

Summary
Plant diseases are an important threat to food production. While major pathogenicity determinants required for disease have been extensively studied, less is known on how pathogens thrive during host colonization, especially at early infection stages.
Here, we used randomly barcoded‐transposon insertion site sequencing (RB‐TnSeq) to perform a genome‐wide screen and identify key bacterial fitness determinants of the vascular pathogen Xanthomonas campestris pv campestris (Xcc) during infection of the cauliflower host plant (Brassica oleracea). This high‐throughput analysis was conducted in hydathodes, the natural entry site of Xcc, in xylem sap and in synthetic media.
Xcc did not face a strong bottleneck during hydathode infection. In total, 181 genes important for fitness were identified in plant‐associated environments with functional enrichment in genes involved in metabolism but only few genes previously known to be involved in virulence. The biological relevance of 12 genes was independently confirmed by phenotyping single mutants. Notably, we show that XC_3388, a protein with no known function (DUF1631), plays a key role in the adaptation and virulence of Xcc possibly through c‐di‐GMP‐mediated regulation.
This study revealed yet unsuspected social behaviors adopted by Xcc individuals when confined inside hydathodes at early infection stages.
Keywords: Brassica oleracea, fitness, hydathodes, RB‐TnSeq, Xanthomonas, xylem
Introduction
Pests and pathogens of crops cause significant losses in yield and quality. The search for new control strategies relies on our understanding of host–pathogen interactions. Xanthomonas bacteria cause disease on more than 400 plant species and are responsible for important losses on various economically important crops worldwide, including rice, citrus, cassava, banana and tomato (Büttner & Bonas, 2010). Among them, Xanthomonas campestris pv campestris (Xcc), the causal agent of black rot disease, is the major bacterial pathogen of Brassica crops such as cauliflower, cabbage, mustard and radish (Vicente & Holub, 2013). Transmissible by seeds, Xcc survives as an epiphyte and enters the plant through leaf organs called hydathodes. Hydathodes are plant organs located at the leaf margins where they mediate guttation of xylem sap‐derived fluid when evapotranspiration is limited (Cerutti et al., 2017, 2019). Hydathodes are composed of three main tissues: stomata‐like water pores within the epidermal layer, an inner epithem composed of small thin‐walled parenchyma and a hypertrophied xylem (Cerutti et al., 2017, 2019). Three days after inoculation, Xcc switches to necrotrophic behavior and destroys the inner epithemal tissue of hydathodes (Cerutti et al., 2017; Luneau et al., 2022a). Simultaneously, Xcc accesses xylem vessels and progressively spreads systemically in the plant.
Xcc deploys a large arsenal of virulence factors to successfully infect the host plant and complete its life cycle. Among them, two secretion systems are essential for pathogenicity: the type II secretion system (T2SS) which exports enzymes such as plant cell wall‐degrading enzymes to the extracellular space and the type III secretion system (T3SS), which translocates type III effector (T3E) proteins into the host cells to suppress immune responses and hijack host metabolism (Büttner & Bonas, 2010; Tang et al., 2021). In addition, Xcc produces lipopolysaccharides and exopolysaccharides (EPS) named xanthan that protect bacterial cells against environmental stresses and support biofilm formation (An et al., 2020). Quorum sensing coordinates bacterial behavior, including biofilm dispersal, and is required for disease (An et al., 2020). Xcc also possesses a wide range of two‐component systems that ensure perception of environmental signals and initiation of adaptive response (Qian et al., 2008). Together, these traits contribute to the fitness of the bacterium during the infection. However, while previous studies have investigated in depth the virulence factors deployed by Xcc to implement its pathogenic lifestyle at late stages of infection, very few have looked at the genetic determinants of fitness at early stages of plant infection (An et al., 2020; Timilsina et al., 2020; Luneau et al., 2022a). Xcc colonization of hydathodes is associated with sedentary behavior, activation of pathogenicity determinants (T3SS) and expression of high‐affinity transporters for nutrient uptake (e.g. phosphate, sulfate, nitrate; Luneau et al., 2022a). Nevertheless, transcriptome profiling only provides gene expression levels without evaluating their in planta contribution to bacterial fitness.
Transposon insertion mutagenesis coupled with next‐generation sequencing technologies now allow the study of bacterial fitness at a genomic scale (Cain et al., 2020; van Opijnen & Levin, 2020). Randomly barcoded‐transposon insertion site sequencing (RB‐TnSeq) allows the high‐throughput evaluation of gene contributions to fitness using a saturated library of bacteria mutagenized by barcoded transposons (Wetmore et al., 2015). Such TnSeq approaches have been widely used to study animal pathogens and have led to the discovery of infectious processes including virulence factors, genes required for transmission between hosts or antibiotic resistance (reviewed in Cain et al., 2020; van Opijnen & Levin, 2020) but have only recently been applied to plant pathogens. To date, TnSeq screens of genes contributing to in planta fitness have been performed in Pantoea stewartii (on corn; Duong et al., 2018), Dickeya dadantii (on chicory; Royet et al., 2019), Agrobacterium tumefaciens (on tomato; Gonzalez‐Mula et al., 2019; Torres et al., 2022), Pseudomonas syringae (on bean and pepper; Helmann et al., 2019, 2020), Ralstonia solanacearum (on tomato; Su et al., 2021) and Xanthomonas hortorum (on lettuce; Morinière et al., 2022). While these studies provided diverse insights specific to each pathosystem, they all highlighted the importance of metabolic capacities and secretion systems for optimal growth in planta.
Here, RB‐TnSeq was used to identify the genetic basis of Xcc adaptation to the environments it colonizes within the host, including hydathodes, xylem sap and synthetic media. We identified essential genes for Xcc, as well as genes that contribute specifically to fitness in planta. We show that XC_3388 encoding a hypothetical protein is important for adaptation and pathogenicity of Xcc inside cauliflower hydathodes, probably through c‐di‐GMP regulation, which highlights the importance of social behaviors during plant infection.
Materials and Methods
Bacterial strains and culture conditions
The Xcc 8004∷GUS‐GFP strain (Cerutti et al., 2017) was used as a recipient for the RB‐TnSeq library and all deletion mutants (Supporting Information Table S1). For competition assays, we used the Xcc 8004::GUS*‐GFP* variant, which contains point mutations inactivating the catalytic sites of both reporter proteins as described (Luneau et al., 2022b). Xcc was cultivated in MOKA rich medium (Blanvillain et al., 2007), MME glucose‐poor medium (Arlat et al., 1991) or MME glucose complemented with dropout – leucine/tryptophan/histidine (LTH) supplements instead of casamino acids (10.5 g l−1 K2HPO4, 4.5 g l−1 KH2PO4, 1 g l−1 (NH4)2SO4, 1 mM MgSO4, 0.15% (w/v) DropOut‐LTH, 20 mM glucose) at 28°C under agitation at 200 rpm or on MOKA‐agar plates.
Deletion mutants were constructed with the SacB double recombination method using pK18 derivatives (Table S4; Schäfer et al., 1994). Complemented strains were obtained by genomic integration in trans of selected genes under the constitutive ptac promoter using pK18_CompR3 plasmid derivatives (Table S4; Luneau et al., 2022a). Primers designed for all constructions are listed in Table S4. Escherichia coli strain TG1 was used as the plasmid donor and strain TG1 pRK2073 as helper for triparental plasmid conjugation into Xcc (Figurski & Helinski, 1979; Ditta et al., 1980). Escherichia coli TG1 strains were cultivated at 37°C on LB‐agar or in LB under shaking at 200 rpm. When appropriate, we used the antibiotics rifampicin (50 μg ml−1), kanamycin (50 μg ml−1) and spectinomycin (40 μg ml−1) and the fungicide pimaricin (30 μg ml−1).
Plant material and xylem sap harvest
Four‐week‐old Brassica oleracea var botrytis cv Clovis (L.) F1 cauliflower plants grown under glasswhouse conditions were inoculated in the second true leaf and placed in growth chambers (8 h light; 22°C; 70% relative humidity). Xylem sap was collected from decapitated cauliflower stems as described (Dugé de Bernonville et al., 2014).
Construction of the RB‐TnSeq library and transposon insertion site sequencing
The RB‐TnSeq barcoded transposon insertion library was constructed by conjugating the E. coli APA752 donor library containing the barcoded mariner plasmid pKMW3 into Xcc 8004::GUS‐GFP. Mating was performed overnight at a 1 : 1 ratio on nutrient agar (2 g l−1 yeast extract, 5 g l−1 peptone, 5 g l−1 NaCl containing 300 μM diaminopimelic acid). The mixture was pooled into a single sample, resuspended in MOKA, plated on MOKA + 50 μg ml−1 kanamycin plates and incubated for 2 d at 28°C to select for insertion mutants. Approximately 600 000 mutant clones were obtained, each one representing one transposition event. These clones were pooled together in 120 ml MOKA kanamycin and 40 ml of 80% glycerol, aliquoted in 1 ml samples and frozen at −80°C. Genomic DNA from Xcc mutant library samples was sequenced and barcodes were mapped as previously described (Wetmore et al., 2015).
Identification of essential genes
The genome of Xcc strain 8004 (NCBI accession no. CP000050, Qian et al., 2005) was annotated using transcriptomic datasets (Luneau et al., 2022a) and used for all RB‐TnSeq analyses (https://doi.org/10.25794/reference/id52ofys).
