

Abstract
Objective
This study was undertaken to gain consensus from experienced physicians and caregivers regarding optimal diagnosis and management of Dravet syndrome (DS), in the context of recently approved, DS‐specific therapies and emerging disease‐modifying treatments.
Methods
A core working group was convened consisting of six physicians with recognized expertise in DS and two representatives of the Dravet Syndrome Foundation. This core group summarized the current literature (focused on clinical presentation, comorbidities, maintenance and rescue therapies, and evolving disease‐modifying therapies) and nominated the 31‐member expert panel (ensuring international representation), which participated in two rounds of a Delphi process to gain consensus on diagnosis and management of DS.
Results
There was strong consensus that infants 2–15 months old, presenting with either a first prolonged hemiclonic seizure or first convulsive status epilepticus with fever or following vaccination, in the absence of another cause, should undergo genetic testing for DS. Panelists agreed on evolution of specific comorbidities with time, but less agreement was achieved on optimal management. There was also agreement on appropriate first‐ to third‐line maintenance therapies, which included the newly approved agents. Whereas there was agreement for recommendation of disease‐modifying therapies, if they are proven safe and efficacious for seizures and/or reduction of comorbidities, there was less consensus for when these should be started, with caregivers being more conservative than physicians.
Significance
This International DS Consensus, informed by both experienced global caregiver and physician voices, provides a strong overview of the impact of DS, therapeutic goals and optimal management strategies incorporating the recent therapeutic advances in DS, and evolving disease‐modifying therapies.
Keywords: cannabidiol, developmental and epileptic encephalopathy, disease‐modifying treatment, fenfluramine, SCN1A, stiripentol
Key Points.
Dravet syndrome is an early onset, developmental and epileptic encephalopathy associated with drug‐resistant seizures and multiple comorbidities
Genetic studies are suggested in developmentally normal, 2–15‐month‐old children presenting with a single prolonged hemiclonic seizure or focal/generalized status epilepticus of unknown etiology in the context of vaccination or fever
Valproic acid, clobazam, stiripentol, and fenfluramine may be considered as first‐ or second‐line maintenance therapies for seizures due to DS
Several disease‐modifying therapies are in clinical development; provided these are safe and efficacious, there is consensus for recommending their use in persons with Dravet syndrome
1. INTRODUCTION
Dravet syndrome (DS) is an infantile onset developmental and epileptic encephalopathy associated with drug‐resistant, lifelong seizures and comorbidities including intellectual disability, behavior concerns, sleep disorders, and gait problems. Nearly all cases are due to pathogenic variants in SCN1A that result in haploinsufficiency of Nav1.1, the alpha‐1 subunit of the sodium channel. 1
Prior consensus papers from North America 2 and Europe 3 summarized treatment options; however, these preceded the recent approval of three DS‐specific therapies, pharmaceutical‐grade cannabidiol (Epidiolex, Epydiolex) 4 by the US Food and Drug Administration (FDA) in June 2018 and the European Medicines Agency (EMA) in September 2019, fenfluramine 5 , 6 by the FDA in June 2020 and the EMA in December 2020, and stiripentol, 7 which has been available in many European countries but was approved by the FDA in August 2018.
Furthermore, disease‐modifying therapies (DMTs) are on the horizon. STK‐001, an antisense oligonucleotide that restored Nav1.1 to wild‐type levels and decreased both seizures and mortality in Dravet mice, 8 is currently in human trials. Genetic therapies are also being pursued. ETX‐101, an adenovirus vector containing an engineered transcription factor designed to upregulate SCN1A coupled with a highly conserved, human regulatory sequence to constrain expression to γ‐aminobutyric acid (GABA)ergic inhibitory interneurons, led to significant seizure reduction and reduced risk of sudden death in Dravet mice 9 , 10 and will likely start human trials shortly.
The aims of our study were to gain international consensus from both physicians and caregivers with extensive expertise in DS regarding optimal diagnosis and management, in the context of newly approved therapies. We also aimed to determine the potential role for DMTs, if these are shown to be both safe and highly efficacious.
2. MATERIALS AND METHODS
The concept of this study was proposed by the Executive and Scientific Directors of the Dravet Syndrome Foundation (DSF; M.A.M. and V.H.) and discussed with E.C.W.
2.1. Identification of the core working group
A core working group was convened, consisting of six physicians with recognized expertise in DS, four of whom were members of the DSF Medical Advisory Board (K.G.K., I.E.S., J.S., E.C.W.), and three of whom practiced outside of North America (R.N., I.E.S., J.W.), and two individuals representing the DSF (M.A.M., V.H.). This core working group reviewed the existing literature, created the initial Delphi survey, and nominated physicians and caregivers from around the world for the expert panel.
2.2. Literature review
Five focus areas were identified: (1) clinical presentation (including seizure semiology, electroencephalography [EEG], magnetic resonance imaging [MRI], and genetic studies), (2) comorbidities (cognition, behavior, autism, gait, sleep, other medical concerns, sudden unexpected death in epilepsy [SUDEP] and mortality, vaccinations), (3) maintenance therapies (medications, diet, surgery, neuromodulation), (4) DMTs, and (5) rescue therapies, management of status epilepticus, and transition to adult care. Two physician members of the core working group summarized the literature in each area, through April 2021, and a collated, referenced literature review was sent to the entire core group for feedback. Following revisions, the final summary was provided to each expert panelist before completion of the surveys.
