

Abstract
Aim
To characterize cabotegravir population pharmacokinetics using data from phase 1, 2 and 3 studies and evaluate the association of intrinsic and extrinsic factors with pharmacokinetic variability.
Methods
Analyses were implemented in NONMEM and R. Concentrations below the quantitation limit were modelled with likelihood‐based approaches. Covariate relationships were evaluated using forward addition (P < .01) and backward elimination (P < .001) approaches. The impact of each covariate on trough and peak concentrations was evaluated through simulations. External validation was performed using prediction‐corrected visual predictive checks.
Results
The model‐building dataset included 23 926 plasma concentrations from 1647 adult HIV‐1‐infected (72%) and uninfected (28%) subjects in 16 studies at seven dose levels (oral 10‐60 mg, long‐acting [LA] intramuscular injection 200‐800 mg). A two‐compartment model with first‐order oral and LA absorption and elimination adequately described the data. Clearances and volumes were scaled to body weight. Estimated relative bioavailability of oral to LA was 75.6%. Race and age were not significant covariates. LA absorption rate constant (KALA) was 50.9% lower in females and 47.8% higher if the LA dose was given as two split injections. KALA decreased with increasing BMI and decreasing needle length. Clearance was 17.4% higher in current smokers. The impact of any covariate was ≤32% on trough and peak concentrations following LA administration. The final model adequately predicted 5097 plasma concentrations from 647 subjects who were not included in the model‐building dataset.
Conclusions
A cabotegravir population pharmacokinetic model was developed that can be used to inform dosing strategies and future study design. No dose adjustment based on subject covariates is recommended.
Keywords: cabotegravir, HIV, integrase inhibitor, long‐acting, population pharmacokinetics
What is already known about this subject
Long‐acting cabotegravir demonstrated superior efficacy vs standard of care for treatment and pre‐exposure prophylaxis of HIV‐1 infection.
Cabotegravir population pharmacokinetics have not been comprehensively evaluated with external validation.
What this study adds
Cabotegravir clearances and volumes increase with weight. Longer terminal half‐life (slower long‐acting absorption) is associated with female sex, unsplit injection, higher body mass index and shorter needles. Clearance is higher in current smokers. Race and age are not significant covariates.
Simulations demonstrated that no dose adjustment based on subject covariates is recommended.
1. INTRODUCTION
Cabotegravir (CAB, GSK1265744; ViiV Healthcare, Research Triangle Park, NC, USA) is a small molecule human immunodeficiency virus (HIV) integrase strand transfer inhibitor (INSTI) and an analogue of dolutegravir (DTG). 1 , 2 , 3 , 4 , 5 , 6 It has been developed as a once‐daily oral tablet for short‐term use and as a long‐acting (LA) intramuscular (IM) injection that has demonstrated efficacy for both treatment 7 , 8 , 9 and prevention of HIV‐1 infection. 10 , 11
Adherence to daily oral antiretroviral therapy (ART) remains a challenge among individuals living with HIV, and LA administration of medication may remove the daily dosing burden. The combination of CAB LA and rilpivirine (RPV) LA (Cabenuva; ViiV Healthcare) has been approved for HIV‐1 treatment after phase 3 studies demonstrated that the IM injection regimen of once every month (QM) was noninferior to standard oral therapy in maintaining HIV‐1 suppression, 7 , 8 and the IM injection regimen of once every 2 months (Q2M) was noninferior to the QM regimen. 9 CAB LA as a single agent (Apretude; ViiV Healthcare) administered Q2M has been approved in the United States for use in adults and adolescents ≥35 kg after phase 3 studies demonstrated superiority to daily standard of care of oral tenofovir disoproxil fumarate/emtricitabine for pre‐exposure prophylaxis in cisgender men and transgender women who have sex with men 10 and in cisgender women. 11
CAB is classified as a compound with high permeability and low solubility. 1 , 2 , 3 , 4 Median oral time to maximum plasma concentration (T max) is 2‐3 hours post‐dose following oral dosing. CAB is primarily bound to albumin in human plasma with a binding of >99%. CAB is primarily eliminated by hepatic metabolism via UGT1A1 with a minor component by UGT1A9. Following oral dosing, CAB apparent clearance, apparent volume of distribution and terminal half‐life have been estimated to be 0.21 L/h, 12.3 L and 35‐42 hours, respectively. In contrast, CAB LA exhibits absorption‐limited (flip‐flop) kinetics with a terminal half‐life of 18‐50 days and an observed T max of approximately 1 week post‐injection based on a limited sampling schedule. 3 , 4 The in vitro protein‐adjusted concentration resulting in 90% of the maximum inhibition (PA‐IC90) of viral growth for CAB is 0.166 μg/mL. 1
Population pharmacokinetic (PK) models for CAB following administration of oral tablet and LA IM injection were previously explored. 12 , 13 , 14 A two‐compartment model with first‐order absorption and elimination adequately characterized the observed data. Scaling of clearances and volumes of distribution to body size improved model fitting. Female sex and high body mass index (BMI) were associated with slower IM absorption. The current analysis has improved on prior analyses with a higher number of subjects (especially female HIV‐1‐infected subjects), evaluation of more potential covariates (eg, laboratory tests and injection‐related information), evaluation of data below the quantitation limit (BQL) following CAB LA IM injections and the addition of data from seven key phase 1 studies.
The objectives of the current analysis were to develop a population PK model of CAB in adult HIV‐1‐infected and uninfected subjects following administration of oral tablet and LA IM injection of CAB using data from phase 1, 2 and 3 studies and evaluate the association of intrinsic and extrinsic factors with PK variability.
2. METHODS
2.1. Study description
The design, dosing regimen and sample size of the 16 studies included in the analysis are summarized in Table 1. The oral dose was administered in the fasted state in all phase 1 studies. All studies complied with the Declaration of Helsinki and Good Clinical Practice guidelines and were approved by the Institutional Review Board (IRB) of participating institutions. All study participants provided written informed consent.
TABLE 1.
Summary of studies included in the analysis
| Study number | Study description | CAB dose and administration | HIV‐1‐infected | Number a of subjects | Number a of concentrations |
|---|---|---|---|---|---|
| Model‐building dataset | |||||
| 116585 | Oral relative bioavailability study | 30 mg (SST) PO SD | No | 18 (10 M, 8 F) | 238 |
| 117010 | Rifampin DDI study, period 1 only (without rifampin) | 30 mg (SST) PO SD | No | 15 (10 M, 5 F) | 194 |
| 117011 | OC DDI study | 30 mg (SST) PO QD × 11 days | No | 19 (19 F) | 186 |
| 117020 | Oral relative bioavailability study | 30 mg (SST) PO SD | No | 22 (13 M, 9 F) | 286 |
| 201741 | Oral relative bioavailability study | 30 mg (SST) PO SD | No | 37 (27 M, 10 F) | 712 |
| 201479 | Hepatic impairment study | 30 mg (SST) PO SD | No | 16 (12 M, 4 F) | 206 |
| 201480 | Renal impairment study | 30 mg (SST) PO SD | No | 16 (12 M, 4 F) | 202 |
| 205696 | Food effect on CAB PK, fasted period | 30 mg (SST) PO SD | No | 23 (16 M, 7 F) | 301 |
| 115428 | Phase 1 repeat dose escalation of CAB LA, oral lead‐in phase only | 30 mg PO QD x 14 days | No | 43 (27 M, 16 F) | 301 |
| 116815 | LA relative bioavailability study (200 nm particle size only) |
Oral: 30 mg QD to SS. LA: 400 mg SD. |
No | 21 (15 M, 6 F) | 225 |
| 116482 (LATTE) | Phase 2b dose‐ranging study in HIV‐infected subjects with two NRTIs, induction phase |
10 mg tablet PO QD × 24 wk 30 mg tablet PO QD × 24 wk 60 mg (2 × 30 mg) PO QD × 24 wk |
Yes |
175 10 mg (57 M, 3 F) 30 mg (54 M, 2 F) 60 mg (55 M, 4 F) |
1478 |
| 200056 (LATTE‐2) | Phase 2b treatment study in HIV‐infected subjects |
30 mg PO QD × 20 wk, then 800 mg IM, then: Arm 1: 800 mg IM, then 400 mg IM Q4W Arm 2: 800 mg IM, 600 mg IM at wk 4, then Q8W |
Yes |
274 (252 M, 22 F) |
5884 |
| 201103 (HPTN 077) | Phase 2a PrEP study of safety, tolerability and PK of CAB LA in HS |
30 mg PO QD × 4 wk, 1‐wk washout, then: Arm 1: 800 mg IM Q12W × 3 Arm 2: 600 mg IM at wk 5, wk 9, then Q8W × 3 |
No |
134 (45 M, 89 F) |
2246 |
| 201120 (ECLAIR) | Phase 2a PrEP enabling study of CAB LA in male HS at low risk of contracting HIV | 30 mg QD × 4 wk, 1‐wk washout, 800 mg IM Q12W × 3 | No | 94 (94 M) | 1324 |
| 201584 (FLAIR) | Phase 3 treatment study b | 30 mg PO QD × 4 wk, 600 mg IM, then 400 mg IM Q4W | Yes |
278 (217 M, 61 F) |
4781 |
| 201585 (ATLAS) | Phase 3 treatment study c | 30 mg PO QD × 4 wk, 600 mg IM, then 400 mg IM Q4W | Yes |
462 (307 M, 155 F) |
5362 |
| External validation dataset d | |||||
| 207966 (ATLAS‐2M) | Phase 3 treatment study c |
Arm 1: 30 mg PO QD × 4 wk, 600 mg IM, then 400 mg IM Q4W Arm 2: 30 mg PO QD × 4 wk, 600 mg IM, 600 mg IM at wk 4, then Q8W |
Yes |
647 (507 M, 140 F) |
5097 |
Abbreviations: CAB, cabotegravir; DDI, drug‐drug interaction; F, female; HIV‐1, human immunodeficiency virus type‐1; INI, integrase inhibitor; IM, intramuscular; LA, long‐acting; M, male; NNRTI, non‐nucleoside reverse transcriptase inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; OC, oral contraceptives; PI, protease inhibitor; PK, pharmacokinetic(s); PO, oral; PrEP, pre‐exposure prophylaxis; QD, once daily; Q4W, every 4 weeks; Q8W, every 8 weeks; Q12W, every 12 weeks; SD, single dose; SS, steady state; SST, sodium salt tablet; wk, week.