Essential genes were predicted as described (Price et al., 2018). Genes that possess insertion sites (TA site) but that lack insertions in our library are likely to be essential in the condition used to build the library (MOKA‐rich medium). Briefly, for each protein‐coding gene, we computed the total read density in TnSeq (reads/nucleotides across the entire gene) and the density of insertion sites within the central 10–90% of each gene (sites/nucleotides). We did not consider the DNA barcodes in this analysis of essential genes. Genes that could have no insertion by chance because of their short length (375 nucleotides or shorter) were excluded from the study.
Fitness screening in hydathodes
For each biological replicate, a 2 ml aliquot of the library was thawed and grown overnight at 28°C with shaking at 200 rpm in MOKA + 50 μg ml−1 kanamycin. The culture was washed twice by serial centrifugations (6000 g , 10 min) and resuspended in 1 mM MgCl2. After measuring the OD600, we pelleted 109 Xcc cells by centrifugation at 13 000 g for 5 min and stored them at −80°C to be used as T0 samples. Hydathode inoculation was achieved using a protocol adapted from Cerutti et al. (2017). Briefly, cauliflower leaves were dipped for 15 s inside the Xcc suspension adjusted to an OD600 = 0.1 (108 cfu ml−1) in 1 mM MgCl2 + 0.5% (v/v) Tween 80. Plants were then placed in growth chamber conditions and covered for 24 h with a miniglasshouse lid to reach 100% relative humidity. The lid was removed and the infection was allowed to proceed for 3 or 6 d. Xcc populations were retrieved by macrodissection of hydathodes (16 per leaf) using a 1.5 mm diameter Biopsy Punch (Miltex®; Electron Microscopy Sciences, Hatfield, PA, USA) and bead‐ground in 1 mM MgCl2 for 2 min at 30 Hz. In total, we collected and pooled c. 1650 hydathodes for each of the four biological replicates. With an average of 35 000 barcodes per hydathode, this theoretically represents an average of 57 × 106 mutants per replicate. Each experiment covers the library c. 100 times. DNA was extracted using the Promega Wizard® Genomic DNA Purification Kit in which we added a protein degradation step by incubating the samples with 400 μg ml of proteinase K at 37°C for 1 h before protein precipitation. Samples were stored at −80°C until preparation for barcode sequencing.
Bottleneck assays in hydathodes
Hydathode inoculation and sampling were performed as described above. After 3 d in the plant, eight hydathodes per leaf were harvested and individually bead‐ground in 200 μl of 1 mM MgCl2 for 2 min at 30 Hz. From this suspension, 10 μl was used to perform serial dilution, plated on MOKA‐agar + kanamycin 50 μg ml−1 and counted to check that each hydathode was well infected and contained the same amount of bacterial cells (c. 105 CFU per hydathode). We performed four biological replicates of three plants each for a total of 96 hydathodes. Individual hydathode homogenates were spotted on sterile filters, placed on MOKA‐agar + kanamycin 50 μg ml−1 and incubated for 3 d at 28°C. Grown populations were individually resuspended by mixing filters with 1.5 ml of MOKA and pelleted (6000 g , 10 min). DNA was extracted from each pellet and used for barcode sequencing as described above.
Barcode sequencing and calculation of gene fitness
Barcode sequencing, mapping and analysis to calculate the relative abundance of barcodes were done using the RB‐TnSeq methodology and computational pipeline developed by Wetmore et al. (2015); the code is available at https://bitbucket.org/berkeleylab/feba/. For each experiment, fitness values (¦f¦) for each gene were calculated as a log2 ratio of relative barcode abundance after library growth in a given condition divided by relative abundance in the time0 sample. Fitness values were normalized across the genome so the typical gene has a fitness value of 0. Insertions outside coding sequences were not considered.
We considered genes to be important for Xcc fitness when the mutants were strongly affected (i.e. gene fitness ¦f¦ ≥ 1), and when this phenotype was deemed robust (i.e. ¦t‐score¦ ≥ 3) (Wetmore et al., 2015; Helmann et al., 2019). We retained genes that match those criteria in at least three of the four independent biological replicates. We verified the correlation between biological replicates of RB‐TnSeq assays to show experimental reproducibility (Fig. S1). Enrichment analysis considering gene ontology was conducted with R (v.4.0.4) using the topgo package v.2.40.0 using Fisher’s exact test and P‐value < 0.01 (Alexa et al., 2006).
Competition assays
We mixed the Xcc strain 8004∷GUS*‐GFP* (hereafter WT*) and deletion mutants in the 8004∷GUS‐GFP background to assess the fitness of the latter as previously described (Luneau et al., 2022b). Briefly, we inoculated cauliflower hydathodes with the same protocol as for the RB‐TnSeq assay except that we adjusted the inoculum suspensions to OD600 = 0.05 for each strain (final OD600 = 0.1) in 1 mM MgCl2 + 0.5% (v/v) Tween 80. To maximize the throughput of the validation assays, we chose to inoculate only one plant for each of the mixtures. We plated inoculation suspensions to use as T0 reference samples as well as 6 d postinoculation (dpi) samples consisting of 16 pooled and ground hydathodes. The competitive index (CI) of each strain was calculated as the ratio of the relative frequencies of mutant vs reference strains in the 6 dpi infected sample compared to the T0 inoculum (Taylor et al., 1987; Macho et al., 2007). Hence, we consider a CI > 1 as a gain of fitness of the mutant compared to the WT* reference strain and a CI < 1 as a loss of fitness. We performed at least four independent biological replicates of the competition assays for all strains tested.
Pathogenicity assays
We evaluated bacterial aggressiveness of the mutant strains by piercing‐inoculation in the midrib of cauliflower leaves using a needle dipped in a Xcc inoculum adjusted at OD600 = 0.1. We assessed disease progression at 10 dpi according to the annotation scale presented by Luneau et al. (2022a) in three independent biological replicates of five individual plants each.
Internal growth curves in hydathodes
Bacterial populations in hydathodes were determined after dip‐inoculation as described above. After 6 d, hydathodes were sampled using a cork borer (1.5 mm in diameter), individually ground using a Tissue Lyser MM 400 grinder (Retsch, Haan, Germany), twice for 30 s at a frequency of 30 Hz, in 1.2 ml deepwell plates containing five or six glass beads (2 mm in diameter) per well and 200 μl of sterile water. The homogenates were serially diluted in sterile water and 5 μl drops were spotted three times on MOKA plates supplemented with rifampicin and pimaricin. Plates were incubated at 28°C for 48 h and colonies were enumerated in spots containing one to 30 colonies. Bacterial densities in leaves were calculated as log CFU cm−2.
Phenotyping assays
Motility, exopolysaccharide production and protease activity were assessed as previously described (Luneau et al., 2022a). For amylase activity assays, 5 μl of an overnight culture adjusted to 4 × 108 CFU ml−1 was spotted on plates containing 15 ml of MOKA agar with 0.125% potato starch (Prolabo; VWR, Radnor, PA, USA) supplemented with 30 μg ml−1 pimaricin. Plates were incubated at 28°C and imaged 24 h after inoculation. Diameters of colonies and halos of degradation were measured by washing plates using distilled water and staining using lugol solution until a strong blue coloration and starch degradation halo appeared. Calculation of enzymatic activity was performed as follows: PrA = [π(r halo)2 − π(r colo)2]/[π(r colo)2] where r is the radius. Each experiment was biologically replicated at least three times.
For biofilm visualization, strains were grown overnight in MOKA‐rich medium, washed twice with sterile water before being resuspended in MME minimal medium supplemented with 20 mM glucose at a final concentration of 108 cells ml−1. Five milliliters of each strain suspension was distributed in six‐well plates (VWR tissue culture plate) containing a sterile borosilicate coverslip in the bottom. One plate per time point is required. Plates were incubated without agitation at room temperature. Before imaging, 3 μl of propidium iodide was added to each well and incubated for 10 min. Biofilms were visualized using a spinning disk microscope with a 60× immersion lens.
Results
A saturated library of Xcc transposon‐insertion mutants identifies 365 essential genes
To screen for genes involved in the adaptation of Xcc strain 8004 to environments encountered during its life cycle, a library of randomly barcoded insertion mutants was constructed using a mariner transposon (Wetmore et al., 2015; Fig. 1a). High‐throughput transposon insertion site sequencing allowed the mapping of each insertion site in the Xcc genome and their association with specific barcode sequences. After filtering out chimeric reads and nonunique transposon barcodes and computationally excluding insertions located outside the main body of the gene (the central 10–90% of the coding sequence length), we identified 192 986 mutants for BarSeq‐based fitness analysis (see Materials and Methods section). The mutant library encompassed 51 853 of the 77 290 possible mariner insertion sites (TA dinucleotides) in the Xcc 8004 genome (NCBI accession no. CP000050, Qian et al., 2005). Because TnSeq analyses depend on the quality of the annotations, we used an improved annotation of the genome (Luneau et al., 2022a).
Fig. 1.