2.3. Establishing the expert panel
Members of the core working group provided nominations for physicians who were clinically recognized for their expertise in the management of DS for the expert panel. Nominees were divided into specific regions with quotas for each region as follows: Europe/UK (n = 6), North America (n = 5), South/Central America (n = 2), Asia (n = 4), Africa (n = 1), and Australia/New Zealand (n = 2). Core working group members anonymously ranked physician nominees from each region, and the top‐ranked candidates were invited to join the expert panel. To enhance diversity, no more than one panelist from each center was included, and we ensured representation from different countries within each region. All physicians accepted the invitation to participate. Members of the core working group (excepting study facilitators E.C.W. and V.H.) were also included as members of the expert panel.
Caregiver expert panelists were selected by the DSF, through their connections with other international DS patient advocacy groups, and included three from Europe/UK, three from North America, two from Asia, and one each from South America, Africa, and Australia/New Zealand.
In the first Delphi round, each expert panelist was asked how long they had worked with/cared for persons with DS. Physicians were asked the number of persons with DS they had ever and were currently managing and whether they saw only children, only adults, or both. Caregivers were asked how many people with DS they were familiar with.
2.4. Delphi questionnaires
A two‐round Delphi process 11 was utilized. The first questionnaire was created by the study facilitators based on literature review and feedback from the core working group and was sent to the expert panel using a Survey Monkey link. Panelists were instructed to answer questions based on both the literature review and their own expertise and were given 4 weeks to complete each round with two reminders sent, as needed.
Caregivers and physicians received similar Delphi questionnaires; however, topics focusing on specific laboratory study results were sent to physicians only.
The first round was comprised of:
Statements where the literature suggested consensus. Panelists rated their overall agreement ranging from 1 to 9, where 1 is strongly disagree and 9 is strongly agree, with an option of "no opinion." Free text comments were encouraged, particularly for any statements rated as 6 or lower.
Open‐ended questions. Panelists were asked to estimate the proportion of cases they were aware of that manifested specific features, and to provide free text answers to specific questions (criteria for defining seizure control, when medication should be changed, optimal first‐ and second‐line therapies, experience with surgical therapies, neuromodulation, anticipated benefits of DMTs, transition to adult care).
Rating of specific medications based on their efficacy for certain seizure types, tolerability, and durability of response.
The study facilitators collated results, evaluated areas where consensus was not achieved in Round 1, refined statements based on panelist comments (where appropriate), and included these in Round 2. Additionally, based on the open‐ended questions from Round 1, they proposed several additional statements in Round 2.
Consensus was determined only for statements when more than half of the target group provided responses, defined as at least 11 physicians and at least five caregivers. Absent responses or “no opinion” were grouped and considered as "no response." Consensus was defined as Strong if 80% or more of panelists providing an opinion rated the statement as 7 or higher and as Moderate if 67% or more of panelists rated the statement as 7 or higher. Statements that did not reach this level of agreement were interpreted as "no consensus."
3. RESULTS
All physicians (n = 20) and nine of 11 caregivers participated in both Delphi rounds. One caregiver participated in the first round only, and another did not participate in either. Ninety percent of physicians and 89% of caregivers had >10 years, and all had >5 years of experience with DS. The proportion of cases that panelists cared for is shown in Figure 1. Of physicians, 15 (75%) saw predominantly children, four (20%) saw both adults and children, and one (5%) saw only adults.
FIGURE 1.
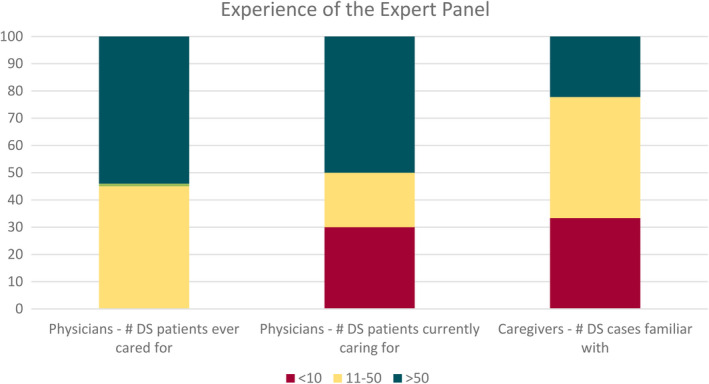
Expertise of the physician and caregiver panel. Shown are the percentage of physicians who have ever cared for or are currently caring for <10, 11–50, or >50 persons with Dravet syndrome (DS) and the percentage of caregivers who are familiar with <10, 11–50, or >50 persons with DS
3.1. Clinical presentation, seizures, and diagnostic testing
Table 1 summarizes where consensus was reached regarding clinical presentation of DS. Genetic studies should be performed in a developmentally normal, 2–15‐month‐old child presenting with a single prolonged (5–29 min) hemiclonic seizure or focal/generalized status epilepticus (≥30 min) of unknown etiology in the context of vaccination or fever (Strong). There was Moderate consensus for genetic testing with (1) a single prolonged generalized tonic–clonic seizure in a child aged 2–5 months associated with fever or vaccination; (2) a single, prolonged generalized convulsive seizure (5–29 min) in a child aged 6–15 months following vaccination; or (3) a single episode of afebrile convulsive status epilepticus or a single, prolonged afebrile hemiclonic seizure in a child aged 2–15 months.