Actual numbers of subjects and CAB plasma concentrations included in the analysis. All participants from the study were included in this analysis unless otherwise specified.
Antiretroviral‐naive subjects, switch from an INI single tablet regimen.
Virologically suppressed, treatment‐experienced subjects, switch current INI‐ NNRTI‐, or PI‐based antiretroviral regimen.
Only the subjects who were not included in the model‐building dataset are summarized in this table.
Human plasma samples were analysed for CAB concentration using a validated analytical method 1 , 2 , 3 , 4 , 5 , 6 based on protein precipitation, followed by high‐performance liquid chromatography with tandem mass spectrometry analysis. Across the program, the assay of CAB in plasma has had 1000‐fold linear range with a lower limit of quantification of 0.01 μg/mL (earlier studies) or 0.025 μg/mL (later studies) and an upper limit of quantification of 10 μg/mL or 25 μg/mL, respectively. The maximum within‐run and between‐run precision was ≤6.8% and ≤9.1%, respectively. Accuracy ranged from 8.8% to 8.0% bias.
The covariates collected for evaluation included those related to demographics laboratory tests, and injection‐related factors. Laboratory tests included albumin, total bilirubin, direct bilirubin, alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma glutamyl transferase (GGT), lactate dehydrogenase (LDH), creatinine, creatinine clearance, urea, UGT1A1 genotype and HIV‐1 viral load (log‐transformed and nontransformed both as continuous and categorical variable). Both baseline and time‐varying covariates were collected.
2.2. Base model
Nonlinear mixed‐effects modelling was performed with NONMEM (version 7.3; ICON Development Solutions, Ellicott City, Maryland, USA) 15 using the FOCE method with interaction, Perl‐speaks‐NONMEM (version 3.5.3; Uppsala University, Uppsala, Sweden) 16 and R. 17 The starting point was the base model from the previous CAB population PK analyses, 12 , 13 , 14 a two‐compartment model with first‐order oral and IM absorption and elimination, as shown in Figure 1. PK parameters included absorption rate constant for oral tablet (KAoral) and LA IM (KALA) formulation, apparent central clearance (CL/F, where F represents the absolute bioavailability of CAB LA IM injection), apparent central volume of distribution (V2/F), apparent intercompartmental clearance (Q/F), apparent peripheral compartment volume of distribution (V3/F) and oral bioavailability relative to the LA IM formulation (F1). This starter model was evaluated and refined to obtain the optimal base model.
FIGURE 1.
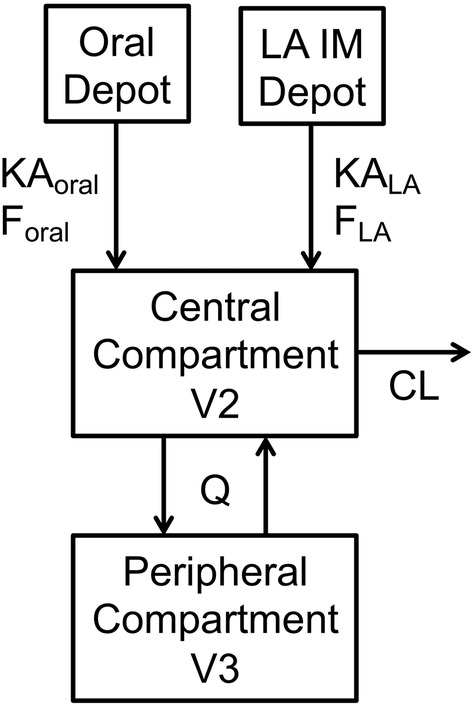
Cabotegravir pharmacokinetic model: two‐compartment model with first‐order oral and LA IM absorption and elimination. CL, central clearance; Foral, absolute bioavailability of oral tablet; FLA, absolute bioavailability of LA IM injection; IM, intramuscular; KAoral, absorption rate constant of oral tablet; KALA, absorption rate constant of LA IM injection; LA, long‐acting; Q, intercompartment clearance; V2, central compartment volume of distribution; V3, peripheral compartment volume of distribution
The base model included a power function of body weight (WT) on clearances and volumes of distribution: , where WT i is WT in the ith subject, P i is the typical value of PK parameter P in the ith subject (ie, subjects with WT i ), P is the typical value of PK parameter P in subjects with median WT (WTmedian), θ P is the estimated exponent for PK parameter P and θ P was set to be the same for CL/F and Q/F, and the same for V2/F and V3/F. Both baseline WT (BWT) and time‐varying WT were evaluated using the same equation. Last observation carried forward (LOCF) was used for time‐varying BWT.
Concentrations BQL following LA IM injections were modelled using a likelihood‐based method. 18 , 19 , 20 The degree of interoccasion variability (IOV) was evaluated to help explain some of the variability in KALA and F1 among injections. The occasion was set to 1 for all oral dosing intervals, the very first LA dosing interval with at least one non‐BQL concentration and all LA dosing intervals with only two or fewer non‐BQL concentrations. The occasion was set to 2, 3, 4 and so on for an LA dosing interval with three or more non‐BQL concentrations within the same subject.
2.3. Covariate evaluation and final model
Only the covariates whose values were missing in ≤20% of subjects were included in covariate evaluation. For continuous covariates, missing values were imputed as the sex‐specific median value in the study. For categorical covariates, missing values were imputed either as the most common category or as a separate category. Both baseline and time‐varying covariates were evaluated using the same equations. LOCF was used for time‐varying covariates. Subjects from race categories with an insufficient number for analysis were grouped into one category.
The covariate relationships were evaluated in a stepwise manner by forward addition followed by backward elimination with significance levels of P < .01 and P < .001, respectively (objective function value [OFV] decrease of 6.63 and 10.83 for one degree of freedom, respectively). The covariate relationship was defined by a power function for continuous covariates: , and defined as fractional change for categorical covariates: , where COV i is the value of a continuous covariate in the ith subject, IND i is an indicator of a categorical covariate equal to 0 or 1 in the ith subject, P i is the typical value of PK parameter P in the ith subject (ie, subjects with COV i or IND i ), P is the typical value of PK parameter P in subjects with the median value of a continuous covariate (COVmedian) or in the absence of a categorical covariate (IND i = 0) and θ P is the estimated exponent or fractional change for PK parameter P.
