

Abstract
We disclose a successive π‐expansion of pyracylene towards boat‐shaped polycyclic scaffolds. The unique structural features of the resulting compounds were revealed by X‐ray crystallographic analysis. Depending on the extent of π‐expansion the compounds display intense bathochromically shifted absorption bands in their UV/Vis spectra and are prone to several redox events as documented by cyclic voltammetry. The experimental observations are in line with the computational studies based on density functional theory, suggesting progressive narrowing of the HOMO–LUMO gap and distinct evolution of the electronic structure and aromaticity.
Keywords: π-expansion, curved PAH, electron acceptor, odd-membered, pyracylene
Curve the flat! A reliable synthetic strategy for the π‐expansion of pyracylene towards boat‐shaped polycyclic scaffolds was established. The evolution of the structural, optoelectronic and redox properties in relation to the progressive π‐expansion was comprehensively studied both in experiment and theory. The results identify the compounds as promising chromophores with multi‐stage amphoteric redox properties.
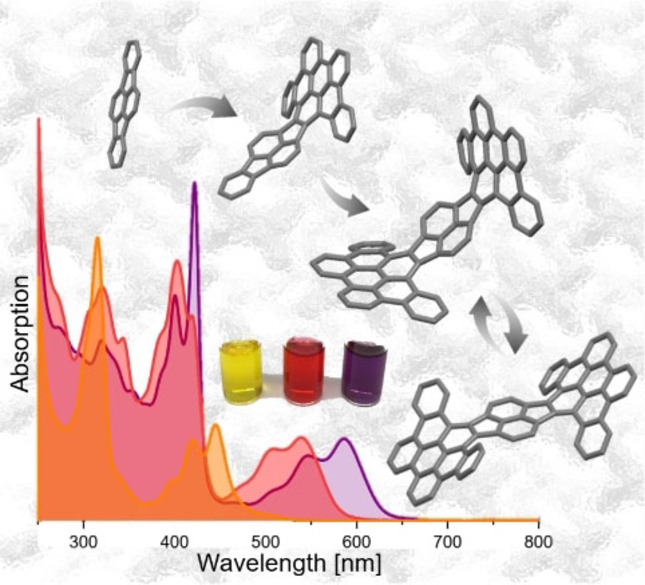
Introduction
The unique structural features associated with the electron‐deficient nature of fullerenes [1] stimulated vigorous research efforts directed towards the development of non‐planar carbonaceous electron acceptors based on polycyclic aromatic hydrocarbons (PAHs).[ 2 , 3 ] Such compounds both in their neutral and reduced states serve as versatile model systems for fundamental studies of electron delocalization and aromaticity [4] and, at the same time, constitute versatile building blocks for tunable n‐type organic materials. [5] The incorporation of nonbenzenoid rings into the sp2‐carbon framework is a particularly attractive concept to endow PAHs with electron acceptor properties and non‐planarity.[ 2 , 6 ] Hence, the incorporation of π‐conjugated cyclopentadiene moieties, which are prone to facilitated electron uptake driven by the aromaticity gain, resulted in highly interesting electron‐deficient systems.[ 3 , 7 ] By this approach not only the electron affinity of the PAH scaffold increases but also its geometry can efficiently be modulated towards curved systems, [8] which, in turn, has a pronounced effect on aggregation behavior and reactivity.[ 2 , 9 ]
Pyracylene (cyclopenta[fg]acenaphthylene, 1), initially reported back in the late 1960s, [10] can be considered as a paradigmatic example of an electron‐accepting PAH comprising 5‐membered rings (Figure 1). [11] Compound 1 is planar and with its 12π‐electron periphery and the localized central C=C bond represents a formally antiaromatic fragment of fullerene C60. [12] Through further π‐expansion of 1, the next, yet considerably less explored, homologue dibenzopyracylene (indeno[1,2,3‐cd]fluoranthene, 2) was realized. [13] A molecular wheelbarrow was assembled around the pyracylene platform and its behavior on a copper surface was investigated by scanning tunneling microscopy. [14] Recently, pyracylene was synthetically married with azaacenes [15] or pyrene units to realize interesting fluorophores. [16] To the best of our knowledge, further π‐expanded derivatives of 1 have not been realized to date.
Figure 1.