TABLE 1.
Clinical presentation: in a developmentally normal child, who presents with seizures of unknown cause (normal magnetic resonance imaging, normal laboratory studies, ± normal cerebrospinal fluid studies), genetic testing to exclude DS should be performed with the following seizure types
| Seizure | Age 2–5 months | Age 6–15 months | ||||
|---|---|---|---|---|---|---|
| Without fever | With fever | After vaccination | Without fever | With fever | After vaccination | |
| Single seizure | ||||||
| Prolonged (5–29 min) GTCS | 63% a | 74% b | 74% b | 63% a | 58% a | 68% b |
| Prolonged (5–29 min) hemiclonic seizure | 68% b | 84% c | 95% c | 68% b | 84% c | 89% c |
| Focal or generalized convulsive status epilepticus (≥30 min) | 74% b | 84% c | 89% c | 74% b | 84% c | 89% c |
| Recurrent seizures | ||||||
| Recurrent brief (<5 min) convulsive seizures | 63% a | 58% a | 58% a | 58% a | ||
| Recurrent brief (<5 min) hemiclonic seizures | 68% b | 74% b | 79% b | 79% b | ||
| Recurrent prolonged focal or generalized convulsive seizures (5–29 min) | 89% c | 100% c | 84% c | 95% c | ||
| Recurrent focal or generalized convulsive status epilepticus (≥30 min) | 89% c | 95% c | 89% c | 95% c | ||
Based on 19 physician responses.
Abbreviations: DS, Dravet syndrome; GTCS, generalized tonic–clonic seizure.
Responses indicate no consensus for genetic testing for DS.
Responses indicate Moderate consensus for genetic testing for DS.
Responses indicate Strong consensus for genetic testing for DS.
In children presenting with recurrent seizures of unknown etiology, genetic testing is indicated in those 2–15 months old with recurrent prolonged focal or generalized convulsive seizures with or without fever (including status epilepticus; Strong), in children 6–15 months old with recurrent brief hemiclonic seizures without fever (Strong), and in infants 2–15 months old with recurrent, brief, hemiclonic seizures with or without fever (Moderate).
Table 2 summarizes consensus regarding seizure types, evolution with time, and diagnostic studies. Myoclonic and focal impaired awareness seizures are seen in more than half of cases before age 5 years (Strong to Moderate). Although some seizures abate with time, brief generalized tonic–clonic seizures persist in most adults (Strong).
TABLE 2.
Consensus regarding seizure types and evolution with time and diagnostic testing
|
Other seizure types: frequency and age at presentation Myoclonic seizures
Absence seizures
Focal impaired awareness seizures
Atonic seizures
Tonic seizures
Nonconvulsive (obtundation) status epilepticus
|
|
Persistence of seizure types into adulthood
|
|
Genetic testing
|
|
Neuroimaging
|
|
EEG
|
Bold and all‐capital text indicates Strong consensus; bold and italic text indicates Moderate consensus; nonbold and italic text indicates no consensus.
Abbreviations: DS, Dravet syndrome; EEG, electroencephalography; MRI, magnetic resonance imaging.
Consensus was not determined for statements where >50% of the group did not provide a response.
SCN1A pathogenic variants are present in >85% of cases (Strong). The initial MRI is normal, but a minority show variable degrees of cortical atrophy or hippocampal sclerosis with time (Strong). The EEG is often normal before 12 months of age but demonstrates background slowing and epileptiform discharges in most cases by age 5 years (Strong). “Falsely generalized” and “unstable” recorded seizures are unique ictal patterns (Strong).
3.2. Comorbidities, vaccination recommendations, and transition to adult care
Development is considered normal before 18 months, although subtle delays may be appreciated prior to that time (physicians: Strong; caregivers: Moderate), but intellectual disability is usually present by 3 years of age and becomes more apparent with time (physicians and caregivers: Strong; Table 3).
TABLE 3.
Comorbidities, development, vaccination recommendations, and transition
|
Development
|
|
Attention problems, autism, and behavior problems
|
|
Gait problems and parkinsonian features
|
|
Speech
|
|
Sleep problems
|
|
Autonomic dysfunction
|
|
SUDEP
|
|
Vaccinations
|
|
Transition of care to adult neurology
|
Bold and all‐capital text indicates Strong consensus; bold and italic text indicates Moderate consensus; nonbold and italic text indicates no consensus.
Abbreviations: DS, Dravet syndrome; EEG, electroencephalogram; IQR, interquartile range; SUDEP, sudden unexpected death in epilepsy.
Consensus was not determined for statements where >50% of the group did not provide a response.
Attention problems are present in most children by school age (physicians and caregivers: Strong) and psychostimulants are considered both safe (physicians: Strong) and effective (physicians: Moderate). Internalizing problems such as depression and anxiety are more prevalent with increasing age and present in most adults (physicians and caregivers: Strong). There was disagreement between physicians and caregivers regarding the prevalence of autistic features; caregivers reported such symptoms in a majority of children (Strong), whereas physicians indicated that most lacked autistic features (Moderate).
