

ABSTRACT
Acute myeloid leukemia (AML) is a highly cancerous and aggressive hematologic disease with elevated levels of drug resistance and relapse resulting in high mortality. Recently, bromodomains and extra-terminal (BET) protein inhibitors have been extensively researched in hematological tumors as potential anticancer agents. MZ1 is a novel BET inhibitor that mediates selective proteins degradation and suppression of tumor growth through proteolysis-targeting chimeras (PROTAC) technology. Accordingly, this study aimed to investigate the role and therapeutic potential of MZ1 in AML. In this study, we first identified that AML patients with high BRD4 expression had poor overall survival than those with low expression group. MZ1 inhibited AML cell growth and induced apoptosis and cycle arrest in vitro. MZ1 induced degradation of BRD4, BRD3 and BRD2 in AML cell strains. Additionally, MZ1 also initiated the cleavage of poly-ADP-ribose polymerase (PARP), which showed cytotoxic effects on NB4 (PML-RARa), K562 (BCR-ABL), Kasumi-1 (AML1-ETO), and MV4-11 (MLL-AF4) cell lines representing different molecular subtypes of AML. In AML mouse leukemia model, MZ1 significantly decreased leukemia cell growth and increased the mouse survival time. According to the RNA-sequencing analysis, MZ1 led to c-Myc and ANP32B genes significant downregulation in AML cell lines. Knockdown of ANP32B promoted AML cell apoptosis and inhibited cell growth. Overall, our data indicated that MZ1 had broad anti-cancer effects on AML cell lines with different molecular lesions, which might be exploited as a novel therapeutic strategy for AML patients.
KEYWORDS: BRD4, MZ1, acute myeloid leukemia, c-Myc, ANP32B
Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy associated with a typically poorer prognosis except for acute promyelocytic leukemia (APL).1,2 Both adults and children can develop the disease. With the implementation of first-line intensive therapy, supportive care and salvage therapy, the prognosis of AML has improved significantly, with about approximately 60% of patients progressing to remission and approximately 30% of patients achieving long-term remission,2 approximately 50% of these children experience disease relapse.3,4 When the preliminary treatment regimens fail, other treatment options (e.g., targeted therapies and cellular therapies) are available; however, even with stem cell transplantation (SCT), only a third of the children survive for 3 years.3 Moreover, there is little room to further intensify therapy due to the treatment-related mortality.5
While the treatment of adult AML patients is rapidly evolving, for instance, the use of the US Food and Drug Administration (FDA) approved FLT3 inhibitors to treat early and relapsed/refractory FLT3-ITD AML,6 the advancements in the treatment of pediatric AML are going rather slowly due to a scarcity of pediatric drugs. Therefore, developing novel drugs and new therapies suitable for AML population is a desperate need to enhance the treatment response, reduce the risk of relapse and improve the prognosis.
Bromodomain-containing protein 4 (BRD4) is the most important protein and most widely studied protein in the bromodomain and extra-terminal domain (BET) family compared to the other three members, BRD2, BRD3 and BRDT.7 It is known that BRD4 is overexpressed in a variety of tumor cells.8,9 It can recognize acetylated lysine of histone, bind it, and recruit chromatin regulators to promote gene transcription and promote tumor occurrence, development and metastasis.10 Previous studies have shown that the overexpression of BRD4 in AML is associated with poor prognosis,11 but the mechanism is not clear. Therefore, a comprehensive analysis to understand the mechanism of BRD4 in AML and identifying BRD4 inhibitors is essential to investigate new therapy targets in AML.
As an important anticancer target, recent studies10–14 have investigated small molecule inhibitors of the BET family. JQ1, the most recent and most intensively studied BRD4 inhibitor, is a monovalent inhibitor against the development and progression of hematologic tumors by inducing cell apoptosis and cell cycle inhibition.12 However, JQ1 has a short half-life and can increase BRD4 protein accumulation in cancer cells.13–15 BRD4 inhibitors have not been yet applied to the clinical trials.
Proteolysis-targeting chimeras (PROTACs) are heterobifunctional molecules which induce the degradation of the specific endogenous proteins via the ubiquitin E3 ligase pathway.16 It is proposed that the potential advantages of PROTAC technology may overcome the shortcomings of conventional drug therapies.17 PROTAC contains two ligands, one binds to the target proteins and the other binds to E3 ubiquitin ligase (e.g., VHL, CRBN), which triggers the ubiquitination and degradation of the target proteins.18 MZ1 is one such novel BRD4 degrader.15 As one ligand of MZ1, E3 ubiquitin ligase VHL could achieve ubiquitination and degradation of the target proteins.19 MZ1 demonstrated a strong inhibitory effect on tumor growth and suppressed BRD4 expression in a mouse model of JQ1-resistant triple-negative breast cancer.20 The efficacy of MZ1 has been evaluated in the AML cell lines, MV4-11 and HL-60,21 but the mechanism is not clear. AML is a genetically complex disease, composed of different molecular subtypes. Therefore, we investigated the anticancer activity of MZ1 in several AML cell lines (i.e., NB4, K562, Kasumi-1 and MV4-11), representing different molecular subtypes (i.e., PML-RARa, BCR-ABL, AML1-ETO, and MLL-AF4, respectively) of AML and aimed to delineate the effective strategy for AML patients.
Materials and methods
Cell culture
Human AML cell lines, namely NB4, K562, Kasumi-1 and MV4-11 and mouse leukemia cell line P388-D1, were purchased from the cell bank of the Chinese Academy of Sciences. All AML cell lines were authenticated by short tandem repeat analysis within 3 years. Cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (Biological Industries, CT, USA) and 1% penicillin-streptomycin (Millipore, Billerica, MA, USA) and were routinely tested for mycoplasma. The cells were maintained in a 37°C humidified incubator with 5% CO2.
Cell viability assay
The AML cells were seeded in 96-well plates at a density of 2 × 104 cells per well; MZ1, JQ1, dBET1 (cat. NO. HY-107425, HY-13030, HY-101838, MedChemExpress) at various concentrations (i.e., 0.25 μM, 0.5 μM, 1 μM, 2 μM, 4 μM) were added to each well and treated continuously for 48 h. MZ1 was dissolved in 100% dimethyl sulfoxide (DMSO) to obtain a 10 mM stock solution and kept at −20°C before use. AML cells were plated at the density of 1 × 105 cells per well in 96-well plate. MZ1 at different concentrations and 0.05% DMSO were added to the wells for 7 days, respectively. A cell Counting kit-8 (CCK8) (Dojindo Molecular Technologies, Tokyo, Japan) was added to each well according to the manufacturer’s specifications, and cell viability was measured as absorbance at 450 nm with the reader (Thermo Fisher Scientific). Cell proliferation was calculated as a percentage of the cell proliferation rate in the control medium. Each concentration was tested in triplicate and independently performed at least three times. The half-maximal inhibitory concentrations (IC50) of MZ1, JQ1, and dBET1 were analyzed and calculated using GraphPad Prism software version 8.3.0 (GraphPad Software Inc., San Diego, CA, USA).
Cell cycle analysis
The AML cells were seeded at the specified density of 2 × 106 cells per well in 6-well plates, exposed to different concentrations of MZ1 for 12–24 h. The cells were collected, centrifuged at 3000 rpm for 5 min, washed, fixed overnight at 4°C in 70% ethanol. They were then washed with cold phosphate-buffered saline (PBS) and resuspended in 0.5 mL PI/RNase Staining Buffer (cat. No. 550825; BD Pharmingen™, San Diego, CA, USA) at room temperature for 15 min, dark area. After flow cytometry analysis using a Beckman Gallios ™ Flow Cytometer (Beckman, Krefeld, Germany), the cell cycle distribution was analyzed with Modfit analysis software.
Cell apoptosis assay
As previously described,22 the AML cells were treated with MZ1 in 0.5 μM and 2 μM for 24 h. The collected cells were washed with cold 1xPBS and resuspended in 1 × binding buffer, and stained with fluorescein isothiocyanate FITC-Annexin V antibody and PI solution using a FITC-Annexin V apoptosis kit (cat. No. 556420; BD Biosciences, Franklin Lakes, NJ, USA) according to manufacturer’s instructions. Cell apoptosis was analyzed using the Beckman Gallios™ flow cytometer.
Western blotting analysis
The AML cells were seeded in 6-well plates at a density of 1 × 106 cells per well, and harvested after adding MZ1 at different concentrations to each well and treating them continuously for 24–48 h. MG132 (cat. No. 474787, Sigma-Aldrich, St. Louis, MO, USA) is an inhibitor of proteasome activity. After treating AML cells with 0.25 μM MZ1 and different concentrations of MG132 for 24 h, cells were harvested, centrifuged at 1000 rpm for 5 min and extracted by adding a RIPA lysed protein. The collected supernatant was added with loading buffer (Cat. No. B1012-100, Applygen Technologies) and boiled for 10 min at 100°C. The proteins were separated electrophoretically by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene fluoride (PVDF) membranes for 1 hour. Incubated the membrane with appropriate dilutions of primary antibody in blocking buffer overnight and followed by the secondary antibodies incubation for one hour. The primary antibodies include BRD2 (cat. No. 5848S, Cell Signaling Technology), BRD3 (cat. No. 11859-1-AP, Proteintech), BRD4 (cat. No. 13440S, Cell Signaling Technology), c-Myc (cat. No. 9402, Cell Signaling Technology), ANP32B (cat. No. ab200836, Abcam), GAPDH (cat. No. MA3374, Millipore), VHL (cat. No. 68547S, Cell Signaling Technology), and PARP (cat. No. 9542; Cell Signaling Technology). Target proteins were detected and analyzed using AI600image (GE, USA).