Successive π‐expansion from pyracylene 1 to dibenzopyracylene 2 and the HBC‐pyracylene hybrid HPH.
We reasoned that a new type of carbonaceous electron acceptors with appealing optoelectronic and structural properties shall be in reach through merging the electron deficient pyracylene moiety with fully benzenoid hexa‐peri‐hexabenzocoronene (HBC). [17] While HBC has been decorated with a broad range of lateral functionalities to adjust its properties for particular applications, [18] its fusion with electron deficient polycyclic π‐systems remains comparably less explored. [19]
Hence, we report herein a successive π‐expansion of pyracylene to afford the very first example of an HBC‐pyracylene hybrid (HPH, Figure 1). The compound, which is accessible through several high‐yielding steps from commercially available acenaphthene, features two facilitated reversible reduction steps and strongly bathochromically shifted absorption bands. As revealed by X‐ray crystallographic analysis the HPH forms a unique boat‐shaped geometry, which is rather surprising, considering the absence of 7‐ or 8‐membered rings in its polycyclic framework.
Results and Discussion
The dibrominated fluoranthene derivative 3 was synthesized in several steps from commercially available acenaphthene and used as the key building block in this work (for synthetic details, see the Supporting Information).[ 20 , 21 ] A two‐fold Pd‐catalyzed ethynylation of 3 with 4‐tert‐butylphenylacetylene gave 4 in 61 % yield (Scheme 1). [22] Subsequent Rh‐catalyzed [(2+2)+2] cyclotrimerization [23] of 4 with bis(4‐tert‐butylphenyl)acetylene at elevated temperature in o‐xylene delivered octaphenyl‐substituted dibenzopyracylene (OPP) derivative as an orange solid in an excellent yield of 87 %. In parallel, compound 3 was subjected to Brønsted acid‐mediated oxidative cyclodehydrogenation with DDQ/TfOH to achieve 3‐fold cyclized 5 in nearly quantitative yield. [24] In analogy to the synthesis of 4, the dibromo compound 5 was ethynylated towards 6 and successfully converted in a Rh‐catalyzed cyclotrimerization to the tetraphenyl‐substituted dibenzopyracylene (TPP) derivative. Finally, both compounds OPP and TPP underwent cyclodehydrogenation upon treatment with DDQ/TfOH in CH2Cl2 at 0 °C to afford the title compound HPH in quantitative yields, which highlights the complementarity of both routes. More importantly, the successive approach via 5 selectively provides the partially cyclized model compound TPP for further studies of the optoelectronic and structural properties.
Scheme 1.

Synthetic strategy towards HPH and its congeners OPP and TPP. a) DDQ, TfOH, CH2Cl2, 0 °C, 15 min, N2; b) 4‐tert‐butylphenylacetylene, CuI, [Pd(PPh3)4], NEt3, 100 °C, 18 h, N2; c) 4‐tert‐butylphenylacetylene, CuI, [Pd(PPh3)4], NEt3/THF/toluene (5 : 1 : 1), 100 °C, 18 h, N2; d) bis(4‐tert‐butylphenyl)acetylene, [RhCl(PPh3)3], o‐xylene, 135 °C, 52 h, N2. DDQ=2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone; TfOH=trifluoromethanesulfonic acid; THF=tetrahydrofuran.
The π‐expanded pyracylenes OPP, TPP and HPH are orange, red and purple solids, respectively, which are soluble in common organic solvents. None of the compounds shows fluorescence both in the solution and the solid state, which is in agreement with the previous studies on pyracylene‐based scaffolds. [25]
Further information about the solid‐state properties of the compounds was provided by X‐ray crystallography (Figure 2). Hence, single crystals were grown via slow gas phase diffusion of MeOH into a saturated solution of the compound in toluene (for OPP) or 1,2‐dichlorobenzene (for HPH and TPP) at rt. The successive cyclization when going from OPP to HPH increasingly induces a boat‐shaped conformation of the polycyclic scaffold (Figure 2A). This observation is in accordance with the previously reported flexibility of dibenzopyracylene (2). [26] While the pyracylene moiety in OPP is nearly planar and the whole molecule 17.4 Å wide, a significant bowl depth of 3.79 Å arises in HPH which is, due to the bent geometry, just 15.6 Å wide (for the definition of the structural elements used to estimate the discussed parameters, see the Supporting Information).