Gait problems (ataxia or crouch gait) are seen in half of school‐aged children and most teens and young adults (Strong) and may resemble Parkinsonian features in adulthood. There was limited consensus on optimal management, with only modest benefits reported for physiotherapy or carbidopa–levodopa.
Sleep problems occur in most persons (Strong); however, optimal management is less clear. Most panelists had experience using melatonin, a minority with clonidine, and very small numbers with other agents. Both melatonin and clonidine were reported to be modestly effective by most respondents.
Families of persons with DS must be counseled about the significant risk of SUDEP at the time of diagnosis (Strong). Caregivers reported higher patient use of seizure‐monitoring devices than physicians. The effectiveness of such devices to detect seizures was rated as 7 (interquartile range [IQR] = 6–9) and 6 (IQR = 5–7) by caregivers and physicians, respectively, on a scale of 1–9, where 1 is ineffective and 9 is highly effective.
Persons with DS should receive all routine vaccines (physicians: Strong; caregivers: Moderate), an annual influenza vaccine (physicians and caregivers: Moderate), and the COVID‐19 vaccination (physicians: Strong; caregivers: Moderate). Antipyretics are recommended to reduce risk of vaccine‐associated fever (Strong), but there was no consensus among physicians on the use of additional seizure medications around vaccination, and moderate consensus among caregivers against this practice.
Regarding transition of care to an adult provider, both physicians and caregivers identified the importance of a knowledgeable adult provider, clear communication between pediatric and adult providers around the time of transition, family education with a focus on progressive transition, and a comprehensive transition document prepared by the pediatric provider (Strong). Barriers to successful transition included lack of adult providers with expertise in DS (Strong), lack of appropriate structure in the adult setting to provide holistic care (Strong), limited involvement of parents or caregivers in clinical decisions on the adult side (caregivers: Moderate; physicians: no consensus), and reluctance of families to transition as they are bonded to the pediatric team (physicians: Moderate; caregivers: no consensus).
3.3. Seizure control, maintenance therapies, disease‐modifying treatment, and management of seizure emergencies
3.3.1. Goals for seizure control
Important goals of seizure control are to maximize quality of life for the patient and their family (Strong) and to limit side effects of medication (Strong; Table 4). Control of convulsive seizures should be prioritized over nonconvulsive seizures, given their greater impact on quality of life and higher association with SUDEP (physicians: Strong; caregivers: no consensus).
TABLE 4.
Seizure control endpoints, maintenance therapies, and management of status epilepticus
|
Goals for seizure control
|
|
Factors that should prompt consideration of a new therapy
|
|
Factors that should prompt consideration of tapering off medication, other than intolerable side effects
|
|
Maintenance ASMs
Lamotrigine
Non‐pharmaceutical grade medical marijuana
Drug–drug interactions
Fenfluramine
|
|
Medications that impact appetite
|
|
Bloodwork monitoring while on ASMs Valproic acid
Cannabidiol
Stiripentol
|
|
Dietary therapy
|
|
Vagus nerve stimulation and epilepsy surgery
Other epilepsy surgical procedures
|
|
Disease‐modifying therapies Assuming a disease‐modifying therapy was proven to be safe, there were no barriers to access, and cost was not prohibitive:
|
|
Seizure emergencies
Status epilepticus (physicians only)
|
Bold and all‐capital text indicates Strong consensus; bold and italic text indicates Moderate consensus; nonbold and italic text indicates no consensus.
Abbreviations: ASM, antiseizure medication; CBC, complete blood count; CBD, cannabidiol; DS, Dravet syndrome; IQR, interquartile range; IV, intravenous; SUDEP, sudden unexpected death in epilepsy; THC, tetrahydrocannabinol.
3/9 caregivers who did not agree commented that certain ASMs may lead to improvement in comorbidities, and that alone may be reason to continue them even if seizures are not improved.
Consensus was not determined for statements where >50% of the group did not provide a response.
There was general agreement between physicians and caregivers regarding when a new therapy should be considered. A medication deemed to be less effective or to have led to greater side effects should be tapered if a prolonged period of seizure freedom is achieved (physicians: Strong; caregivers: no consensus).
3.3.2. Maintenance therapies
Valproic acid is an appropriate first‐line drug, and clobazam can be considered as either the initial or second antiseizure medication (ASM; Strong). Further consensus for other first‐line therapies included fenfluramine (physicians: Strong; caregivers: Moderate), and stiripentol (physicians: Moderate; caregivers: Strong). Pharmaceutical‐grade cannabidiol was supported either as first‐ or second‐line treatment (caregivers: Strong; physicians: no consensus). There was modest consensus among caregivers, but no consensus among physicians to support topiramate as first‐, second‐, or third‐line therapy. Lamotrigine is contraindicated in children with DS (Moderate). Figure 2 summarizes consensus regarding therapy.
FIGURE 2.
