

Abstract
Oxygen is a critical gas for medical and industrial settings. Much of today's global oxygen supply is via inefficient technologies such as cryogenic distillation, membranes or zeolites. Metal–organic frameworks (MOFs) promise a superior alternative for oxygen separation, as their fundamental chemistry can in principle be tailored for reversible and selective oxygen capture. We evaluate the characteristics for reversible and selective uptake of oxygen by MOFs, focussing on redox‐active sites. Key characteristics for separation can also be seen in MOFs for oxygen storage roles. Engineering solutions to release adsorbed oxygen from the MOFs are discussed including Temperature Swing Adsorption (TSA), Pressure Swing Adsorption (PSA) and the highly efficient Magnetic Induction Swing Adsorption (MISA). We conclude with the applications and outlooks for oxygen capture, storage and release, and the likely impacts the next generation of MOFs will have on industry and the broader community.
Keywords: Adsorption, Metal–Organic Frameworks, Oxygen, Separation, Storage
Metal–organic frameworks (MOFs) are emerging materials for oxygen capture and storage with significant benefits for the broader community. This Minireview assesses and discusses the fundamental chemistry for both selective and reversible MOFs for the role of oxygen capture from air. In addition, we describe the current state of play for oxygen storage by MOFs, as well as engineering solutions to aid in the release of oxygen once captured.

1. Introduction
The isolation of pure oxygen has been a critical goal for the medical, chemical and water industries for over a century. Presently, more than 100 million Tons are isolated annually, [1] and most commonly employ cryogenic separation methods, exploiting the small difference in boiling point between liquid oxygen and nitrogen to concentrate oxygen directly from the atmosphere. [2] Whilst cryogenic separations are very mature and their energy requirements optimised, the requisite temperatures place fundamental limitations upon the operating efficiencies and portability of oxygen concentration processes. Future technologies to replace cryogenic separations would ideally be low cost and low power consumption in addition to scalable. In this respect membranes offer a promising route to the delivery of oxygen. [3] Ceramic membranes offer exceptional selectivity for oxygen over nitrogen, but at elevated temperatures,[ 4 , 5 , 6 ] and polymer membranes offer moderate selectivity under ambient conditions.[ 7 , 8 , 9 ] Separations with polymer membranes have the potential to be very energy efficient. Adsorptive separation techniques have emerged, principally employing zeolites as porous separation media.[ 10 , 11 , 12 ] At elevated pressures, these materials adsorb nitrogen over oxygen, which is released through the adsorbent bed for use. Adsorption of the major component in air places a higher limit on the overall efficiency of such a separation, with more adsorbent and more compression required to deliver the same amount of oxygen when compared to what might be attainable should the minor component, oxygen, be able to be captured in preference. Metal‐organic frameworks (MOFs)[ 13 , 14 , 15 , 16 ] offer a new approach to oxygen concentration. The tunable surface chemistry within their pores offers the potential to selectivity adsorb oxygen, reversing the current state of the art in zeolites and lowering the work requirement on the adsorbent bed. In concert with the higher surface areas delivering increased working capacity, a suitable MOF should deliver a step change in oxygen capture performance and make several new applications feasible. Recent advances in the mass manufacture and shaping of MOFs also brings to the fore industrial applications, with availability and cost beginning to approach desirable values. [17] The primary mechanism by which oxygen can be selectively adsorbed within a MOF is by a redox reaction within the pores. However, efforts over recent years have illustrated the challenge of controlling this interaction. Strong redox interactions deliver a MOF from which oxygen cannot be readily desorbed,[ 18 , 19 ] often resulting in a working capacity significantly below the adsorption capacity as a result. Other MOFs that are not redox‐active may be far more stable to cycling but do not exhibit meaningful selectivity over nitrogen, and commonly have very small sorption capacities unless high pressure or low temperatures are deployed.[ 20 , 21 ]
Herein we summarise and analyse the latest efforts to develop metal–organic frameworks for use in oxygen concentration. These analyses indicate the key performance criteria for a suitable oxygen concentration MOF are as follows:
Modulated redox chemistry for capture and release of oxygen at close to room temperature.
Resistance to heat and water vapour.
Reversible sorption without collapse of the reticular structure.
A composition amenable to large scale production at accessible cost.
This Minireview will examine the synthetic approaches taken to addressing these performance criteria with a view to how viable the potential applications are in the coming years.
2. Capture
There are many factors to consider when designing a framework for oxygen capture. One critical challenge is selective oxygen uptake. Selectivity reflects the ability of the framework to separate one gas species from another; in the case of oxygen, this is largely considered to be nitrogen. Although it should be noted that other species, comprising a significantly smaller component of air, must also be addressed when designing an oxygen selective MOF. These species include argon, carbon dioxide and water vapour. A difference in the interaction between two gas species and the framework is required to achieve selectivity. Table 1 highlights selected physical and electronic properties of some common gases. Oxygen has a significantly smaller reduction energy than most common gases, including N2, a property commonly utilised for producing oxygen selective MOFs.
Table 1.
Selected physical and electronic properties of some common gases.
|
Property |
O2 [a] |
N2 [a] |
CO2 [a] |
Ar[a] |
|---|---|---|---|---|
|
Kinetic diameter [Å] |
3.46 |
3.64 |
3.3 |
3.4 |
|
Dipole moment |
0 |
0 |
0 |
0 |
|
Quadrupole moment 1040 θ [cm2] |
1.3 |
4.7 |
13.4 |
0 |
|
Polarisability [Å] |
1.60 |
1.76 |
2.65 |
1.66 |
|
Reduction energy [kJ mol−1] |
4.1 |
241.5 |
351.4 |
331.6 |
[a] Nat. Chem. 2010, 2, 633–637.
Selected examples of MOFs that demonstrate selective oxygen capture can be separated in two main types; via redox‐active sites (metal centres or ligands) or via redox inert interactions (i.e. size‐exclusion). Examples of both are discussed below, further Table 2 lists MOFs reported in the literature which selectively capture oxygen.
Table 2.