Figure 2.

(A) Successive induction of the boat‐shaped geometry evolving from OPP (top) over TPP (center) to HPH (bottom) as revealed by X‐ray crystallographic analysis (50 % probability level, H‐atoms and solvent molecules omitted). (B) Top view of HPH. (C) Short contacts in the solid state of HPH highlighted in red. (D) Relevant bond angles within the central pyracylene moiety of HPH. (E) Solid‐state packing of HPH.
Interestingly, exclusively the boat‐shaped HPH molecules were found in the unit cell of HPH, while the conceivable chair‐shaped conformation with the HBC flanks pointing to the opposite directions with respect to the pyracylene core were not observed. This finding is in agreement with the same relative energy of both conformations as revealed by density functional theory (DFT) calculations at the CAM‐B3LYP level of theory (for details, see the Supporting Information). Hence, the exclusive occurrence of the boat‐shaped conformation in the solid state can be most likely ascribed to the crystal packing forces. In the crystal packing the individual molecules of HPH interact through their convex surfaces with the shortest π⋅⋅⋅π distance of 3.34 Å between the central naphthalene moieties. Further Caryl−H⋅⋅⋅π interactions with a distance of 3.61 Å are observed at the periphery of the molecules (Figure 2C). [27] Within the unit cell the adjacent molecules are entangled in groups along the b axis to form a unique network‐like pattern (Figure 2E).
The carbon‐carbon bonds in HPH can be separated into two categories regarding their lengths. The vast majority of the bonds are in the range from 1.37 Å to 1.43 Å, representing the C=C bonds arranged within the aromatic benzenoid units. In contrast, significantly longer bonds with 1.44 Å to 1.50 Å are found between the individual benzenoid units, corresponding to C(sp2)−C(sp2) single bonds. These observations are in accordance with the X‐ray data of parent dibenzopyracylene with the longest bonds located within the 5‐membered rings linking the naphthalene and benzene subunits. [26] The interior C(sp2) carbons of the central naphthalene moiety in HPH are weakly pyramidalized as indicated by the sum of the C−C−C bond angles reaching 358.9° (Figure 2D). Consequently a π‐orbital axis vector (POAV) [28] of 2.10° is determined at these carbon atoms. For comparison, a comparable POAV value of 2.59° was reported for (20,0) single‐walled carbon nanotube with a diameter of 15.6 nm. [29] The POAV analysis of the partially cyclized TPP resulted in two distinct values of 1.37° and 2.04° for the uncyclized and the cyclized half, respectively. As expected, in uncyclized OPP with its planar structure no pyramidalization is observed (0.04°).
To assess the local aromaticity within the fused polycyclic frameworks of OPP, TPP and HPH, nuclear independent chemical shift (NICS) [30] values were determined by using the geometry optimized structures (Table 1 and the Supporting Information). The 6‐membered rings in all compounds are clearly aromatic as documented by the negative NICS values. In contrast, the cyclopentadiene‐like moieties display positive NICS values, which indicates their antiaromatic character. Interestingly, the antiaromatic character of these moieties as expressed by the NICS(0) values slightly increases with the successive π‐expansion when going from OPP (+8.32) over TPP (+10.3) to HPH (+11.1). The considerable difference between the NICS(+1) (+2.47) and NICS(−1) (+6.99) values determined for the 5‐membered rings in fully cyclized HPH is in line with its bent geometry (for further details, see the Supporting Information).
Table 1.
Summary of the NICS(X), X=+1, 0, −1, and HOMA values of HPH.
|
ring[a] |
NICS(X) |
HOMA |
||
|---|---|---|---|---|
|
|
+1 |
0 |
−1 |
|
|
A |
−7.04 |
−3.13 |
−4.36 |
+0.90 |
|
B |
+2.47 |
+11.1 |
+6.99 |
−0.33 |
|
C |
−9.79 |
−6.93 |
−8.27 |
+0.82 |
|
D |
−4.00 |
−0.34 |
−4.42 |
+0.49 |
|
E |
−11.0 |
−9.10 |
−11.3 |
+0.92 |
|
F |
−4.41 |
−1.16 |
−5.30 |
+0.29 |
|
G |
−9.03 |
−7.70 |
−10.6 |
+0.93 |
[a] Ring labels are shown in Figure 2B.