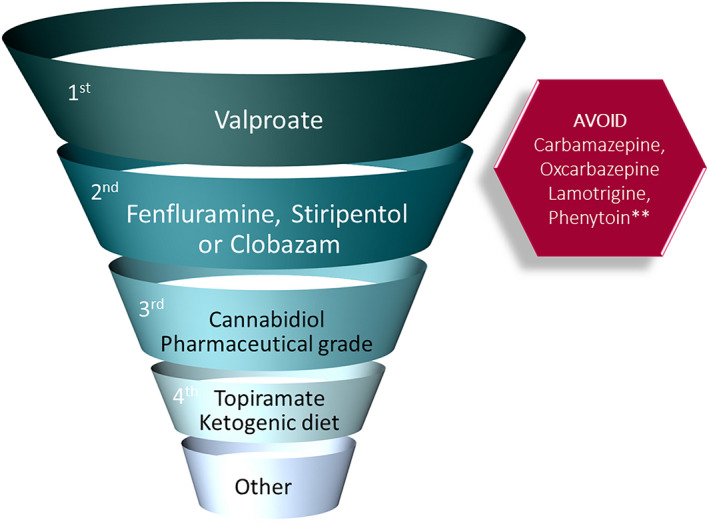
Therapeutic algorithm for maintenance therapies for management of seizures in Dravet syndrome. There was consensus for use of valproic acid as first‐line therapy, and for use of clobazam, fenfluramine, or stiripentol as first‐ or second‐line therapy. There was also consensus for contraindicated medications. **Phenytoin may be helpful for status epilepticus. "Other" includes vagal nerve stimulation, levetiracetam, zonisamide, bromides, clonazepam, and ethosuximide (for absences)
Based on physician ratings (Table S1), valproic acid, clobazam, stiripentol, and fenfluramine were perceived as most efficacious for focal or generalized convulsive seizures. For absence seizures, both valproic acid and ethosuximide rated highly, and for myoclonic seizures, valproate was rated most efficacious. Physicians and caregivers were also asked to rate tolerability of ASMs, on a scale of 1–9, where 1 is poorly tolerable and 9 is highly tolerable (Table S2). Therapies with good tolerability (rated as 7 or higher) by both physicians and caregivers included valproic acid and fenfluramine. Therapies most commonly associated with improved alertness and/or behavior included ketogenic diet (73% improved), fenfluramine (54% improved), and pharmaceutical‐grade cannabidiol (50% improved; Table S3). Conversely, those most correlated with worsening alertness and/or behavior included clobazam (78% worsened), topiramate (68% worsened), clonazepam (61% worsened), and levetiracetam (52% worsened).
Dietary therapy should be considered after failure of three or four ASMs (Strong; Table 4). The classical ketogenic diet was recommended for children 6 years and younger (Moderate) and the modified Atkins diet for teens and adults (Strong).
Interestingly, there was moderate consensus among physicians and caregivers that specific therapies stood out from others due to higher efficacy and/or better tolerability, and among those who responded affirmatively to this statement, fenfluramine was the only therapy with consensus (Strong).
Regarding artisanal marijuana, there was strong consensus from physicians, but no consensus from caregivers against the recommendation of nonpharmaceutical grade cannabidiol. Only a minority of respondents indicated they had experience with the use of low‐dose tetrahydrocannabinol, with none reporting efficacy.
Although vagus nerve stimulation was considered a therapeutic option, there was Strong consensus that valproic acid, clobazam, stiripentol, and ketogenic diet, and Moderate consensus that fenfluramine, cannabidiol, and topiramate should be trialed prior to such therapy. Vagus nerve stimulation typically results in a <50% reduction in seizures (Strong) and use of the magnet has a low to modest impact on stopping seizures. Corpus callosotomy has no therapeutic role in DS (Moderate), and temporal lobectomy should not be considered (Strong).
Figure S1 documents the proportion of physicians who had personal experience using the various therapies for DS.
3.3.3. Disease‐modifying therapies
Assuming DMTs are safe, there was universal consensus among physicians and caregivers for recommendation of a DMT that results in greater seizure reduction than the current best therapy and reduces cognitive and other comorbidities. In such cases, there was consensus from physicians (Moderate), but not caregivers, that this should be started as early as possible.
If a DMT resulted in greater seizure reduction than the current best therapy, but had no impact on comorbidities, there was still consensus for recommendation (physicians: Strong; caregivers: Moderate), but no consensus for first‐line use.
Conversely, if a DMT reduced cognitive and other comorbidities, but had no greater impact on seizure control than the current best therapy, there was still consensus to recommend this therapy (physicians: Strong; caregivers: Moderate), and consensus from only physicians to start as soon as possible (Moderate).
There was consensus that neither current seizure frequency nor degree of intellectual disability in a preschool or early school‐aged child should impact the decision to offer DMTs. Additionally, if DMTs were proven efficacious and safe in clinical trials in younger patients, there was consensus from physicians (Moderate), but not caregivers, to consider their use in persons too old to qualify for the original clinical trial.
3.3.4. Management of seizure emergencies
All persons with DS need a home rescue medication (Strong). For prolonged convulsive seizures that persist despite benzodiazepines, either intravenous valproate or levetiracetam should be the next therapeutic choices (Strong). Intravenous phenytoin or fosphenytoin could be considered after these agents if the seizure persists (Strong).