Lentivirus preparation and infection
IGE Biotechnology, Ltd. (China) constructed lentiviral vectors for overexpression and knockdown. The targeting sequence for VHL was 5’-CCGGGCTCAACTTCGACGGCGAGCCCTCGAGGGCTCGCCGTCGAAGTTGAGCTTTTTGAATT-3’; the targeting sequence for ANP32B#2 was 5’-CCGGGAGGGCTTAACAGCTGAATTTCTCGAGAAATTCAGCTGTTAAGCCCTCTTTTTGAATT-3’; the targeting sequence for ANP32B#3 was 5’- CCGGGCTTACCTACTTGGATGGCTACTCGAGTAGCCATCCAAGTAGGTAAGCTTTTTGAATT-3’. For the lentivirus preparation, the packaging plasmid pMD2.G (Cat. No. 12259, Addgene), psPAX2 (Cat. No. 12260, Addgene) and VHL plasmids were co-transfected into 293 FT cells and the fresh medium was completely replaced after 6 h. Harvested the viral supernatant at 48 h post transfection and filtered through a 0.45 μm filter; and added into AML cell lines afterward with 10 μg/mL Polybrene (Sigma-Aldrich) promoting cell transfection. The stable cell lines were selected with puromycin (Sigma-Aldrich) after 48 h.
RNA-sequencing and data processing
The AML cells (NB4) were treated with DMSO (n = 2) and MZ1 (n = 2) after 32 h. Total RNA from AML cells was first isolate with TRIzol® reagent (Invitrogen), and reverse transcribed to create a cDNA library, which was then sequenced and further analyzed by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). The raw reads were filtered and clean reads were mapped according to HISAT2. The gene expression level (as fragments per kilobase of exon model per million reads mapped) was then calculated. Differentially expressed genes (|log2fold change| > 1 and p < .05) were identified using DESeq2 analysis. Gene Set Enrichment Analysis (GSEA) was carried out using the Java GSEA Desktop Application (http://www.broadinstitute.org/gsea/). RNA-seq data of this study have been deposited in GEO database (https://www.ncbi.nlm.nih.gov/geo, with accession code GSE198011).
ChIP-Seq data collection and analysis
In this study, we analyzed our ChIP-Seq H3K27ac dataset of NB4 cell line (GSE188750). The raw data of ChIP-Seq H3K27ac dataset obtained (GSE188750) was aligned to the reference genome (UCSC hg38) using Bowtie2 (v 2.3.5),23 with alignment parameters -p 4 -q -x. Peaks were identified using MACS2 (v2.0.9),24 with parameters -g hs -n test -B -q 0.01. The bedgraph files generated by MACS2 were converted to bigwig files using the UCSC bed-GraphToBigWig tool, and then bigwig files were visualized by Integrative Genomics Viewer (IGV).25 Then we identified super-enhancers by the ROSE (Rank Order of Super Enhancers) method,26,27 according to the parameters -s 12500 -t 2000 (-s stitching distance; -t TSS exclusion zone size).
RNA preparation and real-time PCR analysis
cDNA was obtained using the procedure described before. Real-time fluorescence quantitative PCR analysis was conducted using LightCycler® 480 SYBR Green I Master mix (cat. No. 04707516001; Roche, Penzberg, Germany), and mRNA expression levels were evaluated by the ΔΔCt method, using GAPDH as internal reference. The real-time PCR primers were as follows: GAPDH: forward: TGCACCACCAACTGCTTAG, reverse: GATGCAGGGATGATGTTC, LMO2: forward: TCTGCCGGAGAGACTATCTCA, reverse: ATAGGCACGAATCCGCTTGTC, ANP32B: forward: CTGTTCGAGAACTTGTCTTGGAC, reverse: AGCTTGGGGAGATTTGAAACTG, NFE2: forward: CGGCGCAGCGAATATGTAGA, reverse: CCGACGTTCATCCCGACTC, IRF2BP2: forward: CCCATGACTCCTACATCCTCTT, reverse: GAGGGCGGACTGTTGCTATTC, ANP32A: forward: CACCTCAATCGCAAACTTACCA, reverse: AACACATTTTCTCGGTAGTCGTT, GRK2: forward: GCTTTGGCGAGGTCTATGGG, reverse: TCTTGATGCGCTTTTTGTCCA, LDB1: forward: CAAACGGCTTCAGAACTGGAC, reverse: TCCGGCCAATGGTATATCTCTT, GATA1: forward: CTGTCCCCAATAGTGCTTATGG, reverse: GAATAGGCTGCTGAATTGAGGG.
In vivo xenografts leukemia model
All our research involving animal experiments were approved and licensed by the Animal Care and Use Committee of the Children’s hospital of Soochow University (CAM-SU-AP#: JP-2018-1). SPF-grade BALB/c mice were purchased from Ling Chang Biotechnology Co., Ltd. (Shanghai, China). Five-week-old female mice were randomly grouped (5 mice per group) after tail vein injection of 3 × 105 luciferase-labeled P388-D1 cells. Bioluminescence imaging tests were performed using an in-vivo imaging system (Berthold, Germany) to obtain a successful leukemia model. The leukemic mice were then given a daily intraperitoneal injection of 12.5 mg/Kg MZ1 or the same dosages of vehicle (5% Kolliphor®HS15). The mice’s body weight was measured every three days.
Immunohistochemistry
Immunohistochemical staining was carried out using Ultra-Sensitive SP (Mouse/Rabbit) IHC Kit (MXB Biotechnologies, China) and the DAB Plus Kit (MXB Biotechnologies, China). BRD4 (cat. No. 13440s, Cell Signaling Technology), Ki-67 (cat. No. GB111499, Servicebio), and cleaved-caspase 3 (cat. No. GB11009-1, Servicebio, Boston, MA, USA) antibodies were used.
Statistical analysis of data
GraphPad Prism version 8.3.0 (GraphPad Software, Inc, USA) was used for graph plotting and statistical analysis. Comparison between two groups was carried out using the Student’s t-test or the Mann-Whitney u test. All experiments were conducted at least three times independently. A p-values of 0.05 or lower was considered statistically significant, for which *p < .05, **p < .01, ***p < .001, **** p < .0001.
Results
BRD4 overexpression is associated with poor prognosis in AML patients
Cancer Cell Line Encyclopedia (CCLE, https://depmap.org/portal/download/, Figure 1a) analysis showed that the expression of BRD4 gene was highly increased in AML, ALL and CML compared with that in other cancer cell lines. According to the R2 (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi, Figure 1b) and GEPIA2 databases (http://gepia2.cancer-pku.cn/#index, Figure 1c), the expected overall survival was lower in AML patients with high BRD4 expression than in those with low expression. Based on these data we further explored the role of BRD4 inhibitors in AML as a potential therapeutic target.
Figure 1.
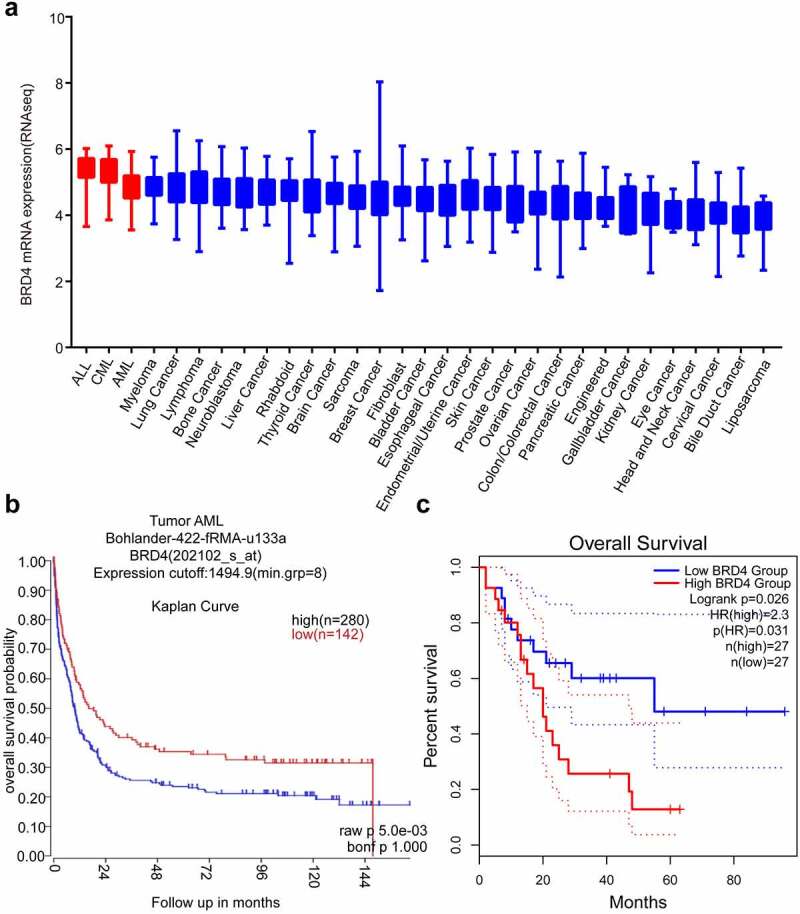
BRD4 could be a good target for Acute Myeloid Leukemia (AML). (a) BRD4 mRNA expression was characterized in different cancer cell lines (generated from the web site: Broad Cancer Cell Line Encyclopedia: https://depmap.org/portal/download/). High expression of BRD4 was observed in AML cell lines. (b) According to the R2 database (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi, Bohlander-422-fRMA-u133a; source:GEO ID, gse37642), the overall survival rate of high BRD4 expression with AML is lower than that of patients with low BRD4 expression. (c)According to the GEPIA2 database (http://gepia2.cancer-pku.cn/#index), the overall survival rate of high BRD4 expression in patients with AML is lower than that of patients with low BRD4 expression.