Based on the atomic positions derived from X‐ray crystallographic analysis the harmonic oscillator model of aromaticity (HOMA) [31] was applied to the individual rings of OPP, TPP, and HPH. In this model the experimental C−C bond lengths are compared with the optimized bond lengths of aromatic reference compounds. Hence, the HOMA value for a particular ring ranges between 0 and ±1, whereas +1 represents perfect aromaticity and lower values indicate decreased aromatic character. Furthermore, negative HOMA values suggest antiaromaticity. The obtained HOMA values for fully cyclized HPH are summarized in Table 1 (for OPP and TPP, see the Supporting Information). As already suggested by the NICS values, the hexagonal rings A, C, E and G in HPH with their formal Clar sextets possess an increased aromatic character as suggested by the HOMA values between +0.82 and +0.93. The remaining hexagons are basically non‐aromatic according to their HOMA values ranging from +0.29 to +0.49. Interestingly, the 5‐membered ring appears antiaromatic according to its HOMA value of −0.33, which nicely corresponds to its positive NICS value. From the HOMA values of OPP, TPP, and HPH it can be concluded that the antiaromatic character of the fused cyclopentadiene moiety continuously increases with the progressing cyclization of the surrounding π‐systems (see the Supporting Information). This evolution is in line with the trends suggested by the NICS values.
The UV/Vis absorption spectra of OPP, TPP and HPH in CH2Cl2 feature progressive redshift reflecting the impact of the consecutive cyclization and π‐expansion of the polycyclic system (Figure 3A). Hence, λ max evolves from 445 nm (for OPP) over 539 nm (for TPP) to 587 nm (for HPH), which indicates a continuous narrowing of the optical band gap from 2.64 eV to 1.98 eV (Table 2). In comparison, the lowest energy absorption of parent dibenzopyracylene in ethanol is reported to occur at 410 nm with a clearly visible vibronic structure. [13]
Figure 3.

UV/Vis absorption spectra of OPP (blue), TPP (black) and HPH (red). A: experimental (CH2Cl2, rt). B: TD‐DFT calculated (CAM‐B3LYP/6‐31G(d)).
Table 2.
Experimental and optoelectronic data for OPP, TPP and HPH.
|
|
λ [nm] |
Eg opt [eV][a] |
E 1/2 [V][b] |
E CV [eV] |
E DFT [eV][e] |
band gap [eV][e] |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
λ max |
λ end |
|
E (−1/−2) |
E (−1/0) |
E (0/+1) |
HOMO[c] |
LUMO[d] |
EA |
IP |
|
|
OPP |
445 |
470 |
2.64 |
– |
– |
– |
– |
– |
2.22 |
5.95 |
3.73 |
|
TPP |
539 |
582 |
2.13 |
– (−2.06) |
−1.59 (−1.66) |
+0.70 (+0.80) |
−5.50 |
−3.21 |
2.36 |
5.46 |
3.10 |
|
HPH |
587 |
627 |
1.98 |
−1.82 (−1.86) |
−1.46 (−1.51) |
+0.61 (+0.72) |
−5.41 |
−3.34 |
2.46 |
5.47 |
3.01 |
[a] Calculated according to hcλ end −1, λ end estimated from the intersection of a tangent line to the lowest energy absorption band of the UV/Vis spectrum with the x‐axis. [37] [b] measured in CH2Cl2 (in THF). [c] HOMO energies calculated from E HOMO,CV=−(E (0/+)+4.8) eV. [d] LUMO energies calculated from E LUMO, CV=−4.8 eV‐E (−/0), under the assumption that the potential of Fc/Fc+ is −4.8 eV vs. vacuum. [38] [e] DFT calculated values (CAM−B3LYP/6‐31G(d)).
To gain insight into the origin of the observed absorption bands, a time‐dependent density functional theory (TD‐DFT) study was performed at the CAM‐B3LYP level of theory. The calculations overall nicely reproduce the experimental spectra (Figure 3B) and verify that the lowest energy absorptions of OPP, TPP and HPH are characterized by a major contribution of the HOMO→LUMO transition (for further details, see the Supporting Information). The continuous redshift when going from uncyclized OPP to fully π‐expanded HPH originates from the increased spatial extension of the orbitals which leads to the observed narrowing of the HOMO‐LUMO gap from 3.15 eV to 2.42 eV (Figure 4).
