's health 2018. Canberra: AIHW; 2018. [Google Scholar]
- 52. Schumacher TL, Burrows TL, Neubeck L, Redfern J, Callister R, Collins CE. How dietary evidence for the prevention and treatment of CVD is translated into practice in those with or at high risk of CVD: a systematic review. Public Health Nutr. 2017;20:30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. National Vascular Disease Prevention Alliance . Guidelines for the management of absolute cardiovascular disease risk: National Health and Medical Research Council. 2012.
- 54. Australian Institute of Health Welfare . Medicare‐subsidised GP, allied health and specialist health care across local areas: 2013–14 to 2018–19. https://www.aihw.gov.au/reports/primary-health-care/medicare-subsidised-health-local-areas-2019/contents/allied-health-services/allied-health-services (2020). Accessed 23 August 2021.
- 55. Sikand G, Cole RE, Handu D, deWaal D, Christaldi J, Johnson EQ, et al. Clinical and cost benefits of medical nutrition therapy by registered dietitian nutritionists for management of dyslipidemia: A systematic review and meta‐analysis. J Clin Lipidol. 2018;12:1113–22. [DOI] [PubMed] [Google Scholar]
- 56. Bulik‐Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet. 2015;47:1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.