MZ1 suppresses the viability of AML cells
The effect of a 48 h graded concentration MZ1-treatment on cell viability of NB4, Kasumi-1, MV4-11 and K562 cell lines was examined by CCK8 assay. The results found that the viability in all AML cell lines decreased by MZ1 in a dose-dependent manner (Figure 2a), and all cell lines were sensitive to MZ1 treatment (IC50 values: NB4: 0.279 μM; Kasumi-1: 0.074 μM; MV4-11: 0.110 μM and K562: 0.403 μM (Figure 2a).
Figure 2.
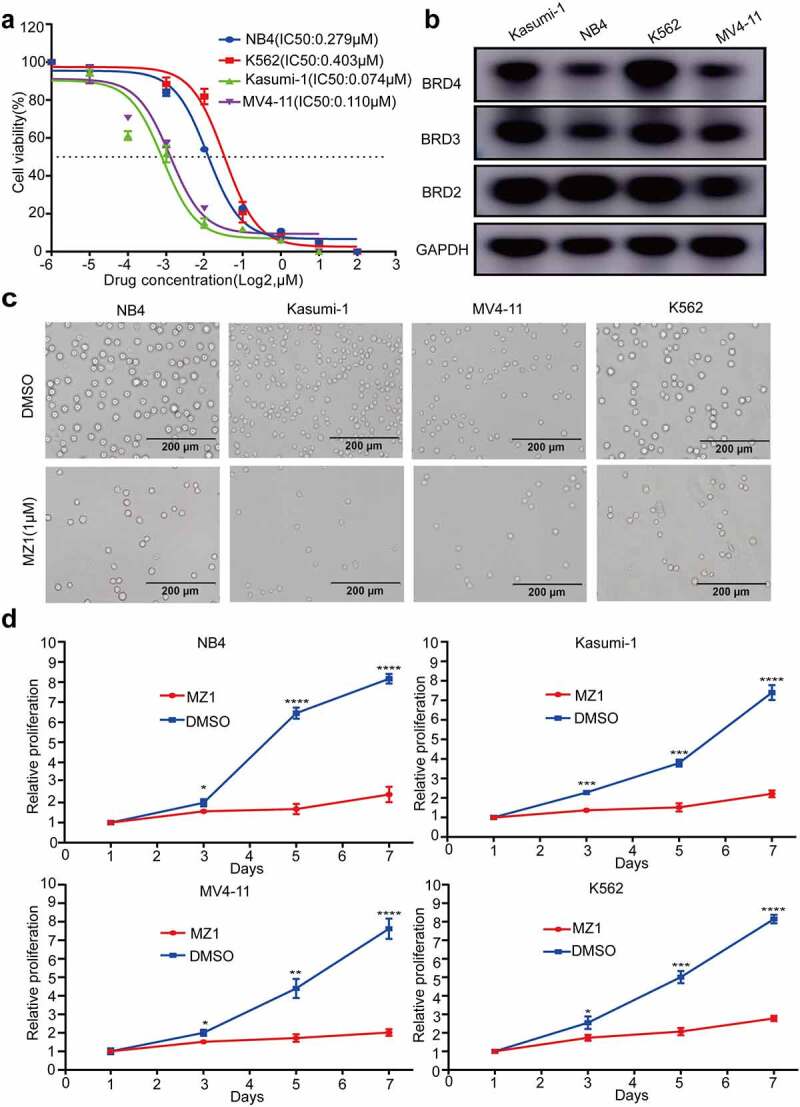
MZ1 suppresses the proliferation of AML cells. (a) Cell viability and IC50 values of NB4, Kasumi-1, MV4-11, and K562 cells after treatment with different concentrations of MZ1 for 48 h. (b) Basal BET protein levels of NB4, Kasumi-1, MV4-11, and K562 cells. (c) Morphology of NB4, Kasumi-1, MV4-11, and K562 cells treated with DMSO or MZ1. (d) Cell growth curves of NB4, Kasumi-1, MV4-11, K562 cells treated with DMSO or MZ1 for 7 days. Each concentration was tested in triplicate and independently performed at least three times. *p < .05, **p < .01, ***p < .001, ****p < .0001.
Meanwhile, all four AML cell lines are positive for BRD2, BRD3 and BRD4 expression (Figure 2b), indicating that BET proteins were also widely expressed. We further explored the cytotoxic effect of MZ1 on AML cell lines by examining the effect of 1 μM MZ1 and DMSO on the viability of the four AML cell lines and found that the number of cells under the white slice was significantly reduced in the MZ1 group (Figure 2c). Also, the viability decreased significantly in response to MZ1 in all four AML cell lines in a time-dependent manner (Figure 2d). These results indicate that MZ1 can inhibit the proliferation ability of AML cell lines.
MZ1 degrades BET protein expression in AML cell lines
We found that under the same conditions, MZ1 had lower IC50 values for antitumor activity and better suppression effect on AML cell proliferation compared with JQ1 and dBET1 (Figure 3a). The western blotting analysis revealed that BRD4 protein was almost completely degraded in four AML cell lines treated with MZ1 (Figure 3b). In addition, besides BRD4, MZ1 could simultaneously decrease the expression of both BRD2 and BRD3 proteins (Figure 3b). Also, AML cell lines treated with MZ1 resulted in the cleavage of PARP. These data suggest that MZ1 is an effective BET protein degrader which can downregulate BET protein expression in AML cell lines.
Figure 3.
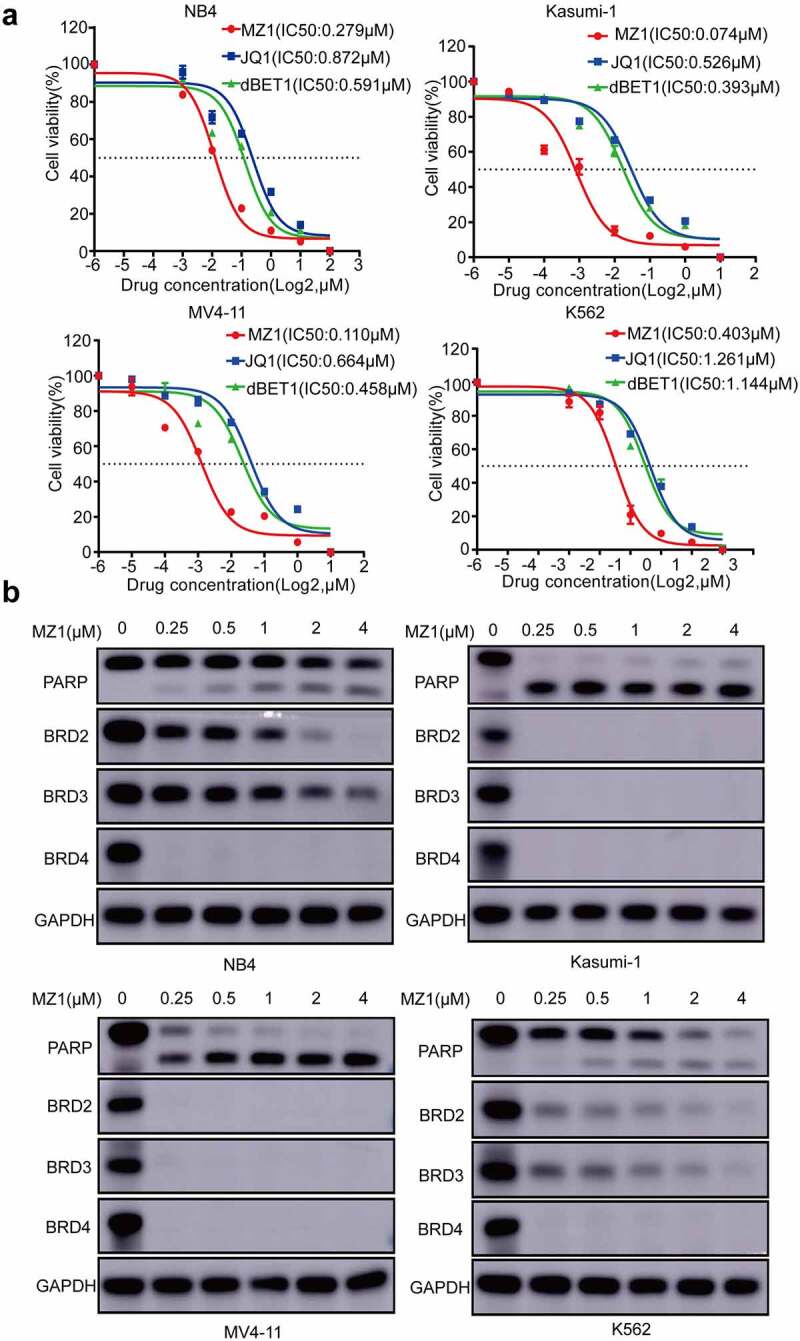
MZ1 clears BET protein expression in AML cell lines. (a) Cell viability of NB4, Kasumi-1, MV4-11 and K562 cell lines treated with different concentrations of MZ1, JQ1 and dBET1 for 48 h. (b) Western blot results showed that MZ1 induced degradation of BET protein and induced PARP cleavage in NB4, Kasumi-1, MV4-11 and K562 cells. Each concentration was tested in triplicate and independently performed at least three times.
MZ1 induces cell cycle block and apoptosis in AML cell lines
Previous studies have reported that the BET family is crucial for cell cycle regulation.28 We therefore examined whether MZ1 could regulate the cell cycle of AML cell lines. Cell cycle analysis by PI staining showed that the ratio of G1 phase cells increased in the MZ1 treatment group compared to the DMSO treated control group (Figure 4a), suggesting that MZ1 is capable of interfering with AML cell proliferation by inhibiting the cell cycle. Furthermore, apoptosis of AML cells was also observed after MZ1 treatment. The Annexin V/PI staining analysis demonstrated that MZ1 induced apoptosis of AML cells in a dose-dependence manner. (Figure 4b). Collectively, these results revealed that MZ1 could block cell cycle progression and induce apoptosis of AML cell lines.