Summary data for MOFs for potential O2 separation reported in the literature.
|
Metal–organic frameworks |
Surface area [m2 g−1] |
O2 adsorption [mmol g−1] |
Pressure [bar] |
Heat of adsorption, Qst [kJ mol−1] |
T [K] |
Ref. |
|
|---|---|---|---|---|---|---|---|
|
BET |
Langmuir |
||||||
|
Cd(bpndc)(4,4′‐bpy) |
– |
– |
6.7 |
1 |
– |
90 |
|
|
Co2(OH)2(BBTA) |
1360 |
– |
1.2 |
1 |
45 |
298 |
|
|
Co2Cl2(BBTA) |
1280 |
– |
≈ 0.26 |
1 |
19 |
298 |
|
|
Co‐BTTri |
1595 |
1853 |
4.8 |
1 |
34 |
195 |
|
|
Co‐BDTriP |
1332 |
1517 |
4.8 |
1 |
47 |
195 |
|
|
Co‐MOF‐74 |
– |
1417 |
18 |
0.015 P/P 0 |
– |
77 |
|
|
Co‐MOF‐74 Composite |
1008 |
1150 |
4.8 |
1.2 |
19 |
204 |
|
|
Cr3(BTC)2 |
1810 |
2040 |
0.73 |
0.21 |
– |
298 |
|
|
Cr3(BTC)2 |
1403 |
– |
4.46 |
1 |
– |
298 |
|
|
Cr‐BTT |
2030 |
2300 |
2.5 |
1 |
65 |
298 |
|
|
Cu(BDT) |
200 |
– |
14 |
1.0 P/P 0 |
|
77 |
|
|
Cu(BDTri)L (L=DMF) |
– |
1160 |
17.8 |
0.2 |
– |
77 |
|
|
Cu(BDTri)L (L=DEF) |
– |
240 |
15.9 |
0.2 |
– |
77 |
|
|
Cu3(BTC)2 |
1206 |
– |
0.2 |
1 |
– |
298 |
|
|
Cu3(BTC)2 |
– |
2141 |
25.8 |
0.015 P/P 0 |
– |
77 |
|
|
Cu‐BTC |
– |
2237 |
0.3 |
1 |
10.7 |
298 |
|
|
Cu‐BTC Composite |
1143 |
– |
0.34 |
1 |
15.3 |
298 |
|
|
Fe‐BTTri |
1630 |
1930 |
5.9 |
1 |
51 |
195 |
|
|
Fe‐MOF‐74 |
1360 |
1535 |
5.33 |
0.21 |
41 |
226 |
|
|
Mg3(NDC)3 |
190 |
– |
3.5 |
1.0 P/P 0 |
– |
77 |
|
|
MIL‐100 (Fe) |
– |
1900 |
0.25 |
1 |
8.5 |
298 |
|
|
MIL‐100 (Sc) |
– |
1635 |
0.28 |
1 |
15.1 |
298 |
|
|
MIL‐101 (Cr) Composite |
264 |
424 |
2.0 |
1 |
– |
298 |
|
|
MIL‐101 (Ti) |
2970 |
4440 |
0.85 |
9x10−4 |
– |
298 |
|
|
MOF‐177 |
3100 |
≈4300 |
0.18 |
1 |
– |
298 |
|
|
Ni2(cyclam)2(mtb) |
141 |
154 |
1.2 |
0.196 |
– |
77 |
|
|
Ni‐MOF‐74 |
– |
1234 |
15 |
0.015 P/P 0 |
– |
77 |
|
|
PCN‐13 |
150 |
– |
3.0 |
1.0 P/P0 P/P 0 |
– |
77 |
|
|
PCN‐17 |
820 |
– |
9.3 |
1.0 P/P 0 |
|
77 |
|
|
PCN‐224FeII |
2901 |
– |
2.4 |
1 |
34 |
195 |
|
|
UMCM‐1 |
4100 |
6500 |
0.23 |
0.96 |
4.9 |
298 |
|
|
Zn(TCNQ‐TCNQ)bpy |
– |
– |
12.0 |
1.0 P/P 0 |
– |
77 |
|
|
K0.82Fe2(bdp)3 |
– |
1700 |
0.53 |
1 |
– |
298 |
|
|
1.43 |
1 |
– |
453 |
||||
|
K1.09Fe2(bdp)3 |
– |
750 |
0.68 |
1 |
– |
298 |
|
|
1.32 |
1 |
– |
473 |
||||
|
K1.88Fe2(bdp)3 |
– |
70 |
0.87 |
1 |
– |
298 |
|
|
1.64 |
1 |
– |
473 |
||||
|
K2.07Fe2(bpeb)3 |
– |
600 |
0.65 |
1 |
– |
298 |
|
|
2.3 |
1 |
– |
453 |
||||
2.1. Redox‐Active Sites (Metal Centres or Ligands)
One possible mechanism for selective oxygen absorption in MOFs is via redox‐active sites, which can take the form of metal centres or ligands that participate in charge‐transfer interactions with oxygen. Due to oxygen's low reduction energy in contrast with that of nitrogen, these redox‐active sites provide specificity for oxygen binding over nitrogen. To date, numerous frameworks have employed this approach and key examples are discussed herein.
In 2010, Long and co‐workers described the metal–organic framework Cr3(BTC)2 (BTC3−=1,3,5‐benzenetricarboxylate), which exhibited selectivity for O2 over N2. [22] Its structure features a porous three‐dimensional network with paddle‐wheel units as depicted in Figure 1. The Cr centres of the units in Figure 1 bind dimethylformamide (DMF) molecules in the as‐synthesised framework which can be vacated to produce redox‐active open metal sites. Isotherms of the activated framework at 298 K showed the material sorbs 0.73 mmol g−1 O2 at 0.21 bar. At the time it was reported it had an outstanding O2/N2 selectivity ratio of 22, based on the uptake of O2 at 0.21 bar and 0.033 mmol g−1 N2 at 0.78 bar (reflecting the corresponding partial pressures in air). This ambient pressure selectively was attributed to the CrII centres forming favourable charge‐transfer interaction with O2 species. Structures from neutron diffraction support this assertion that shows oxygen binds to the Cr metal sites (Figure 1). Infrared and X‐ray absorption spectroscopy further indicate a partial charge‐transfer interaction but not necessarily a complete charge transfer to give a CrIII‐superoxide adduct. Reversibility of oxygen adsorbed at 298 K was shown, however the oxygen‐adsorbed material was heated under vacuum for the prolonged period of 48 hrs at 323 K.
Figure 1.
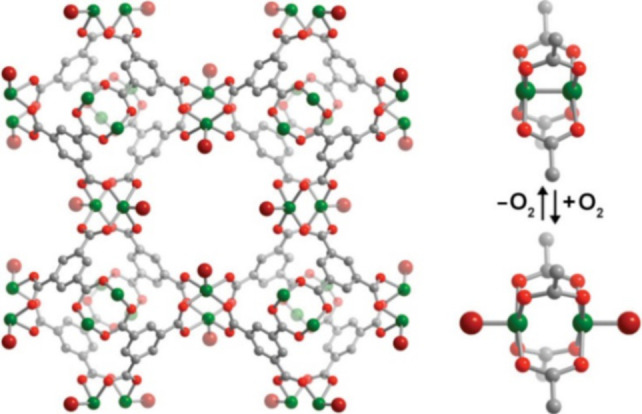
The structure of the metal–organic framework Cr3(BTC)2, where green, red, and grey spheres represent Cr, O, and C atoms, respectively, while large red spheres represent either bound DMF molecules in the solvated structure or bound O2 molecules upon desolvation and exposure to O2. Reprinted (adapted) with permission from J. Am. Chem. Soc. 2010, 132(23), 7856–7857. Copyright 2021 American Chemical Society.