Figure 4.

HOMO and LUMO of OPP (left), TPP (center), HPH (right) with their corresponding energy values (B3LYP/6‐31G(d)). [32]
Cyclic voltammetry (CV) was used to evaluate the redox properties of the π‐expanded pyracylenes OPP, TPP and HPH (Figure 5 and Table 2). For uncyclized compound OPP no redox events were observed within the potential range from −2.5 V to +1.5 V in CH2Cl2 or THF. In contrast, the partially cyclized species TPP exhibited a single reversible oxidation at +0.70 V (vs. ferrocene/ferrocenium (Fc/Fc+)) in CH2Cl2. For fully cyclized HPH two reversible oxidations at +0.61 V and +0.84 V were observed (Figure 5A). Further electrochemical analysis of both compounds HPH and TPP in THF revealed two reversible reduction processes for each derivative. While the partially cyclized compound TPP is reversibly reduced at −1.66 V and −2.06 V, its fully cyclized congener HPH undergoes two facilitated electron uptakes at −1.51 V and −1.86 V. In other words, the π‐expansion when going from TPP to HPH results in an anodic shift of 150 mV and 200 mV of the first and second reduction event, respectively (Figure 5B). In comparison, the reversible oxidations and reductions of hexa‐tert‐butyl‐substituted HBC were reported to occur at +0.99 V, +1.42 V (in CH2Cl2) and −2.10 V, −2.40 V (in THF), respectively. [33] Parent pyracylene (1) with its formally antiaromatic 12π‐electron system undergoes under comparable conditions two reductions at −1.56 V and −2.14 V accompanied by a single oxidation at +0.75 V.[ 34 , 35 ] From these data it can be concluded that upon π‐expansion the redox features of the pyracylene core remain largely unaffected, which is also documented by the predominant localization of the LUMO on the pyracylene moiety in OPP, TPP and HPH (Figure 4).
Figure 5.

Cyclic voltammograms of TPP (black) and HPH (red) (∼2 mM) at rt (vs. Fc/Fc+, n Bu4NPF6 as supporting electrolyte, scan rate 149 mVs−1). Recorded in A: CH2Cl2 B: THF.
More importantly, the π‐expansion leads to the occurrence of a second oxidation step in HPH and considerably reduced potential difference between the two reduction events from 580 mV (for 1) to 350 mV (for HPH), which points towards better charge stabilization within the π‐expanded polycyclic framework. The evolution of the redox properties is in good agreement with the calculated vertical ionization potentials (IPs) and electron affinities (EAs) summarized in Table 2. For example, an increase of 0.24 eV is observed for the EA values when going from uncyclized OPP to HPH. Overall, both compounds HPH and TPP can be considered as moderate electron acceptors comparable to, for instance, 9,10‐anthraquinone (Ered,1: −1.27 V; Ered,2: −1.80 V) or cyclopenta[hi]aceanthrylene (Ered: −1.56 V).[ 3d , 36 ]
Conclusion
In conclusion, we established a reliable, high yielding synthetic route towards π‐expanded pyracylenes. As revealed by X‐ray crystallographic analysis, the successive π‐expansion leads to a pronounced boat‐shaped geometry of the polycyclic scaffold, which has a considerable impact on the electronic structure and the aromaticity. In the same time a progressive bathochromic shift of the UV/Vis absorption bands accompanied by a consecutive narrowing of the HOMO‐LUMO gap is observed. The compounds undergo several redox events which become facilitated with the increasing extent of π‐expansion. The experimental observations are in line with the computational studies based on density functional theory and identify the compounds as promising chromophores with multi‐stage redox amphotheric properties.
Experimental Section
Experimental details and characterization data can be found in the Supporting Information. Deposition Number(s) 2165129 (3, for structure see the Supporting Information), 2165130 (4, for structure see the Supporting Information), 2165131 (OPP), 2165132 (TPP), and 2165133 (HPH) contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The generous funding by the Deutsche Forschungsgemeinschaft (DFG) – Project number 182849149‐SFB 953 is acknowledged. Open Access funding enabled and organized by Projekt DEAL.