Figure 4.
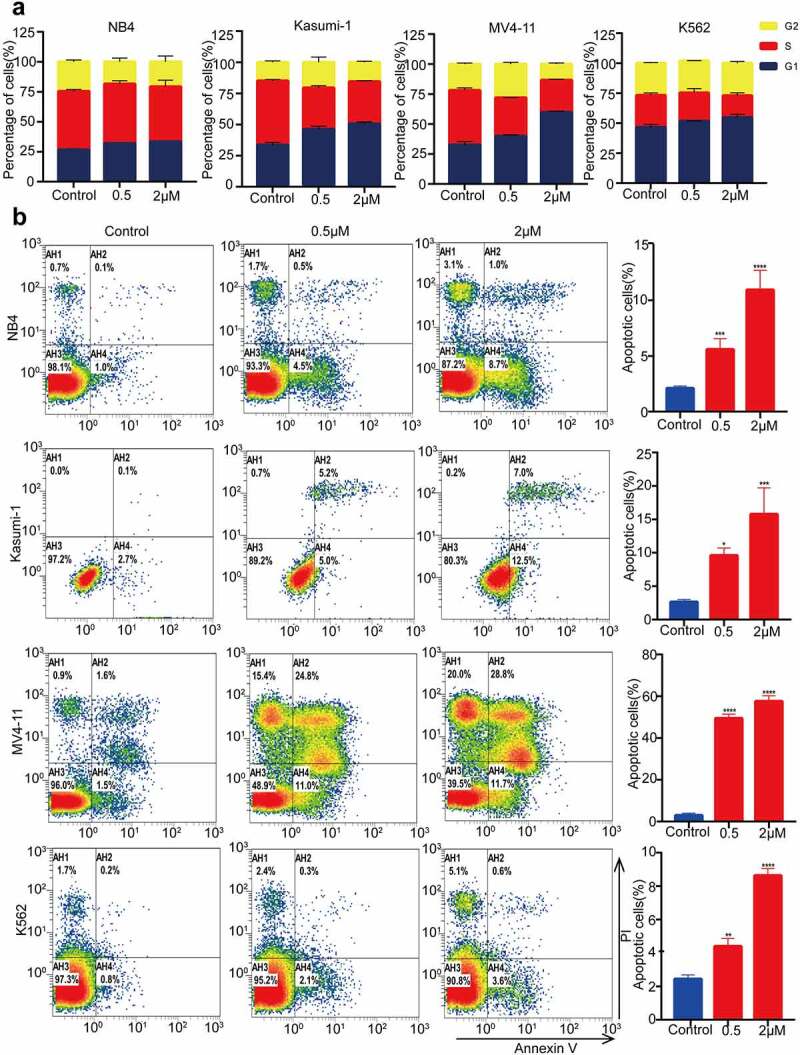
MZ1 blocks the AML cells cycle and promotes apoptosis. (a) PI-labeled cell cycle analysis after 12–24 h treatment of NB4, Kasumi-1, MV4-11 and K562 cell lines with DMSO or MZ1 revealed that AML cells were distributed in the G1/S phase and the cell population in the G1 phase was significantly increased. (b) A dose-dependent increase in apoptosis was observed in AML cell lines after 24 h of MZ1 treatment. Each concentration was tested in triplicate and independently performed at least three times. *p < .05, **p < .01, ***p < .001, ****p < .0001.
VHL is a powerful assistant in MZ1 treatment
By western blotting, we observed that the expression levels of VHL were higher in NB4 and Kasumi-1 cells and lower in MV4-11 and K562 cells (Figure 5a). Also, since MZ1 consists of JQ1, E3 ubiquitin ligase ligand-VHL and PEG3 (Figure 5b), we investigated the relationship between VHL and MZ1 sensitivity to confirm whether VHL mediate the MZ1 induced degradation of BET family protein. The VHL-overexpression and VHL-knockdown vectors were successfully transfected into NB4 and Kasumi-1 cells respectively, and the expression of VHL in transfected cells was detected using Western blotting (Figure 5c). The CCK8 assay showed that VHL-knockdown increased the IC50 of MZ1 in NB4 and Kasumi-1 cells compared with the empty vector, and VHL overexpression significantly decreased the IC50 of MZ1 in these cells (Figure 5d). These data suggest that VHL is essential for MZ1 to enhance its anticancer effects in AML cell lines.
Figure 5.
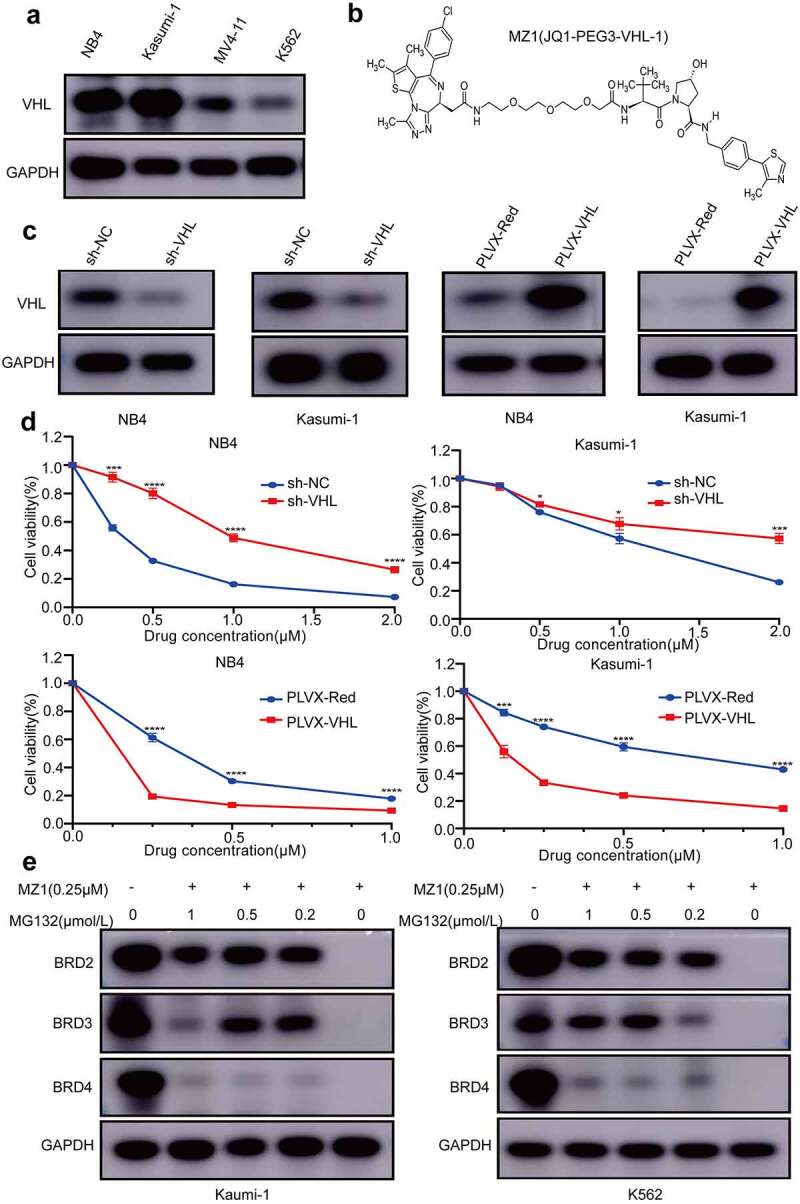
VHL is a powerful helper for MZ1 in AML cell lines; BRD4 protein degradation is dependent on the ubiquitin-proteasome system. (a) Western blot analysis of VHL protein expression in AML cell lines. (b) Schematic illustration of the bifunctional PROTAC molecule (MZ1). (c) sh-VHL or PLVX-VHL lentivirus was transfected into NB4 and Kasumi-1 cells, and the expression level of VHL was detected by Western blot. (d) Comparison of the sensitivity of VHL knockdown or overexpressed NB4 and Kasumi-1 cells. VHL downregulation increased the IC50 of MZ1 in NB4 and Kasumi-1 cells; VHL overexpression decreased the IC50 of MZ1 in NB4 and Kasumi-1 cells. (e) After MZ1 treatment (0.25μΜ) of Kasumi-1 and K562 cells and different concentrations of MG132 for 12 h, Western blot showed that MG132 inhibited BRD2, BRD3 and BRD4 protein degradation in a dose-dependent manner. Each concentration was tested in triplicate and independently performed at least three times. *p < .05, **p < .01, ***p < .001, ****p < .0001.
It is known that MG132 is widely used for the inhibition of proteasome activity. In Figure 2d, we measured the IC50 values of four AML cell lines to MZ1 treatment by CCK-8 assay. For comparison, we selected K562 with the highest and Kasumi-1 with the lowest IC50 values for the following MG132 experiments. We treated Kasumi-1 and K562 cells with 0.25μΜ MZ1 and different concentrations of MG132 for 12 h to identify the role of the proteasome in MZ1-induced BET family proteins degradation. Western blot was performed to detect BRD2, BRD3 and BRD4 protein expression (Figure 5e). These results revealed that MG132 adequately inhibited the degradation of BRD4 family proteins in a dose-dependent manner.