The metal–organic framework, Fe2(dobdc) (dobdc4−=2,5‐dioxido‐1,4‐benzenedicarboxylate) described in 2011 was similarly shown to have the ability to separate O2 from N2 (Figure 2). [18] Long and co‐workers ascribed this to the presence of an iron open metal site that is redox‐active. At low temperatures (211 K & 226 K) reversible oxygen adsorption is associated with partial oxidation of the Fe centres to FeII/FeIII and a partial reduction of the oxygen species to the near superoxide evidenced by Mössbauer spectroscopy. In contrast, oxygen adsorption at 298 K occurs with complete transformation of the Fe centres to FeIII, which is accompanied by irreversible adsorption.
Figure 2.
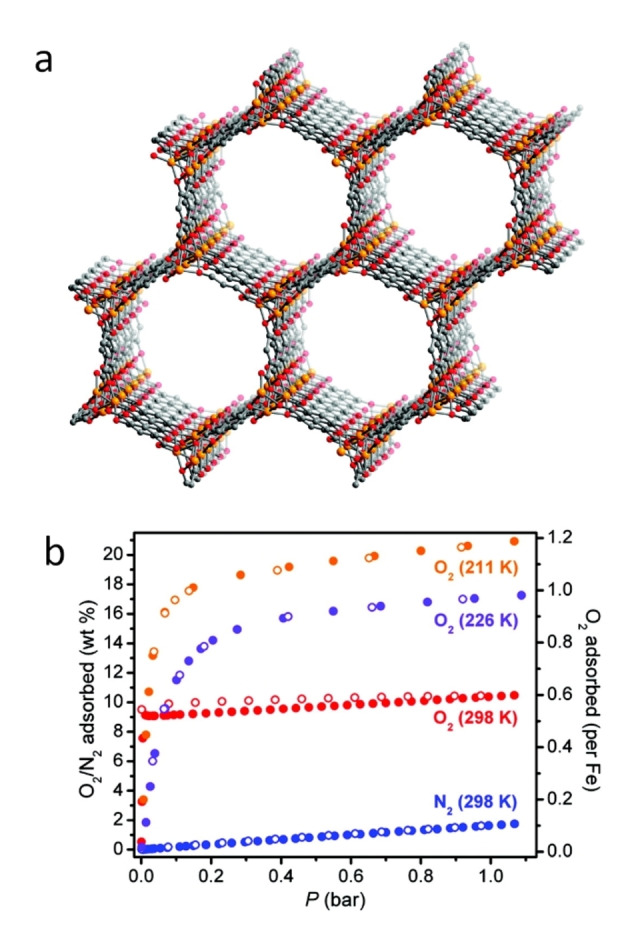
a) The structure of Fe2(dobdc) viewed along the [001] plane; orange, red and grey correspond to Fe, O, and C atoms. b) Gas sorption isotherms for Fe2(dobdc); blue corresponds to N2 at 298 K, whilst yellow, purple and red correspond to O2 at 211 K, 226 K and 298 K, respectively. Desorption isotherm is represented by open circles. Reprinted (adapted) with permission from J. Am. Chem. Soc. 2011, 133(37), 14814–14822. Copyright 2021 American Chemical Society.
In 2016, Long and co‐workers described the metal–organic framework Cr‐BTT (BTT3−=1,3,5‐benzenetristetrazolate). [23] Open metal sites which are redox‐active are generated via an activation process that removes coordinated DMF molecules. The authors describe very strong isosteric heat of adsorption for O2 (Qst=−65 kJ mol−1) and relatively low isosteric heat of adsorption for N2 (Qst=−15.3 kJ mol−1) for the activated material. With such a significant difference in host–guest interactions, Cr‐BTT displays an exceptional selectivity of 2570 as calculated by ideal adsorbed solution theory (IAST) for a gas mixture of 1 : 4 O2:N2 at 298 K and 1 bar. Notably, Cr‐BTT displays reversible oxygen adsorption at 298 K across 15 cycles with a shorter regeneration time than that of Cr3(BTC)2 (dynamic vacuum, 423 K, 30 min).
As we have detailed, selectivity for O2 over N2 in MOFs has been reported with redox‐active centres in the Cr3(BTC)2, Fe2(dobdc) and Cr‐BTT frameworks. A problem, however, with these reported Fe‐ and Cr‐based frameworks is they lose crystallinity and adsorption capacity upon unregulated exposure to the atmosphere, in part due to their strong interactions with oxygen. [24]
To this end, Long and co‐workers in 2016 examined a Co‐based framework, Co‐BTTri (H3BTTri=1,3,5‐tri(1H1,2,3‐triazol‐5‐yl)benzene), as shown in Figure 3. [24] Cobalt(II) centres within this framework form a cobalt(II)‐dioxygen species with partial electron transfer. This framework showed selective sorption of O2 over N2, with 3.3 mmol g−1 O2 at 0.21 bar (at 195 K). The authors described a moderate isosteric heat of adsorption for O2 (Qst=−34(1) kJ mol−1) coupled with very low N2 isosteric heat of adsorption (Qst=−12(1) kJ mol−1). Using an IAST model with a mixture of 0.21 bar O2 and 0.79 bar N2, the selectivity for oxygen was 41 at 195 K. Significantly, no loss in adsorption is observed when the framework is exposed to room temperature air at 90 % relative humidity and reactivated at 423 K.
Figure 3.
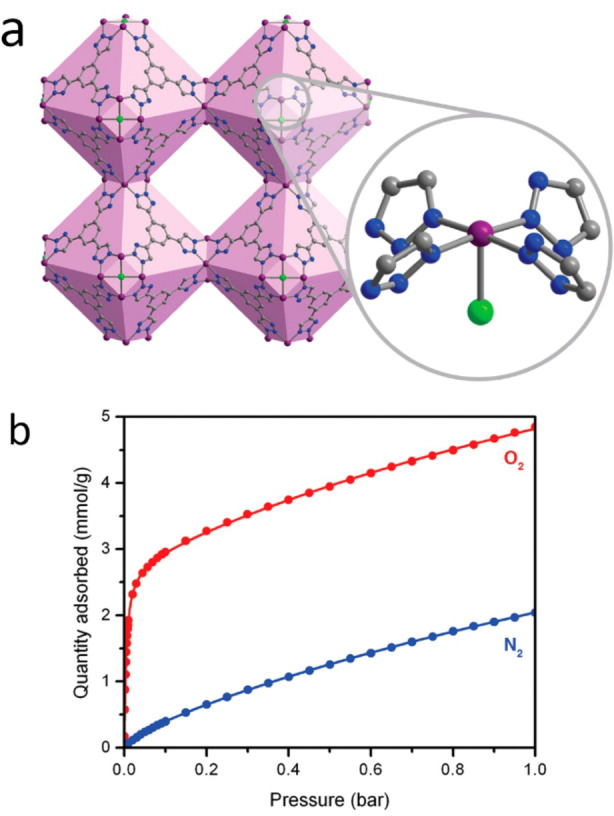
a) Structure of the Co‐BTTri framework, with zoomed in section highlighting the coordination geometry around the Co metal centre, note the vacant metal site. b) Sorption isotherms of O2 (red) and N2 (blue) in the Co‐BTTri framework. Reprinted (adapted) with permission from J. Am. Chem. Soc. 2016, 138, 7161–7170. Further permissions related to the material should be directed to the ACS.