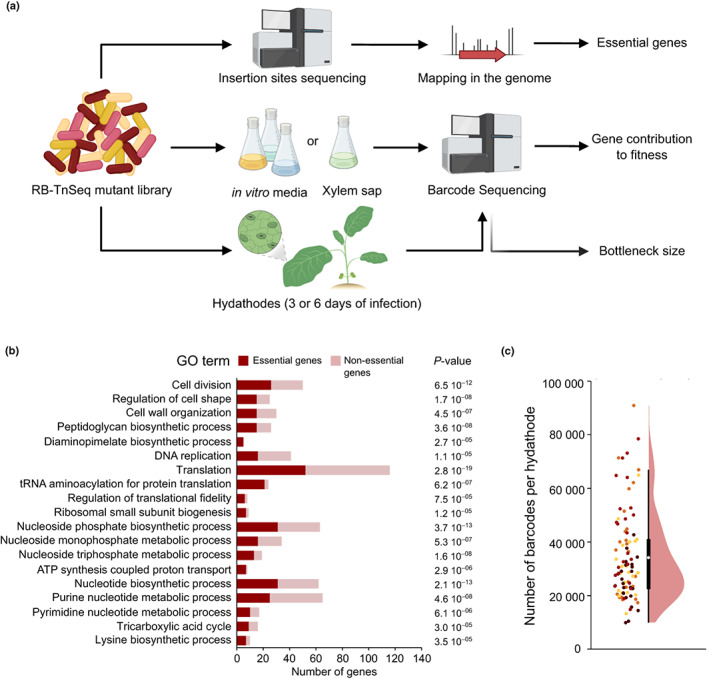
RB‐TnSeq approach to identify Xanthomonas campestris pathovar campestris (Xcc) fitness determinants. (a) Experimental workflow. Barcoded‐transposon insertion sites in the Xcc 8004::GUS‐GFP genome were determined by transposon insertion site sequencing of the RB‐TnSeq mutant library and used to identify essential genes. Barcode sequencing of the bacterial population was performed to quantify the relative abundance of all mutants before and after growth of the library in synthetic media, xylem sap and hydathodes of Brassica oleracea var botrytis cv Clovis F1 cauliflower plants. Individual gene fitness values were obtained in each condition. Counting the number of distinct barcodes present within individual hydathodes indicates the bottleneck size for hydathode infection. Created with BioRender.com. (b) Summary of Gene Ontology (GO) functional categories enriched among Xcc essential genes (red) compared to nonessential genes (pink). Enrichment significance (P‐value) was calculated using Fisher’s exact test. (c) Infection bottleneck in hydathodes determined by counting the number of distinct barcodes found within single cauliflower hydathodes (n = 96) 3 d after dip inoculation with the RB‐TnSeq library. Each dot represents a single hydathode. The color represents the four biological repetitions. The boxplot (black) and densityplot (pink) show the distribution of barcode counts across all samples. The white dot indicates the mean value. The central box of the boxplot shows the central 50% of values (from the first quartile to the third quartile) while the whiskers indicate the values located within 1.5 times the interquartile range.
With a four‐fold coverage and one insertion every 100 bp on average, the mutagenesis was saturated and allowed the study of 3665 of 4617 annotated cosing sequences (CDS) (Luneau et al., 2022a).
Among those 4617 Xcc genes, 80 do not have a TA site in their sequence and cannot be disrupted by a mariner transposon. In total, 365 genes were identified as essential for viability in Xcc strain 8004 based on both the presence of TA sites localized in the gene body and the absence or very low abundance of transposon insertions (see Methods S1; Table S2). A Gene Ontology (GO) enrichment analysis highlighted that core cellular functions related to DNA replication, translation, cell envelope, cell division and nucleotide metabolism were most significantly enriched among those essential genes (Fig. 1b). The lysine biosynthesis pathway was the only essential amino acid biosynthetic pathway for Xcc growth in the rich medium used to construct the library. The use of such TnSeq library data for essentiality of genes in defined environmental conditions significantly improved functional annotation of the genome of the Xcc strain 8004 and identified potential targets for disease control.
Hydathode pores do not impose a biologically significant infection bottleneck for Xcc
Before any TnSeq analysis, it was important to ensure that a significant part of the mutant library can enter host tissues to minimize sampling artifacts. We thus determined the inoculation bottleneck in Brassica oleracea var. botrytis (cauliflower), the host of isolation of strain 8004 (Turner et al., 1984). Robust hydathode infection was achieved by dip inoculation in the suspension of the mutant library, and the number of distinct barcodes present in each hydathodes was determined by BarSeq (see Materials and Methods section). A mean of 34 582 distinct barcodes was found per hydathode (Fig. 1c), giving a low‐end estimate of the infection bottleneck. This result indicates that, in our experimental setup, the entry into hydathodes is not limiting infection as only a few Xcc cells per hydathode are sufficient to initiate an infection that leads to disease (Robeson et al., 1989).
In total, 181 genes impact Xcc multiplication in xylem sap and hydathodes
Because Xcc is a vascular pathogen colonizing xylem vessels, we first screened for fitness determinants supporting Xcc growth in xylem sap (see Methods S1). Xylem sap contains low concentrations of mineral ions, amino acids, organic acids and simple sugars (Dugé de Bernonville et al., 2014) and was thus used as a proxy for the environment encountered in the xylem. This ex planta RB‐TnSeq fitness analysis in cauliflower xylem sap identified 53 Xcc genes (Fig. 2; Table S3a). Fifty‐one genes had negative fitness scores, indicating that the corresponding genes are needed for growth in xylem sap, while only two had positive fitness scores. These 51 genes are mainly involved in amino acid and sugar metabolism consistent with the limited nutritional value of xylem sap. Comparing ‘plant‐associated environments’ together, only 11 of the 53 genes important for fitness in xylem sap were specific to this condition (Fig. 2b; Table S3). Most of these 11 genes are involved in central metabolic pathways and are important for growth in synthetic media (Table S3). (p)ppGpp metabolism was specifically important in xylem sap, where Xcc probably faces nutrient‐scarce conditions requiring a stringent response (Irving et al., 2021; Bai et al., 2022).
Fig. 2.
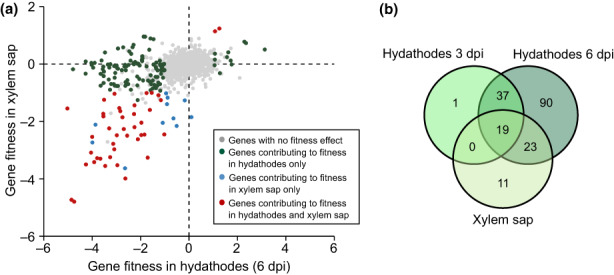
RB‐TnSeq screening of Xanthomonas campestris pathovar campestris (Xcc) fitness determinants during growth in plant‐associated environments. (a) Comparison of gene contribution to fitness during growth within hydathodes and in xylem sap of Brassica oleracea var botrytis cv Clovis F1 cauliflower plants. Genes with no fitness effect or that do not fit our robustness requirement are in gray (¦f¦ ≥ 1, ¦t‐score¦ ≥ 3). Genes found as differentially represented in hydathodes only are in green, in xylem sap only are in blue and in both conditions are in red. Values used here are the mean of four biological replicates. (b) Venn diagram showing the number of genes contributing to Xcc fitness during growth in hydathodes at 3 or 6 d postinoculation and in xylem sap.
Before reaching the xylem, Xcc infects hydathodes where it resides as a biotroph for 3 d and becomes a necrotroph at 6 dpi (Cerutti et al., 2017). An in planta RB‐TnSeq fitness analysis was conducted at 3 and 6 dpi to dissect early infection processes and adaptive responses inside hydathodes (Fig. 1a). Together, 170 genes significantly contributed to Xcc fitness (Fig. 2; Table S3b,c). Despite the low generation number at 3 dpi (three generations), all but one of those early fitness determinants were again identified at 6 dpi, indicating that similar selective forces apply during those two infection stages (0–3 dpi and 4–6 dpi; Fig. 2b). A GO term enrichment analysis revealed that functions related to the biosynthesis of cofactors, purine (inosine monophosphate), lipopolysaccharides (LPS) and multiple amino acids as well as phosphate uptake and gluconeogenesis were significantly associated with Xcc fitness in hydathodes.
To distinguish genes specifically important for growth in planta from those important for growth in vitro, a fitness analysis was performed in MME minimal medium supplemented with glucose as the carbon source as well as in MOKA‐rich medium, originally used to build the library (Fig. S2a; Table S3e). Among the 170 genes identified to be important for fitness in hydathodes, 51 are also important for in vitro growth, including the 19 genes identified to be important for Xcc fitness in both hydathodes and xylem sap (Fig. 2b; Table S3d). These genes are therefore not specific to in planta environments (Table S3e). Those 19 genes were associated with biosynthesis of purine (inosine monophosphate) and vitamins (NADH and folate), and TCA cycle‐related pathways. Among amino acid biosynthetic pathways, all except cysteine, methionine, proline and valine were required for optimal fitness in hydathodes (Table S3b). In xylem sap, only proline biosynthesis was important.