The model was then refined to obtain the final model: a covariate was removed if the PK parameter estimate of a typical person differed by <15% from a person with an extreme covariate value (10th and 90th percentiles for a continuous covariate or the extreme categories for a categorical covariate).
2.4. Model evaluation
In each step of the model‐building process, models were assessed based on collective evaluation of physiological relevance and plausibility, diagnostic plots, 21 , 22 decrease in OFV, visual predictive check (VPC), 22 , 23 adequate parameter estimates, decrease in variability, increase in precision and shrinkage 24 assessments. Bootstrapping 25 and prediction‐corrected VPC (pcVPC) 26 were performed using 500 replicates based on the final model.
The relative impact of each covariate retained in the final model on CAB trough concentration following the initiation injection (Ctau‐1) and steady‐state trough (Ctau‐SS) and peak (Cmax‐SS) concentration was evaluated through a series of simulations following CAB QM and Q2M regimens. Both the QM and Q2M regimens started with 4 weeks of oral lead‐in with CAB 30‐mg tablet once daily, followed by a CAB LA IM injection of 600 mg (initiation injection) at 2 hours after the last oral dose. For the QM regimen, CAB LA IM injection of 400 mg was administered QM after the initiation injection. For the Q2M regimen, CAB LA IM injection of 600 mg was administered 1 month after the initiation injection and then Q2M. Ctau‐SS and Cmax‐SS were defined as Ctau and Cmax following the 11th injection of the QM regimen or the sixth injection of the Q2M regimen. In each covariate subgroup, 2000 virtual subjects were simulated.
2.5. External validation
After the final model was built, data from phase 3 study 207 966 (ATLAS‐2 M) 9 became available. Data from ATLAS‐2M subjects who were not included in the model‐building dataset (Table 1) were used to validate the model using pcVPC and numerical predictive check (NPC) with 500 replications. The pcVPC and NPC were also stratified by the covariates retained in the final model.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22. 27
3. RESULTS
3.1. Model‐building dataset
A total of 23 926 plasma concentrations from 1647 adult subjects across 16 studies were included in the model‐building dataset. No subject was transgender. The dosing records included three oral dose levels (10, 30 and 60 mg) and a total of 12 169 CAB LA IM injections (200, 400, 600 and 800 mg). A total of 537 BQL concentrations were observed following LA IM injections, approximately 3.1% of all post‐injection concentrations. Subject characteristics are summarized in Table 2 and Supporting Information Figure S1. Values of albumin, direct bilirubin, GGT, LDH, urea and UGT1A1 genotype were missing in 134 (8.1%), 506 (30.7%), 1387 (84.2%), 1630 (99.0%), 175 (10.6%) and 634 (38.5%) subjects. There were no missing values for other continuous covariates. Missing values for categorical covariates are summarized in Table 2. Individual concentration versus time profiles are displayed in Figure 2.
TABLE 2.
Summary of baseline characteristics of subjects included in the analysis
| Model‐building dataset | External validation dataset | ||
|---|---|---|---|
| Number of subjects | 1647 | 647 | |
| Number of concentrations | 23 926 | 5097 | |
| Mean (SD), median [range] a | |||
| Age (year) | 37.9 (11.5), 36 [18‐74] | 42.2 (11.1), 42 [19‐83] | |
| Total body weight (kg) | 78.5 (16.1), 76.6 [41.2‐168.3] | 79.4 (16.7), 77.1 [41.8‐138.9] | |
| Body mass index (kg/m2) | 26.17 (5.01), 25.37 [15.3‐69.51] | 26.24 (5.17), 25.26 [16.57‐54.02] | |
| Albumin (g/L) | 43.8 (3.4), 44 [28‐56] | 45.0 (2.9), 45 [28‐53] | |
| Total bilirubin (μmol/L) | 9.64 (4.90), 8.00 [1.71‐42.75] | 10.12 (7.57), 8.00 [2.00‐106.00] | |
| ALT (IU/L, alanine aminotransferase) | 23.5 (15.0), 20 [2‐153] | 22.8 (12.5), 20 [4‐121] | |
| AST (IU/L, aspartate aminotransferase) | 24.2 (13.6), 22 [9‐352] | 23.0 (9.2), 21 [8‐105] | |
| Number of subjects (percentage) | |||
| Female | 424 (25.7%) | 140 (21.6%) | |
| HIV‐1‐infected subjects | 1189 (72.2%) | 647 (100%) | |
| Race | White b | 1102 (66.9%) | 491 (75.9%) |
| Black or African American | 394 (23.9%) | 100 (15.5%) | |
| Asian | 56 (3.4%) | 27 (4.2%) | |
| Other | 93 (5.6%) | 29 (4.5%) | |
| Unknown | 2 (0.12%) | 0 (0%) | |
| Smoker status | Never smoked | 590 (35.8%) | 340 (52.6%) |
| Former smoker | 210 (12.8%) | 103 (15.9%) | |
| Current smoker | 447 (27.1%) | 204 (31.5%) | |
| Not current smoker c | 120 (7.3%) | 0 (0%) | |
| Unknown | 280 (17.0%) | 0 (0%) | |
| Number of injections (percentage) | |||
| Total d | 12 169 (100%) | 5367 (100%) | |
| Injection type d | Unsplit | 11 443 (94.0%) | 5367 (100%) |
| Split | 726 (6.0%) | 0 (0%) | |
| Injection needle gauge d | 20 | 7 (0.1%) | 0 (0%) |
| 21 | 3053 (25.1%) | 1914 (35.7%) | |
| 22 | 230 (1.9%) | 200 (3.7%) | |
| 23 | 2586 (21.3%) | 2381 (44.4%) | |
| 24 | 1 (0.0%) | 1 (0.02%) | |
| 25 | 1545 (12.7%) | 871 (16.2%) | |
| Unknown | 4747 (39.0%) | 0 (0%) | |
| Injection volume d (mL) | 1 | 8 (0.1%) | 0 (0%) |
| 2 | 9187 (75.5%) | 3142 (58.5%) | |
| 3 | 2547 (20.9%) | 2225 (41.5%) | |
| 4 | 427 (3.5%) | 0 (0%) | |
| Injection needle length d (inch) | 1 | 151 (1.2%) | 50 (0.9%) |
| 1.5 | 10 766 (88.5%) | 5042 (93.9%) | |
| 2 | 766 (6.3%) | 275 (5.1%) | |
| 2.5 | 1 (0.0%) | 0 (0%) | |
| Unknown | 485 (4.0%) | 0 (0%) | |
Abbreviations: HIV‐1, human immunodeficiency virus type‐1; IU, international unit; SD, standard deviation.
Albumin was missing in 8.1% of the subjects in the model‐building dataset, otherwise no missing value was observed.
Included two subcategories: White/Caucasian/European Heritage and Arabic/North African Heritage.
Not current smoker could be “never smoked” or “former smoker” due to the lack of detailed specification in the source data.
Not baseline values. Each subject may have multiple injection types, volumes, needle gauge or needle lengths for different injections.
FIGURE 2.