We highlighted water stability as a key feature for oxygen enrichment by MOFs and this cobalt‐based framework displays exceptional characteristics in this regard. As water is a minor but significant component in air, the chemistry of the MOF must withstand the presence of water.
Following work with Co‐BTTri, Long and co‐workers reported a framework with biomimetic O2 adsorption. [19] The framework, Fe‐BTTri, possesses an iron coordination environment similar to that found in haemoglobin. Once the framework is activated its metal's sixth coordination site is unoccupied, and the redox‐active iron centre can bind oxygen. Unfortunately, like other iron based frameworks the FeII species binds oxygen irreversibly above 258 K. Nonetheless, the framework is able to sorb 3.3 mmol g−1 of O2 at 0.21 bar and 195 K. Furthermore, the isosteric heat of adsorption was found to be −51 kJ mol−1, consistent with measurements on heme‐based systems.
Given the temperature dependent performance of Fe‐BTTri, it is worth discussing the effect of temperature on adsorbent performance. Ideally, for oxygen capture, uptake should occur at ambient temperatures to maximise energy savings. However, it can be challenging to modulate the redox chemistry such that the MOF performs ideally, i.e. moderate and reversible uptake.
Low or cryogenic temperatures are associated with greater adsorption capacity and generally more reversible. The increased capacity is an effect of Le Chatelier's principle, wherein adsorption is an exothermic process and additional heat to the system favours the desorption of the gaseous species. In terms of reversibility, Fe‐based systems seem particularly sensitive to adsorption temperature. At low or cryogenic temperatures, they show reversibility but at room temperature they show irreversibility. In Fe2(dobdc) this can be rationalised as the activation energy requirements being fulfilled to allow the FeII species to be irreversibly oxidised to FeIII along with the formation of a bound peroxide anion.
Recently, a significant set of computational studies examined the O2 selectivity over N2 in several related metal–organic frameworks with differing bridging ligands (i.e. μ‐Br−, μ‐Cl−, μ‐F−, μ‐SH−, or μ‐OH−). [25] Based on the results of the theoretical study, the authors explored two key candidates, Co2(OH)2(BBTA) and Co2Cl2(BBTA). As expected from the theoretical results the Co2(OH)2(BBTA) framework showed superior O2 selectively with the redox activity of the open CoII metal site enhanced by the greater electron‐donating character of the μ‐OH−. At 298 K and 0.21 bar O2 and 0.79 bar N2, IAST predicts an O2/N2 selectivity of 49.
Other types of MOFs being studied for potential O2/N2 separation in computational simulations are those containing metal catecholates.[ 26 , 27 ] These systems are redox‐active and generally result in greater binding of O2 than N2, allowing for potential selectivity. From an extensive study of transition row metal catecholates, Fe2+ and Zn2+ were found to have the best potential for air separation. This was due to them possessing an O2 binding energy that would be conducive to oxygen reversibility, whilst maintaining a good O2/N2 selectivity.
Further, in 2020, Long and co‐workers reported a high‐temperature selective oxygen adsorption in a metal–organic framework. [28] This redox‐active iron‐pyrazolate network, Fe2(bdp)3 (bdp2−=1,4‐benzenedipyrazolate), can be reduced with potassium naphthalenide to give an oxygen sorbing material. The reduced FeIII framework reduces the dioxygen species to superoxide, with the electron transfer occurring via an outer‐sphere mechanism. The framework adsorbed O2 significantly better at an elevated temperature. For example, at 298 K, the framework adsorbed 0.68 mmol g−1 at 1 bar compared to 1.32 mmol g−1 at 1 bar for 473 K. The authors propose the difference between the two uptakes is a kinetic effect, where there is an energy cost associated with rearrangement of the alkali metal cations.
In contrast to the redox‐active metal centres discussed for the above frameworks, Kitagawa and co‐workers in 2010 relied on the redox‐active ligand species in Zn(TCNQ‐TCNQ)bpy (TCNQ=7,7,8,8‐tetracyanoquinodimethane and bipy=4,4′‐bipyridine). [29] The framework showed high selectivity for O2 (77 K) and NO (121 K) over a range of other gases including N2 (77 K), CO (77 K), CO2 (193 K) and C2H2 (193 K). Whilst the authors did not report room temperature measurements, this framework shows an elegant alternative approach to redox‐active metal sites.
2.2. Redox‐Inert (i.e. Size Exclusion)
Whilst redox‐active metal centres and ligands have been utilised to much success for the separation of oxygen in MOFs, others have adopted an alternative approach using redox‐inert methods. Perhaps the most common approach involves construction of pores that only allow specific guests (i.e. oxygen). This can be challenging due to the extremely similar kinetic diameters of O2 (3.46 Å) and N2 (3.64 Å). An example of this approach is described by Zhou and co‐workers, who reported a porous coordination network, PCN‐13, in 2007. [30] From crystallographic studies the framework has a pore aperture of 3.5×3.5 Å—intermediate in size of O2 and N2. Isotherms with H2, O2, N2, and CO at 77 K to 1.0 P/P 0 reveal the framework uptakes only significant amounts of H2 (2.1 mmol g−1) and O2 (3.0 mmol g−1).
Similarly, a doubly interpenetrated Yb‐based porous coordination network, PCN‐17, reported by Zhou and co‐workers, had a pore size of around 3.5 Å. [31] Again, the authors explored the gas selectivity of the framework, with H2, O2 and N2 and CO at 77 K to 1.0 P/P 0. PCN‐17 can adsorb large amounts of O2 (9.4 mmol g−1), moderate amounts of H2 (4.7 mmol g−1), whilst only small amounts of both N2 and CO (≈0.9 mmol g−1). The large difference in the gas uptakes between oxygen and nitrogen was attributed to the kinetic diameters of the respective gases.
3. Storage
In contrast to oxygen capture, there have been fewer reports on oxygen storage in MOFs; nonetheless, MOFs are considered excellent candidates for oxygen storage. Table 3 lists MOFs reported in the literature which have the potential to store oxygen. Oxygen storage has many applications, including first responders, medical, and aerospace industries. Typically, there is also a desire to increase the amount of stored oxygen or decrease the pressure at which the oxygen is stored, due to safety concerns.
