

Abstract
Background
Neurofibromatosis type 2 (NF2) is a genetic disease characterized by the appearance of multiple tumours in the nervous system. Cutaneous lesions are common and may provide useful diagnostic and prognostic information, but they have not been widely studied.
Objectives
To characterize cutaneous lesions in a Spanish cohort of patients with NF2 and investigate associations with clinical and genetic severity.
Methods
We studied the clinical and histologic characteristics of cutaneous lesions in 49 patients with NF2 and analysed correlations with phenotype‐ and genotype‐based severity scores. We collected information on the presence/absence of cutaneous lesions, location, age at onset, type of lesion, and histologic features. We also studied level of systemic involvement and genetic mutations involved.
Results
Forty‐nine patients (31 women [63.3%] and 18 men [36.7%]) were analysed, and 33 (67.3%) had cutaneous lesions presumed to be schwannomas. According to their clinical form, they were distributed as follows: 24 patients (48%) had deep tumours, 21 (42%) had plaque‐like lesions, and 3 (6%) had superficial tumours. Histologic examination from 27 lesions analysed out 23 patients showed classic schwannoma or hybrid schwannoma‐neurofibroma features in the 8 deep tumours biopsied and plexiform schwannoma features in the 17 plaque‐like lesions and the 2 superficial tumours analysed. Early onset (first 2 decades of life) was reported by all patients with plaques and superficial tumours. In our cohort, 100% of the patients with plaque‐like lesions and superficial tumours with microscopic features of plexiform schwannoma were in the 2 groups with the most severe clinical phenotypes, and 82.6% of them were in the 3 most severe genotype‐based classes.
Conclusions and Relevance
Cutaneous lesions, specially plexiform schwannomas, are common in NF2, and they usually appear at an early age providing useful diagnostic and prognostic information. These tumours are part of the spectrum of cutaneous manifestations in this disease. Although its diagnostic and prognostic value has been pointed out, there are few studies focussed on their analysis.
Introduction
Neurofibromatosis type 2 (NF2) (MIM101000) is an autosomal dominant disorder caused by mutations in the NF2 gene, which codes for a tumour‐suppressor protein known as merlin. It is characterized by the development of schwannomas, meningiomas, ependymomas, mononeuropathy, eye disorders, and cutaneous lesions. 1 , 2
Patients have traditionally been classified into 2 main groups according to clinical severity: those with Gardner‐type NF2, a less severe form of disease with onset in adulthood, low tumour burden and low mortality, and those with Wishart‐type NF2, a more severe form of disease with onset in childhood or adolescence, high tumour burden and high mortality. 1 , 2 It is known that these differences can be largely due to the type of constitutional mutation carried, which constitute a fairly good predictor of clinical severity. 3 , 4 , 5 The UK NF2 Genetic Severity Score (GSS) was designed to predict clinical phenotype based on type of mutation detected and considering several clinical severity factors. 6 GSS classifies patients into 3 severity groups—tissue mosaic, classic, and severe—based on the pathogenic germline NF2 variant present. Our research group recently validated the GSS in NF2 patients seen at the Spanish National Reference Center (CSUR) for phakomatoses and proposed a revised version incorporating data from functional assays and the predicted mutational effect on merlin. 7 This new version, called the Functional Genetic Severity Score (FGSS), may have greater prognostic accuracy as it was found to reduce intragroup variability and offer enhanced stratification of mosaic patients. 7
Different cutaneous lesions have been described in association with NF2, although little has been published on their clinical utility for early diagnosis or on their association with disease severity. The largest series in the literature describe café‐au‐lait (CAL) macules and cutaneous schwannomas, but none of them provide detailed information on clinical or histologic findings or possible associations with NF2 severity. 1 , 2 , 8 In 2019, Halliday et al. did publish a large series of 86 patients in which they stratified different clinical variables, including skin lesions, according to genetic severity by GSS. Nevertheless, this study only included paediatric patients and was not focussed on cutaneous findings. 9 The largest study to focus exclusively on cutaneous abnormalities in NF2 was published in 1997 by Mautner et al., 10 who provided a detailed clinical picture of cutaneous lesions and their association with disease phenotypical severity. To our knowledge, no studies to date have analysed associations between cutaneous lesions and genetic severity in adult patients with NF2.