4. DISCUSSION
The recommendations in this paper reflect those of the first International DS Consensus, which included both expert physicians and caregivers, from all continents. Our methodology utilized the Delphi methodology, a rigorous process to gather consensus regarding a disease. 11
Over the past 10 years, DS has been diagnosed at younger ages, due to improved awareness by child neurologists and increased access to genetic testing, which is becoming part of routine clinical care in many regions. 12 , 13 Expedient diagnosis is critical to avoid contraindicated therapies that may exacerbate seizures and negatively impact development, 14 and importantly, to allow timely access to DMTs, if these are shown to be efficacious and safe in clinical trials. SCN1A variants can present with a range of epilepsy phenotypes, and several prediction models combining both clinical and genetic information have been developed that allow more confident early diagnosis of DS. 1 , 15 There was consensus from our expert panel regarding clinical presentations that should mandate genetic testing for DS, including infants 2–15 months old with either a first prolonged hemiclonic seizure or convulsive status epilepticus of unknown cause, with fever or following vaccination. Although we found strong consensus for where a diagnosis of DS should be considered, this does not preclude testing in other settings, as DS can present up to 19 months of age, with afebrile brief seizures and in the setting of developmental delay. 16
Although there was strong consensus that specific comorbidities evolve with time, including intellectual disability, attention problems, gait abnormalities, and sleep problems, agreement on their optimal management was limited. There was only moderate consensus from physicians, and no consensus from caregivers, that psychostimulants were effective for attention problems. Whereas most panelists reported modest benefit with melatonin for sleep disorders, 17 the majority lacked experience with other agents. Further work is needed to define optimal management strategies for these comorbidities, which markedly impact quality of life.
We found several areas in which caregivers and physicians expressed discordant opinions, with caregivers reporting higher rates of both autistic features and externalizing behaviors than physicians. The prevalence of autism in studies of children with DS that employed formal autism‐specific instruments ranges from 22% to 46%. 18 However, autism appears to be underrecognized, as in a recent study, clinically significant social communication deficits were noted in 67%, of whom only 44% had been diagnosed with autism. 19 These findings suggest that behavioral comorbidities in children with DS may be underdiagnosed and untreated, despite their significant negative impact on quality of life. These findings support the need for periodic formal evaluation by neuropsychology, developmental pediatrics, or child psychiatry for children with DS.
In the 5 years since the North American consensus publication, there have been three new ASMs approved for the treatment of seizures associated with DS, each with robust, class 1 evidence documenting efficacy and well‐described side effect profiles. 4 , 5 , 6 , 7 These studies allow clinicians to have data‐driven discussions with families about expected outcomes regarding seizure reduction and long‐term durability of this response. The demonstrated efficacy of these “DS‐specific” medications strongly supports their use earlier in the treatment paradigm.
Most young children with suspected or genetically proven DS are still started on a more conventional ASM as opposed to one of these “DS‐specific” medications. In many ways, this is justified and practical due to lack of access to and the expense of such therapies. It can be challenging to start one of the newer DS‐specific ASMs, as they only have labeling as adjunctive therapy from 1 or 2 years of age. Patients with DS often have some beneficial response to a conventional ASM; the decision to add or change therapy should be predicated on the overall goal of minimizing seizures and side effects, acknowledging that for most patients, complete seizure freedom remains unrealistic. To automatically switch to one of the DS‐specific medications may not be indicated if the patient is controlled on their current regimen. However, we should redefine our expectations of seizure control, and no longer accept seizures every 1–2 months as the best we can do.
A rescue plan for seizure emergencies remains an important part of a comprehensive treatment plan. Although there was widespread consensus regarding the early use of benzodiazepines for convulsive seizures, the specific agents and formulations should be tailored to each patient. Some patients may respond to one benzodiazepine more favorably than another, and these observations remain important in arriving at an individualized seizure action plan. This should include a home rescue plan followed by an emergency department plan, and for the latter, despite phenytoin's primary mechanism of action being a sodium channel blocker, there was consensus that phenytoin/fosphenytoin is not contraindicated as a treatment for status epilepticus.
Although treatment is often focused on seizure reduction, both clinicians and caregivers agree that non‐seizure‐related comorbidities must also be addressed. Thankfully, some of these recent DS‐specific therapy trials have also demonstrated improvements in non‐seizure‐related outcomes such as executive function, 20 and it remains to be seen whether earlier use of “DS‐specific” ASMs will have more favorable impact on long‐term outcomes. Although a recent study suggested that fenfluramine may be associated with reduced risk of SUDEP, 21 further research is needed.
As comorbidities remain an important determinant of overall quality of life, DMTs that go beyond seizure management are needed. These types of therapeutics are in various stages of development, and although their perceived role is speculative, our findings highlight the current climate and state of the art looking forward. One clinical trial is underway with an antisense oligonucleotide (ASO) that, in an SCN1A mouse model, restored the haploinsufficient state to that of wild type, resulting in reduced incidence of seizures and SUDEP. 8 This specific ASO is being studied in multiple ascending doses in children 2–18 years of age with DS caused by pathogenic SCN1A variants (ClinicalTrials.gov identifier: NCT04442295) and has the potential not only to reduce seizures but also to improve other comorbidities. There was universal consensus to recommend a DMT if it addresses both comorbidities and seizures; however, in that scenario, there was only Moderate consensus from physicians to prescribe such therapy as soon as possible. This is likely due to the lack of clinical safety and efficacy outcome data in this first in‐human trial; perceptions and opinions will likely evolve as more data become available. Future data will also shed light on whether there is an optimal age to intervene with a DMT. Within the DS community, there has been concern regarding the intrathecal route of administration and potential exclusion from participation in future trials should gene therapy not work, which may have led to greater hesitation from caregivers. Taken altogether, these developments are moving toward a precision‐based treatment approach for persons with DS, and importantly today, clinicians and caregivers should never feel it is too late to optimize care using all available treatments.