Table 3.
Summary data for MOFs for potential O2 storage reported in the literature.
|
Metal–organic frameworks |
Surface area [m2 g−1] |
Deliverable O2 (mmol g−1] |
Pressure (bar] |
Heat of adsorption, Qst (kJ mol−1] |
T [K] |
Ref. |
|
|---|---|---|---|---|---|---|---|
|
BET |
Langmuir |
||||||
|
Al‐Soc‐MOF‐1 |
5585 |
6530 |
13.2 |
50 |
10 |
298 |
|
|
Al‐soc‐MOF‐2 |
5162 |
5976 |
12.7 |
50 |
– |
298 |
|
|
Al‐soc‐MOF‐3 |
4849 |
5212 |
12.3 |
50 |
– |
298 |
|
|
HKUST‐1 |
1880 |
– |
8.9 |
140 |
9.7 |
298 |
|
|
NU‐125 |
2880 |
– |
10.1 |
140 |
6.9 |
298 |
|
|
UiO‐66 |
– |
– |
3.5 |
30 |
8.3 |
298 |
|
|
UMCM‐152 |
3760 |
– |
11.8 |
140 |
11–16 |
298 |
|
In 2014, Farha and co‐workers reported a systematic study of MOFs for oxygen storage. [32] From 10 000 candidates using grand canonical Monte Carlo simulations, they identified two prime targets for oxygen storage. These frameworks were HKUST‐1 and NU‐125. At 30 bar and 298 K HKUST‐1 and NU‐125 have 5.0 mol kg−1 and 8.3 mol kg−1 oxygen capacities respectively (Figure 4). Whilst both frameworks have coordinatively unsaturated Cu sites, these do not appear to play a significant role in adsorbing oxygen at high pressures.
Figure 4.
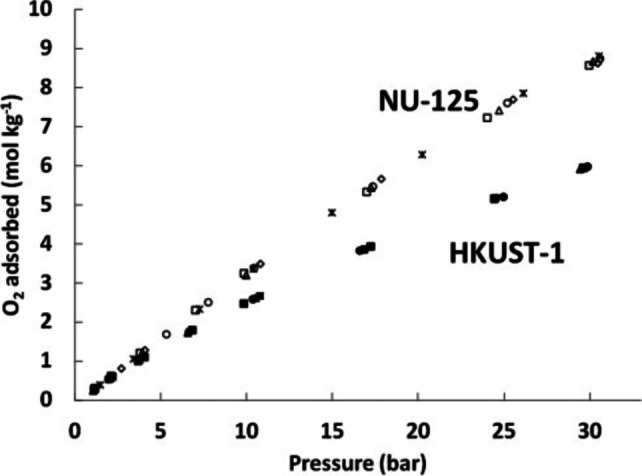
Excess oxygen adsorption isotherms measured at 1 (square), 5 (diamond), 10 (triangle), 20 (star), and 50 (circle) cycles at 298 K and pressures up to 30 bar for NU‐125 (hollow symbols) and HKUST‐1 (solid symbols). Reprinted (adapted) with permission from Angew. Chem. Int. Ed. 2014, 53, 14092–14095; Angew. Chem. 2014, 126, 14316–14319. Copyright 2021.
In 2015, Eddaoudi and co‐workers reported the novel framework, Al‐soc‐MOF‐1, which has an apparent Langmuir surface area of 6000 m2 g−1. [33] At high pressures, the framework outperforms both HKUST‐1 and NU‐125, being able to sorb 29 mmol g−1 at 140 bar.
4. Release
The performance of adsorbents used in gas capture and storage applications depends on the amount of adsorbate released from the adsorbent during the regeneration process. Depending on the regeneration method, a high working capacity [34] of the material can be achieved when most or all of the adsorbed molecules are released. Several different but related processes have been developed for the separation and purification of gas mixtures utilising adsorbent materials. These are Temperature Swing Adsorption (TSA), Pressure Swing Adsorption (PSA) and Vacuum Swing Adsorption (VSA). These processes allow for the separation of gas species based on the molecular affinity of adsorbent. In the case of oxygen separation from air, a selective adsorbent such as a MOF would preferentially adsorb oxygen. A swing process in the form of temperature, pressure or vacuum would subsequentially release the oxygen from the adsorbent material. TSA typically operates at ambient pressures; however, the temperature is varied between the adsorb and release swing, with a higher temperature driving the adsorbed gases from the adsorbent material. PSA in contrast operates at ambient temperatures, with a high pressure used for gas adsorption, which swings to a low pressure for release. Finally, VSA works on a similar concept to PSA, however the gas species are adsorbed at ambient pressures and a vacuum swing is applied for the release. Over the years, TSA, PSA and VSA are relatively energy‐intensive processes that have been deployed to regenerate and reuse adsorbents for gas storage and separation applications.[ 35 , 36 , 37 , 38 , 39 ] Consequently, recent publications on new materials for oxygen separation or storage have demonstrated the performance of their materials by employing one of these methods. However, the very strong isosteric heat of desorption of oxygen over nitrogen in new MOF materials[ 19 , 23 , 24 ] resulting in higher O2 selectivity over N2 relative to commercially available zeolites with N2 selectivity over O2[ 40 , 41 ] would require better and energy‐efficient processes to achieve high working capacities.
In 2016, our group reported an energy‐efficient adsorbent regeneration process known as the Magnetic Induction Swing Adsorption (MISA) process[ 42 , 43 ] capable of completely regenerating MOFs to achieve high working capacities. MISA is comparable to TSA but is significantly more efficient. MOFs are generally poor thermal conductors. [44] As such, the efficiency comes from the direct heating of the absorbent as compared to conventional heating. In MISA the material needs to be magnetic. An example material could be a magnetic nanocomposite‐MOF. Under an alternating magnetic field these types of materials undergo efficient and rapid heating to trigger the release of the adsorbed species. We evaluated the feasibility of efficient delivery of oxygen from MOFs through the use of MISA. The work evaluated the use of a composite MOF fabricated from M‐MOF‐74 (M=Co), a MOF known for its high density of unsaturated open metal sites [45] and magnetic Fe3O4 nanoparticles in a process that resulted in oxygen uptake of 4.8 mmol g−1 at 1.2 bar and 204 K with 100 % release of adsorbed molecules achieved during regeneration. [46] Our group also evaluated the effect of MISA on the cycling performance of CuBTC MOF for oxygen storage applications. CuBTC and MgFe2O4 nanoparticles were combined with the aid of a binder to make pellets that were then exposed to multiple cycles of oxygen adsorption at room temperature and MISA desorption. [47] The composite achieved an uptake capacity of 0.34 mmol g−1 at 298 K and was regenerated via MISA at a temperature of 359 K achieving 100 % oxygen release efficiency (Figure 5).