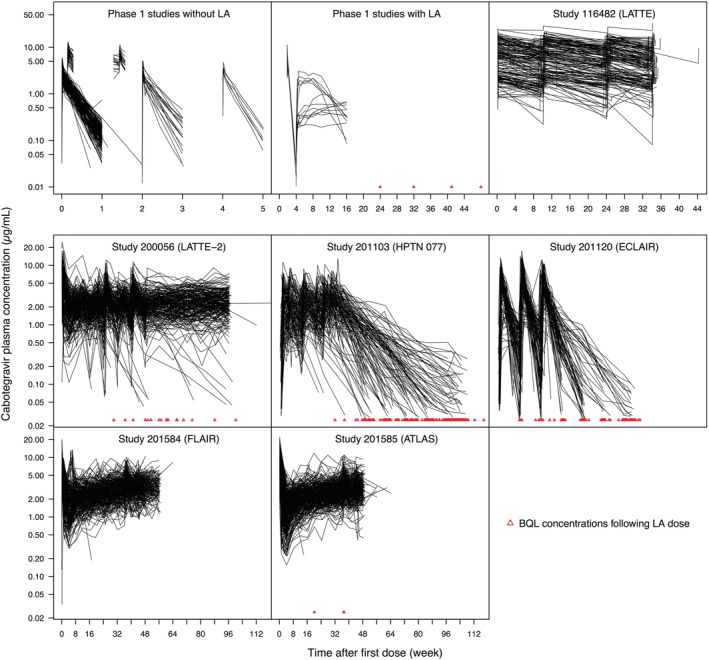
Individual concentration versus time profiles in the model‐building dataset. Phase 1 studies without LA data: studies 116585, 117010, 117011, 117020, 201741, 201479, 201480, 205696 and 115428. Phase 1 study with LA data: study 116815. For cosmetic purposes, one concentration in study 200056 (LATTE‐2) at 152 weeks after the first dose is not displayed. BQL, below the quantification limit; LA, long‐acting
3.2. Population pharmacokinetic modelling
The optimal base model was a linear two‐compartment model with first‐order oral and IM absorption and elimination (Figure 1) with exponents estimated on BWT. Interindividual variability (IIV) was estimated on all PK parameters except for Q/F and V3/F. Off‐diagonal covariance terms were estimated for IIV on KALA, CL/F and V2/F. Combined additive and proportional residual errors were estimated and assumed to be the same for both oral and LA dosing. IOV did not significantly improve the model fitting.
In the base model, the typical estimates of KAoral, KALA, CL/F, V2/F, Q/F, V3/F and F1 in a typical subject of 76.6 kg were 1.12 h−1, 0.000642 h−1, 0.16 L/h, 5.5 L, 0.563 L/h, 2.45 L and 74.6% (Table 3). The M3 method was used to model BQL data by maximizing the likelihood for the non‐BQL data treating BQL data as censored. 20 Parameter estimates of the final model are summarized in Table 3 and displayed in the equations below:
where “split” indicates that the LA IM injection was given as two split injections and NDL denotes needle length in inches.
TABLE 3.
Parameter estimates of the base model and final model
| Parameter | Base model | Final model | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | RSE (%) | IIV (%) | RSE of IIV (%) | Shrinkage (%) | Estimate | RSE (%) | 90% CI (bootstrap) | IIV (%) | RSE of IIV (%) | Shrinkage (%) | |
| KAoral (h−1) | 1.12 | 5.9 | 61.4 | 7.7 | 67.9 | 1.41 | 4 | 1.23,1.52 | 89.4 | 4.34 | 69.1 |
| KALA (h−1) | 0.000642 | 2.2 | 71.4 | 2 | 17 | 0.000733 | 2.3 | 0.000705, 0.000761 | 57.9 | 2.46 | 17.9 |
| CL/F (L/h) | 0.16 | 0.8 | 24.9 | 2.4 | 10.5 | 0.151 | 0.9 | 0.148, 0.153 | 23.3 | 2.58 | 10.3 |
| V2/F (L) | 5.5 | 2.5 | 22.6 | 8.9 | 29.9 | 5.27 | 2 | 5.07, 5.45 | 20.3 | 9.38 | 31.1 |
| Q/F (L/h) | 0.563 | 7.8 | 0.507 | 6.6 | 0.455, 0.579 | ||||||
| V3/F (L) | 2.45 | 5.7 | 2.43 | 4.8 | 2.26, 2.66 | ||||||
| F1 | 0.746 | 1 | 17.2 | 5.9 | 42.3 | 75.6% | 0.9 | 74.4%, 76.8% | 17.4 | 5.70 | 40 |
| Add err (μg/mL) | 0.0405 | 18.3 | 0.0319 | 19.4 | 0.0202, 0.0496 | ||||||
| Prop err | 27.3% | 1.1 | 27.3% | 1.1 | 26.7%, 27.8% | ||||||
| BWT on CL/F and Q/F | 0.611 | 5.9 | 0.618 | 5.6 | 0.562, 0.68 | ||||||
| BWT on V2/F and V3/F | 0.675 | 7.5 | 0.702 | 7.1 | 0.622, 0.79 | ||||||
| Smoke on CL/F | 17.4% | 9.3 | 14.7%, 20% | ||||||||
| Female on KALA | −50.9% | 4.4 | −54.1%, −47.1% | ||||||||
| BMI exponent on KALA | −0.766 | 13 | −0.922, −0.611 | ||||||||
| Split on KALA | 47.8% | 14.2 | 36.9%, 60.2% | ||||||||
| NDL exponent on KALA | 0.478 | 36.2 | 0.184, 0.747 | ||||||||
Abbreviations: 90% CI (bootstrap), 5th and 95th percentiles of the bootstrap replicates; Add Err, additive component of residual variability; BMI, body mass index (kg/m2); BWT, baseline body weight (kg); CI, confidence interval; CL/F, apparent central clearance; F1, relative bioavailability of the oral tablet relative to LA IM formulation; IIV, interindividual variability; IM, intramuscular; KAoral, absorption rate constant for oral tablet; KALA, absorption rate constant for LA IM injection; LA, long‐acting; NDL, needle length (inch); Prop Err, proportional component of residual variability; Q/F, apparent intercompartmental clearance; RSE, relative standard error; Smoke, current smoker status; Split, split injection; V2/F, apparent central compartment volume of distribution; V3/F, apparent peripheral compartment volume of distribution.
As compared to the base model, IIV in the final model was reduced by 19% for KALA and 6% for CL/F (Table 3). Based on the final model, the terminal half‐life of the CAB oral tablet was calculated to be 36.5 hours in a nonsmoker and 31.2 hours in a current smoker with a body weight of 76.6 kg. The terminal half‐life of CAB LA IM injection was calculated to be 5.6 weeks in males and 11.5 weeks in females, who had the population median BMI (25.4 kg/m2) and received unsplit injections using a 1.5‐inch needle. Based on the base model, the terminal half‐life of CAB LA was calculated to be 6.4 weeks in the population described in the model‐building dataset (25.7% female).
3.3. Model evaluation
Goodness‐of‐fit plots showed good agreement between predicted and observed concentrations with no apparent bias in residual (Figure 3). Based on pcVPC (Figure 4 and Supporting Information Figure S2), the model predictions adequately captured the prediction‐corrected concentration versus time profiles within the 90% prediction interval (PI, 5th and 95th percentiles). Similar results were observed when the pcVPC was stratified by sex, BMI (BMI ≥ 30 kg/m2 vs <30 kg/m2), smoker status (current smoker vs not current smoker) and body weight. Bootstrapping (Table 3) resulted in median parameter estimates and 90% CIs (5th and 95th percentiles of the bootstrap replicates) similar to the estimates from the model‐building dataset.
FIGURE 3.
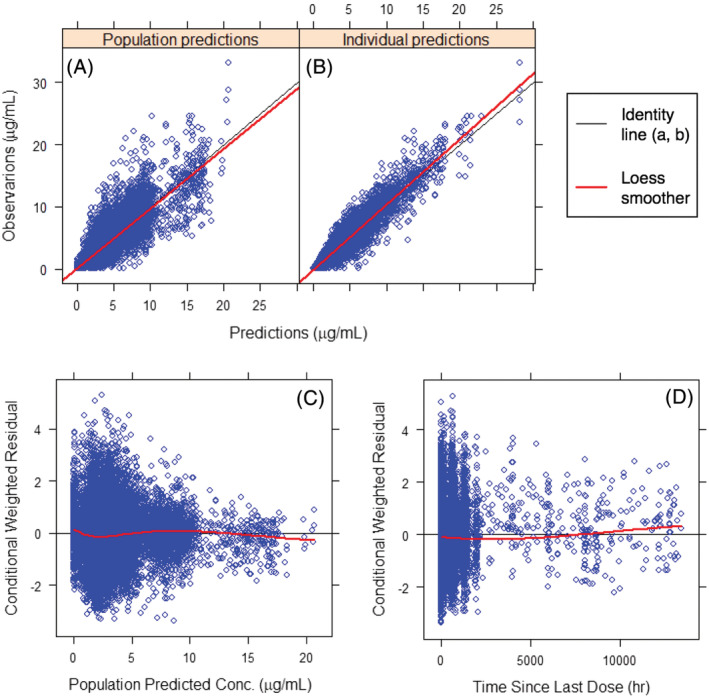
Goodness‐of‐fit plots of the final model. Good agreement is shown between predicted and observed concentrations with no apparent bias in residual. Conc., concentration
FIGURE 4.