Expression of virulence determinants represents a measurable fitness cost during hydathode infection
Very few genes important for Xcc fitness in hydathodes are related to virulence functions (Y‐Q. He et al., 2007; Fig. S3). Those genes are involved in EPS biosynthesis and the T2SS, but genes for the T3SS, T3Es or quorum sensing signal biosynthesis were not identified. A gain of fitness was observed in mutants for major virulence regulators, including the HrpG and HrpX transcriptional activators of the T3SS, the RpfC and RpfG two‐component system involved in quorum sensing signal perception, and many transcriptional regulators involved in adaptive responses to the environment such as XibR (iron metabolism; Pandey et al., 2016), RavR (oxygen tension; He et al., 2009), HpaR (extracellular proteases production; Wei et al., 2007), VemR (motility and EPS production; Tao & He, 2010) and Clp (EPS and extracellular enzyme production; Y‐W. He et al., 2007) (Table 1). Such cross‐complementation at the population level highlights the fitness cost of producing virulence factors for the individual and reveals social behaviors of Xcc within hydathodes.
Table 1.
Genes whose mutation results in a gain of fitness inside hydathodes.
| Gene ID a | Locus tag b | Fitness browser ID c | Gene name | Hyd 3 dpi | Hyd 6 dpi | Xylem sap | MME Glucose DROPOUt –LTH | MME glucose | MOKA |
|---|---|---|---|---|---|---|---|---|---|
| XC_0486 | XCC8004_a06231 | Xcc‐8004.624.1 | clp | 1.13 | 2.29 | 0.78 | 0.24 | 0.2 | 0.29 |
| XC_0522 | XCC8004_a06761 | Xcc‐8004.673.1 | – | 0.2 | 1.07 | 0.15 | 0.04 | −0.05 | 0.04 |
| – | XCC8004_a27231 | Xcc‐8004.2699.1 | – | 1.23 | 1.25 | 1.23 | 0.03 | 0.02 | 0.28 |
| XC_2228 | XCC8004_a27921 | Xcc‐8004.2768.1 | ravR | 0.03 | 1.06 | −0.04 | 0.13 | 0.06 | 0.02 |
| XC_2252 | XCC8004_a28231 | Xcc‐8004.2799.1 | vemR | 0.35 | 1.07 | 0.36 | −0.42 | −0.28 | −0.1 |
| XC_2272 | XCC8004_a28471 | Xcc‐8004.2823.1 | fliQ | 1.39 | 1.57 | 0.24 | 0.07 | 0.02 | 0.02 |
| XC_2333 | XCC8004_a29211 | Xcc‐8004.2898.1 | rpfC | 1.25 | 2.35 | 0.73 | −0.28 | −0.07 | 0.26 |
| XC_2335 | XCC8004_a29231 | Xcc‐8004.2900.1 | rpfG | 1.65 | 3.13 | 0.5 | −0.12 | 0.06 | 0.21 |
| XC_2827 | XCC8004_a35451 | Xcc‐8004.3512.1 | hpaR | 0.73 | 1.62 | 0.41 | 0.04 | −0.01 | 0.27 |
| XC_3076 | XCC8004_a38371 | Xcc‐8004.3806.1 | hrpX | 0.31 | 1.72 | 0.12 | −0.11 | −0.06 | 0 |
| XC_3077 | XCC8004_a38401 | Xcc‐8004.3809.1 | hrpG | 0.77 | 1.76 | −0.02 | −0.03 | −0.18 | 0.01 |
| XC_3388 | XCC8004_a42701 | Xcc‐8004.4210.1 | – | 0.3 | 1.27 | −0.32 | −1.24 | −0.9 | −0.61 |
| XC_3760 | XCC8004_a47281 | Xcc‐8004.4669.1 | xibR (xerR) | 0.75 | 1.08 | 1.14 | 0.18 | 0.02 | 0.31 |
Fitness values in red for mutants with a gain of fitness and in blue for a loss of fitness; complete fitness data can be found in Supporting Information Table S2. Hyd, hydathodes.
Gene ID corresponds to Xanthomonas campestris pathovar campestris (Xcc) 8004’s CDS annotation given by Qian et al. (2005).
Locus_tag corresponds to the identification tag attributed when the new annotation was performed using RNAseq data published by Luneau et al. (2022a).
Fitness browser ID corresponds to the gene name used for fitness analysis.
RB‐TnSeq enables the identification of genes with subtle and significant contributions to in planta fitness or pathogenicity
We assessed individually the fitness of seven mutants corresponding to eight genes with reduced fitness in the RB‐TnSeq experiment (Fig. 3; Table 2). The fitness deficit measured by RB‐TnSeq was independently confirmed and complemented upon coinoculation in cauliflower hydathodes with the WT strain (1 : 1 ratio) for six mutants except for XC_1113 (Fig. 3a). This experiment supports the RB‐TnSeq results (Spearman correlation coefficient r = 0.92, P = 0.0011 for genes with reduced fitness). Alone, five mutants also reached reduced population levels in hydathodes at 6 dpi, while the ΔXC_1431 and ΔXC_1113 mutants behaved similarly to the WT (Fig. 3b). Upon wound‐inoculation into cauliflower leaves, five mutants but not ΔglgB1 also showed reduced pathogenicity at 6 dpi which could be complemented (Fig. 4a).
Fig. 3.
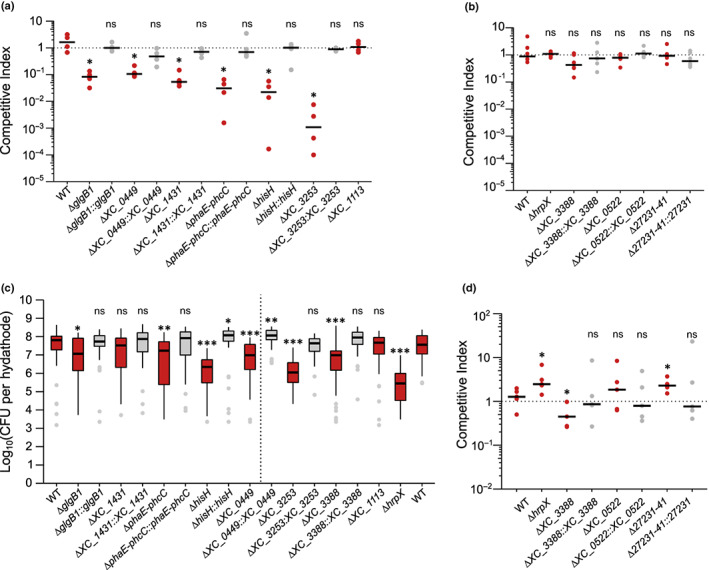
Contribution of Xanthomonas campestris pathovar campestris (Xcc) genes identified by RB‐TnSeq to fitness during infection of cauliflower hydathodes. (a, b) Competition assays at a 1 : 1 ratio of mutants (red boxes) associated with a loss (a) or gain (b) of fitness in RB‐TnSeq or complemented mutants (gray boxes) against the Xcc strain 8004∷GUS*‐GFP* (hereafter WT*). Dots represent independent biological replicates (N ≥ 4) consisting of 16 pooled hydathodes of Brassica oleracea var botrytis cv Clovis F1 cauliflower plants each. (c) Internal growth curves in individual hydathodes 6 d after dip‐inoculation (three independent biological replicates, n ≥ 37 hydathodes in total per strain). Strains separated by the dotted line were inoculated in different experiments. The central box of the boxplot shows the central 50% of values (from the first quartile to the third quartile) while the whiskers indicate the values located within 1.5 times the interquartile range. The median is represented as a black horizontal. Outliers are shown as grey dots. (d) Competition assays at a 1 : 100 ratio (Mutant/WT*) of Xcc mutants associated with a gain of fitness in RB‐TnSeq. Dots represent independent biological replicates (N ≥ 4) consisting of 16 pooled hydathodes each. Statistical significance of growth differences between each strain and the WT was assessed with the Wilcoxon test (ns: not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
Table 2.
Candidate genes selected for investigation based on the RB‐TnSeq data.
| Gene ID a | Locus tag b | Fitness browser ID c | Predicted function | Gene name | Hyd 3 dpi | Hyd 6 dpi | Xylem sap | MME glucose dropout –LTH | MME glucose | MOKA |
|---|---|---|---|---|---|---|---|---|---|---|
| XC_0143 | XCC8004_a01841 | Xcc‐8004.186.1 | Glycogen branching protein | glgB1 | −1.97 | −2.67 | −0.09 | −0.08 | −0.15 | −0.08 |
| XC_0449 | XCC8004_a05761 | Xcc‐8004.577.1 | MarR family transcriptional regulator | – | −1.51 | −2.14 | −0.44 | −0.24 | −0.22 | 0.11 |
| XC_0522 | XCC8004_a06761 | Xcc‐8004.673.1 | PbsX family transcriptional regulator | – | 0.20 | 1.07 | 0.15 | 0.04 | −0.05 | 0.04 |
| XC_1113 | XCC8004_a14101 | Xcc‐8004.1413.1 | Ferric enterobactin receptor | bfeA | −0.47 | −1.94 | 0.10 | −0.01 | −0.05 | 0.09 |
| XC_1431 | XCC8004_a17891 | Xcc‐8004.1792.1 | Transcriptional regulator | – | −0.98 | −2.04 | −0.05 | −0.48 | −0.18 | 0.10 |
| XC_1958 | XCC8004_a24301 | Xcc‐8004.2420.1 | PHA synthase subunit | phaE | −1.99 | −2.94 | −0.77 | 0.44 | 0.31 | 0.10 |
| XC_1959 | XCC8004_a24311 | Xcc‐8004.2421.1 | Poly (3‐hydroxybutyric acid) synthase | phcC | −1.79 | −3.26 | −0.90 | 0.44 | 0.22 | 0.03 |
| – | XCC8004_a27231 | Xcc‐8004.2699.1 | Hypothetical protein | – | 1.23 | 1.25 | 1.23 | 0.03 | 0.02 | 0.28 |
| XC_2377 | XCC8004_a29681 | Xcc‐8004.2945.1 | Imidazole glycerol phosphate synthase subunitHisH | hishH | −2.07 | −4.03 | 0.04 | −4.30 | 0.18 | 0.20 |
| XC_3076 | XCC8004_a38371 | Xcc‐8004.3806.1 | HrpX protein | hrpX | 0.31 | 1.72 | 0.12 | −0.11 | −0.06 | 0.00 |
| XC_3253 | XCC8004_a40621 | Xcc‐8004.4026.1 | Hypothetical protein | – | −1.61 | −4.04 | −3.09 | −0.63 | −0.16 | 0.05 |
| XC_3388 | XCC8004_a42701 | Xcc‐8004.4210.1 | Hypothetical protein | – | 0.30 | 1.27 | −0.32 | −1.24 | −0.90 | −0.61 |
Fitness values in red for mutants with a gain of fitness and in blue for a loss of fitness; complete fitness data can be found in Supporting Information Table S2. Hyd, hydathodes.