We believe that a greater understanding of the cutaneous manifestations of NF2 might facilitate early diagnosis and provide useful prognostic information, as on reviewing the literature we observed that skin lesions are a common presenting symptom in children with NF2 11 , 12 , 13 and that all cases of cutaneous plexiform schwannomas described to date have occurred in young patients (4–28 years) or patients with a severe phenotype. 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Moreover, in 2018, our group described a series of 7 patients with cutaneous plexiform schwannomas who all later developed a severe NF2 phenotype. 22
The aim of this study was to describe the clinical and histologic characteristics of cutaneous lesions in a cohort of patients with NF2 and to explore associations with phenotype and genotype based on the FGSS.
Material and methods
All the procedures were performed in accordance with the principles of the Declaration of Helsinki and met the ethical standards of the Institutional Review Board at Germans Trias i Pujol Research Center, which approved this study.
From 2010 to 2020, we prospectively studied all patients with a clinical and genetic diagnosis of NF2 evaluated by the NF2 Multidisciplinary Clinics in the Spanish National Reference Centre (CSUR) of Phacomatoses HGTP‐ICO‐IGTP at Hospital Universitari Germans Trias i Pujol in Badalona, Spain. Informed consent was obtained from all patients. We collected information on age at the time of this evaluation, sex, age at diagnosis, and presence of schwannomas and other NF2‐related tumours and cutaneous lesions. Data regarding the degree of systemic involvement of 45 of our patients were previously collected in the article published recently by Catasus et al. 7
The following information was noted for cutaneous lesions: location (head and neck, upper or lower extremities, trunk), number and type of lesions and age at onset. The lesions were classified as CAL macules or cutaneous schwannomas. Cutaneous schwannomas were further divided into 3 groups based on their clinical morphology: (i) deep tumours, defined as firm, nodular lesions attached to the deep planes with a smooth surface and no skin changes; (ii) superficial tumours, defined as soft superficial papules or nodules, sometimes with a wrinkled surface; and (iii) plaques, defined as soft, slightly raised, roughened lesions with hypertrichosis and a darker colour than the surrounding skin.
We also analysed the histologic features of cutaneous lesions that had been excised over the 10‐year study period for diagnostic purposes or at the patient's request. This study was based solely on light microscopy of haematoxylin and eosin–stained slides. For the immunohistochemical analysis, fixed cells were stained with primary polyclonal rabbit anti‐S100 (Dako) and mouse monoclonal anti‐Melan A (Ventana Medical Systems). Neurofilament antibody staining was performed in some cases.
Genetic testing of blood, or tissue when available, was performed using a custom NGS Panel (I2HCP), which comprises 135 genes associated with cancer predisposition syndromes and RASopathies, 23 as described by Catasus et al. 7
Patients were grouped into 6 classes using the FGSS, which predicts clinical phenotype based on the type of mutation detected, ranging from class 1 (less severe) to class 6 (most severe) (Table S1). Patients were also grouped according to phenotype severity scores calculated using well‐known prognostic markers 24 , 25 at the time of evaluation, as described by Catasus et al. 7 Phenotype severity was classified into 4 categories, where group 1 represented the most severe form of disease and group 4 the less severe (Table S2).
All data were recorded on a spreadsheet (Microsoft Excel 2019) and expressed as numbers (n) and percentages (%) or means ± standard deviation (SD). Categorical phenotype variables (e.g. sex and cutaneous features) were compared using the χ2 test. Trends with severity were investigated using the Mantel–Haenszel linear‐by‐linear χ2 test of association. Spearman's rank‐order correlation analysis was used to assess correlations between disease severity and patient age and number of skin lesions. The preliminary analysis (visual inspection of scatterplots) showed the relationships to be monotonic. Statistical analyses were performed in SPSS V.25 (IBM Statistics). Standard summary statistics are reported throughout with statistical significance of inferences set at P < 0.05.
Results
Description of NF2 cohort
The study population included 49 patients diagnosed of NF2 according to Manchester NF2 diagnostic criteria, 1 , 2 45 of them previously described in Catasus 2021. 7 There were 31 women (63.3%) and 18 men (36.7%) (female‐to‐male ratio, 1.7 : 1). Mean (SD) age was 45.48 years at the time of analysis and 29.49 years at the time of diagnosis; 51% of patients had been diagnosed with NF2 before the age of 25 years. Bilateral schwannomas were found in 89.8% of the patients, 55.1% presented spinal schwannomas, and 32.7% had peripheral schwannomas (Table 1).
Table 1.