Transition to adult care is important; adult patients with DS develop additional adult medical issues that pediatricians are poorly equipped to manage. However, panelists noted numerous barriers to this process. There is a critical need for knowledgeable adult providers and specialized clinics focused on adults with developmental and epileptic encephalopathies, such as DS, to assume this role. A recent transition guide to help with transition from pediatric to adult practice for persons with DS has been published. 22
One of the most significant limitations of our study was that not all new therapies are approved worldwide and current trials of DMTs are being performed in a limited number of settings. The use and order of selection of ASMs is based on which drugs are available in each expert's country and patient population, so each expert's experience is understandably colored by accessibility issues. Furthermore, as DMTs are still in clinical trials, opinions on their utility are purely theoretical. A further limitation, implicit to all Delphi processes, is that conclusions may not be based on the most recent evidence, but rather the consensus view. It takes time for novel scientific insights to filter to the clinical domain if they differ from current teaching.
We believe that this International DS Consensus, informed by both experienced global caregiver and physician voices, provides a strong overview of the impact of DS, therapeutic goals, and optimal management strategies, taking into account the recent therapeutic advances and evolving DMTs. We hope these results will impact clinical practice by identifying who to screen and how to manage seizures and comorbidities to improve outcomes in persons with DS.
CONFLICT OF INTEREST
E.C.W. has served as a paid consultant for Encoded Therapeutics, Eisai, Epygenix, and BioMarin. She is Editor‐in‐Chief of Epilepsy.com. K.G.K. has received research funding from Zogenix, Encoded, Eisai, and West Pharmaceuticals. She has participated on data and safety monitoring boards for GW Pharmaceuticals and Epygenix, and has received consulting funds from BioMarin, Zogenix, Encoded, Eisai, Stoke, and Biocodex. R.N. has served as principal investigator in clinical trials for Novartis, Nutricia, Eisai, UCB, GW Pharma, and LivaNova. She has received consulting and lecturer honoraria from Biogene, BioMarin, Praxis, GW Pharma, Zogenix, Novartis, Nutricia, Stoke, Ionis, Targeon, Neuraxpharma, Takeda, Nutricia, Biocodex, Advicennes, and Eisai. She has received unrestricted research grants from Eisai, UCB, LivaNova, and GW Pharma and academic research grants from EJP‐RD (Horizons 2020). I.S. has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Chiesi, Encoded Therapeutics, Knopp Biosciences, and Xenon Pharmaceuticals; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, Chiesi, LivaNova, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, BioMarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Xenon Pharmaceuticals, Anavex Life Sciences, Ovid Therapeutics, Epygenix, Encoded Therapeutics, and Marinus; has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Care Beyond Diagnosis, Epilepsy Consortium, and UCB; and is a Non‐Executive Director of Bellberry. She may accrue future revenue on pending patent WO61/010176 (filed 2008): Therapeutic Compound; has a patent for SCN1A testing held by Bionomics and licensed to various diagnostic companies; has a Patent Molecular Diagnostic/Theranostic Target for Benign Familial Infantile Epilepsy (BFIE) (PRRT2) 2011904493 & 2012900190 and PCT/AU2012/001321 (TECH ID: 2012‐009). J.W. has received an honorarium for activities as Associate Editor for Epilepsia. J.S. has served as a paid consultant for the Epilepsy Study Consortium, Encoded Therapeutics, Greenwich Biosciences, Epygenix Therapeutics, Invitae, and Longboard, and has stock options in Epygenix. Neither of the other authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Supporting information
Fig S1
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
This study was supported by the Dravet Syndrome Foundation. We thank our caregiver experts, including Galia Wilson (UK), Simona Borroni (Italy), Renata Jungman (Switzerland), Sally Sun (China), Felipe Carniero (Brazil), Jessica Reekie (South Africa), Guida Clozza (Canada), Mary Anne Meskis (USA), and Nicole Villas (USA). We thank our physician expert panelists, including Drs J. H. Cross (Pediatric Epilepsy, UK), A. Brunklaus (Pediatric Epilepsy, Genetics, UK), L. Lagae (Pediatric Epilepsy, Belgium), N. Specchio (Pediatric Epilepsy, Italy), A. Strelczyk (Pediatric and Adult Epilepsy, Germany), R. Caraballo (Pediatric Epilepsy, Argentina), A. Coan (Pediatric Epilepsy, Brazil), K. P. Vinayan (Pediatric and Adult Epilepsy, India), S. Sabbagh (Pediatric Epilepsy, Lebanon), M. Kato (Pediatric Epilepsy, Japan), H. Zhang (Pediatric Epilepsy, China), L. Sadleir (Pediatric Epilepsy, New Zealand), D. Andrade (Adult Epilepsy with focus on Developmental and Epileptic Encephalopathies, Canada), L. Laux (Pediatric and Adult Epilepsy, USA), and S. Perry (Pediatric Epilepsy, USA).