Gene ID corresponds to Xanthomonas campestris pathovar campestris (Xcc) 8004’s CDS annotation given by Qian et al. (2005).
Locus_tag corresponds to the identification tag attributed when the new annotation was performed using RNAseq data published by Luneau et al. (2022a).
Fitness browser ID corresponds to the gene name used for fitness analysis.
Fig. 4.
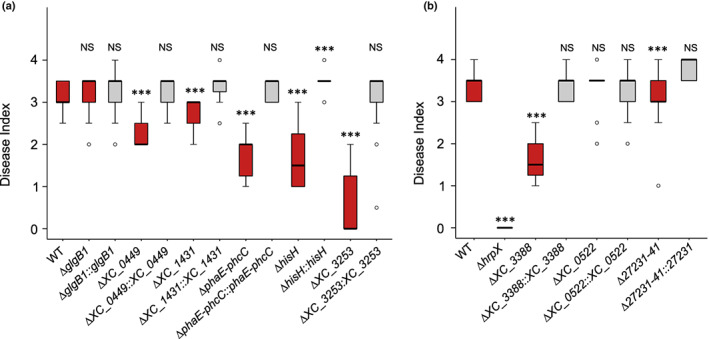
Pathogenicity of Xanthomonas campestris pathovar campestris (Xcc) mutants in genes identified by RB‐TnSeq during infection of cauliflower hydathodes. Severity of disease symptoms caused by knockout (red boxes) and complemented (gray boxes) strains of candidate genes 10 d after piercing inoculation into Brassica oleracea var botrytis cv Clovis F1 cauliflower leaf midvein. Strains are grouped based on the phenotype of associated mutants that displayed either a loss (a) or a gain (b) of fitness by RB‐TnSeq analysis at 6 dpi in hydathodes. Statistical significance of differences in symptoms severity between each strain and the 8004 WT was determined with the Wilcoxon test (***, P < 0.001; ns, not significant). The central box of the boxplot shows the central 50% of values (from the first quartile to the third quartile) while the whiskers indicate the values located within 1.5 times the interquartile range (note that sometimes the whiskers are not visible because the first or third quartile coincides with the minimum or maximum value, respectively). The median is represented as a black horizontal (note that sometimes the box and whiskers are not visible because almost all values are equal to the median). Outliers are shown as empty dots.
We also focused on three genes of unknown function (XC_0522, XC_3388 and XCC8004_a27231‐41) whose mutation conferred a fitness gain in the RB‐TnSeq screen at 6 dpi. Competition assays in a 1 : 1 ratio against the WT strain failed to reproduce the gain of fitness observed in RB‐TnSeq (Fig. 3c). However, a significant gain of fitness for the ΔhrpX and Δ27231‐41 (knockout of the overlapping XCC8004_a27231 and XCC8004_a27241 genes) mutants could be measured at a 1 : 100 ratio (mutant : WT, Fig. 3d) indicating that these mutants can indeed behave like cheaters. The failure to reproduce the gain of fitness for ΔXC_0522 and ΔXC_3388 mutants may thus be due to the 1 : 100 ratio because it remains much higher than the ratio of these mutants inside our RB‐TnSeq mutant library. Among those three genes, Δ27231‐41 and ΔXC_3388 deletion mutants had a reduced pathogenicity, though to a lesser extent than the hrpX mutant which was avirulent (Fig. 4b).
Overall, these results indicate that a gene’s contribution to fitness does not strictly correlate with a measurable contribution to virulence nor in planta growth (Figs 3, S4b) and that the RB‐TnSeq screen was able to robustly identify genes with subtle contributions to fitness and pathogenicity.
Functional analysis of genes encoding proteins of unknown function involved in pathogenicity
Two putative transcriptional MarR and TetR family regulators (XC_0449 and XC_1431 respectively) and two proteins of unknown function (XC_3253 and XC_3388) which contribute to fitness and pathogenicity during plant infection were selected for their potential roles in the regulation of gene expression. Transcriptome analyses of the mutants and complemented strains grown in MOKA‐rich medium were conducted. The XC_0449 regulon includes only 11 genes with function involved mainly in amino acid biosynthesis whereas XC_1431 seems to regulate only five genes. Most of these genes belong to the same operon (XC_1432 to XC_1436) coding for a multidrug efflux system, suggesting that XC_1431 is involved in stress responses (Table S4). RNAseq profiling of ΔXC_3253 showed a deregulation of 35 genes, in part associated with stress responses and motility, which was confirmed by plate motility assays (Fig. S5a). Also, 10 genes code for hypothetical proteins and seven genes are involved in sulfate metabolism and biosynthesis of sulfur amino acids such as methionine and cysteine (Table S4). The ΔXC_3253 strain also shows a decrease of extracellular protease activity (Fig. S5b). All of these results partially explain the strong phenotype of this mutant in pathogenicity and fitness. However, the exact function of this protein remains unclear. Because the XC_3253 gene is located downstream of an LPS biosynthesis operon, we hypothesize that this gene could be involved in cell envelope formation.
The ΔXC_3388 mutant displayed pleiotropic physiological alterations including hypermotility on swimming plates, reduced extracellular amylase activity, low EPS production and disturbed biofilm formation (Fig. 5). These observations are consistent with the ‘rough’ colony phenotype visible on agar plates (Fig. S6a). In addition, metabolic fingerprinting on Biolog plates of the ΔXC_3388 mutant identified altered metabolic signatures on several carbon sources including glucose, glutamate, maltose and arbutin (Fig. S6b). These observations were independently confirmed by growth experiments in minimal medium supplemented with these carbon sources (Fig. S6c). These results indicate a broad physiological role for XC_3388 in Xcc adaptation to environmental conditions.
Fig. 5.
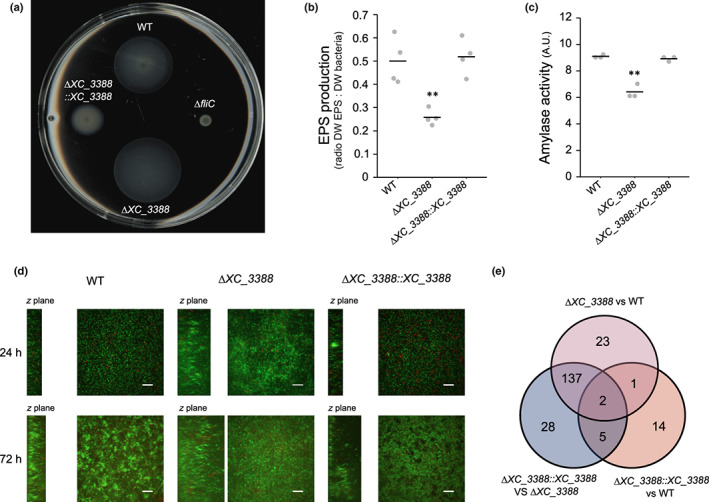
In vitro phenotypes of the ΔXC_3388 mutant strain. (a) Motility assay showing swimming abilities after 48 h on soft 0.3% agar plates. The WT strain and the ΔfliC nonmotile flagellin mutant are positive and negative controls, respectively. (b) Mean exopolysaccharide (EPS) production (ratio dry weight EPS/dry weight bacteria) after 24 h of growth in MOKA‐rich medium. (c) Mean amylase activity measured after 24 h on MOKA plates supplemented with 0.125% potato starch. (b, c) Statistical significance of differences was assessed with the Wilcoxon test (**, P < 0.01). (d) Xcc biofilm architecture using spinning disk confocal microscopy after 24 h (upper panels) and 72 h (lower panels). Representative images are shown. Cells were grown in chambers with a coverslip containing MME minimal medium supplemented with 20 mM glucose. Propidium iodide staining was used to visualize dead cells (red) among live Xcc 8004::GUS‐GFP cells (green). Square panels show the biofilm on the x and y planes, while left rectangles show the biofilm on the z plane. Side panel size varies depending on the thickness of the biofilm. Scale bar, 50 μm. (e) Number of differentially expressed genes identified by RNAseq (¦log2FC¦ ≥ 1.5 and FDR ≤ 0.05) during exponential growth in MOKA.