Clinical characteristics and patient distribution according to severity classifications
| Patient characteristics | |
|---|---|
| Demographic | |
| Sex (N [%]) | |
| Male | 18 (36.7%) |
| Female | 31 (63.3%) |
| Current age (years), (mean [±SD]) | 45.84 (18.62) |
| Age at diagnosis (years), (mean [±SD]) | 29.49 (19.08) |
| Diagnosis before age 25 (N [%]) | 25 (51%) |
| Familiar history of NF2 (N [%]) | |
| Familiar NF2 | 4 (8.2%) |
| Sporadic/de novo NF2 | 45 (91.8%) |
| Classification of NF2 patients by phenotype severity (N [%]) | |
| Group 1 | 21 (42.9%) |
| Group 2 | 13 (26.5%) |
| Group 3 | 4 (8.2%) |
| Group 4 | 11 (22.4%) |
| Classification of NF2 patients by FGSS categories (N [%]) | |
| Class 1 | 17 (34.7%) |
| Class 2 | 0 (0) |
| Class 3 | 3 (6.1%) |
| Class 4 | 8 (16.3%) |
| Class 5 | 9 (18.4%) |
| Class 6 | 12 (24.5%) |
| Cutaneous manifestations | |
| Cutaneous lesions (N [%]) | |
| Present | 33 (67.3%) |
| Absent | 16 (32.7%) |
| Café‐au‐lait macules (CAL macules) | |
| Presence of CAL macules (N [%]) | 19 (38.8%) |
| Proportion of patients by number of CAL macules | |
| 0 | 30 (61.2%) |
| 1 | 6 (12.2%) |
| 2 | 2 (4.1%) |
| 3 | 2 (4.1%) |
| 4 | 6 (12.2%) |
| 5 | 1 (2%) |
| 6 | 1 (2%) |
| 7 | 1 (2%) |
| Deep tumours | |
| Presence of cutaneous schwannomas (N [%]) | 24 (49%) |
| Plaque‐like lesions | |
| Presence of plaques (N [%]) | 21 (42.9%) |
| Superficial tumours | |
| Presence of tumours (N [%]) | 3 (6.1%) |
| Plaque‐like lesions + Superficial tumours | |
| Presence of plaques + tumours (N [%]) | 23 (46.9%) |
| Non‐cutaneous manifestations | |
| Vestibular schwannomas (total N [%]) | 48 (98%) |
| Bilateral (N [%]) | 44 (89.8%) |
| Unilateral (N [%]) | 4 (8.2%) |
| Age at diagnosis (years) (mean [±SD]) | 32 (±17.89) |
| Meningiomas (total N [%]) | 28 (57.1%) |
| Intracranial (N [%]) | 27 (55.1%) |
| Spinal (N [%]) | 15 (30.6%) |
| Ependymomas (total N [%]) | 11 (22.4%) |
| Extravestibular schwannomas (total N [%]) | 33 (67.3%) |
| Intracranial schwannomas (N [%]) | 15 (30.6%) |
| Spinal schwannomas (N [%]) | 27 (55.1%) |
| Peripheral schwannomas (N [%]) | 16 (32.7%) |
Cutaneous lesions
Thirty‐three out of 49 NF2 patients (67.3%) had cutaneous manifestations (Table 2). Almost half of them (38.8%) had at least one CAL macule (mean [SD], 1.2 [1.8]; range, 0–7). The macules were all light brown, and 58.6% were located on the trunk. They had been present at birth or had appeared in the first years of life.
Table 2.