MZ1 has potent anticancer effects in AML mice models
To further investigate the in vivo efficacy and safety of MZ1, we established a luciferase-labeled AML mice model by tail vein injection of P388-D1 cells (Figure 6a). Either 12.5 mg/kg of MZ1 or equivalent vehicle were given intraperitoneally every day and fluorescence imaging tests were performed regularly (Figure 6b). On the other hand, as determined by fluorescence imaging of liver and spleen, MZ1 treatment group showed a significantly reduction of tumor burden rather than the control group (Figure 6c). The histogram of tumor luminous flux showed that the MZ1 group was much lower than the control group (Figure 6d). These results showed that the leukemia luminescence signal in mice treated with MZ1 was significantly reduced, while that in the control group was sharply increased. It is proved that MZ1 could prolong the survival time of mice when compared with the control group (Figure 6e). No statistically significant difference in overall body weight was observed between the treatment and control groups (Figure 6f). Also, the MZ1 treatment group showed a significant reduction in the weight of the liver and spleen (Figure 6g). The H&E staining analysis of the mice’s liver, spleen and bone marrow showed that tumor cells were significantly reduced in the treated group compared with the control group (Figure 6h). Furthermore, the Immunohistochemical (IHC) analysis showed that MZ1 treatment downregulated the BDR4 expression in mice, which was consistent with the results from the in vitro experiments. Also, the amount of Ki-67 positive cells was significantly decreased in tumors from the MZ1-treated mice, while the proportion of cleaved-caspase 3 positive cells was increased, indicating the dysregulation in proliferation and apoptosis (Figure 6i).
Figure 6.
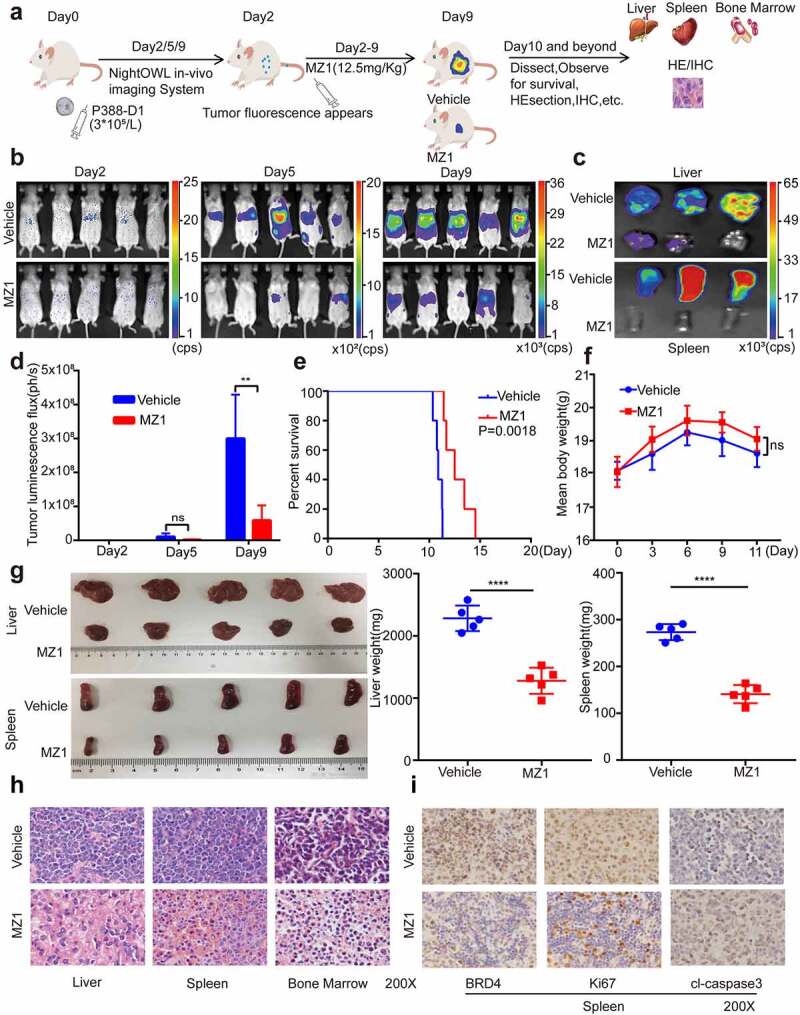
MZ1 suppresses AML cells growth in vivo. (a) Schematic diagram of in vivo experimental design. (b) Representative bioluminescence imaging of mice at different time points in the MZ1 treatment group and the vehicle control group. (c) Bioluminescence imaging of the endpoint liver and spleen in both groups of mice. The color scale indicates the bioluminescence intensity of the counts. (d) Statistical analysis of the bioluminescence imaging values of the two groups of mice at different time points. (e) Survival curves of the two groups of mice. (f) Monitoring of body weight of the two groups of mice. (g) Endpoint liver and spleen size and weight of the two groups of mice. (h) H&E staining analysis of liver, spleen and bone marrow in two groups of mice. (i) Immunohistochemical analysis of spleen tissue sections for BRD4, Ki67 and cleaved-caspase 3 (cl-caspase 3) in the two groups. NS: not significant, **p < .01, **** p < .0001.
MZ1 reduces the expression of c-Myc and ANP32B in AML cells
To further investigate the mechanism of action of MZ1 in AML cell lines, we treated the NB4 cells with 2 μM MZ1 for 32 h and then performed RNA-seq studies. We found that 8075 genes were differentially expressed in the MZ1 group against the DMSO control group including 3577 upregulated and 4498 downregulated genes. (Figure 7a). The heatmap showed the top downregulated genes in the MZ1 group (Figure 7b). We also identified super-enhancer associated genes in NB4 cell line (Figure 7c). When enriching the RNA-seq downregulated genes with super-enhancer associated genes in the NB4 cell line, we found that MZ1 significantly downregulated the expression of super-enhancer associated gene ANP32B in NB4 cell line (Figure 7d). The GSEA plots also revealed gene enrichment in HALLMARK_APOPTOSIS, HALLMARK_P53 and HALLMARK_TGF_BETA signaling pathway in RNA-Seq after MZ1 treatment in the NB4 cells (Figure 7e). As c-Myc and ANP32B are important downstream targets of BRD4 inhibitor, western blot was used to detected the expression of c-Myc and ANP32B in AML cell lines (Figure 7f). And as expected, after MZ1 treatment, the protein levels of c-Myc and ANP32B in NB4 and Kasumi-1 cells decreased significantly (Figure 7g), and the cell proliferation was inhibited (Figure S1). These results suggest that MZ1 interferes with BRD4-mediated transcription of c-Myc and ANP32B, resulting in a decrease in c-Myc and ANP32B protein levels.
Figure 7.
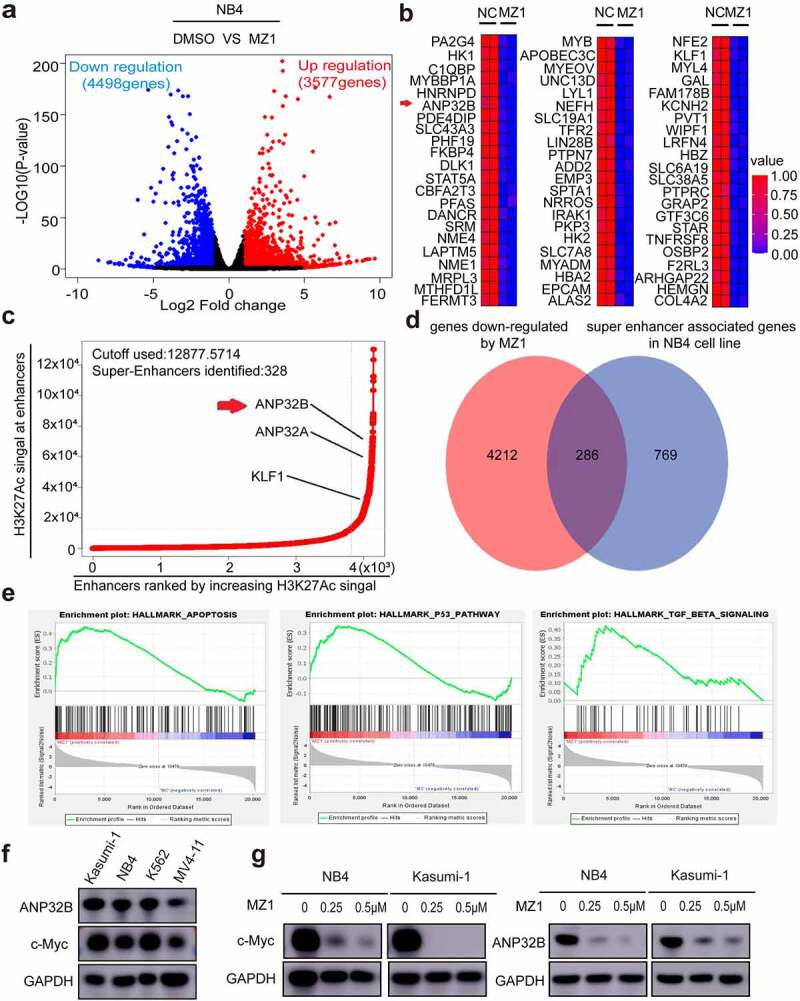
MZ1 decreases c-Myc and ANP32B expression in AML cell lines. (a) RNA-sequencing volcano plot analysis illustrates the expression changes of genes in NB4 cells between the MZ1 treatment and the control groups. Genes highlighted in red were upregulated, and those in blue were downregulated. (b) Heatmap view displayed the top downregulated genes in NB4 cells treated with 2 μM MZ1 for 32 h, including ANP32B. (c) The NB4 cell line chip-seq obtained 328 super enhancers, including super-enhancer associated gene ANP32B, ANP32A and KLF1. (d) After MZ1-treated NB4 cell down-regulated genes and NB4 cell chip-seq obtained super-enhancer associated genes were enriched, 286 common genes were obtained, including ANP32B. (e) GSEA plots displayed gene enrichment in HALLMARK_APOPTOSIS, HALLMARK_P53 and HALLMARK_TGF_BETA signaling pathways in NB4 cells treated with MZ1. (f) Western blot assay for c-Myc and ANP32B expression in the four AML cell lines. (g) Western blot analysis showed that the c-Myc and ANP32B protein levels were downregulated in NB4 and Kasumi-1 cells after treatment with different concentrations of MZ1.