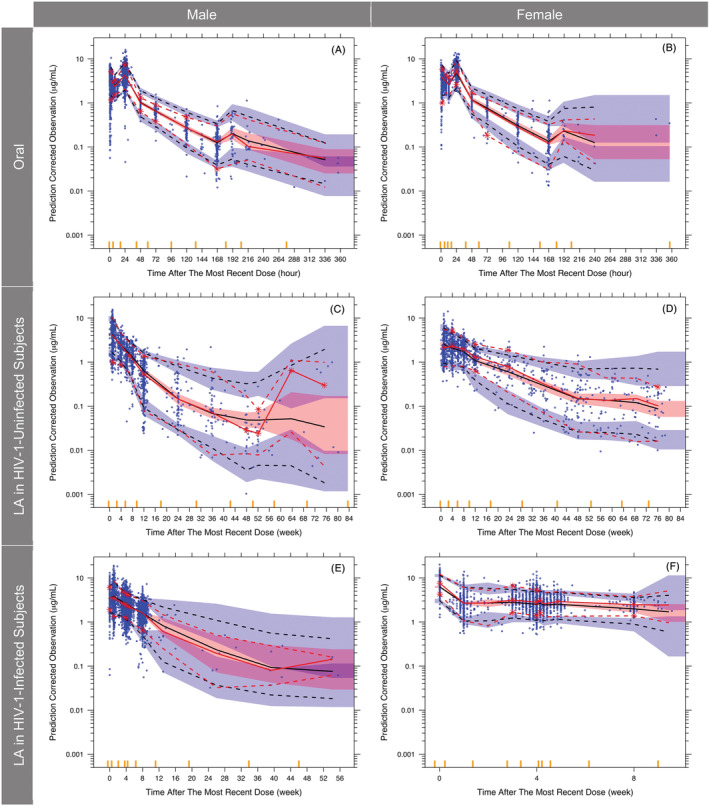
Prediction‐corrected visual predictive check of the final model stratified by route, sex and HIV‐1 status. Red solid line, median of prediction‐corrected observations; red dashed lines, 5th and 95th percentiles of prediction‐corrected observations; black dashed lines, predicted median, 5th and 95th percentiles; shaded regions, 95% confidence interval of the predicted median (pink), 5th and 95th percentiles (blue); blue dots, prediction‐corrected observations; red asterisks, prediction‐corrected observation lines were not contained within the shaded region; yellow bars on the x axis, boundaries between bins; HIV, human immunodeficiency virus; LA, long‐acting. For cosmetic purpose, four data points between 1700 and 8000 hours in females receiving LA dose (F) are not displayed
The relative impact of covariates retained in the final model on CAB exposure is demonstrated in Figure 5. Sex and BMI had the greatest impact on Ctau‐1 among all covariates or covariate combinations (Figure 5A). Median Ctau‐1 was 28% lower in females than males, and 30% lower in subjects with BMI ≥ 30 kg/m2 than in subjects with BMI < 30 kg/m2. BWT, split injection, needle length and smoker status led to a differential of ≤16% on Ctau‐1. The impact of all covariates was ≤15% on Ctau‐SS (Figure 5B) and ≤21% on Cmax‐SS (Figure 5C) following the QM regimen. Sex had the greatest impact on Ctau‐SS (Figure 5D) following the Q2M regimen. Median Q2M Ctau‐SS was 32% higher in females than males. The impact of all other covariates was ≤19% on Ctau‐SS (Figure 5D) and ≤24% on Cmax‐SS (Figure 5E) following the Q2M regimen.
FIGURE 5.
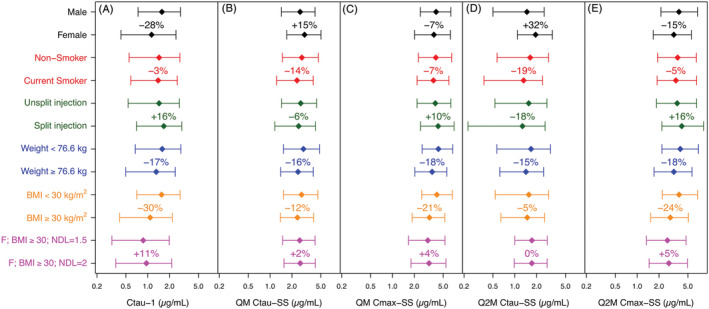
Relative impact of covariates retained in the final model on cabotegravir plasma concentrations: (A) Ctau‐1, (B) Ctau‐SS following QM regimen, (C) Cmax‐SS following QM regimen, (D) Ctau‐SS following Q2M regimen, (E) Cmax‐SS following Q2M regimen. Each bar represents 5th to 95th percentile with median (diamond) in each subgroup within the covariate or covariate combination. Percentage numbers next to each bar represent the percentage change in median versus the median of the reference subgroup within the same covariate or covariate combination (ie, versus the bar of the same colour above it). BMI, body mass index (kg/m 2 ); Cmax, maximum concentration; Ctau, trough concentration at the end of the dosing interval; Ctau‐1, Ctau following the initiation injection; F, female; NDL, needle length (inch); QM, once every month; Q2M, once every 2 months; SS, steady state (following the 11th injection of the QM regimen or the 6th injection of the Q2M regimen)
The predicted concentration versus time profiles following the QM and Q2M regimens for 1 year of CAB LA IM injections in a blended population of 50% female is presented in Figure 6. After the final CAB LA IM injection, CAB plasma concentration was predicted to fall below the quantification limit of 0.025 μg/mL in approximately 12‐14 months in 50% of the population, and to fall below the PA‐IC90 of 0.166 μg/mL in approximately 7‐9 months in 50% of the population.
FIGURE 6.
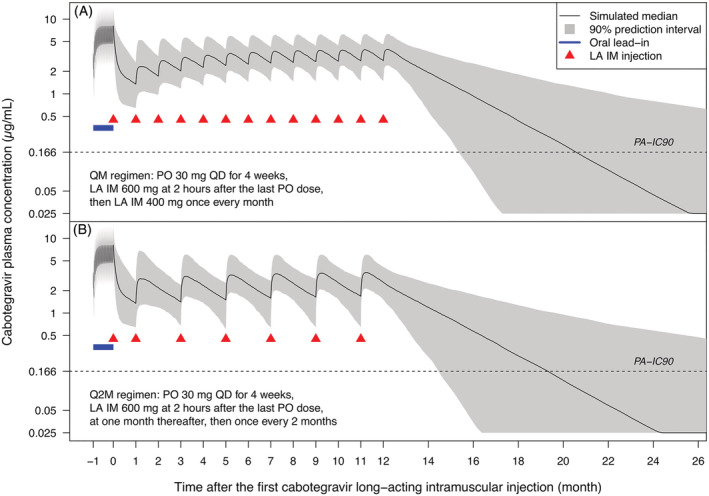
Predicted concentration versus time profiles following QM (A) and Q2M (B) regimens. The final injection is administered at (A) 12 or (B) 11 months after the initiation injection. Residual variability was not included in the simulation. Negative time on x axis (−1) indicates that oral dosing starts approximately 1 month prior to the initiation injection. IM, intramuscular; LA, long‐acting; PA‐IC90, in vitro protein‐adjusted concentration resulting in 90% of the maximum inhibition of viral growth for CAB (0.166 μg/mL); PO, oral; QM, once every month; Q2M, once every 2 months; QD, once daily
3.4. External validation
A total of 5097 plasma concentrations from 647 adult subjects from ATLAS‐2M (Table 1) were included in the external validation dataset (Table 2 and Supporting Information Figure S1). The model predictions adequately captured the prediction‐corrected concentration versus time profiles within the 90% PI (Figure 7 and Supporting Information Figure S3). Overall, 6.2% of the prediction‐corrected observations were above the 95th percentile of the model prediction and 3.4% were below the 5th percentile. Overall, 44% and 56% of the prediction‐corrected observations were below and above the median of the model prediction, respectively. Similar results were observed when the pcVPC was stratified by sex, BMI (BMI ≥ 30 kg/m2 vs <30 kg/m2) and smoker status (current smoker vs not current smoker).