J. Bergner, C. Walla, F. Rominger, A. Dreuw, M. Kivala, Chem. Eur. J. 2022, 28, e202201554.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Xie Q., Perez-Cordero E., Echegoyen L., J. Am. Chem. Soc. 1992, 114, 3978; [Google Scholar]
- 1b. Guldi D. M., Chem. Commun. 2000, 321; [Google Scholar]
- 1c. Liu T., Troisi A., Adv. Mater. 2013, 25, 1038. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Li X., Kang F., Inagaki M., Small 2016, 12, 3206; [DOI] [PubMed] [Google Scholar]
- 2b. Pun S. H., Miao Q., Acc. Chem. Res. 2018, 51, 1630; [DOI] [PubMed] [Google Scholar]
- 2c. Majewski M. A., Stępień M., Angew. Chem. Int. Ed. 2019, 58, 86; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 90; [Google Scholar]
- 2d. Stuparu M. C., Acc. Chem. Res. 2021, 54, 2858. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Brunetti F. G., Gong X., Tong M., Heeger A. J., Wudl F., Angew. Chem. Int. Ed. 2010, 49, 532; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 542; [Google Scholar]
- 3b. Chase D. T., Fix A. G., Rose B. D., Weber C. D., Nobusue S., Stockwell C. E., Zakharov L. N., Lonergan M. C., Haley M. M., Angew. Chem. Int. Ed. 2011, 50, 11103; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11299; [Google Scholar]
- 3c. Wood J. D., Jellison J. L., Finke A. D., Wang L., Plunkett K. N., J. Am. Chem. Soc. 2012, 134, 15783; [DOI] [PubMed] [Google Scholar]
- 3d. Xia H., Liu D., Xu X., Miao Q., Chem. Commun. 2013, 49, 4301; [DOI] [PubMed] [Google Scholar]
- 3e. Chaolumen, Murata M., Sugano Y., Wakamiya A., Murata Y., Angew. Chem. Int. Ed. 2015, 54, 9308; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9440. [Google Scholar]
- 4.
- 4a. Konarev D. V., Kuzmin A. V., Khasanov S. S., Shestakov A. F., Otsuka A., Yamochi H., Kitagawa H., Lyubovskaya R. N., Chem. Asian J. 2020, 15, 2689; [DOI] [PubMed] [Google Scholar]
- 4b. Zhu Y., Zhou Z., Wei Z., Petrukhina M. A., Organometallics 2020, 39, 4688; [Google Scholar]
- 4c. Zhou Z., Zhu Y., Wei Z., Bergner J., Neiß C., Doloczki S., Görling A., Kivala M., Petrukhina M. A., Angew. Chem. Int. Ed. 2021, 60, 3510; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 3552; [Google Scholar]
- 4d. Zhou Z., Zhu Y., Wei Z., Bergner J., Neiß C., Doloczki S., Görling A., Kivala M., Petrukhina M. A., Chem. Commun. 2022, 58, 3206. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Anthony J. E., Facchetti A., Heeney M., Marder S. R., Zhan X., Adv. Mater. 2010, 22, 3876; [DOI] [PubMed] [Google Scholar]
- 5b. Quinn J. T. E., Zhu J., Li X., Wang J., Li Y., J. Mater. Chem. C 2017, 5, 8654. [Google Scholar]
- 6.
- 6a. Márquez I. R., Castro-Fernández S., Millán A., Campaña A. G., Chem. Commun. 2018, 54, 6705; [DOI] [PubMed] [Google Scholar]
- 6b. Chaolumen, Stepek I. A., Yamada K. E., Ito H., Itami K., Angew. Chem. Int. Ed. 2021, 60, 23508; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 23700. [Google Scholar]
- 7. Liu Y.-H., Perepichka D. F., J. Mater. Chem. C 2021, 9, 12448. [Google Scholar]
- 8.