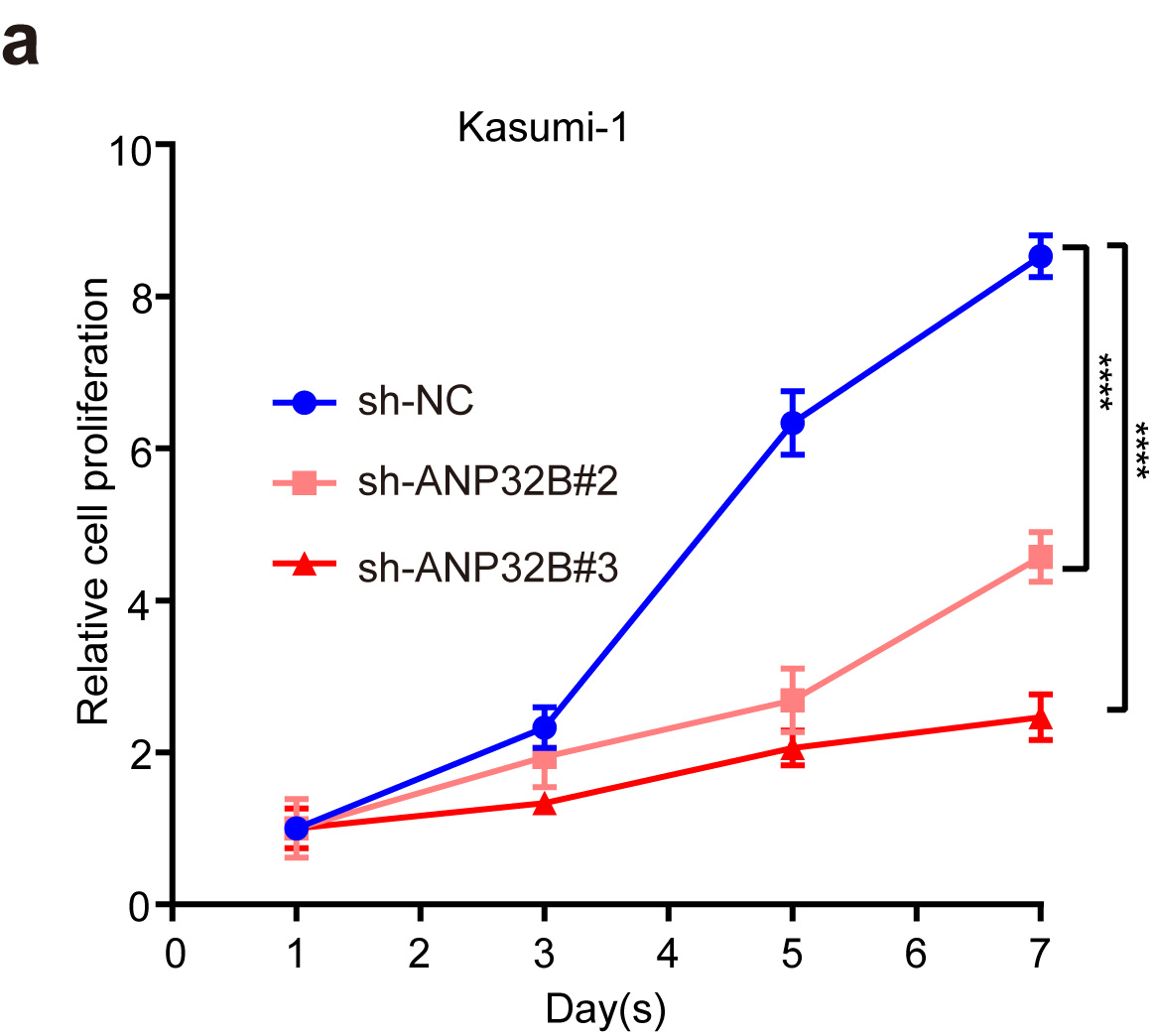
Downregulation of the ANP32B gene in AML cell lines promotes cell apoptosis and inhibits cell proliferation
After we treated the NB4 cells with 2 μM of MZ1, RT-PCR verified that the ANP32B gene was more significantly downregulated among the downregulated genes (Figure 8a). ANP32B expression was analyzed by R2 database (http://gepia.cancer-pku.cn/index.html, Figure 8b). The expected overall survival rate of low ANP32B expression with AML is high than that of patients with high ANP32B expression. ANP32B was knocked down by the sh-RNAs (sh-ANP32B#2 and sh-ANP32B#3) (Figure 8c) to test the potential role of ANP32B in NB4 cells. Both white slice and CCK8 assays showed that ANP32B knockdown significantly inhibited NB4 cell proliferation compared to scramble cells (Figures 8d, e). Also, ANP32B knockdown increased apoptosis in NB4 cells compared with scramble cells (Figure 8f). These data suggest that ANP32B knockdown promoted apoptosis and inhibited the proliferation of AML cell lines and low expression of ANP32B was correlated with the better survival of AML patients.
Figure 8.
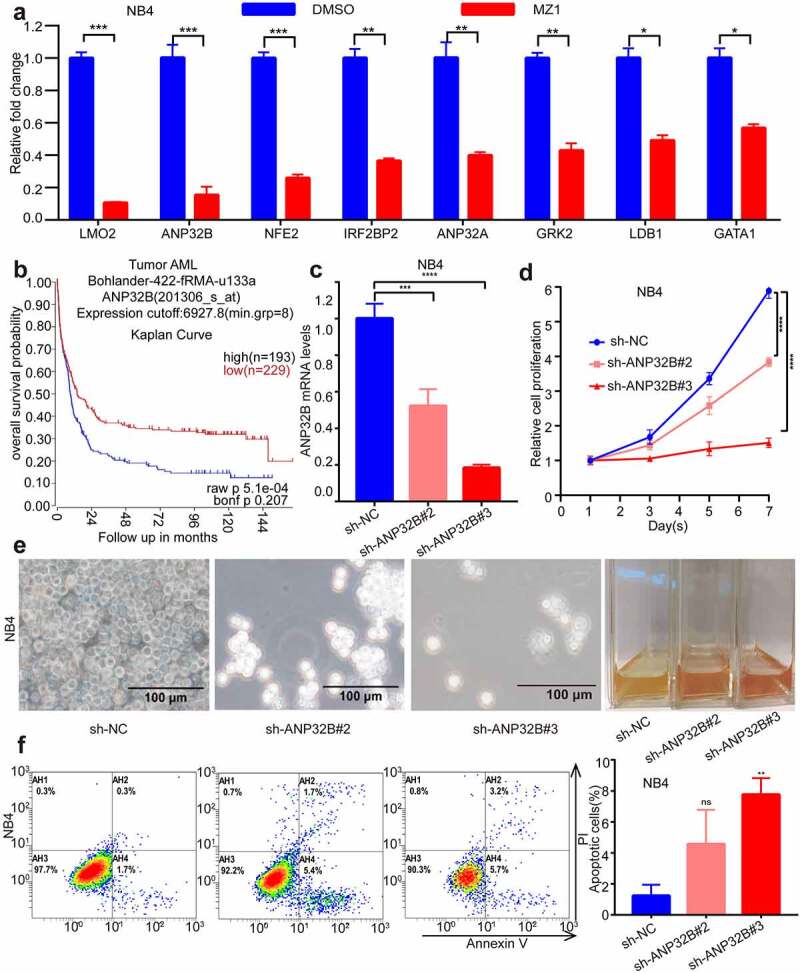
ANP32B knockdown promotes apoptosis of the NB4 cell line and inhibits its proliferation. (a) RT-PCR verified that ANP32B was significantly more downregulated among the downregulated genes after treatment of NB4 with DMSO or MZ1 (2 μM). (b) Patients with high ANP32B expression had lower survival rates than those with low expression. (generated from the web site: https://hgserver1.amc.nl/cgi-bin/r2/main.cgi, Bohlander-422-fRMA-u133a; source:GEO ID, gse37642).(c) RT-PCR verified the ANP32B knockdown by sh-RNAs (sh-ANP32B#2 and sh-ANP32B#3). (d, e) White slice and CCK8 assays showed that ANP32B knockdown significantly inhibited the proliferation of NB4 cells compared to scramble cells. (f) Compared to the scramble cells, apoptosis was increased in NB4 cell after ANP32B knockdown.
Discussion
Acute myeloid leukemia is a rare but fatal hematologic malignancy and represents approximately 20% of all pediatric acute leukemia cases.29 It comprises a heterogeneous group of hematologic tumors with different biological and clinical implications, characterized by a clonal proliferation of myeloid progenitor cells or granulocyte precursor cells that fail to differentiate normally.30 For decades, there has been little substantial progress in the treatment of AML patients, and despite the recent regulatory approval of some new drugs for AML, their prognosis remains unsatisfactory, with more than half of patients relapsing and eventually dying from the disease.3 Therefore, it is of utmost importance to deeply study the mechanisms of occurrence and development of AML, actively seek molecular targets, and search for novel targeted drugs.
The BRD4 is involved in the cell cycle, cell proliferation, immune response and other physiological functions in the human body through the regulation of cellular transcription.31 Many studies have shown that BRD4 is overexpressed in many different tumors, such as melanoma, ovarian cancer, colon cancer, liver cancer, and hematologic malignancies.8,9,32–34 We compared the expression of BRD4 in different tumor cell lines using CCLE database. The results showed that BRD4 was highly expressed in patients with AML, ALL and CML, which signify strong dependence of these cancer cell lines on high expression of BRD4. It is also reported that samples of AML patients showed significantly increased BRD4 expression compared to healthy samples.35 Notably, BRD4 are considered to be a poor prognostic marker for AML according to the survival analysis. It is also reported that BRD4 activates key tumor driver genes, such as c-Myc and BCL-2, thereby promoting the proliferation of leukemia and solid tumors,34,36 and suggesting its potential as an important therapeutic target providing to improve outcomes for AML patients.