Figure 5.
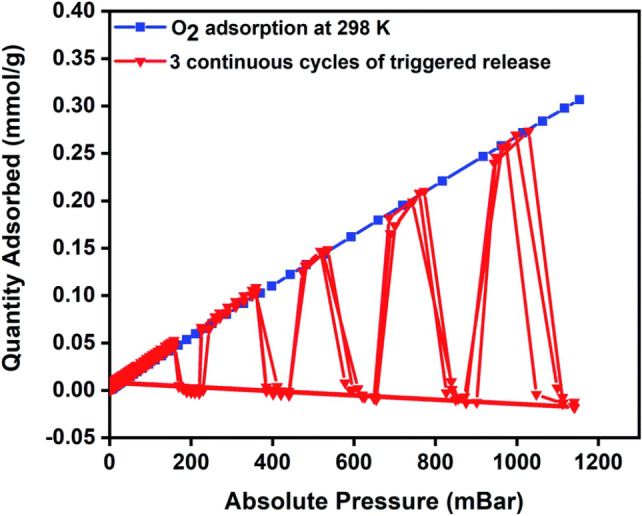
Oxygen cycling experiments for CuBTC MOF composite showing consistent and 100 % release with the aid of MISA. Adsorption isotherm conducted at 298 K with magnetically triggered desorption (31 mT, 359 K) at 200, 400, 600, 800, and 1000 mbar. Reproduced from RSC Adv. 2020, 10, 40960–40968 with permission from the Royal Society of Chemistry
Finally, the reversibility and stability of MOFs during the regeneration process is an important consideration. Thermal stability is particularly important for TSA‐ and MISA‐based processes. A framework which undergoes sequential degradation upon repeated application of heat is of little commercial benefit. The kinetics of desorption should be relatively fast and occur on the timescale of minutes, not hours or days. Overall, the MOF performance should not degrade with cycle count.
5. Applications & Outlook
Commercially produced oxygen has many uses within society. These can be broken down into three main sectors; industry, medical and life support (diving and space). We will examine the applications and outlook of MOFs for oxygen capture, storage and release with respect to these three key sectors.
Industry is the largest consumer, accounting for over 80 % of the 100 million tons of oxygen produced each year. [1] Smelting of iron ore accounts for 55 % of all commercially used oxygen, whilst the chemical industry consumes about 25 %. Further uses of oxygen in industry include metal cutting and welding, as an oxidiser in rocket fuel and water treatment. Many of these important industrial applications currently rely on the energy‐intensive and inefficient cryogenic distillation process for high purity oxygen production, whilst the MOFs described in this Minireview are not likely to be adopted for the replacement of cryogenic distillation due to their inability to provide selective oxygen adsorption at room temperature and poor release performances. It may be reasonable to suggest next‐generation MOFs coupled with efficient Swing Adsorption (SA) technologies that may be able to provide high‐purity oxygen to such industry users. However, it is unlikely we will see a complete shift to MOF‐based SA technologies, as Air Separation Units (ASUs) are critical for supplying other commodity‐based gases such as N2 and argon. [2] In‐depth feasibility studies are still required to reveal the magnitude of energy savings that MOFs with SA technology can deliver over standard cryogenic distillation.
Medical‐based oxygen is increasingly portable, with the aid of portable oxygen concentrators. These systems currently rely on absorbent‐based SA technologies using zeolites. [63] Zeolites, as we discussed in the introduction, are nitrogen selective and thus inefficient for oxygen separation. Here, MOFs with oxygen selectivity will outperform zeolites for oxygen capture. This will result in portable concentrators with decreased size and lower energy use (and hence longer runtime on batteries). Oxygen‐selective MOFs will likely see adoption in applications such as portable oxygen concentrators first as they utilise SA technology. Further enhancements in energy savings could be seen with the adoption of MISA technologies.
Life support systems (space and diving) rely upon oxygen in tanks to perform critical activities in extreme environments. Unfortunately, these oxygen tanks can be bulky, heavy and operated at hazardously high pressures. MOF‐based materials offer an attractive alternative for oxygen storage. The adsorption‐based storage enables oxygen at much lower pressures and higher amounts than in a conventional tank.
The potential applications of MOF‐based oxygen adsorbents are extensive. However, their cost and scalability will be a significant factor in their success for widespread adoption. Currently, high synthetic costs have impeded the development of practical industrial uses of MOFs in general. A reduction in cost via scale‐up and switching to continuous processing will likely be required. [64] Flow chemistry has been shown to be applicable to the scalable synthesis of some of the MOFs outlined in this Minireview. [65] This provides significant promise that next‐generation MOF‐based oxygen adsorbents may well be delivered at low cost and at scale.
Future work should allow MOF‐based materials to adsorb significant amounts of oxygen at relatively low pressures. We encourage researchers to examine their MOF‐based adsorbents not only for O2/N2 selectivity but also for O2/CO2, O2/Ar and O2/H2O selectivity as these are important components of air. Adsorption measurements with mixed gases and/or breakthrough experiments to supplement IAST calculations are also warranted. Overall, the outlook for MOFs as oxygen capture, storage and release materials is favourable. Future research can be anticipated to bring us MOF materials that can selectively adsorb oxygen at room temperature and store large quantities of oxygen at low pressure. These features will see MOF materials utilised in commercial applications in industry, medical and life support settings.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Ashley Sutton received his PhD from The University of Melbourne in 2020 under the supervision of Prof. Brendan Abrahams. Prior to this he pursued a BSc(Hons) in Chemistry also from The University of Melbourne. From late 2020, he joined Prof. Matthew Hill at the Commonwealth Scientific and Industrial Research Organisation (CSIRO) as a postdoctoral fellow working on metal–organic frameworks (MOFs) for reversible oxygen capture.

Biographical Information
Leena Melag is an associate research fellow at Institute for Frontier Materials (IFM) Deakin University. She received her PhD in Chemical engineering from Monash University in 2021 under the supervision of Prof. Matthew Hill and Prof. Kiyonori Suzuki. She pursued her bachelor's and master's studies in Materials engineering from COEP, Pune, India. She joined Prof. Jenny Pringle's group at Deakin University in 2021 and her current work is on the development of organic ionic plastic crystals (OIPCs) for applications in clean energy technologies.

Biographical Information
Muhammad Munir Sadiq received his BEng. (2006) and MEng. (2011) in Chemical Engineering from Abubakar Tafawa Balewa University Bauchi, Nigeria and Universiti Teknologi Malaysia, respectively. He completed his PhD in Materials Science and Engineering at Monash University, Australia and is currently a research fellow in the Chemical and Biological Engineering Department. His research interests include capture, storage and triggered release of gases from porous materials/composites, direct air capture, prototype development and process integration.

