

Abstract
Pulmonary metastases pose significant treatment challenges for many cancers, including triple-negative breast cancer (TNBC). We developed and tested a novel suicide gene and therapeutic microRNAs (miRs) combination therapy against lung metastases in vivo in mouse models after intranasal delivery using nontoxic gold nanoparticles (AuNPs) formulated to carry these molecular therapeutics. We used AuNPs coated with chitosan-β-cyclodextrin (CS-CD) and functionalized with a urokinase plasminogen activator (uPA) peptide to carry triple cancer suicide genes (thymidine kinase-p53-nitroreductase: TK-p53-NTR) plus therapeutic miRNAs (antimiR-21, antimiR-10b and miR-100). We synthesized three AuNPs: 20nm nanodots (AuND), and 20nm or 50nm nanostars (AuNS), then surface coated these with CS-CD using a microfluidic-optimized method. We sequentially coated the resulting positively charged AuNP-CS-CD core with synthetic miRNAs followed by TK-p53-NTR via electrostatic interactions, and added uPA peptide through CD-adamantane host-guest chemistry. A comparison of transfection efficiencies for different AuNPs showed that the 50nm AuNS allowed ∼4.16-fold higher gene transfection than other NPs. The intranasal delivery of uPA-AuNS-TK-p53-NTR-microRNAs NPs (pAuNS@TK-p53-NTR-miRs) in mice predominantly accumulated in lungs and facilitated ganciclovir and CB1954 prodrug-mediated gene therapy against TNBC lung metastases. This new nanosystem may serve as an adaptable-across-cancer-type, facile, and clinically scalable platform to allow future inhalational suicide gene-miR combination therapy for patients harboring pulmonary metastases.
Keywords: Genetic therapy, Lung neoplasms, Neoplasms, multiple primaries
Graphical Abstract
This research describes a novel approach to treating pulmonary metastases using a p53-targeted dual suicide gene therapy (TK-NTR) plus therapeutic microRNAs (miR-100, antimiR-21, and antimiR-10b) layered onto biocompatible chitosan-cyclodextrin di-block polymer coated gold nanostars (AuNS-CS-CD) after intranasal delivery. We describe the preclinical development and evaluation of this novel approach to treating lung metastases in mouse models.
Introduction
The lungs are the most common sites for metastases from extrathoracic cancers because of the unique anatomical and biological characteristics of the pulmonary system [1]. Unfortunately, the most common malignant tumors in the lungs are metastases [2], and up to half of the patients who die of malignant cancers have pulmonary metastases at autopsy [1]. In particular, the incidence of lung metastases is highest among patients with triple-negative breast cancer (TNBC), accounting for 10–15% of all breast cancer. Only 23% of patients with lung metastatic TNBC survive two years or longer [3]. The poor prognosis for patients harboring lung metastases from most cancers despite available treatment options (chemotherapy and immunotherapy), illustrated in particular by those with TNBC primaries, drives a crucial need to explore novel theranostic approaches and delivery methods that might improve treatment outcomes in patients having breast (or other systemic) cancers with pulmonary metastases.
Suicide gene therapy has been extensively investigated in the past for the cancer treatment [4–6]. Nanotechnology-based gene therapy applications have shown promising outcomes [7]. Recently, we developed a herpes simplex virus type-1 thymidine kinase sr39 mutant (HSV1-sr39TK, or, here simply, TK)-Escherichia coli nitroreductase (NTR) enzyme dual prodrug combination suicide gene therapy for effective killing of cancer cells [8, 9]. The TK-catalyzed products of the ganciclovir (GCV) prodrug cause cell death through inhibiting DNA synthesis, and the NTR-catalyzed product of the prodrug 5-(aziridine-1-yl)-2,4-dinitrobenzamide (CB1954) kill cancer cells by DNA alkylation, which produces a cumulative anticancer response. Furthermore, we fused p53, an important tumor suppressor gene that is mutated in most cancers, into TK-NTR as a triple block cancer suicide gene (TK-p53-NTR) to augment the anti-cancer therapeutic efficacy by replenishing the endogenous p53-mediated apoptotic pathway [9, 10]. MicroRNA (miR)-10b, miR-21, and miR-100 have been well documented and studied for their effects on cancer cell invasion, proliferation, and chemoresistance in multiple cancers including breast cancer. Interfering with the cellular functions of oncogenic miRs can improve chemotherapy outcomes [11–15]. Our previous studies have demonstrated the potentials of antimiR-21, antimiR-10b or miR-100 in TNBC therapy by sensitization of chemotherapeutic drugs [16–19]. By retrieving the anti-oncogenic and tumor suppressor functions of endogenous miRs through the delivery of synthetic miR mimics and/or antisense-miRs (antimiR-21 antimiR-10b and miR-100), we can augment the therapeutic effects against TNBC cells (here used as an appropriate model for many other cancer cell types that can metastasize to the lungs) by sensitizing them to ensuing TK-p53-NTR triple gene therapy in the presence of prodrugs GCV and CB1954. This therapeutic approach was designed to target both proliferating and non-proliferating populations of TNBC cells using TK-NTR and the accompanying cellular pre-sensitization by supplementing the p53 gene as well as antimiR-21, antimiR-10b, and miR-100, to achieve an enhanced anticancer effect.