FIGURE 7.
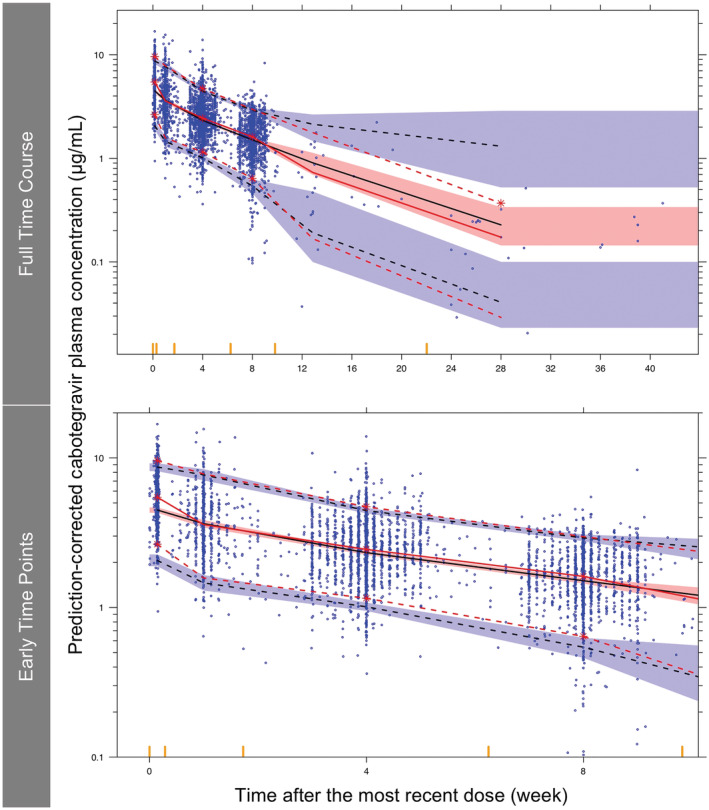
External validation of the final model using prediction‐corrected visual predictive check. Red solid line, median of prediction‐corrected observations; red dashed lines, 5th and 95th percentiles of prediction‐corrected observations; black dashed lines, predicted median, 5th and 95th percentiles; shaded regions, 95% confidence interval of the predicted median (pink), 5th and 95th percentiles (blue); blue dots, prediction‐corrected observations; red asterisks, prediction‐corrected observation lines were not contained within the shaded region; yellow bars on the x axis, boundaries between bins
4. DISCUSSION
This analysis is a comprehensive population PK evaluation of CAB in both HIV‐1‐infected and uninfected adult subjects in phase 1, 2 and 3 studies. The model‐building dataset included both single‐dose and multiple‐dose administrations, both oral tablet and LA IM injection, and both intensively and sparsely sampled CAB plasma concentration data. A population PK model was built based on a large PK population of 1647 subjects from 16 studies with three oral dose levels ranging from 10 to 60 mg and four LA dose levels ranging from 200 to 800 mg. The analysis confirmed that CAB exposure was dose proportional across these dose ranges. The results were consistent with previous analyses. 12 , 13 , 14 A two‐compartment model with first‐order oral and IM absorption and elimination adequately described the PK data. The pcVPC demonstrated accurate model fitting of the final model across a wide range of dosing regimens and time courses. Bootstrapping indicated good precision for parameter estimation. External validation confirmed that the final model was able to predict data that were not included in the model‐building dataset. The terminal half‐life of 5.6‐11.5 weeks for CAB LA IM injection was substantially longer than the terminal half‐life of 31.2‐36.5 hours for CAB oral tablets, characterizing the flip‐flop nature of CAB PK following LA IM injection.
Scaling of clearances and volumes of distribution to body size improved model fitting. Scaling using BWT, baseline fat‐free mass (FFM), lean body mass and ideal WT was similar based on overall evaluation of goodness‐of‐fit plots, drop in OFV, VPC, parameter estimates, decrease in variability and increase in precision. BWT was chosen based on its practicality and ease of use in the clinical setting (eg, potential dose adjustment for children) and for simulations. The exponents on BWT were estimated in this analysis. As a sensitivity test, PK parameters were also obtained by fixing the exponents on BWT to 0.75 for clearances and 1 for volumes of distribution, and comparable parameter estimates were obtained, while the model with estimated exponents yielded better OFV (P < .001).
Statistically significant covariates obtained from the forward addition and backward elimination procedure initially included BMI, needle length and split injection on KALA, sex on KALA, CL/F and V2/F, albumin and smoker status on CL/F and V2/F, race on CL/F and F1, total bilirubin on CL/F, and HIV‐1 status on F1. Following the predefined criteria, covariate effects with low impact on PK parameters were removed during model refinement. Cabotegravir data with co‐administered UGT1A1 inducer rifampin (Study 117 010; Table 1) were not included in this analysis because Cabenuva and Apretude are contraindicated in individuals receiving rifampin.
In the final model, KALA was 50.9% lower in females than males, and 28.6% lower in subjects with BMI of 32.5 kg/m2 (90th percentile) than those with BMI of 20.9 kg/m2 (10th percentile), possibly due to body composition and fat thickness (skin to muscle depth) as failure to deliver gluteal IM injections into the muscle was previously reported to be associated with female sex and higher BMI. 28 , 29 No transgender subjects participated in the studies. Although correlated with BMI, sex had the most profound effect on KALA even after adjusting for BMI, possibly due to the different correlations between BMI and fat thickness within each sex 28 , 29 and unknown sex‐associated factors other than BMI. KALA was higher when split injection (47.8% higher than nonsplit injection) or longer needle length were used, possibly due to greater surface area for absorption and penetration. KALA has significant impact on trough and peak concentrations, but has no impact on steady‐state AUC (Supporting Information Figure S4). With all other PK parameter values being equal, a higher KALA value results in higher Cmax and Ctau‐1, but lower Ctau‐SS. CL/F was 17.4% higher in current smokers, possibly due to the potential induction of hepatic UGT1A1 activity by smoking. 30 , 31
The current analysis improved in several areas compared to prior population PK analyses. 12 , 13 , 14 First, more subjects were included in this analysis (n = 2294 vs 346 12 and 766 13 , 14 ). In particular, more HIV‐1‐infected females (n = 387 vs 11 12 and 34 13 , 14 ) and a higher percentage of HIV‐1‐infected females (16.9% vs 3.2% 12 and 4.4% 13 , 14 ) were included. This improved the estimation of population PK parameters and the evaluation of covariate relationships, especially the impact of the combination of sex and HIV‐1 status (eg, similarity in PK between HIV‐1‐infected and HIV‐1‐uninfected females). This is relevant because CAB is developed for both treatment and presentation of HIV‐1 infection, and sex has substantial impact on PK. Furthermore, covariates evaluated in prior analyses were mostly limited to demographics while covariates evaluated in this analysis also included laboratory tests (eg, albumin, liver function tests, etc), injection‐related information (eg, needle length and gauge) and other potential covariates (eg, smoking status). Needle length and smoking status remained significant in the final model. In addition, it was important to consider BQL concentrations due to the long‐acting nature of CAB LA and the relatively high percentage of BQL concentrations of up to 19.5% in some studies. BQL concentrations were evaluated in this analysis and significantly improved model fitting. Finally, seven key phase 1 studies (studies 117 010, 117 011, 117 020, 201 741, 201 479, 201 480 and 205 696) were added in this analysis (Table 1). These phase 1 studies contained intensive PK sampling that helped characterize CAB PK.
Although CAB and DTG are structural analogues with similar metabolic pathways, there are differences between the CAB and DTG population PK analyses. 32 Firstly, DTG PK was described by a one‐compartment model with oral absorption lag time, while CAB PK was described by a two‐compartment model without oral absorption lag time. Secondly, age and total bilirubin were associated with DTG CL/F, while adult age was not found correlated with CAB CL/F, and total bilirubin was found correlated with CAB CL/F but removed during model refinement. Finally, the estimated absolute oral bioavailability of DTG was 21% higher in females than in males, while the relative bioavailability of CAB oral tablet to the LA IM formulation (equal to the absolute oral bioavailability if LA IM injection is assumed to be 100% bioavailable) was not correlated with sex. Smoking status had a similar impact in both models, with CAB and DTG CL/F being 17.4% and 16% higher in current smokers, respectively.