Cutaneous lesions
| Cutaneous manifestations | |||||
|---|---|---|---|---|---|
| Type of lesion | No. of patients | Location | Appearance | Average per patient | Histopathology |
| Café‐au‐lait macules (CAL macules) | 19 (38.8%) | Trunk 58.6%; Extremities 39.6%; Head and neck 1.7% | 31.5% congenital o during 1st year; 68.4% from 1 year to 12 years old | 1.2 CLM per patient (3.05 per patient counting only patients with macules) | None of the were biopsied |
| Deep tumours | 24 (49%) | Trunk 15.3%; 20.5% Head and Neck; 64% Extremities | 0% Congenital o during 1st year; 25% from 1 year old to 12 years old; 29% Adolescence; 37.5% from 18 years old to 29 years old; 8.3% after 30 years old | 1.59 deep tumour per patient (3.2 per patient counting only patients with deep tumours) | 6 cases with classic schwannoma features; 2 cases with a hybrid schwannoma‐neurofibroma pattern |
| Plaque‐like lesions | 21 (42.9%) | Trunk 65%; Head and neck 8.9%; Extremities 25.3% | 4.7% Congenital o during 1st year; 57.1% from 1 year old to 12 years old; 28.5% Adolescence; 4.7% After adolescence but before 20 years old | 1.36 per patient (3.1 per patient counting only patients with plaque‐like lesions) | 17 cases with plexiform schwannoma features |
| Superficial tumours | 3 (6.1%) | Extremities 71.4%; Trunk 28.5% | 100% from 1 year to 12 years old | 0.14 per patient (2.3 per patient counting only patients with superficial tumours) | 2 cases with plexiform schwannoma features |
Twenty‐four patients (49%) had deep tumours presumed to be schwannomas (Fig. 1). Most of them (64%) were located on the limbs and followed the course of the nerves. They had appeared in the first 2 decades of life in 57% of patients. Histology of the 8 deep tumours that had been biopsied or excised over the study period confirmed a diagnosis of schwannoma in all cases. The tumours exhibited 2 distinct histologic patterns: the classic schwannoma pattern (6 cases) and a hybrid schwannoma‐neurofibroma pattern (2 cases). The classic pattern showed a round, very well‐circumscribed, encapsulated, dermal tumour formed by spindle cells in cellular areas (some of which contained nuclear palisading and Verocay bodies) together with a number of hypocellular areas with a loose myxoid stroma and blood vessels. The hybrid pattern was characterized by an area with classic schwannoma features and an adjacent area with neurofibroma features comprising a mixed proliferation of haphazardly arranged nonencapsulated Schwann cells, fibroblasts and collagen with dispersed axons. Immunohistochemical staining was positive for s‐100 and negative for neurofilaments in the schwannoma area and positive for s‐100 and neurofilaments in the neurofibroma area.
Figure 1.
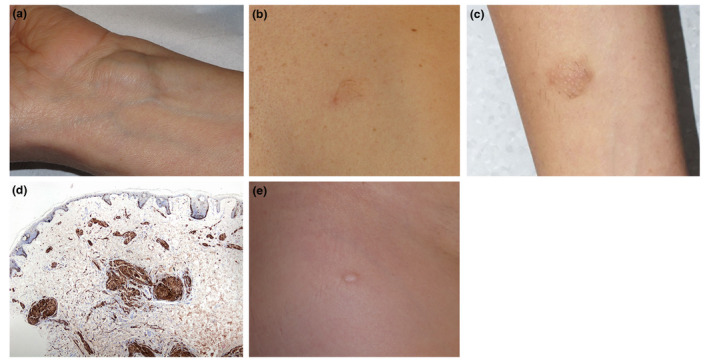
(a) Deep schwannoma, located on the wrist of a patient with NF2. (b and c) Plaque‐like plexiform schwannoma, defined as soft, slightly raised, roughened lesions with hypertrichosis and a darker colour than the surrounding skin, in a patient with NF2. (d) Plexiform schwannoma characterized by bundles of spindle cells surrounded by a thin fibrous capsule in the superficial dermis. The cells are positive for s‐100. (e) Superficial schwannoma in a patient with NF2. [Colour figure can be viewed at wileyonlinelibrary.com]
Similarly, nearly half of the patients included in this study (42.9%) had at least one plaque‐like lesion defined as soft, slightly raised, roughened lesions with hypertrichosis and a darker colour than the surrounding skin (Fig. 1b,c). Furthermore, 65% of them were located on the trunk. All the patients said their plaques had always been present, suggesting they were congenital or had appeared in childhood or adolescence. Deep tumours, by contrast, were much less likely to develop in early life (just 57% of cases). One plaque from seventeen patients, for a total of seventeen plaques, had been excised and biopsied over the 10‐year study period, and they all showed the same plexiform schwannoma pattern characterized by bundles of spindle cells surrounded by a thin fibrous capsule and Verocay bodies and Antoni A areas scattered throughout the superficial and deep dermis. There were no Antoni B areas. Immunohistochemically, the cells were positive for s‐100 and negative for neurofilaments (Fig. 1d).
Finally, very few patients (6%) had superficial tumours (Fig. 1e). These were mainly located on the extremities and had all appeared during childhood. From them, two of these tumours had been biopsied and showed the typical plexiform schwannoma pattern observed in the plaque‐like lesions.
NF2 cutaneous manifestations and association with predicted NF2 severity
To study the association between cutaneous manifestations and NF2 severity predicted by the type of germline mutation and determine the potential prognostic value of these manifestations, all the patients in the cohort were grouped into the 6 FGSS classes. From them, 17 patients (34.7%) were assigned to class 1, the lowest severity class, corresponding to probable or confirmed mosaicism. Of the remaining patients all harbouring a pathogenic germline NF2 variant, 11 were assigned to mild and moderate NF2 phenotype, while seventeen were classified as 5 or 6, the classes associated with severest NF2 phenotype (Tables 1, 3). Patients in the higher severity genotype classes were more likely to have any cutaneous lesion than those in the lower severity classes (Table 3). Twenty‐five of the 33 patients with cutaneous lesions were in the top 3 classes of severity. The opposite trend was observed for patients without these lesions.