To better evaluate the extent and basis of ∆XC_3388 mutant phenotypes, we compared the transcriptomes of the ∆XC_3388 mutant with the Xcc 8004 WT strain and the complemented strain ΔXC_3388::3388 (Fig. 5e; Table S4). In support of the observed phenotypes (Fig. 5a–d), the XC_3388 mutation strongly induces the expression of chemotaxis and flagellar motility genes while repressing EPS biosynthesis genes. We also noted a decreased expression of genes encoding ribosomal proteins, possibly as a stress response. Importantly, several signaling pathways were significantly deregulated in ΔXC_3388, including the higher expression of many genes encoding GGDEF domain‐containing proteins and diguanylate cyclases, which mediate synthesis of c‐di‐GMP (Table S4). XC_3388 might thus be involved in the metabolism of this important intracellular messenger, providing a plausible explanation for the pleiotropic phenotypes of the ΔXC_3388 mutant. XC_3388 possesses a domain of unknown function, DUF1631, for which no information is available. Structure prediction and comparisons with alphafold2 (Jumper et al., 2021) or rosettafold (Baek et al., 2021) could not provide additional insights into the biological roles of XC_3388. blastp analysis revealed that XC_3388 is highly conserved among the Xanthomonadales and plant‐associated γ‐proteobacteria (for conservation and structure predictions, see Methods S1). Together, these results suggest a c‐di‐GMP‐mediated regulatory function for XC_3388, which is probably conserved in a broad range of γ‐proteobacteria. Accordingly, XC_3388 was renamed pacR (for pathogenicity and adaptation through c‐di‐GMP regulation).
Discussion
Colonization of hydathodes is constrained by postinvasive selection
By counting the number of barcoded strains entering individual hydathodes, we showed that hydathode pores do not constitute a significant bottleneck with our dip inoculation procedure. It is consistent with the observation that hydathode pores cannot fully close in response to biotic and abiotic stimuli (Cerutti et al., 2017) and that a single Xcc cell can be sufficient for successful hydathode infection (Robeson et al., 1989). Therefore, hydathodes could represent easy infection routes for any microbe. Yet, very few pathogens are reported to infect hydathodes (Cerutti et al., 2019). This paradox suggests that hydathode immunity supported by hostile growth conditions exists and that postinvasive in planta selection rather than infection bottlenecks drives the evolution of hydathode pathogens. The drivers of this infectious behavior are unknown but could include the microbial ability to sustain growth in low‐nutrient conditions or to overcome hydathode‐specific immune responses.
Metabolic adaptation of Xcc to plant environments relies on high‐affinity scavenging of mineral and organic matter from the xylem sap and guttation fluid
Our genetic screens highlighted the importance of several amino acid biosynthetic pathways for Xcc fitness in hydathodes and xylem sap. We hypothesize that the apoplastic plant tissue environment is limiting for such nutrients. Consistently, P. syringae mutants in the tryptophan, histidine and leucine/isoleucine/valine biosynthetic pathways have reduced growth in bean leaf mesophyll (Helmann et al., 2019). Sulfur‐containing amino acids methionine and cysteine biosynthetic pathways were important during Dickeya dadantii infection of chicory (Royet et al., 2019) in contrast to our study and others (Duong et al., 2018; Gonzalez‐Mula et al., 2019; Helmann et al., 2019, 2020; Royet et al., 2019; Torres et al., 2022). These TnSeq results suggest that, even though amino acids are key nutrients, their requirements for fitness vary among pathogenic bacteria and are probably dependent on microbial metabolic features as well as individual amino acid availability within plant tissues.
Cabbage xylem sap contains amino acids, organic acids and sugars in the low millimolar range and most of these compounds are actively metabolized by Xcc (Dugé de Bernonville et al., 2014). However, the abundance or the utilization of these amino acids present in xylem sap did not correlate with the gene fitness analysis in xylem sap, suggesting that Xcc feeds on multiple nutrients. Interestingly, relA (XC_1173), which is important for (p)ppGpp biosynthesis, contributes to fitness in xylem sap. Since accumulation of the (p)ppGpp alarmone triggers the stringent response and allows bacterial cells to face nutrient starvation, it indicates that Xcc faces starvation in xylem sap (Bai et al., 2022). Guttation fluid in hydathodes is derived from xylem sap and is presumably depleted from most mineral and organic components, as reported for a wide range of plants (Cerutti et al., 2019). In support of this observation, Xcc expresses a diversity of nutrient‐scavenging systems inside hydathodes (Luneau et al., 2022a) including high‐affinity transporters for phosphate, nitrate and sulfate. Future metabolomic analyses of guttation fluids and xylem sap will be needed to fully characterize the nutritional environment of Xcc in planta. These metabolomic and RB‐TnSeq results will be instrumental to construct metabolic networks of vascular pathogens and understand their adaptation to a vascular lifestyle (Kim et al., 2019; Gerlin et al., 2020).
RB‐TnSeq identifies genes of unknown functions relevant for pathogenicity
RB‐TnSeq allows high‐throughput genome‐wide screens and provides phenotypic information about individual gene contributions to bacterial fitness. Price et al. (2018) exploited this feature to investigate the adaptive value of genes from over 30 ecologically distant bacteria in a wide variety of conditions. This analysis identified condition‐dependent cofitness relationships for more than 11 000 previously uncharacterized genes, showing that this approach is particularly powerful to unravel biological functions of hypothetical proteins. Based on our RB‐TnSeq screening in planta, two proteins with unknown function, XC_3253 and XC_3388, were studied in greater detail. In agreement with a preliminary report (Qian et al., 2005), we showed that XC_3388‐PacR contributes to adaptation to environmental conditions and pathogenicity. Our study also showed that PacR could be a master regulator of Xcc physiology and virulence. The strong deregulation of c‐di‐GMP‐related genes is consistent with the impact on biofilm formation and motility, which are known to be regulated by c‐di‐GMP (Ryan et al., 2007). Hypothetically, PacR could be involved in c‐di‐GMP homeostasis, but direct evidence for this is missing. Further studies will be required to elucidate the functions of PacR‐ and DUF1631‐containing proteins at the molecular and cellular levels.
RB‐TnSeq analysis reveals Xcc social behaviors inside hydathodes
A key result of our TnSeq study is the gain of fitness displayed by mutants in several major regulators of pathogenicity during hydathode infection. In TnSeq approaches, the fitness of individual cells is measured in a population context. Therefore, mutations associated with the production of public goods (i.e. secreted resources available for the whole population) can be cross‐complemented in a population. Xcc mutants in those virulence regulators are not only cross‐complemented but also have an increased fitness. These results are evidence of a fitness cost associated with the expression of virulence‐related genes during hydathode infection. Because Xcc cells are confined within hydathodes, we suggest that hydathodes constitute a conducive environment for cheater‐like behaviors and that RB‐TnSeq can finely quantify the cost of virulence during infection.
There is a growing amount of evidence for spatio‐temporal heterogeneity in the dynamics of microbial populations (Ackermann, 2015; Friesen, 2020). Infection strategies sometimes result in very few bacterial cells entering plant tissues, resulting in the direct challenge of their intrinsic fitness. For instance, P. syringae infection of tomato leaf mesophyll counter‐selects cheaters at low inoculum density but promotes their fitness at high population density (Rufián et al., 2016, 2018). Bistable phenotypic heterogeneity of P. syringae was also evidenced in a clonal population leading to the coexistence of two subpopulations that either express or do not express the T3SS (Rufián et al., 2016). Such phenotypic heterogeneity was also reported in the population of phytopathogenic Ralstonia pseudosolanacearum during tomato infection (Poussier et al., 2003; Perrier et al., 2019). Based on frequency‐dependent gain of fitness for the Xcc ∆hrpX mutant in competition assays, bistable T3SS expression could occur theoretically in Xcc during hydathode colonization. Further studies will be required to test this hypothesis.
Mechanistically, faster growth of the hrpX mutants could be explained by lower expression cost for the T3S regulon in an environment where immunosuppression is established by cooperators, as described for Salmonella typhimurium (Sturm et al., 2011). In addition to dividing labor at the population scale, bistable expression of the T3SS in Salmonella maintains virulence traits fixed in the population and limits the emergence of T3SS‐deficient mutants (Diard et al., 2013). From an evolutionary standpoint, a high abundance of Xcc T3SS‐deficient mutants could lead to the collapse of infection in proximal and distal tissues. Indeed, according to the Black Queen Hypothesis, emergence of T3SS‐deficient mutants would be favored during hydathode infection because it confers local short‐term fitness advantages (Morris et al., 2012) allowing mutations occurring early in the infection cycle to be fixed in the population and drive the evolution of Xcc. This mechanism may explain how nonpathogenic Xanthomonas strains lacking a T3SS and/or T3E are routinely isolated in the phyllosphere (Vorholt, 2012; Merda et al., 2017). Nevertheless, in the larger context of the Xcc life cycle, losing the ability to repress host immunity is a weakness compromising Xcc systemic spread from the hydathode, seed‐mediated propagation of the disease and the infection of neighboring host plants.
Competing interests
None declared.