Currently available BET inhibitors, such as JQ1 is reported to be effective in AML,34,37,38 however, the short half-life of JQ1 and the development of drug resistance with continued dosing significantly limit its clinical application prospects. To address this problem, PROTAC technology has emerged as an effective tool for endogenous protein degradation. These are bifunctional PROTAC molecules consisting of ligands for proteins of interest (mainly small molecule inhibitors) and covalently linked ligands for E3 ubiquitin ligases (E3), which can suppress tumor growth through ubiquitinate target proteins via the ubiquitin-proteasome system (UPS).15,17 MZ1 is one such novel PROTAC-based BRD4 degraders examined in this study. Our results showed that MZ1 can achieve the desired therapeutic effect at a lower concentration with significantly lower toxicity compared to JQ1 and dBET1 (other PROTAC BRD4 inhibitor). MZ1 can inhibit cell proliferation, promote cell apoptosis, caused cell cycle arrest at G1, and effectively inhibit the growth of AML cell lines both in vivo and in vitro.
It has been reported that MZ1 has toxic effects on MV4-11 and HL-60 cells, but its potential molecular mechanism is not clear.21 In our study, we detected the therapeutic response of MZ1 in more AML cell lines and found the mechanism of MZ1. Our study confirmed that levels of BRD4, along with BRD2 and BRD3 were decreased after MZ1 treatment. Similar findings have been observed in other studies using MZ1 treatment.14,20 The most probable explanation for this involves the highly homologous structural domains of the BET family members.39 MZ1 is formed by linking JQ1 and VH-032, and as an E3 ubiquitin ligase, VHL can recruit target proteins and effectively increase target proteins degradation through the ubiquitin-proteasome pathway. In this study, we observed that VHL expression was essential in the sensitivity of AML cell lines to MZ1: knockdown of VHL decreased the inhibition of MZ1 on AML cell lines, and VHL overexpression increased the inhibition of MZ1 on AML cell lines. Moreover, MG132 blocked the proteasome and significantly inhibited the MZ1-induced degradation of BRDs. These findings were in accordance with previous studies suggesting that the antitumor effect of MZ1 is closely related to VHL.19
In this study, RNA-seq and Western blotting analysis showed how MZ1 influenced c-Myc and ANP32B expression at both mRNA and protein levels in AML cell lines by inhibiting BRD4 expression. The transcription factor c-Myc is essential in normal non-transformed cells and regulates cell proliferation, metabolism and survival.40 Additionally, the oncogene function of c-Myc leads to excessive cell proliferation, cell cycle progression and metastasis,41 thus, dysregulation of c-Myc promotes the development of multiple cancers. Accordingly, inhibition of c-Myc expression has been considered as a therapeutic option for malignancies. The RNA-seq results in this study showed that both c-Myc and ANP32B were downregulated after MZ1 treatment of NB4 cells. c-Myc is an important downstream target gene of BRD4 inhibitor.42 The correlation between the occurrence and development of AML and c-Myc has been reported.43 Our result confirmed the decrease of c-Myc expression after MZ1 treatment. Therefore, we speculate that c-Myc is likely to participate in the treatment of AML patients by MZ1. C. Otto et al. also reported that MZ1 suppressed colorectal cancer through c-Myc pathway genes, which is consistent with findings regarding AML. ANP32B is another important target gene, which is a highly conserved acidic leucine-rich nuclear member of the acidic leucine-rich nuclear phosphoprotein 32 kDa (ANP32) family. Our studies found that knockdown of ANP32B can significantly inhibit the proliferation and promote apoptosis of NB4 cells, but the mechanism is not clear. ANP32 family proteins are mainly involved in the regulation of gene transcription. Previous studies have found that ANP32 can affect cell apoptosis by regulating the activity of protein phosphatase in cancers.44 ANP32B is highly expressed in human breast cancer and ANP32B affects the proliferation of breast cancer cells in vivo and in vitro by regulating AKT phosphorylation.45 The mechanism through which ANP32 functions in AML needs further research. ANP32 members have multiple physiological functions, including caspase regulation, chromatin modification and remodeling, protein phosphatase inhibition, and intracellular transport regulation.46 Our study is the first report essentially describing that MZ1 inhibits AML by regulating ANP32B expression.
Lastly, MZ1 significantly inhibited the growth of AML cell lines, while there was no statistically significant difference of body weight between the MZ1-treated and control groups in the AML mouse model. These results suggested that MZ1 has good efficacy and safety in controlling the progress of AML.
Conclusions
In summary, the PROTAC BET inhibitor MZ1 has potential anticancer activity against AML both in vitro and in vivo. MZ1 acts by degrading BET proteins, effectively inhibiting the growth of AML cell lines, promoting their apoptosis, and downregulating the transcription of c-MYC and ANP32B. These findings suggest that MZ1 should be considered as a promising therapeutic strategy for AML.
Supplementary Material
{kind=link}
Acknowledgments
We would like to thank the department of Hematology, Children’s Hospital of Soochow University and Institute of Pediatric Research, Children’s Hospital of Soochow University for the support in this study.
Biographies
Li Ma, associate chief physician of Pediatrics, The Affiliated Huaian NO.1 People’s Hospital of Nanjing Medical University, major pediatric hematology and oncology. Currently, she is studying for doctorate in the Children’s Hospital of Soochow University.
Jianwei Wang, research assistant of Institute of Pediatric Research, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Yongping Zhang, Ph.D., Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Fang Fang, Associate Research Fellow, Institute of Pediatric Research, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Jing Ling, associate chief physician, Department of Hematology, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Xinran Chu, resident physician, Department of Hematology, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Zimu Zhang, research assistant of Institute of Pediatric Research, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Yanfang Tao, associate chief physician, Department of Hematology, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Xiaolu Li, research assistant of Institute of Pediatric Research, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Yuanyuan Tian, research assistant of Institute of Pediatric Research, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Zhiheng Li, research assistant of Institute of Pediatric Research, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Xu Sang, Ph.D., Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Kunlong Zhang, Ph.D., Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Lihui Lu, Ph.D., Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Xiaomei Wan, Ph.D., Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Yanling Chen, Ph.D., Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Juanjuan Yu, Ph.D., Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Ran Zhuo, Ph.D., Children’s Hospital of Soochow University, major pediatric oncology.
Shuiyan Wu, associate chief physician, Department of Intensive Care Unit, Children’s Hospital of Soochow University, major pediatric hematology and severe disease.
Jun Lu, professor of Hematology, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Jian Pan, professor of Institute of Pediatric Research, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Shaoyan Hu, professor of Hematology, Children’s Hospital of Soochow University, major pediatric hematology and oncology.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was supported by grants from the National Natural Science Foundation (81971867, 81970163, 81902534, 81902027, 82072767, 52003183, 82141110, 82171797); Natural Science Foundation of Jiangsu Province (SBK2019021442, BK20190185, BK20190186, BK20191175); the Universities Natural Science Foundation of Jiangsu Province (No.16KJB310014); Jiangsu province’s science and technology support program (Social Development) project (BE2021657, BE2021654); Jiangsu Province Key R&D Program (Social Development) Projects (BE2020659); Department of Pediatrics Clinical Center of Suzhou (Szzx201504); Gusu Health Talents program of Soochow city (2020-104); the Applied Foundational Research of Medical and Health Care of Suzhou City (SYS2019080, SYS2019082, SYS2019077, SYS2020150, SYS2020151, GSWS2020039, GSWS2021028); the Science and Technology Development Project of Suzhou City (SKJY2021111, SKJY2021112); the Science and Technology Project of Soochow(SS2019011). The Science and Technology Project of Soochow (SS2019064; National Natural Science Foundation of China National Natural Science Foundation of China [81971867].
Ethical approval
This study was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki). The Animal Care Committee of Suzhou College approved all animal studies (approval number: CAM-SU-AP#: JP-2018-1).
Disclosure statement
The authors have declared that no conflict of interest exists.
Author contributions
Jian Pan and Shaoyan Hu designed and directed the study; Li Ma performed most of the experiments, analyzed the data, and wrote the paper; Jianwei Wang and Fang Fang helped with statistical analysis and guided the writing of paper; Yongping Zhang performed part of the experiments; Jing Ling, Xinran Chu, Yan-Fang Tao and Jun Lu helped with statistical analysis; Xiaolu Li, Yuanyuan Tian and Zhiheng Li helped with the apoptosis and cell cycle analysis; Xu Sang, Kunlong Zhang, Lihui Lu and Xiaomei Wan participated in western blotting; CCK8 assay and PCR; Yongping Zhang and Xu Sang participated in vitro experiments; Yanling Chen supported the design of primers for real-time PCR; Zimu Zhang and Juanjuan Yu participated in plasmid construction; Ran Zhuo and Shuiyan Wu performed lentivirus preparation and transfection. All authors read and approved the final manuscript.