Biographical Information
Matthew Hill is a Professor, and Deputy Head of Chemical Engineering at Monash University, and a Senior Prinicipal Research Scientist at CSIRO. His research into advanced porous polymers and Metal‐Organic Frameworks (MOFs) aims to solve both fundamental scientific questions and incorporate these into devices. He has published more than 140 referred journal articles, 19 patents (6 licenced) with over 7700 citations.

A. L. Sutton, L. Melag, M. M. Sadiq, M. R. Hill, Angew. Chem. Int. Ed. 2022, 61, e202208305; Angew. Chem. 2022, 134, e202208305.
References
- 1. Emsley J., Nature's Building Blocks: An A–Z Guide to the Elements, Oxford University Press, Oxford, 2001. [Google Scholar]
- 2. Castle W. F., Int. J. Refrig. 2002, 25, 158–172. [Google Scholar]
- 3. Chong K. C., Lai S. O., Thiam H. S., Teoh H. C., Heng S. L., J. Eng. Sci. Technol. 2016, 11, 1016–1030. [Google Scholar]
- 4. Xu D., Dong F., Chen Y., Zhao B., Liu S., Tade M. O., Shao Z., J. Membr. Sci. 2014, 455, 75–82. [Google Scholar]
- 5. Bi X., Meng X., Liu P., Yang N., Zhu Z., Ran R., J. Membr. Sci. 2017, 522, 91–99. [Google Scholar]
- 6. Song J., Feng B., Chu Y., Tan X., Gao J., Han N., Liu S., Ceram. Int. 2019, 45, 12579–12585. [Google Scholar]
- 7. Qiao Z., Chai S., Nelson K., Bi Z., Chen J., Mahurin S. M., Xiang Z., Sheng D., Nat. Commun. 2014, 5, 3705. [DOI] [PubMed] [Google Scholar]
- 8. Himma N. F., Wardani A. K., Prasetya N., Aryanti P. T. P., Wenten I. G., Rev. Chem. Eng. 2019, 35, 591. [Google Scholar]
- 9. Sidhikku Kandath Valappil R., Ghasem N., Al-Marzouqi M., J. Ind. Eng. Chem. 2021, 98, 103–129. [Google Scholar]
- 10. Talu O., Li J., Kumar R., Mathias P. M., Moyer J. D., Schork J. M., Gas Sep. Purif. 1996, 10, 149–159. [Google Scholar]
- 11. Jensen N. K., Ru T. E., Watson G., Zhang D. K., Chan K. I., May E. F., J. Chem. Eng. Data 2012, 57, 106–113. [Google Scholar]
- 12. White J. C., Dutta P. K., Shqau K., Verweij H., Langmuir 2010, 26, 10287–10293. [DOI] [PubMed] [Google Scholar]
- 13. Batten S. R., Champness N. R., Chen X., Garcia-martinez J., Kitagawa S., Öhrström L., O'Keeffe M., Suh M. P., Reedijk J., Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar]
- 14. James S. L., Chem. Soc. Rev. 2003, 32, 276–288. [DOI] [PubMed] [Google Scholar]
- 15. Furukawa H., Cordova K. E., O'Keeffe M., Yaghi O. M., Science 2013, 341, 1230444. [DOI] [PubMed] [Google Scholar]
- 16. Zhou H., Kitagawa S., Chem. Soc. Rev. 2014, 43, 5415–5418. [DOI] [PubMed] [Google Scholar]
- 17. Rubio-Martinez M., Avci-Camur C., Thornton A. W., Imaz I., Maspoch D., Hill M. R., Chem. Soc. Rev. 2017, 46, 3453–3480. [DOI] [PubMed] [Google Scholar]
- 18. Bloch E. D., Murray L. J., Queen W. L., Chavan S., Maximoff S. N., Bigi J. P., Krishna R., Peterson V. K., Grandjean F., Long G. J., Smit B., Bordiga S., Brown C. M., Long J. R., J. Am. Chem. Soc. 2011, 133, 14814–14822. [DOI] [PubMed] [Google Scholar]
- 19. Reed D. A., Xiao D. J., Jiang H. Z. H., Chakarawet K., Oktawiec J., Long J. R., Chem. Sci. 2020, 11, 1698–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mu B., Schoenecker P. M., Walton K. S., J. Phys. Chem. C 2010, 114, 6464–6471. [Google Scholar]
- 21. Li Y., Yang R. T., Langmuir 2007, 23, 12937–12944. [DOI] [PubMed] [Google Scholar]
- 22. Murray L. J., Dinca M., Yano J., Chavan S., Bordiga S., Brown C. M., Long J. R., J. Am. Chem. Soc. 2010, 132, 7856–7857. [DOI] [PubMed] [Google Scholar]
- 23. Bloch E. D., Queen W. L., Hudson M. R., Mason J. A., Xiao D. J., Murray L. J., Flacau R., Brown C. M., Long J. R., Angew. Chem. Int. Ed. 2016, 55, 8605–8609; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8747–8751. [Google Scholar]
- 24. Xiao D. J., Gonzalez M. I., Darago L. E., Vogiatzis K. D., Haldoupis E., Gagliardi L., Long J. R., J. Am. Chem. Soc. 2016, 138, 7161–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosen A. S., Mian M. R., Islamoglu T., Chen H., Farha O. K., Notestein J. M., Snurr R. Q., J. Am. Chem. Soc. 2020, 142, 4317–4328. [DOI] [PubMed] [Google Scholar]
- 26. Stoneburner S. J., Gagliardi L., J. Phys. Chem. C 2018, 122, 22345–22351. [Google Scholar]
- 27. Demir H., Stoneburner S. J., Jeong W., Ray D., Zhang X., Farha O. K., Cramer C. J., Siepmann J. I., Gagliardi L., J. Phys. Chem. C 2019, 123, 12935–129. [Google Scholar]
- 28. Jaffe A., Ziebel M. E., Halat D. M., Biggins N., Murphy R. A., Chakarawet K., Reimer J. A., Long J. R., J. Am. Chem. Soc. 2020, 142, 14627–14637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shimomura S., Higuchi M., Matsuda R., Yoneda K., Hijikata Y., Kubota Y., Mita Y., Kim J., Takata M., Kitagawa S., Nat. Chem. 2010, 2, 633–637. [DOI] [PubMed] [Google Scholar]
- 30. Ma S., Wang X., Collier C. D., Manis E. S., Zhou H., Inorg. Chem. 2007, 46, 8499–8501. [DOI] [PubMed] [Google Scholar]
- 31. Ma S., Wang X., Yuan D., Zhou H., Angew. Chem. Int. Ed. 2008, 47, 4130–4133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4198–4201. [Google Scholar]