To evaluate the relative impact of covariates retained in the final model on CAB exposure, individual PK parameters of each virtual subject were calculated from the population parameter estimates and subject‐specific covariates of BWT, sex, BMI and smoker status, which were sampled with replacement from the subjects within this subgroup in the model‐building dataset. This maintained the correlations between covariates and the physiologically relevant spread of the other covariates (the intercovariate variance relationships). Therefore, each subgroup reflected not only the effect of the covariate of choice but also the influence of any covarying covariates. All injections were assigned with the most common needle length of 1.5 inches.
Ctau‐1 represents the lowest concentration during CAB LA administration. Based on the final model, KALA was predicted to be the lowest in females with high BMI (eg, BMI ≥ 30 kg/m2), potentially leading to low Ctau‐1. The predicted median (5th and 95th percentiles) of Ctau‐1 in females with high BMI was 0.87 μg/mL (0.32‐1.98 μg/mL), lower than Ctau‐1 in the overall model‐building dataset of 1.43 μg/mL (0.54‐2.73 μg/mL). Per study protocols, needle length longer than 1.5 inches was recommended for those with BMI ≥ 30 kg/m2, although not mandated. A needle length of 2 inches instead of the standard 1.5‐inch needle length was predicted to increase the median and 5th percentile of Ctau‐1 in this subgroup by 11% and 13%, respectively (Figure 5A). Given that this subgroup was successfully treated in phase 3 studies (using a 1.5‐inch needle in 72% of the 1405 injections with available needle length information) and was not predisposed to virologic failure, no dose adjustment is recommended for this subgroup for HIV‐1 treatment.
No dose adjustment based on subject covariates is recommended. All subgroups were successfully treated in phase 3 studies with favourable safety profiles and were not predisposed to virologic failure. For all subgroups and all individuals included in this analysis following oral, QM and Q2M regimens, Ctau and Cmax‐SS are comparable to those observed in phase 3 studies, which demonstrated favourable efficacy and safety profiles.
Simulations confirmed that a loading strategy can mitigate the risk of low initial CAB exposure. For the QM regimen, the initiation injection of 600 mg (loading dose) helps to achieve higher exposure more rapidly (Figure 6A). For the Q2M regimen, the loading schedule (the second injection is administered 1 month after the initiation injection instead of 2 months) helps achieve steady state more rapidly (Figure 6B).
This analysis has several limitations. First, based on pcVPC of the final model (Figure 4F), concentrations in female subjects and subjects with BMI ≥ 30 kg/m2 (data not shown) were slightly underpredicted. Similar results (Supporting Information Figure S3) were observed during external validation (eg, 3.7% of the prediction‐corrected observations were below the 5th percentile of the model prediction for males, but 2.0% for females). This could be caused by the relatively low proportion of female subjects and subjects with BMI ≥ 30 kg/m2 in the model‐building dataset. However, this underprediction does not have substantial impact on the interpretation of simulation results. Low exposure is a greater concern than high exposure, and underprediction leads to a more conservative interpretation of simulation results. Furthermore, some intrinsic and extrinsic factors that could affect intramuscular injection and absorption could not be collected and evaluated, such as distribution of body fat, muscle density, accumulated scar tissue after prolonged therapy, physical activity and ambient temperature. In addition, no transgender subjects participated in the studies included in this analysis, who are part of the target population for treatment and prevention of HIV‐1 infection. Given that sex and BMI are significantly associated with CAB PK, further analysis is warranted to evaluate whether CAB PK is similar in transgender subjects and whether hormone therapies used by transgender subjects are associated with CAB PK. Finally, the high shrinkage of 69.1% for KAoral was likely caused by the considerable portion of the subjects who did not have data in the oral absorption phase, and the moderate shrinkage of 40% for F1 was likely driven by subjects who only had data following either oral or LA dosing.
One of the main applications of a CAB population PK model is to perform simulations under various scenarios that can inform dosing strategies and study design, such as delayed injections, dosing interruptions, oral bridging, drug‐drug interactions and injection discontinuation. This is particularly important for LA medications because the long terminal half‐life (determined by slow absorption) leads to prolonged time to reach steady state and BQL on discontinuation of dosing. This results in long duration of clinical studies and long washout period (Figure 6), which may impact the study feasibility and may expose study participants to unnecessary risks.
In conclusion, a CAB population PK model was developed in adult HIV‐1‐infected and uninfected subjects following administration of oral tablet and LA IM injection. Race and adult age were not significant covariates. KALA was 50.9% lower in females and 47.8% higher if the LA IM injection was given as two split injections. KALA decreased with increasing BMI and decreasing needle length. CL/F was 17.4% higher in current smokers. No dose adjustment based on subject covariates is recommended. This model adequately characterized cabotegravir pharmacokinetics and sources of variability in exposure to inform dosing strategies and future study design.
POTENTIAL CONFLICT OF INTEREST/DISCLOSURE
K.H., C.S.S. and S.L.F. receive salaries from GSK and hold stocks of GSK. M.B., P.Patel, K.P.M., A.G.C., R.D.D'A., P.D.B. and W.R.S. receive salaries from ViiV Healthcare and hold stocks of GSK. M.L., P.Paul and Y.X. received salaries from Certara Consulting Services when conducting this work. All authors declare no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years, and no other relationships or activities that could appear to have influenced the submitted work.
AUTHOR CONTRIBUTIONS
All authors contributed substantially to the conception and design of the analysis, drafting or revising the paper as well as giving final approval for submission.
PATIENT CONSENT
This is a meta‐analysis involving 17 studies and written informed consent was obtained from all study participants in each study, as stated in the Methods section.
Supporting information
Supporting Information Figure S1 Distribution of baseline characteristics of subjects included in the model‐building dataset (a‐g) and external validation dataset (h‐n)
{kind=link}
Supporting Information Figure S2 Prediction‐corrected visual predictive check of the final model with baseline body weight as independent variable. Red solid line, median of prediction‐corrected observations; red dashed lines, 5th and 95th percentiles of prediction‐corrected observations; black dashed lines, predicted median, 5th and 95th percentiles; shaded regions, 95% confidence interval of the predicted median (pink), 5th and 95th percentiles (blue); blue dots, prediction‐corrected observations; red asterisks, prediction‐corrected observation lines were not contained within the shaded region; yellow bars on the x axis, boundaries between bins
{kind=link}
Supporting Information Figure S3 External validation of the final model using prediction‐corrected visual predictive check stratified by sex. Red solid line, median of prediction‐corrected observations; red dashed lines, 5th and 95th percentiles of prediction‐corrected observations; black dashed lines, predicted median, 5th and 95th percentiles; shaded regions, 95% confidence interval of the predicted median (pink), 5th and 95th percentiles (blue); blue dots, prediction‐corrected observations; red asterisks, prediction‐corrected observation lines were not contained within the shaded region; yellow bars on the x axis, boundaries between bins.
{kind=link}
Supporting Information Figure S4 Illustration of the impact of KALA alone on cabotegravir PK profiles while keeping all other PK parameter values unchanged. A higher KALA value results in a higher Cmax and Ctau‐1, but a lower Ctau‐SS. KALA alone has no impact on steady‐state AUC
{kind=link}
ACKNOWLEDGEMENT
We acknowledge the contribution from David A. Margolis.
Han K, Baker M, Lovern M, et al. Population pharmacokinetics of cabotegravir following administration of oral tablet and long‐acting intramuscular injection in adult HIV‐1‐infected and uninfected subjects. Br J Clin Pharmacol. 2022;88(10):4607‐4622. doi: 10.1111/bcp.15439
Yuan Xiong was affiliated with Certara Consulting Services, Research Triangle Park, NC, USA when the work was conducted.
Principal Investigator statement: Kelong Han is the principal investigator.
Contributor Information
Kelong Han, Email: kelong.x.han@gsk.com.
Susan L. Ford, Email: susan.l.ford@gsk.com
DATA AVAILABILITY STATEMENT
No new data were created for this analysis. The data that support the findings of this analysis may be available from ViiV Healthcare upon request and approval. Restrictions apply to the availability of these data.