Data Availability Statement
The data that support the findings of this study are available on reasonable request from the corresponding author. RNA‑seq and original data have been submitted to the GEO database with Accession Number GSE198011 https://www.ncbi.nlm.nih.gov/geo.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384047.2022.2125748
References
- 1.Creutzig U, Zimmermann M, Bourquin JP, Dworzak MN, Fleischhack G, Graf N, Klingebiel T, Kremens B, Lehrnbecher T, von Neuhoff C, et al. Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: results from Study AML-BFM 2004. Blood. 2013;122(1):37–43. DOI: 10.1182/blood-2013-02-484097. [DOI] [PubMed] [Google Scholar]
- 2.Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. DOI: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aplenc R, Meshinchi S, Sung L, Alonzo T, Choi J, Fisher B, Gerbing R, Hirsch B, Horton T, Kahwash S, et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: a report from the children’s oncology group. Haematologica. 2020;105(7):1879–1886. DOI: 10.3324/haematol.2019.220962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasche M, Zimmermann M, Borschel L, Bourquin JP, Dworzak M, Klingebiel T, Lehrnbecher T, Creutzig U, Klusmann JH, Reinhardt D.. Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia. 2018;32(10):2167–2177. DOI: 10.1038/s41375-018-0071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lange BJ, Smith FO, Feusner J, Barnard DR, Dinndorf P, Feig S, Heerema NA, Arndt C, Arceci RJ, Seibel N, et al. Outcomes in CCG-2961, a children’s oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children’s oncology group. Blood. 2008;111(3):1044–1053. DOI: 10.1182/blood-2007-04-084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, Montesinos P, Baer MR, Larson RA, Ustun C, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728–1740. DOI: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 7.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–736. DOI: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, Gonzalez-Gomez P, Morante M, Jubierre L, Zhang W, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73(20):6264–6276. DOI: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goundiam O, Gestraud P, Popova T, De la Motte Rouge T, Fourchotte V, Gentien D, Hupe P, Becette V, Houdayer C, Roman-Roman S, et al. Histo-genomic stratification reveals the frequent amplification/overexpression of CCNE1 and BRD4 genes in non-BRCAness high grade ovarian carcinoma. Int J Cancer. 2015;137(8):1890–1900. DOI: 10.1002/ijc.29568. [DOI] [PubMed] [Google Scholar]
- 10.Ferri E, Petosa C, McKenna CE, McKenna CE. Bromodomains:Structure,function and pharmacology of inhibition. Biochem Pharmacol. 2016;106:1–18. DOI: 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Chen C, Xu L, Gao R, Wang S, Zhang Y, Wang C, Zeng C, Li Y. Transcriptome-based co-expression of BRD4 and PD-1/PD-L1 predicts poor overall survival in patients with acute myeloid leukemia. Front Pharmacol. 2021;11. DOI: 10.3389/fphar.2020.582955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang G, Deng W, Liu Y, Wang C. General mechanism of JQ1 in inhibiting various types of cancer. Mol Med Rep. 2020;21(3):1021–1034. DOI: 10.3892/mmr.2020.10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Q, Ding X, Huang T, Zhang S, Li Y, Feng Z, Wang L, Zou X, Wang Y, Feng Z. BRD4 degrader ARV-825 produces long-lasting loss of BRD4 protein and exhibits potent efficacy against cholangiocarcinoma cells. Am J Transl Res. 2019;11(9):5728–5739. [PMC free article] [PubMed] [Google Scholar]
- 14.Lim SL, Damnernsawad A, Shyamsunder P, Chng WJ, Han BC, Xu L, Pan J, Pravin DP, Alkan S, Tyner JW, et al. Proteolysis targeting chimeric molecules as therapy for multiple myeloma: efficacy, biomarker and drug combinations. Haematologica. 2019;104(6):1209–1220. DOI: 10.3324/haematol.2018.201483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J Hematol Oncol. 2020;13(1):50. DOI: 10.1186/s13045-020-00885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potjewyd F, Turner AW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, Margolis DM, Pearce KH, Herring LE, James LI. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem Biol. 2020;27(1):47–56 e15. DOI: 10.1016/j.chembiol.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.S-M Q, Dong J, Z-Y X, Cheng X-D, Zhang W-D, Qin -J-J. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front Pharmacol. 2021;12:692574. DOI: 10.3389/fphar.2021.692574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Testa A, Lucas X, Castro GV, Chan K-H, Wright JE, Runcie AC, Gadd MS, Harrison WTA, E-J K, Fletcher D, et al. 3-Fluoro-4-hydroxyprolines: synthesis, conformational analysis, and stereoselective recognition by the VHL E3 ubiquitin ligase for targeted protein degradation. J Am Chem Soc. 2018;140(29):9299–9313. DOI: 10.1021/jacs.8b05807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otto C, Schmidt S, Kastner C, Denk S, Kettler J, Müller N, Germer CT, Wolf E, Gallant P, Wiegering A. Targeting bromodomain-containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia. 2019;21(11):1110–1120. DOI: 10.1016/j.neo.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noblejas-López M, Nieto-Jimenez C, Burgos M, Gómez-Juárez M, Montero JC, Esparís-Ogando A, Pandiella A, Galán-Moya EM, Ocaña A. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. 2019;38(1):383. DOI: 10.1186/s13046-019-1387-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan K-H, Zengerle M, Testa A, Ciulli A. Impact of target warhead and linkage vector on inducing protein degradation: comparison of Bromodomain and Extra-Terminal (BET) degraders derived from Triazolodiazepine (JQ1) and tetrahydroquinoline (I-BET726) BET inhibitor scaffolds. J Med Chem. 2017;61(2):504–513. DOI: 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, Yang C, Li X, Du X, Tao Y, Ren J, Fang F, Xie Y, Li M, Qian G, et al. The dual role of BI 2536, a small-molecule inhibitor that targets PLK1, in induction of apoptosis and attenuation of autophagy in neuroblastoma cells. J Cancer. 2020;11(11):3274–3287. DOI: 10.7150/jca.33110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. DOI: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. DOI: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. DOI: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. DOI: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. DOI: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devaiah BN, Singer DS. Two faces of brd4: mitotic bookmark and transcriptional lynchpin. Transcription. 2013;4(1):13–17. DOI: 10.4161/trans.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.BC V, A G, C A. Actual biological diagnosis of acute myeloblastic leukemia in children. J Med Life. 2014;7(2):291–295. [PMC free article] [PubMed] [Google Scholar]
- 30.Newell LF, Cook RJ. Advances in acute myeloid leukemia. BMJ. 2021;375:n2026. DOI: 10.1136/bmj.n2026. [DOI] [PubMed] [Google Scholar]
- 31.Borck PC, Guo LW, Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res. 2020;126(9):1190–1208. DOI: 10.1161/CIRCRESAHA.120.315929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Y, Zhou J, Ye F, Xiong H, Peng L, Zheng Z, Xu F, Cui M, Wei C, Wang X, et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int J Mol Sci. 2015;16(1):1928–1948. DOI: 10.3390/ijms16011928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.G-Q L, Guo W-Z, Zhang Y, X-X M, Zhang G, Li J, Yan B, Wang D, Zhang S-J, Tang H-W. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget. 2016;7(3):2462–2474. DOI: 10.18632/oncotarget.6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang S, Zhao Y, Heaster TM, Fischer MA, Stengel KR, Zhou X, Ramsey H, Zhou -M-M, Savona MR, Skala MC, et al. BET inhibitors reduce cell size and induce reversible cell cycle arrest in AML. J Cell Biochem. 2018. DOI: 10.1002/jcb.28005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sang X, Zhang Y, Fang F, Gao L, Tao Y, Li X, Zhang Z, Wang J, Tian Y, Li Z, et al. BRD4 inhibitor GNE-987 exerts anticancer effects by targeting super-enhancer-related gene LYL1 in acute myeloid leukemia. J Immunology Res. 2022;2022:1–18. DOI: 10.1155/2022/7912484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. 2017;28(8):1776–1787. DOI: 10.1093/annonc/mdx157. [DOI] [PubMed] [Google Scholar]
- 37.Dong X, Hu X, Chen J, Hu D, Chen LF. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell Death Dis. 2018;9(2):203. DOI: 10.1038/s41419-017-0181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou S, Zhang S, Wang L, Huang S, Yuan Y, Yang J, Wang H, Li X, Wang P, Zhou L, et al. BET protein inhibitor JQ1 downregulates chromatin accessibility and suppresses metastasis of gastric cancer via inactivating RUNX2/NID1 signaling. Oncogenesis. 2020;9(3):33. DOI: 10.1038/s41389-020-0218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang CY, Qin C, Bai L, Wang S. Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins - A review. Drug Discov Today Technol. 2019;31:43–51. DOI: 10.1016/j.ddtec.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. DOI: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22(20):2755–2766. DOI: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pang Y, Bai G, Zhao J, Wei X, Li R, Li J, Hu S, Peng L, Liu P, Mao H. The BRD4 inhibitor JQ1 suppresses tumor growth by reducing c-Myc expression in endometrial cancer. J Transl Med. 2022;20(1). DOI: 10.1186/s12967-022-03545-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goswami S, Mani R, Nunes J, Chiang C-L, Zapolnik K, Hu E, Frissora F, Mo X, Walker LA, Yan P, et al. PP2A is a therapeutically targetable driver of cell fate decisions via a c-Myc/p21 axis in human and murine acute myeloid leukemia. Blood. 2022;139(9):1340–1358. DOI: 10.1182/blood.2020010344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh Y-H, Ohno Y, Koizumi M, Nakayama H, Watanabe T, Hirooka M, Tokumoto Y, Kuroda T, Abe M, Fukuda S, et al. Downregulation of ANP32B exerts anti-apoptotic effects in hepatocellular carcinoma. Plos One. 2017;12(5). DOI: 10.1371/journal.pone.0177343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang S, Zhou L, Reilly PT, Shen SM, He P, Zhu XN, Li CX, Wang LS, Mak TW, Chen GQ, et al. ANP32B deficiency impairs proliferation and suppresses tumor progression by regulating AKT phosphorylation. Cell Death Dis. 2016;7(2):e2082. DOI: 10.1038/cddis.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reilly PT, Yu Y, Hamiche A, Wang L. Cracking the ANP32 whips: important functions, unequal requirement, and hints at disease implications. Bioessays. 2014;36(11):1062–1071. DOI: 10.1002/bies.201400058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on reasonable request from the corresponding author. RNA‑seq and original data have been submitted to the GEO database with Accession Number GSE198011 https://www.ncbi.nlm.nih.gov/geo.