- 32. DeCoste J. B., Weston M. H., Fuller P. E., Tovar T. M., Peterson G. W., LeVan M. D., Farha O. K., Angew. Chem. Int. Ed. 2014, 53, 14092–14095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14316–14319. [Google Scholar]
- 33. Alezi D., Belmabkhout Y., Suyetin M., Bhatt P. M., Weseliński L. J., Solovyeva V., Adil K., Spanopoulos I., Trikalitis P. N., Emwas A. H., Eddaoudi M., J. Am. Chem. Soc. 2015, 137, 13308–13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mason J. A., Sumida K., Herm Z. R., Krishna R., Long J. R., Energy Environ. Sci. 2011, 4, 3030–3040. [Google Scholar]
- 35. Raganati F., Chirone R., Ammendola P., Ind. Eng. Chem. Res. 2020, 59, 3593–3605. [Google Scholar]
- 36. Ntiamoah A., Ling J., Xiao P., Webley P. A., Zhai Y., Ind. Eng. Chem. Res. 2016, 55, 703–713. [Google Scholar]
- 37. Mofarahi M., Towfighi J., Fathi L., Ind. Eng. Chem. Res. 2009, 48, 5439–5444. [Google Scholar]
- 38. Santos J. C., Cruz P., Regala T., Magalhaes F. D., Mendes A., Ind. Eng. Chem. Res. 2007, 46, 591–599. [Google Scholar]
- 39. Hedin N., Andersson L., Bergström L., Yan J., Appl. Energy 2013, 104, 418–433. [Google Scholar]
- 40. Pan M., Omar H. M., Rohani S., Nanomaterials 2017, 7, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watanabe K., Austin N., Stapleton M. R., Mol. Simul. 1995, 15, 197–221. [Google Scholar]
- 42. Sadiq M. M., Li H., Hill A. J., Falcaro P., Hill M. R., Suzuki K., Chem. Mater. 2016, 28, 6219–6226. [DOI] [PubMed] [Google Scholar]
- 43. Li H., Sadiq M. M., Suzuki K., Ricco R., Doblin C., Hill A. J., Lim S., Falcaro P., Hill M. R., Adv. Mater. 2016, 28, 1839–1844. [DOI] [PubMed] [Google Scholar]
- 44. Huang B. L., Ni Z., Millward A., Mcgaughey A. J. H., Uher C., Kaviany M., Yaghi O., Int. J. Heat Mass Transfer 2007, 50, 405–411. [Google Scholar]
- 45. Märcz M., Johnsen R. E., Dietzel P. D. C., Fjellvåg H., Microporous Mesoporous Mater. 2012, 157, 62–74. [Google Scholar]
- 46. Melag L., Sadiq M. M., Smith S. J. D., Suzuki K., Hill M. R., J. Mater. Chem. A 2019, 7, 3790–3796. [Google Scholar]
- 47. Melag L., Sadiq M. M., Konstas K., Zadehahmadi F., Suzuki K., Hill M. R., RSC Adv. 2020, 10, 40960–40968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanaka D., Nakagawa K., Higuchi M., Horike S., Kubota Y., Kobayashi T. C., Takata M., Kitagawa S., Angew. Chem. Int. Ed. 2008, 47, 3914–3918; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3978–3982. [Google Scholar]
- 49. Parkes M. V., Gallis D. F. S., Greathouse J. A., Nenoff T. M., J. Phys. Chem. C 2015, 119, 6556–6567. [Google Scholar]
- 50. Zhang Z., Wang Y., Jia X., Yang J., Li J., Dalton Trans. 2017, 46, 15573–15581. [DOI] [PubMed] [Google Scholar]
- 51. Dincǎ M., Yu A. F., Long J. R., J. Am. Chem. Soc. 2006, 128, 8904–8913. [DOI] [PubMed] [Google Scholar]
- 52. Demessence A., Long J. R., Chem. Eur. J. 2010, 16, 5902–5908. [DOI] [PubMed] [Google Scholar]
- 53. Sava Gallis D. F., Parkes M. V., Greathouse J. A., Zhang X., Nenoff T. M., Chem. Mater. 2015, 27, 2018–2025. [Google Scholar]
- 54. Reed D. A., Xiao D. J., Gonzalez M. I., Darago L. E., Herm Z. R., Grandjean F., Long R., J. Am. Chem. Soc. 2016, 138, 5594–5602. [DOI] [PubMed] [Google Scholar]
- 55. Dincǎ M., Long J. R., J. Am. Chem. Soc. 2005, 127, 9376–9377. [DOI] [PubMed] [Google Scholar]
- 56. Sava Gallis D. F., Chapman K. W., Rodriguez M. A., Greathouse J. A., Parkes M. V., Nenoff T. M., Chem. Mater. 2016, 28, 3327–3336. [Google Scholar]
- 57. Zhang W., Banerjee D., Liu J., Schaef H. T., Crum J. V., Fernandez C. A., Kukkadapu R. K., Nie Z., Nune S. K., Motkuri R. K., Chapman K. W., Engelhard M. H., Hayes J. C., Silvers K. L., Krishna R., Mcgrail B. P., Liu J., Thallapally P. K., Adv. Mater. 2016, 28, 3572–3577. [DOI] [PubMed] [Google Scholar]
- 58. Mason J. A., Darago L. E., Lukens W. W., Long J. R., Inorg. Chem. 2015, 54, 10096–10104. [DOI] [PubMed] [Google Scholar]
- 59. Mcintyre S. M., Shan B., Wang R., Zhong C., Liu J., Mu B., Ind. Eng. Chem. Res. 2018, 57, 9240–9253. [Google Scholar]
- 60. Cheon Y. E., Suh M. P., Chem. Eur. J. 2008, 14, 3961–3967. [DOI] [PubMed] [Google Scholar]
- 61. Anderson J. S., Gallagher A. T., Mason J. A., Harris T. D., J. Am. Chem. Soc. 2014, 136, 16489–16492. [DOI] [PubMed] [Google Scholar]
- 62. Moghadam P. Z., Islamoglu T., Goswami S., Exley J., Fantham M., Kaminski C. F., Snurr R. Q., Farha O. K., Fairen-jimenez D., Nat. Commun. 2018, 9, 1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chai S. W., Kothare M. V., Sircar S., Ind. Eng. Chem. Res. 2011, 50, 8703–8710. [Google Scholar]
- 64. Mckinstry C., Cathcart R. J., Cussen E. J., Fletcher A. J., Patwardhan S. V., Sefcik J., Chem. Eng. J. 2016, 285, 718–725. [Google Scholar]
- 65. Rubio-Martinez M., Batten M. P., Polyzos A., Carey K., Mardel J. I., Lim K., Hill M. R., Sci. Rep. 2014, 4, 5443. [DOI] [PMC free article] [PubMed] [Google Scholar]