- 8a. Scott L. T., Pure Appl. Chem. 1996, 68, 291; [Google Scholar]
- 8b. Tsefrikas V. M., Scott L. T., Chem. Rev. 2006, 106, 4868; [DOI] [PubMed] [Google Scholar]
- 8c. Scott L. T., Jackson E. A., Zhang Q., Steinberg B. D., Bancu M., Li B., J. Am. Chem. Soc. 2012, 134, 107; [DOI] [PubMed] [Google Scholar]
- 8d. Rickhaus M., Mayor M., Juríček M., Chem. Soc. Rev. 2017, 46, 1643. [DOI] [PubMed] [Google Scholar]
- 9. Scott L. T., Chem. Soc. Rev. 2015, 44, 6464. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Trost B. M., Bright G. M., J. Am. Chem. Soc. 1967, 4244; [Google Scholar]
- 10b. Trost B. M., Bright G. M., Frihart C., Brittelli D., J. Am. Chem. Soc. 1971, 93, 737; [Google Scholar]
- 10c. Coulson C. A., Mallion R. B., J. Am. Chem. Soc. 1976, 98, 592. Please note that in recent literature pyracylene is also denoted as pyracyclene. [Google Scholar]
- 11. Lo D. H., Whitehead M. A., Chem. Commun. 1968, 771. [Google Scholar]
- 12. Diogo H. P., Kiyobayashi T., da Piedade M. E. M., Burlak N., Rogers D. W., McMasters D., Persy G., Wirz J., Liebman J. F., J. Am. Chem. Soc. 2002, 124, 2065. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Clar E., Stubbs H. W. D., Tucker S. H., Nature 1950, 166, 1075; [DOI] [PubMed] [Google Scholar]
- 13b. Stubbs H. W. D., Tucker S. H., J. Chem. Soc. 1951, 2936; [Google Scholar]
- 13c. Wegner H. A., Scott L. T., de Meijere A., J. Org. Chem. 2003, 68, 883. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Jimenez-Bueno G., Rapenne G., Tetrahedron Lett. 2003, 44, 6261; [Google Scholar]
- 14b. Rapenne G., Grill L., Zambelli T., Stojkovic S. M., Ample F., Moresco F., Joachim C., Chem. Phys. Lett. 2006, 431, 219; [Google Scholar]
- 14c. Rapenne G., Jimenez-Bueno G., Tetrahedron 2007, 63, 7018. [Google Scholar]
- 15. Ganschow M., Koser S., Hodecker M., Rominger F., Freudenberg J., Dreuw A., Bunz U. H. F., Chem. Eur. J. 2018, 24, 13667. [DOI] [PubMed] [Google Scholar]
- 16. Deng X., Liu X., Wei L., Ye T., Yu X., Zhang C., Xiao J., J. Org. Chem. 2021, 86, 9961. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Clar E., Ironside C. T., Proc. Chem. Soc. 1958, 125; [Google Scholar]
- 17b. Clar E., Ironside C. T., Zander M., J. Chem. Soc. 1959, 142. [Google Scholar]
- 18.
- 18a. Ito S., Wehmeier M., Brand J. D., Kübel C., Epsch R., Rabe J. P., Müllen K., Chem. Eur. J. 2000, 6, 4327; [DOI] [PubMed] [Google Scholar]
- 18b. Wasserfallen D., Fischbach I., Chebotareva N., Kastler M., Pisula W., Jäckel F., Watson M. D., Schnell I., Rabe J. P., Spiess H. W., Müllen K., Adv. Funct. Mater. 2005, 15, 1585; [Google Scholar]
- 18c. Wong W. W. H., Singh T. B., Vak D., Pisula W., Yan C., Feng X., Williams E. L., Chan K. L., Mao Q., Jones D. J., Ma C.-Q., Müllen K., Bäuerle P., Holmes A. B., Adv. Funct. Mater. 2010, 20, 927; [Google Scholar]
- 18d. Seyler H., Purushothaman B., Jones D. J., Holmes A. B., Wong W. W. H., Pure Appl. Chem. 2012, 84, 1047. [Google Scholar]
- 19. Fernández-García J. M., Evans P. J., Rivero S. M., Fernández I., García-Fresnadillo D., Perles J., Casado J., Martín N., J. Am. Chem. Soc. 2018, 140, 17188. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Lewis I. K., Topsom R. D., Vaughan J., Wright G. J., J. Org. Chem. 1968, 33, 1497; [Google Scholar]
- 20b. Constantine P. R., Deady L. W., Topsom R. D., J. Org. Chem. 1969, 34, 1113; [Google Scholar]
- 20c. Tesmer M., Vahrenkamp H., Eur. J. Inorg. Chem. 2001, 1183. [DOI] [PubMed] [Google Scholar]
- 21. Ogliaruso M. A., Romanelli M. G., Becker E. I., Chem. Rev. 1965, 65, 261. [Google Scholar]
- 22. Chinchilla R., Nájera C., Chem. Rev. 2007, 107, 874. [DOI] [PubMed] [Google Scholar]
- 23. Shibata Y., Tanaka K., Synthesis 2012, 44, 323. [Google Scholar]
- 24.