Table 3.
Demographic and clinical data according to FGSS classification
|
|
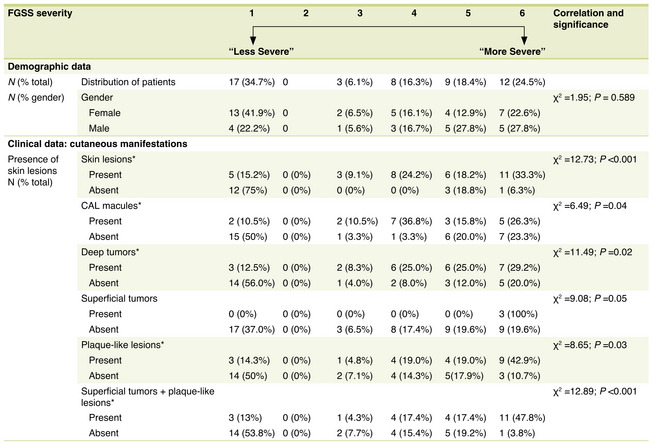
Asterisk (*) indicates statistical significance (P < 0.05) in trends (X2) and correlations (r s ) of measures with phenotypical severity classification.
CAL macules, Café‐au‐lait macules; N, Number; NF2, neurofibromatosis type 2; SD, standard deviation.
The association between cutaneous lesions and predicted severity based on the causative germline mutation was more evident for tumours than for CAL macules, although it was statistically significant in both cases (Table 3). Of the 19 patients with CAL macules, 15 (78.9%) were in classes 4 to 6, 2 (10.5%) were in class 3, and another 2 were in class 1. The majority of patients with deep tumours (n = 19, 79.2%) were also in classes 4 to 6, while those without these tumours were more evenly distributed across the classes. Finally, 19 (82.6%) of the 23 patients with superficial tumours or plaques were in classes 4 to 6, while 16 (61.5%) of those without these lesions were in classes 1 to 3. The differences were statistically significant (Table 3).
The above results indicate that the presence of cutaneous lesions in patients with NF2 is associated with severity predicted by the type of NF2 variant present.
NF2 cutaneous manifestations and association with phenotype severity
To study the correlation between cutaneous lesions and phenotype at the time of this study, we quantified phenotype severity using a numeric nine‐scale based on the sum of transformed quantitative variables based on well‐known NF2 prognosis markers and grouped into four different groups, ranging from severe phenotype (group 1) to very mild phenotype (group 4) (Table S2).
There were 21 patients (42.9%) in group 1 (early onset/high tumour burden/high mortality) and 11 (22.4%) in group 4 (late onset/low tumour burden/low mortality). Of the remaining patients, 13 (26.5%) were in group 2 and 4 (8.2%) in group 3 (Table 1).
Cutaneous lesions were more common in patients with severe phenotypes (groups 1/2) than in those with milder disease (groups 3/4) (P < 0.001) (Table 4). Twenty of the 33 patients (60.6%) were in group 1, 10 (30.3%) were in group 2, and 3 (9.1%) were in group 3. The distribution of patients without cutaneous lesions followed the opposite trend. Similar distribution patterns were observed on analysing the cutaneous lesions by category, particularly for lesions assumed to be cutaneous schwannomas, regardless of their clinical form.
Table 4.
Demographic data and presence of skin lesions according to phenotypical severity classification
|
|

Asterisk (*) indicates statistical significance (P < 0.05) in trends (X2) and correlations (r s ) of measures with phenotypical severity classification.
CLM, Café‐au‐lait macules; N, Number; NF2, neurofibromatosis type 2; SD, standard deviation.
Most of the patients with CAL macules had a severe clinical phenotype, with 11 patients (57.9%) in group 1 and 6 (31.6%) in group 2. The distribution of patients without CAL macules was more uniform. The presence of CAL macules was significantly associated with a more severe phenotype (P < 0.05). Tumour lesions were even more strongly associated with a severe phenotype. All the patients with deep tumours were either in group 1 (17 patients, 70.8%) or group 2 (5 patients, 20.8%). The presence of cutaneous tumours was significantly associated with more severe disease (P < 0.001).