Author contributions
AB, LDN, AMD and JDL conceived the project. AB supervised the project. JSL performed most of the experiments. MB built the Xanthomonas RB‐TnSeq library. JF and CG constructed some mutant strains. TQM and AB performed some competitive index experiments. EL performed in vitro and in planta bacterial growth assays. MA and BT performed phylogenetic analysis on some selected genes. SC conducted bioinformatic analyses. M‐FJ conducted biostatistic analyses. OB contributed to the bottleneck analysis. JR performed the library mapping. The manuscript was written by JSL, LDN and AB. All authors read and approved the final manuscript.
Supporting information
Fig. S1 Biological reproducibility of RB‐TnSeq assays.
Fig. S2 Growth of the RB‐TnSeq library in vitro identifies genes contributing to multiplication in plant‐independent conditions.
Fig. S3 Xanthomonas campestris pathovar campestris (Xcc) fitness measured by RB‐TnSeq for virulence‐associated genes (as defined by Y‐W. He et al., 2007) in plant‐associated conditions.
Fig. S4 Correlation between RB‐TnSeq fitness results, competitiveness and pathogenicity during hydathode infection with Xanthomonas campestris pathovar campestris (Xcc) mutant strains in genes identified by RB‐TnSeq during infection of cauliflower hydathodes.
Fig. S5 In vitro phenotypes of the ΔXC_3253 mutant strain.
Fig. S6 Importance of XC_3388 for Xanthomonas campestris pathovar campestris (Xcc) metabolism.
Methods S1 Supplementary material and methods for fitness screening in vitro; carbon and nitrogen substrate phenotyping; growth measurements in vitro.
Table S1 Strains, plasmids and oligos used in this study.
Table S2 List of essential genes of Xanthomonas campestris pathovar campestris (Xcc).
Table S3 Genes affected in fitness under each tested condition.
Table S4 RNAseq data.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
JSL was funded by a PhD grant from the French Ministry of Higher Education, Research and Innovation. JSL, M‐FJ, SC, EL, MA, LDN and AB were funded by grants from the Agence Nationale de la Recherche XBOX (ANR‐19‐CE20‐JCJC‐0014‐01). JSL was funded by the Fédération de Recherche Agrobiosciences, Interactions et Biodiversité through project CHACCCAL. AB and OB were funded by the Fédération de Recherche Agrobiosciences, Interactions et Biodiversité and TULIP Labex through project 31000509_FRT4. This study is set within the framework of the ‘Laboratoires d’Excellences’ (LABEX) TULIP (ANR‐10‐LABX‐41) and of the ‘École Universitaire de Recherche’ (EUR) TULIP‐GS (ANR‐18‐EURE‐0019). Research in JDL’s lab is supported by USDA ARS Grants 2030‐21000‐046‐00D and 2030‐21000‐050‐00D, and NSF Directorate for Biological Sciences Grant IOS‐1557661. All authors benefited from the COST action CA16107 EuroXanth. Thanks to Aurélie LE RU from TRI‐FRAIB Imaging platform facilities, FRAIB 3450 CNRS‐UTIII for biofilm imaging.
Contributor Information
Laurent D. Noël, Email: laurent.noel@inrae.fr.
Alice Boulanger, Email: alice.boulanger@inrae.fr.
Data availability
The fitness data are available from the Fitness Browser (http://fit.genomics.lbl.gov). Sequence read archive accession numbers for RNA‐Seq libraries are openly available using accession no. SRP363574. Sequence read archive accession numbers for bottleneck analysis are openly available using accession no. SRP363580.
References
- Ackermann M. 2015. A functional perspective on phenotypic heterogeneity in microorganisms. Nature Reviews Microbiology 13: 497–508. [DOI] [PubMed] [Google Scholar]
- Alexa A, Rahnenführer J, Lengauer T. 2006. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22: 1600–1607. [DOI] [PubMed] [Google Scholar]
- An S‐Q, Potnis N, Dow M, Vorhölter F‐J, He Y‐Q, Becker A, Teper D, Li Y, Wang N, Bleris L et al. 2020. Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas . FEMS Microbiology Reviews 44: 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlat M, Gough C, Barber CE, Boucher C, Daniels M. 1991. Xanthomonas campestris contains a cluster of hrp genes related to the larger hrp cluster of Pseudomonas solanacearum . Molecular Plant–Microbe Interactions 4: 593. [DOI] [PubMed] [Google Scholar]
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD et al. 2021. Accurate prediction of protein structures and interactions using a three‐track neural network. Science 373: 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai K, Yan H, Chen X, Lyu Q, Jiang N, Li J, Luo L. 2022. The role of RelA and SpoT on ppGpp production, stress response, growth regulation, and pathogenicity in Xanthomonas campestris pv. campestris . Microbiology Spectrum 9: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denancé N, Vasse J, Lauber E, Arlat M. 2007. Plant carbohydrate scavenging through TonB‐dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS ONE 2: e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner D, Bonas U. 2010. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiology Reviews 34: 107–133. [DOI] [PubMed] [Google Scholar]
- Cain AK, Barquist L, Goodman AL, Paulsen IT, Parkhill J, van Opijnen T. 2020. A decade of advances in transposon‐insertion sequencing. Nature Reviews Genetics 21: 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti A, Jauneau A, Auriac M‐C, Lauber E, Martinez Y, Chiarenza S, Leonhardt N, Berthomé R, Noël LD. 2017. Immunity at cauliflower hydathodes controls systemic infection by Xanthomonas campestris pv campestris . Plant Physiology 174: 700–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti A, Jauneau A, Laufs P, Leonhardt N, Schattat MH, Berthomé R, Routaboul J‐M, Noël LD. 2019. Mangroves in the leaves: anatomy, physiology, and immunity of epithemal hydathodes. Annual Review of Phytopathology 57: 91–116. [DOI] [PubMed] [Google Scholar]
- Diard M, Garcia V, Maier L, Remus‐Emsermann MNP, Regoes RR, Ackermann M, Hardt W‐D. 2013. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 494: 353–356. [DOI] [PubMed] [Google Scholar]
- Ditta G, Stanfield S, Corbin D, Helinski DR. 1980. Broad host range DNA cloning system for gram‐negative bacteria: construction of a gene bank of Rhizobium meliloti . Proceedings of the National Academy of Sciences, USA 77: 7347–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugé de Bernonville T, Noël LD, SanCristobal M, Danoun S, Becker A, Soreau P, Arlat M, Lauber E. 2014. Transcriptional reprogramming and phenotypical changes associated with growth of Xanthomonas campestris pv. campestris in cabbage xylem sap. FEMS Microbiology Ecology 89: 527–541. [DOI] [PubMed] [Google Scholar]
- Duong DA, Jensen RV, Stevens AM. 2018. Discovery of Pantoea stewartii ssp. stewartii genes important for survival in corn xylem through a Tn‐Seq analysis. Molecular Plant Pathology 19: 1929–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figurski DH, Helinski DR. 1979. Replication of an origin‐containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proceedings of the National Academy of Sciences, USA 76: 1648–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen ML. 2020. Social evolution and cheating in plant pathogens. Annual Review of Phytopathology 58: 55–75. [DOI] [PubMed] [Google Scholar]
- Gerlin L, Cottret L, Cesbron S, Taghouti G, Jacques M‐A, Genin S, Baroukh C. 2020. Genome‐scale investigation of the metabolic determinants generating bacterial fastidious growth. mSystems 5: e00698‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Mula A, Lachat J, Mathias L, Naquin D, Lamouche F, Mergaert P, Faure D. 2019. The biotroph Agrobacterium tumefaciens thrives in tumors by exploiting a wide spectrum of plant host metabolites. New Phytologist 222: 455–467. [DOI] [PubMed] [Google Scholar]
- He Y‐Q, Zhang L, Jiang B‐L, Zhang Z‐C, Xu R‐Q, Tang D‐J, Qin J, Jiang W, Zhang X, Liao J et al. 2007. Comparative and functional genomics reveals genetic diversity and determinants of host specificity among reference strains and a large collection of Chinese isolates of the phytopathogen Xanthomonas campestris pv. Campestris . Genome Biology 8: R218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y‐W, Boon C, Zhou L, Zhang L‐H. 2009. Co‐regulation of Xanthomonas campestris virulence by quorum sensing and a novel two‐component regulatory system RavS/RavR. Molecular Microbiology 71: 1464–1476. [DOI] [PubMed] [Google Scholar]
- He Y‐W, Ng AY‐J, Xu M, Lin K, Wang L‐H, Dong Y‐H, Zhang L‐H. 2007. Xanthomonas campestris cell–cell communication involves a putative nucleotide receptor protein Clp and a hierarchical signalling network. Molecular Microbiology 64: 281–292. [DOI] [PubMed] [Google Scholar]