- 24a. Grzybowski M., Skonieczny K., Butenschön H., Gryko D. T., Angew. Chem. Int. Ed. 2013, 52, 9900; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10084; [Google Scholar]
- 24b. Grzybowski M., Sadowski B., Butenschön H., Gryko D. T., Angew. Chem. Int. Ed. 2020, 59, 2998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3020. [Google Scholar]
- 25. Hodecker M., Ganschow M., Abu-Odeh M., Bunz U. H. F., Dreuw A., ChemPhotoChem 2019, 3, 755. [Google Scholar]
- 26. Halling M. D., Orendt A. M., Strohmeier M., Solum M. S., Tsefrikas V. M., Hirao T., Scott L. T., Pugmire R. J., Grant D. M., Phys. Chem. Chem. Phys. 2010, 12, 7934. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Salonen L. M., Ellermann M., Diederich F., Angew. Chem. Int. Ed. 2011, 50, 4808; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4908. [Google Scholar]
- 28. Haddon R. C., J. Am. Chem. Soc. 1987, 109, 1676. [Google Scholar]
- 29. Niyogi S., Hamon M. A., Hu H., Zhao B., Bhowmik P., Sen R., Itkis M. E., Haddon R. C., Acc. Chem. Res. 2002, 35, 1105. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Schleyer P. v. R., Maerker C., Dransfeld A., Jiao H., van Eikema Hommes N. J. R., J. Am. Chem. Soc. 1996, 118, 6317; [DOI] [PubMed] [Google Scholar]
- 30b. Chen Z., Wannere C. S., Corminboeuf C., Puchta R., Schleyer P. v. R., Chem. Rev. 2005, 105, 3842. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Krygowski T. M., J. Chem. Inf. Comput. Sci. 1993, 33, 70; [Google Scholar]
- 31b. Krygowski T. M., Cyrański M. K., Chem. Rev. 2001, 101, 1385. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Hohenberg P., Kohn W., Phys. Rev. 1964, 136, B864; [Google Scholar]
- 32b. Kohn W., Sham L. J., Phys. Rev. 1965, 140, A1133-A1138; [Google Scholar]
- 32c. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785; [DOI] [PubMed] [Google Scholar]
- 32d. Becke A. D., J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- 33. Herwig P. T., Enkelmann V., Schmelz O., Müllen K., Chem. Eur. J. 2000, 6, 1834. [DOI] [PubMed] [Google Scholar]
- 34.For the sake of comparison, the potentials reported vs. SCE were converted to the Fc/Fc+ reference by subtracting a value of 500 mV. This conversion represents an approximation.
- 34a. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877; [DOI] [PubMed] [Google Scholar]
- 34b. Gisselbrecht J.-P., Moonen N. N. P., Boudon C., Nielsen M. B., Diederich F., Gross M., Eur. J. Org. Chem. 2004, 2959. [Google Scholar]
- 35. Koper C., Sarobe M., Jenneskens L. W., Phys. Chem. Chem. Phys. 2004, 6, 319. [Google Scholar]
- 36. van Dijk E. H., Myles D. J. T., van der Veen M. H., Hummelen J. C., Org. Lett. 2006, 8, 2333. [DOI] [PubMed] [Google Scholar]
- 37. Costa J. C. S., Taveira R. J. S., Lima C. F. R. A. C., Mendes A., Santos L. M. N. B. F., Opt. Mater. 2016, 58, 51. [Google Scholar]
- 38. Pommerehne J., Vestweber H., Guss W., Mahrt R. F., Bässler H., Porsch M., Daub J., Adv. Mater. 1995, 7, 551. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.