Pulmonary delivery of therapeutics facilitates the local presentation of drugs or nanoformulations to the lungs directly through the nasal or oral routes. The advantages of pulmonary over systemic delivery include rapid degradation of therapeutics, the capability to bypass systemic first-pass metabolism, smaller doses of therapeutics to achieve desired effects, and a means to limit non-specific toxicity [20, 21]. Intranasally delivered therapeutics also spread to the lymphatic circulation and reach the distal bronchi, allowing exposure to poorly reachable regions of the lungs [22]. This might be particularly advantageous in cases of lung metastases because most of these tumors are located peripherally in the lungs [23]. For instance, an aerosolized delivery of interleukin-2 liposome in a large animal model has been shown to be effective for lung metastases from osteosarcoma [24]. However, the complex architecture of the bronchial tree and its particular clearance mechanisms can still pose challenges to the deposition of intranasally delivered insoluble particles in the lungs. Mucociliary clearance plays a dominant role in the clearance of the upper airway [25]. The mucus secreted by the ciliated columnar epithelium traps the inhaled particles deposited in the trachea and proximal bronchi. Most inhaled insoluble particles greater than 5 μm in size and deposited in the trachea and proximal bronchi are eliminated by mucociliary clearance [26]. Smaller-sized insoluble particles deposited in the distal bronchi, where mucociliary-clearance is less functional, can be retained for longer periods than larger insoluble particles. The mechanism of clearance of inhaled particles in the distal lungs is comparatively complex. It depends more on the physicochemical properties of the particles, such as their size and dissolution kinetics. Mucoadhesive nanoparticles (MANP) have been well explored and documented for pulmonary drug delivery by capitalizing on their perceived advantages of improving nanoparticle (NP) accumulation in the lungs [27]. Further, chitosan-β-cyclodextrin (CS-CD) has been widely adopted in various drug-delivery applications. It possesses cationic surface charges that interact with negatively-charged glycosylated/cysteine-rich hydrophobic domains of respiratory tract mucin fibers, thus increasing the resident time of nanoparticle-drugs at the location of absorption/action. This provides a sustained drug release profile and minimizes drug degradation [28–30]. Moreover, alveolar macrophage-mediated phagocytosis is considered a major clearance mechanism in this location [31]. This is, for the most part, responsible for the clearance of particle sizes between 1 μm and 5 μm [32, 33]. Particles in the size below 200 nm are less likely to be recognized by macrophages or be rapidly taken up by epithelial cells [34, 35]. Therefore, optimizing the physicochemical properties of drug-carrying nanoparticles is of great importance in effective pulmonary anti-cancer drug deliveries.
Recently, we successfully developed an efficient in vivo gene delivery nanocarrier for intranasal delivery of therapeutic miRNAs to orthotopic glioblastoma (GBM) in a mouse model [36, 37]. We found that the size and surface charge of gold-polymeric NPs allowed preferential trafficking to and accumulation in brain tumors through olfactory and trigeminal nerve pathways. Notably, by controlling the regimen of NP intranasal administration, as well as the degree of anesthesia in mice, the NPs can be diverted in large quantities to the lungs by minimizing their intranasal mucosal adhesion, which, in general, is responsible for brain targeting.