Superficial lesions were markedly distributed in the most severe phenotype groups. All 3 patients with superficial tumours were in group 1, while the 21 patients with plaque‐like lesions were either in group 1 (16 patients, 76.2%) or 2 (5 patients, 23.8%). Patients without plaques were more likely to have milder disease. The 23 patients with superficial lesions (tumours or plaques) all had a severe phenotype (group 1 or 2). Those without superficial lesions were more uniformly distributed. Overall, superficial lesions were significantly associated with a more severe phenotype (P < 0.001).
Number of cutaneous lesions (CAL macules, superficial lesions, and deep tumours) was also associated with greater phenotype severity.
The above results indicate that cutaneous lesions, and superficial tumours in particular, are strongly associated with a severe NF2 phenotype. The association in this case was stronger than that observed using the FGSS.
Discussion
Dermatologists are not usually involved in the diagnosis, treatment, or follow‐up of patients with NF2, meaning that skin changes are often underdiagnosed, poorly defined and characterized, and their importance underestimated. Our findings confirm that accurate diagnosis of cutaneous lesions by dermatologists provides valuable diagnostic and prognostic information in the setting of NF2.
About 70% of patients in our cohort had cutaneous lesions, which is similar to rates observed in other series. 1 , 2 , 10
CAL macules are less common in NF2 than in NF1. This is true in terms of the percentage of patients affected—close to 40% in our cohort and others 1 , 2 , 10 (vs. 95% for NF1 26 )—and the number of macules per patient—1.2 in NF2 vs. 6 or more in NF1. CAL macules are paler and have less distinct borders than those of NF1. Their diagnostic value is thus low, due to their relative infrequency and the fact that they are observed in many other diseases, including RASopathies, Fanconi anaemia and tuberous sclerosis. 27 They are also found in healthy individuals. 28 Nevertheless, NF2 should always be suspected in a patient with CAL macules and other manifestations such as mononeuropathy (in particular facial paralysis), eye disorders or meningiomas.
Most studies of NF2 to date have used different systems (based on lesion depth) to classify cutaneous schwannomas, with some authors using 2 categories (superficial lesions/plaques and deep lesions) 1 , 2 and others using 3 (superficial lesions/plaques, intracutaneous lesions and subcutaneous lesions). 10 Based on our findings, we propose 2 main categories: (i) deep schwannomas with histologic features of classic schwannoma or hybrid schwannoma‐neurofibroma that can appear at different times of life and (ii) superficial schwannomas in the form of plaques, nodules or small soft tumours that appear at birth or in childhood and show typical features of a plexiform schwannoma on histology.
Deep schwannomas were observed in 49% of patients in our cohort. While they are clinically non‐specific (they are similar to other subcutaneous lesions, such as lipomas, cysts or fibrohistiocytic tumours), they should always raise suspicion of NF2, especially in patients with multiple lesions. Deep schwannomas, however, can also occur in isolation or in patients with schwannomatosis. Schwannomatosis is caused by mutations in the SMARCB1 or LZTR1 genes (located on chromosome 22 and near the NF2 gene) and is characterized by schwannomas involving the peripheral or cranial nerves or the spine. Unlike NF2, it does not manifest with bilateral vestibular involvement, meningiomas, ependymomas or eye disorders. 29
Superficial schwannomas would appear to be the most useful diagnostic lesions because of their highly characteristic clinical and histological findings. Clinically, they present as nodules, small soft tumours or plaques that are slightly darker than the surrounding skin and have a wrinkled surface, hypertrichosis and a texture similar to that of an atrophic neurofibroma. Histologically, they exhibit the highly characteristic features of a plexiform schwannoma. Superficial schwannomas are relatively common. In our cohort, they were present in 46% of patients, a rate similar to that seen in other series, 1 , 2 and unlike deep schwannomas, most of them had developed in childhood or adolescence. Because they can be easily identified by an experienced dermatologist and subsequently confirmed by histology, they are practically pathognomonic for NF2. Isolated plexiform schwannomas are very rare and have mostly been described in NF2. When they occur in a healthy individual or in a patient with schwannomatosis, they tend to be solitary, late‐onset lesions with an extracutaneous location. 30 NF2, however, must always be suspected on detection of a cutaneous plexiform schwannoma, even if the patient has no other symptoms or family history. The index of suspicion must be particularly high in cases of multiple lesions or paediatric onset.