REFERENCES
- 1. Spreen WR, Margolis DA, Pottage JC Jr. Long‐acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS. 2013;8(6):565‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spreen W, Min S, Ford SL, et al. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192‐203. [DOI] [PubMed] [Google Scholar]
- 3. Spreen W, Ford SL, Chen S, et al. GSK1265744 pharmacokinetics in plasma and tissue after single‐dose long‐acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr. 2014;67(5):481‐486. [DOI] [PubMed] [Google Scholar]
- 4. Spreen W, Williams P, Margolis D, et al. 2014. Pharmacokinetics, safety, and tolerability with repeat doses of GSK1265744 and rilpivirine (TMC278) long‐acting nanosuspensions in healthy adults. J Acquir Immune Defic Syndr. 2014;67(5):487‐492. [DOI] [PubMed] [Google Scholar]
- 5. Margolis DA, Brinson CC, Smith GHR, et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral‐naive adults with HIV‐1 infection (LATTE): a randomised, phase 2b, dose‐ranging trial. Lancet Infect Dis. 2015;15(10):1145‐1155. [DOI] [PubMed] [Google Scholar]
- 6. Margolis DA, Gonzalez‐Garcia J, Stellbrink HJ, et al. Long‐acting intramuscular cabotegravir and rilpivirine in adults with HIV‐1 infection (LATTE‐2): 96‐week results of a randomised, open‐label, phase 2b, non‐inferiority trial. Lancet. 2017;390(10101):1499‐1510. [DOI] [PubMed] [Google Scholar]
- 7. Orkin C, Arastéh K, Hernández‐Mora MG, et al. Long‐acting cabotegravir + rilpivirine for HIV maintenance: FLAIR week 48 results. Conference on Retroviruses and Opportunistic Infections (CROI). Abstract Number: 140. March 4‐7, 2019, Seattle, WA.
- 8. Swindells S, Andrade‐Villanueva JF, Richmond GJ, et al. Long‐acting cabotegravir + rilpivirine as maintenance therapy: ATLAS week 48 results. Conference on Retroviruses and Opportunistic Infections (CROI). Abstract Number: 139. March 4‐7, 2019, Seattle, WA.
- 9. Overton ET, Richmond G, Rizzardini G, et al. Long‐acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV‐1 infection (ATLAS‐2M), 48‐week results: a randomised, multicentre, open‐label, phase 3b, non‐inferiority study. Lancet. 2021;396(10267):1994‐2005. doi: 10.1016/S0140-6736(20)32666-0 [DOI] [PubMed] [Google Scholar]
- 10. Cabotegravir for HIV Prevention in Cisgender Men and Transgender Women . Landovitz et al. accepted by New England Journal of Medicine. To be updated when publication information becomes available. [DOI] [PMC free article] [PubMed]
- 11. Long acting injectable cabotegravir is safe and effective in preventing HIV infection in cisgender women: results from HPTN 084. S Delany‐Moretlwe, et al. HIV R4P virtual conference, 27 January 2021. LB1479. To be updated when publication information becomes available.
- 12. Ford SL, Chiu J, Lovern M, Spreen W, Kim J. Population PK approach to predict cabotegravir (CAB, GSK1265744) long‐acting injectable doses for phase 2b (Ph2b). Abstract H‐645. 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; September 5‐9, 2014; Washington, DC.
- 13. Han K, Patel P, Baker M, Margolis DA, Spreen WR, Ford SL. Population pharmacokinetics of cabotegravir in adult healthy subjects and HIV‐1 infected patients following administration of oral tablet and long acting intramuscular injection. Abstract WEPDB0205. 22nd International AIDS Conference 23‐27 July 2018, Amsterdam, the Netherlands.
- 14. Han K, Patel P, Baker M, Margolis DA, et al. Cabotegravir (CAB) long‐acting (LA) phase 3 (Ph3) PrEP dose selection based on population pharmacokinetics (PPK) in healthy and HIV‐infected adults. Abstract OA15.05. HIV Research for Prevention 21‐25 October 2018, Madrid, Spain.
- 15. Beal S, Sheiner LB, Boeckmann A, Bauer, RJ. NONMEM User's Guides. (1989‐2009), Icon Development Solutions, Ellicott City, MD, USA, 2009.
- 16. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN)‐‐a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85‐94. [DOI] [PubMed] [Google Scholar]
- 17. R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2014. http://www.R-project.org/
- 18. Beal SL. Ways to fit a model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481‐504. [DOI] [PubMed] [Google Scholar]
- 19. Hing JP, Woolfrey SG, Greenslade D, Wright PMC. Analysis of toxicokinetic data using NONMEM: impact of quantification limit and replacement strategies for censored data. J Pharmacokinet Pharmacodyn. 2001;28:465‐479. [DOI] [PubMed] [Google Scholar]
- 20. Ahn EA, Karlsson MO, Dunne A, et al. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35:401‐421. [DOI] [PubMed] [Google Scholar]
- 21. Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82:17‐20. [DOI] [PubMed] [Google Scholar]
- 22. Guidance for Industry Population Pharmacokinetics, US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), February 1999. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072137.pdf
- 23. Committee for Medicinal Products for Human Use (CHMP) . Guideline on Reporting the Results of Population Pharmacokinetic Analyses, Doc. Ref. CHMP/EWP/185990/06, London, 21 June 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf
- 24. Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37:486‐495. [DOI] [PubMed] [Google Scholar]
- 26. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexander SP, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2021/22: Enzymes. Br J Pharmacol. 2021;178(Suppl 1):S313‐S411. [DOI] [PubMed] [Google Scholar]
- 28. Chan VO, Colville J, Persaud T, Buckley O, Hamilton S, Torreggiani WC. Intramuscular injections into the buttocks: Are they truly intramuscular? Eur J Radiol. 2006. Jun;58(3):480‐484. doi: 10.1016/j.ejrad.2006.01.008 [DOI] [PubMed] [Google Scholar]
- 29. Boyd A, DeFord L, Mares J, et al. Improving the success rate of gluteal intramuscular injections. Pancreas. 2013. Jul;42(5):878‐882. doi: 10.1097/MPA.0b013e318279d552 [DOI] [PubMed] [Google Scholar]
- 30. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36:425‐438. [DOI] [PubMed] [Google Scholar]
- 31. van der Bol JM, Mathijssen RH, Loos WJ, et al. Cigarette smoking and irinotecan treatment: pharmacokinetic interaction and effects on neutropenia. J Clin Oncol. 2007;25:2719‐2726. [DOI] [PubMed] [Google Scholar]
- 32. Zhang J, Hayes S, Sadler BM, et al. Population pharmacokinetics of dolutegravir in HIV‐infected treatment‐naive patients. Br J Clin Pharmacol. 2015;80(3):502‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure S1 Distribution of baseline characteristics of subjects included in the model‐building dataset (a‐g) and external validation dataset (h‐n)
Supporting Information Figure S2 Prediction‐corrected visual predictive check of the final model with baseline body weight as independent variable. Red solid line, median of prediction‐corrected observations; red dashed lines, 5th and 95th percentiles of prediction‐corrected observations; black dashed lines, predicted median, 5th and 95th percentiles; shaded regions, 95% confidence interval of the predicted median (pink), 5th and 95th percentiles (blue); blue dots, prediction‐corrected observations; red asterisks, prediction‐corrected observation lines were not contained within the shaded region; yellow bars on the x axis, boundaries between bins
Supporting Information Figure S3 External validation of the final model using prediction‐corrected visual predictive check stratified by sex. Red solid line, median of prediction‐corrected observations; red dashed lines, 5th and 95th percentiles of prediction‐corrected observations; black dashed lines, predicted median, 5th and 95th percentiles; shaded regions, 95% confidence interval of the predicted median (pink), 5th and 95th percentiles (blue); blue dots, prediction‐corrected observations; red asterisks, prediction‐corrected observation lines were not contained within the shaded region; yellow bars on the x axis, boundaries between bins.
Supporting Information Figure S4 Illustration of the impact of KALA alone on cabotegravir PK profiles while keeping all other PK parameter values unchanged. A higher KALA value results in a higher Cmax and Ctau‐1, but a lower Ctau‐SS. KALA alone has no impact on steady‐state AUC
Data Availability Statement
No new data were created for this analysis. The data that support the findings of this analysis may be available from ViiV Healthcare upon request and approval. Restrictions apply to the availability of these data.