- Helmann TC, Deutschbauer AM, Lindow SE. 2019. Genome‐wide identification of Pseudomonas syringae genes required for fitness during colonization of the leaf surface and apoplast. Proceedings of the National Academy of Sciences, USA 116: 18900–18910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann TC, Deutschbauer AM, Lindow SE. 2020. Distinctiveness of genes contributing to growth of Pseudomonas syringae in diverse host plant species. PLoS ONE 15: e0239998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving SE, Choudhury NR, Corrigan RM. 2021. The stringent response and physiological roles of (pp)pGpp in bacteria. Nature Reviews Microbiology 19: 256–271. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Joe A, Lee M, Yang S, Ma X, Ronald PC, Lee I. 2019. A genome‐scale co‐functional network of Xanthomonas genes can accurately reconstruct regulatory circuits controlled by two‐component signaling systems. Molecules and Cells 42: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luneau JS, Cerutti A, Roux B, Carrère S, Jardinaud M‐F, Gaillac A, Gris C, Lauber E, Berthomé R, Arlat M et al. 2022a. Xanthomonas transcriptome inside cauliflower hydathodes reveals bacterial virulence strategies and physiological adaptations at early infection stages. Molecular Plant Pathology 23: 159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luneau JS, Noël LD, Lauber E, Boulanger A. 2022b. A β‐glucuronidase (GUS) based bacterial competition assay to assess fine differences in fitness during plant infection. Bio‐Protocol 12: e3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macho AP, Zumaquero A, Ortiz‐Martín I, Beuzón CR. 2007. Competitive index in mixed infections: a sensitive and accurate assay for the genetic analysis of Pseudomonas syringae–plant interactions. Molecular Plant Pathology 8: 437–450. [DOI] [PubMed] [Google Scholar]
- Merda D, Briand M, Bosis E, Rousseau C, Portier P, Barret M, Jacques M‐A, Fischer‐Le SM. 2017. Ancestral acquisitions, gene flow and multiple evolutionary trajectories of the type three secretion system and effectors in Xanthomonas plant pathogens. Molecular Ecology 26: 5939–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinière L, Mirabel L, Gueguen E, Bertolla F. 2022. A comprehensive overview of the genes and functions required for lettuce infection by the hemibiotrophic phytopathogen Xanthomonas hortorum pv. vitians. mSystems 7: e0129021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JJ, Lenski RE, Zinser ER. 2012. The black queen hypothesis: evolution of dependencies through adaptive gene loss. mBio 3: e00036‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Levin HL. 2020. Transposon insertion sequencing, a global measure of gene function. Annual Review of Genetics 54: 337–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SS, Patnana PK, Lomada SK, Tomar A, Chatterjee S. 2016. Co‐regulation of iron metabolism and virulence associated functions by iron and XibR, a novel iron binding transcription factor, in the plant pathogen Xanthomonas . PLoS Pathogens 12: e1006019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier A, Barlet X, Rengel D, Prior P, Poussier S, Genin S, Guidot A. 2019. Spontaneous mutations in a regulatory gene induce phenotypic heterogeneity and adaptation of Ralstonia solanacearum to changing environments. Environmental Microbiology 21: 3140–3152. [DOI] [PubMed] [Google Scholar]
- Poussier S, Thoquet P, Trigalet‐Demery D, Barthet S, Meyer D, Arlat M, Trigalet A. 2003. Host plant‐dependent phenotypic reversion of Ralstonia solanacearum from non‐pathogenic to pathogenic forms via alterations in the phcA gene. Molecular Microbiology 49: 991–1003. [DOI] [PubMed] [Google Scholar]
- Price MN, Wetmore KM, Waters RJ, Callaghan M, Ray J, Liu H, Kuehl JV, Melnyk RA, Lamson JS, Suh Y et al. 2018. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 557: 503–509. [DOI] [PubMed] [Google Scholar]
- Qian W, Han Z‐J, He C. 2008. Two‐component signal transduction systems of Xanthomonas spp.: a lesson from genomics. Molecular Plant–Microbe Interactions 21: 151–161. [DOI] [PubMed] [Google Scholar]
- Qian W, Jia Y, Ren S‐X, He Y‐Q, Feng J‐X, Lu L‐F, Sun Q, Ying G, Tang D‐J, Tang H et al. 2005. Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris . Genome Research 15: 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robeson DJ, Bretschneider KE, Gonella MP. 1989. A hydathode inoculation technique for the simulation of natural black rot infection of cabbage by Xanthomonas campestris pv. campestris . Annals of Applied Biology 115: 455–459. [Google Scholar]
- Royet K, Parisot N, Rodrigue A, Gueguen E, Condemine G. 2019. Identification by Tn‐seq of Dickeya dadantii genes required for survival in chicory plants. Molecular Plant Pathology 20: 287–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufián JS, Macho AP, Corry DS, Mansfield JW, Ruiz‐Albert J, Arnold DL, Beuzón CR. 2018. Confocal microscopy reveals in planta dynamic interactions between pathogenic, avirulent and non‐pathogenic Pseudomonas syringae strains. Molecular Plant Pathology 19: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufián JS, Sánchez‐Romero M‐A, López‐Márquez D, Macho AP, Mansfield JW, Arnold DL, Ruiz‐Albert J, Casadesús J, Beuzón CR. 2016. Pseudomonas syringae differentiates into phenotypically distinct subpopulations during colonization of a plant host. Environmental Microbiology 18: 3593–3605. [DOI] [PubMed] [Google Scholar]
- Ryan RP, Fouhy Y, Lucey JF, Jiang B‐L, He Y‐Q, Feng J‐X, Tang J‐L, Dow JM. 2007. Cyclic di‐GMP signalling in the virulence and environmental adaptation of Xanthomonas campestris . Molecular Microbiology 63: 429–442. [DOI] [PubMed] [Google Scholar]
- Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. 1994. Small mobilizable multi‐purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum . Gene 145: 69–73. [DOI] [PubMed] [Google Scholar]
- Sturm A, Heinemann M, Arnoldini M, Benecke A, Ackermann M, Benz M, Dormann J, Hardt W‐D. 2011. The cost of virulence: retarded growth of Salmonella typhimurium cells expressing type III secretion system 1. PLoS Pathogens 7: e1002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Xu Y, Liang H, Yuan G, Wu X, Zheng D. 2021. Genome‐wide identification of Ralstonia solanacearum genes required for survival in tomato plants. mSystems 6: e00838‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Tang D‐J, Dubrow ZE, Bogdanove A, An S. 2021. Xanthomonas campestris Pathovars. Trends in Microbiology 29: 182–183. [DOI] [PubMed] [Google Scholar]
- Tao J, He C. 2010. Response regulator, VemR, positively regulates the virulence and adaptation of Xanthomonas campestris pv. campestris: VemR‐regulated virulence and adaptation in Xcc . FEMS Microbiology Letters 304: 20–28. [DOI] [PubMed] [Google Scholar]
- Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proceedings of the National Academy of Sciences, USA 84: 2833–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timilsina S, Potnis N, Newberry EA, Liyanapathiranage P, Iruegas‐Bocardo F, White FF, Goss EM, Jones JB. 2020. Xanthomonas diversity, virulence and plant–pathogen interactions. Nature Reviews Microbiology 18: 415–427. [DOI] [PubMed] [Google Scholar]
- Torres M, Jiquel A, Jeanne E, Naquin D, Dessaux Y, Faure D. 2022. Agrobacterium tumefaciens fitness genes involved in the colonization of plant tumors and roots. New Phytologist 233: 905–918. [DOI] [PubMed] [Google Scholar]
- Turner P, Barber C, Daniels M. 1984. Behaviour of the transposons Tn5 and Tn7 in Xanthomonas campestris pv. campestris . Molecular and General Genetics MGG 195: 101–107. [Google Scholar]
- Vicente JG, Holub EB. 2013. Xanthomonas campestris pv. campestris (cause of black rot of crucifers) in the genomic era is still a worldwide threat to brassica crops. Molecular Plant Pathology 14: 2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorholt JA. 2012. Microbial life in the phyllosphere. Nature Reviews Microbiology 10: 828–840. [DOI] [PubMed] [Google Scholar]
- Wei K, Tang D‐J, He Y‐Q, Feng J‐X, Jiang B‐L, Lu G‐T, Chen B, Tang J‐L. 2007. hpaR, a Putative marR family transcriptional regulator, is positively controlled by HrpG and HrpX and involved in the pathogenesis, hypersensitive response, and extracellular protease production of Xanthomonas campestris Pathovar campestris . Journal of Bacteriology 189: 2055–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP et al. 2015. Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar‐coded transposons. mBio 6: e00306–e00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Biological reproducibility of RB‐TnSeq assays.
Fig. S2 Growth of the RB‐TnSeq library in vitro identifies genes contributing to multiplication in plant‐independent conditions.
Fig. S3 Xanthomonas campestris pathovar campestris (Xcc) fitness measured by RB‐TnSeq for virulence‐associated genes (as defined by Y‐W. He et al., 2007) in plant‐associated conditions.
Fig. S4 Correlation between RB‐TnSeq fitness results, competitiveness and pathogenicity during hydathode infection with Xanthomonas campestris pathovar campestris (Xcc) mutant strains in genes identified by RB‐TnSeq during infection of cauliflower hydathodes.
Fig. S5 In vitro phenotypes of the ΔXC_3253 mutant strain.
Fig. S6 Importance of XC_3388 for Xanthomonas campestris pathovar campestris (Xcc) metabolism.
Methods S1 Supplementary material and methods for fitness screening in vitro; carbon and nitrogen substrate phenotyping; growth measurements in vitro.
Table S1 Strains, plasmids and oligos used in this study.
Table S2 List of essential genes of Xanthomonas campestris pathovar campestris (Xcc).
Table S3 Genes affected in fitness under each tested condition.
Table S4 RNAseq data.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The fitness data are available from the Fitness Browser (http://fit.genomics.lbl.gov). Sequence read archive accession numbers for RNA‐Seq libraries are openly available using accession no. SRP363574. Sequence read archive accession numbers for bottleneck analysis are openly available using accession no. SRP363580.