Cutaneous schwannomas are useful for guiding diagnosis in NF2, but in some cases, they may be key. Patients with mosaicism, for example, do not always have detectable mutations in the blood, but if they have a skin lesion, tissue can be used for genetic testing, thus facilitating diagnosis. In an earlier study by our group, 7 patients were diagnosed with NF2 following the demonstration of 2‐hit mutations in the tissue of cutaneous tumours. 22
Cutaneous lesions can also provide prognostic information. In our cohort, all the patients with superficial schwannomas and microscopic features of plexiform schwannoma, and 91.6% of those with deep schwannomas were in the top 2 phenotype severity groups, while 82.6% of those with superficial schwannomas and 79.2% of those with deep schwannomas were in the top 3 genotype severity classes. The presence of cutaneous plexiform schwannomas, therefore, is correlated with both clinical and genetic severity. The link with clinical severity, however, is particularly strong, probably because genotype‐based classification systems cannot always accurately predict phenotype, particularly in patients with mosaicism. 7 It is reasonable to assume that a patient with severe NF2—and consequently higher tumour burden—will have more cutaneous lesions. Knowing, however, that these lesions appear at a young age facilitates diagnosis and may have prognostic relevance. Early identification of severe disease is crucial in NF2, as early treatment facilitates the management and significantly improves prognosis.
Our study is limited by its low statistical power due to the relatively small study population. Nonetheless, considering the low prevalence and incidence of NF2, a sample size of 49 is not insignificant. Furthermore, ours is the first study to prospectively collect information on cutaneous lesions in patients with NF2 and to perform a detailed case‐by‐case analysis.
In summary, cutaneous lesions are common in NF2 and are useful from both a diagnostic and prognostic perspective, particularly in the case of superficial plexiform schwannomas, which have highly specific clinical and histologic features.
Informed consent
The patients in this manuscript have given written informed consent to publication of their case details.
Supporting information
Table S1. Functional genetic severity score.
Table S2 Phenotype severity classification.
Funding information
None.
Conflict of interest
The authors have no conflict of interest.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 2009; 4: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asthagiri AR, Parry DM, Butman JA et al. Neurofibromatosis type 2. Lancet 2009; 373: 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Selvanathan SK, Shenton A, Ferner R et al. Further genotype‐phenotype correlations in neurofibromatosis 2. Clin Genet 2010; 77: 163. [DOI] [PubMed] [Google Scholar]
- 4. Smith MJ, Higgs JE, Bowers NL et al. Cranial meningiomas in 411 neurofibromatosis type 2 (NF2) patients with proven gene mutations: clear positional effect of mutations, but absence of female severity effect on age at onset. J Med Genet 2011; 48: 261–265. [DOI] [PubMed] [Google Scholar]
- 5. Hexter A, Jones A, Joe H et al. Clinical and molecular predictors of mortality in neurofibromatosis 2: a UKnational analysis of 1192 patients. J Med Genet 2015; 52: 699–705. [DOI] [PubMed] [Google Scholar]
- 6. Halliday D, Emmanouil B, Pretorius P et al. Genetic severity score predicts clinical phenotype in NF2. J Med Genet 2017; 54: 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Catasus N, Garcia B, Galván‐Femenía I et al. Revisiting the UKgenetic severity score for NF2: a proposal for the addition of a functional genetic component. J Med Genet 2021; jmedgenet‐2020‐107548. [DOI] [PubMed] [Google Scholar]
- 8. Ferner RE. The neurofibromatoses. Pract Neurol 2010; 10: 82–93. [DOI] [PubMed] [Google Scholar]
- 9. Halliday D, Emmanouil B, Vassallo G et al. Trends in phenotype in the English paediatric neurofibromatosis type 2 cohort stratified by genetic severity. Clin Genet 2019; 96: 151–162. [DOI] [PubMed] [Google Scholar]
- 10. Mautner VF, Lindenau M, Baser ME, Kluwe L, Gottshaclk J. Skin abnormalities in neurofibromatosis 2. J Arch Dermatol 1997; 133: 1539–1543. [PubMed] [Google Scholar]