Wirrell EC, Hood V, Knupp KG, Meskis MA, Nabbout R, Scheffer IE, et al. International consensus on diagnosis and management of Dravet syndrome. Epilepsia. 2022;63:1761–1777. 10.1111/epi.17274
REFERENCES
- 1. Cetica V, Chiari S, Mei D, Parrini E, Grisotto L, Marini C, et al. Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology. 2017;88:1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American consensus panel. Pediatr Neurol. 2017;68:18–34.e3. [DOI] [PubMed] [Google Scholar]
- 3. Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, Lagae L. Dravet syndrome: treatment options and management of prolonged seizures. Epilepsia. 2019;60(Suppl 3):S39–48. [DOI] [PubMed] [Google Scholar]
- 4. Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011–20. [DOI] [PubMed] [Google Scholar]
- 5. Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2019;394:2243–54. [DOI] [PubMed] [Google Scholar]
- 6. Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil‐Nagel A, Sanchez‐Carpintero R, et al. Fenfluramine for treatment‐resistant seizures in patients with Dravet syndrome receiving stiripentol‐inclusive regimens: a randomized clinical trial. JAMA Neurol. 2020;77:300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chiron C, Marchand MC, Tran A, Rey E, d'Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo‐controlled syndrome‐dedicated trial. STICLO Study Group. Lancet. 2000;356:1638–42. [DOI] [PubMed] [Google Scholar]
- 8. Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12:eaaz6100. [DOI] [PubMed] [Google Scholar]
- 9. Young AN, Tanenhaus A, Belle A, McLaughlin J, Li J, Lin W. A GABA‐selective AAV vector upregulates endogenous SCN1A expression and reverses multiple phenotypes in a mouse model of Dravet syndrome. Paper presented at: American Epilepsy Society Annual Meeting; December 2019; Baltimore, Maryland.
- 10. Belle A, McLaughlin J, Li J, Lucey G, Chou T. ETX101, a GABAergic interneuron selective AAV‐mediated gene therapy for the treatment of SCN1A+ Dravet syndrome: biodistribution and safety in non‐human primates. Paper presented at: American Epilepsy Society Annual Meeting; December 2020; virtual.
- 11. Dalkey N, Helmer O. An experimental application of the DELPHI method to the use of experts. Manage Sci. 1963;9:458–67. [Google Scholar]
- 12. Howell KB, Freeman JL, Mackay MT, Fahey MC, Archer J, Berkovic SF, et al. The severe epilepsy syndromes of infancy: a population‐based study. Epilepsia. 2021;62:358–70. [DOI] [PubMed] [Google Scholar]
- 13. Symonds JD, Elliott KS, Shetty J, Armstrong M, Brunklaus A, Cutcutache I, et al. Early childhood epilepsies: epidemiology, classification, aetiology, and socio‐economic determinants. Brain. 2021;144:2879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Lange IM, Gunning B, Sonsma ACM, van Gemert L, van Kempen M, Verbeek NE, et al. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A‐related seizure phenotypes. Epilepsia. 2018;59:1154–65. [DOI] [PubMed] [Google Scholar]
- 15. Brunklaus A, Perez‐Palma E, Ghanty I, Xinge J, Brilstra E, Ceulemans B, et al. Development and validation of a prediction model for early diagnosis of SCN1A‐related epilepsies. Neurology. 2022;98:e1163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li W, Schneider AL, Scheffer IE. Defining Dravet syndrome: an essential pre‐requisite for precision medicine trials. Epilepsia. 2021;62:2205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Myers KA, Davey MJ, Ching M, Ellis C, Grinton BE, Roten A, et al. Randomized controlled trial of melatonin for sleep disturbance in Dravet syndrome: the DREAMS study. J Clin Sleep Med. 2018;14:1697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jansson JS, Hallbook T, Reilly C. Intellectual functioning and behavior in Dravet syndrome: a systematic review. Epilepsy Behav. 2020;108:107079. [DOI] [PubMed] [Google Scholar]
- 19. Brown A, Arpone M, Schneider AL, Micallef S, Anderson VA, Scheffer IE. Cognitive, behavioral, and social functioning in children and adults with Dravet syndrome. Epilepsy Behav. 2020;112:107319. [DOI] [PubMed] [Google Scholar]
- 20. Bishop KI, Isquith PK, Gioia GA, Gammaitoni AR, Farfel G, Galer BS, et al. Improved everyday executive functioning following profound reduction in seizure frequency with fenfluramine: analysis from a phase 3 long‐term extension study in children/young adults with Dravet syndrome. Epilepsy Behav. 2021;121:108024. [DOI] [PubMed] [Google Scholar]
- 21. Cross JH, Galer BS, Gil‐Nagel A, Devinsky O, Ceulemans B, Lagae L, et al. Impact of fenfluramine on the expected SUDEP mortality rates in patients with Dravet syndrome. Seizure. 2021;93:154–9. [DOI] [PubMed] [Google Scholar]
- 22. Andrade DM, Berg AT, Hood V, Knupp KG, Koh S, Laux L, et al. Dravet syndrome: a quick transition guide for the adult neurologist. Epilepsy Res. 2021;177:106743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Table S2
Table S3