- 11. Ruggieri M, Lanetti P, Polizzi A et al. Earliest clinical manifestatios and natural history of neurofibromatosis type 2 (NF2) in childhood: a study of 24 patients. Neuropediatrics 2005; 36: 21–34. [DOI] [PubMed] [Google Scholar]
- 12. Tibussek D, Hübsch S, Berger K, Schaper J, Rosenbaum T, Mavatepek E. Infantil onset neurofibromatosis type 2 presenting with peripheral facial palsy, skin patches, retinal hamartoma and foot drop. Klin pediatr 2009; 221: 247–250. [DOI] [PubMed] [Google Scholar]
- 13. Gugel I, Grimm F, Zipfel J et al. Age at onset and presenting symptoms of neurofibromatosis type 2as prognostic factors for clinical course of vestibular schwannomas. Cancers 2020; 12: 2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Val‐Bernal JF, Figols J, Vazquez‐Barquero A. Cutaneous plexiform schwannoma associated with neurofibromatosis type 2. Cancer 1995; 76: 1181–1186. [DOI] [PubMed] [Google Scholar]
- 15. Reith JD, Goldblum JR. Multiple cutaneous plexiform schwannomas. Reporto f a case and review of the literatura with particular reference to the association with types 1 and 2 neurofibromatosis and schwannomatosis. Arch Pathol Lab Med 1996; 120: 399–401. [PubMed] [Google Scholar]
- 16. Ishida T, Kuroda M, Motoi T, Oka T, Imamura T, Machinami R. Phenotypic diversity of neurofibromatosis 2: association with plexiform schwannoma. Histopathology 1998; 32: 264–270. [DOI] [PubMed] [Google Scholar]
- 17. Lim HS, Jung J, Chung KY. Neurofibromatosis type 2 with multiple plexiform schwannomas. Int J Dermatol 2004; 43: 336–340. [DOI] [PubMed] [Google Scholar]
- 18. Miyakawa T, Kamada N, Kobayashi T et al. Neurofibromatosis type 2 in an infant with multiple plexiform schwannomas as first symptom. J Dermatol 2007; 34: 60–64. [DOI] [PubMed] [Google Scholar]
- 19. Yeon Ko J, Eun Kim J, Hoon Kim Y, Suck RY. Cutaneous plexiform schwannomas in a patient with neurofibromatosis type 2. Ann Dermatol 2009; 21: 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. João Cruz M, Baudrier T, Gil‐Da‐Costa MJ, Azevedo F, Mota A. Multiple cutaneous plexiform schwannomas revealing neurofibromatosis type 2 in a child – report of a novel mutation in this rare association. Eur J Dermatol 2011; 21: 1010–1011. [DOI] [PubMed] [Google Scholar]
- 21. Nguyen TV, Matthews MR, Barrera FF, Browning JC. Multiple cutaneous plexiform schwannomas as initial presentation of neurofibromatosis 2 in a 9‐year‐old. Pediatr Dermatol 2012; 29: 536–538. [DOI] [PubMed] [Google Scholar]
- 22. Castellanos E, Plana A, Carrato C et al. Early genetic diagnosis of Neurofibromatosis type 2 from skin plaque plexiform schwannomas in childhood. JAMA Dermatol 2018; 154: 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Castellanos E, Gel B, Rosas I et al. A comprehensive custom panel design for routine hereditary cancer testing: preserving control, improving diagnostics and revealing a complex variation landscape. Sci Rep 2017; 7: 39348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iwatate K, Yokoo T, Iwatate E et al. Population characteristics and progressive disability in neurofibromatosis type 2. World Neurosurg 2017; 106: 653–660. [DOI] [PubMed] [Google Scholar]
- 25. Baser ME, Friedman JM, Aeschliman S et al. Predictors of the risk of mortality in neurofibromatosis type 2. Am J Hum Genet 2002; 71: 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nunley KS, Gao F, Albers AC, Bayliss SJ, Gutmann DH. Predictive value of caféau lait macules at initial consultation in the diagnosis of neurofibromatosis type 1. Arch Dermatol 2009; 145: 883. [DOI] [PubMed] [Google Scholar]
- 27. Hernandez‐Martin A, Duat‐Rodriguez A. An update on neurofibromatosis type 1: not just café‐au‐lait spots, freckling and neurofibromas. An update. Part 1. Dermatological clinical criteria diagnostic of the disease. Actas Dermosifiliogr 2016; 107: 454–464. [DOI] [PubMed] [Google Scholar]
- 28. Landau M, Krafchik BR. The diagnostic value of caféau‐lait macules. J Am Acad Dermatol 1999; 40: 877. [DOI] [PubMed] [Google Scholar]
- 29. Hadfield KD, Newman WG, Bowers NL et al. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet 2008; 45: 332. [DOI] [PubMed] [Google Scholar]
- 30. Berg JC, Scheithauer BW, Spinner RJ, Allen CM, Koutlas IG. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck región. Hum Pathol 2008; 39: 633–640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Functional genetic severity score.
Table S2 Phenotype severity classification.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.