In breast cancer, elevated levels of urokinase-type plasminogen activator (uPA) have been widely studied as a biomarker or therapeutic target [38, 39]. The surface of gold NPs can be modified with different functional compounds, thus broadening their potential therapeutic applications, especially against cancer [40]. Here, based on the above rationale, and guided by the results of our previous studies, we developed and evaluated a novel gold-nanostar-chitosan-β-cyclodextrin (AuNS-CS-CD) NP. We comprehensively optimized the gene loading efficiency of the NPs by first evaluating three different forms of gold NPs. We then established an optimized CS and CD ratio, and used this to create a novel microfluidic-based CS-CD coating strategy during the NP synthesis process. This rendered the AuNS-CS-CD NPs more uniform and scalable during production. The uPA-conjugated AuNS-CS-CD (pAuNS) of 50 nm resulted in ∼4.16-fold higher gene loading and cell transfection efficiency when compared to smaller pAuNP of different structures [36]. Moreover, we tested the feasibility of in vivo pulmonary delivery of pAuNS carrying TK-p53-NTR genes by using the converted CytoCy5S substrate as a reporter to target mouse lungs. We set out to develop and validate this strategy in an animal model prior to future clinical translational applications for the delivery of therapeutic genes to lung metastases. The relatively non-invasive pulmonary delivery of therapeutic genes using this uPA targeted AuNS potentially provides a novel addition to available therapeutic options against lung metastases of many primary origins, including those from advanced TNBC.
Results and Discussion
Synthesis and characterization of CS-β-CD coated gold nanostar (AuNS) and gold nanodot (AuND) NPs, and optimization of gene delivery in cells
We synthesized AuNS using a modified reduction procedure that we outlined previously [36, 41]. In brief, the synthesis process was optimized to generate gold nano-octopods that can increase surface area for coating CS-CD and encapsulating higher payloads [40] (Figure S1). By controlling the reaction conditions, we synthesized three different forms of AuNPs. The surfaces of as-synthesized AuNS/AuND NPs were modified by β-CD-grafted natural cationic CS polymer (CS-CD) using a microfluidic-based coating process to improve their colloidal stability, biocompatibility, uniformity, and scalability, thus providing a cationic surface zeta potential for loading anionic therapeutic genes and miRNAs (Figure 1a). The particle sizes and surface charges were estimated using dynamic light scattering (DLS) (Figure 1b). The size distribution analysis revealed that the size of as-prepared AuNS was in the range of 67.5 ± 3.25 nm. After coating with CS-CD, the size of AuNS increased to 160.5±1.63 nm. This increase in size upon CS-CD and DNA loading could, in part, be owing to the analytical nature of DLS, which measures the hydrodynamic size of the particle and also detects some loosely bound polymer as part of the structure during size calculation. Hence, we further confirmed the physical size of the particles by TEM analysis, which we present later. The change in the NP surface charge measurement confirmed the successful coating of CS-CD and nucleic acids loading on the AuNPs. The pristine surface potential of AuNS was −18.3±1.13 mV, which was then shifted to a cationic surface potential of +32.7±1.84 mV upon coating with CS-CD polymer complex (Figure 1b). We further repeated the NP size evaluation using NP tracking analysis (NTA). The AuNS of 50 nm and 20 nm, and AuND of 20 nm coated with CS-CD were analyzed for the sizes and the concentrations of each particle. We observed the concentration of NPs to be in the range of 6.54 ×109 ± 1.30 ×109/mL (pAuNS-50-CS-CD), 8.86 ×109 ± 9.88 ×108/mL (pAuNS-20-CS-CD), and 8.43×1010 ±1.57 ×1010/mL (pAuND-20-CS-CD), for different AuNPs (Figure S2, supporting information). We also compared the gene loading capacity of all three AuNPs. AuNS with an enlarged surface area had a higher amount of pDNA loading performance than the spherical AuND. We were able to load 1 μg of pDNA in 3.92 ×108 ± 7.80 ×107 and 1.11 ×109 ± 1.24 ×108 particles of pAuNS of 50 nm and 20 nm, respectively, while 2.11 ×1010 ± 3.93 ×109 particles are required for loading the same amount of DNA when we used pAuND of 20 nm. We observed a gene loading capacity of 2.55 ×10−9 ± 1.3 ×10−8 μg/particle for the pAuNS-50 nm, 9.0 ×10−10 ± 8.1 ×10−8 μg/particle for pAuNS-20 nm, and 4.7 ×10−11 ± 2.5 ×10−10 μg/particle for the pAuND-20 nm, indicating that the pAuNS-50 nm with the resultant hydrodynamic size of ~160.5 nm by DLS and 130 nm by NTA had the highest loading capacity (Figure 1c).
Figure 1. Synthesis and characterization of Chitosan-β-Cyclodextrin (CS-CD) di-block copolymer coated gold NPs.
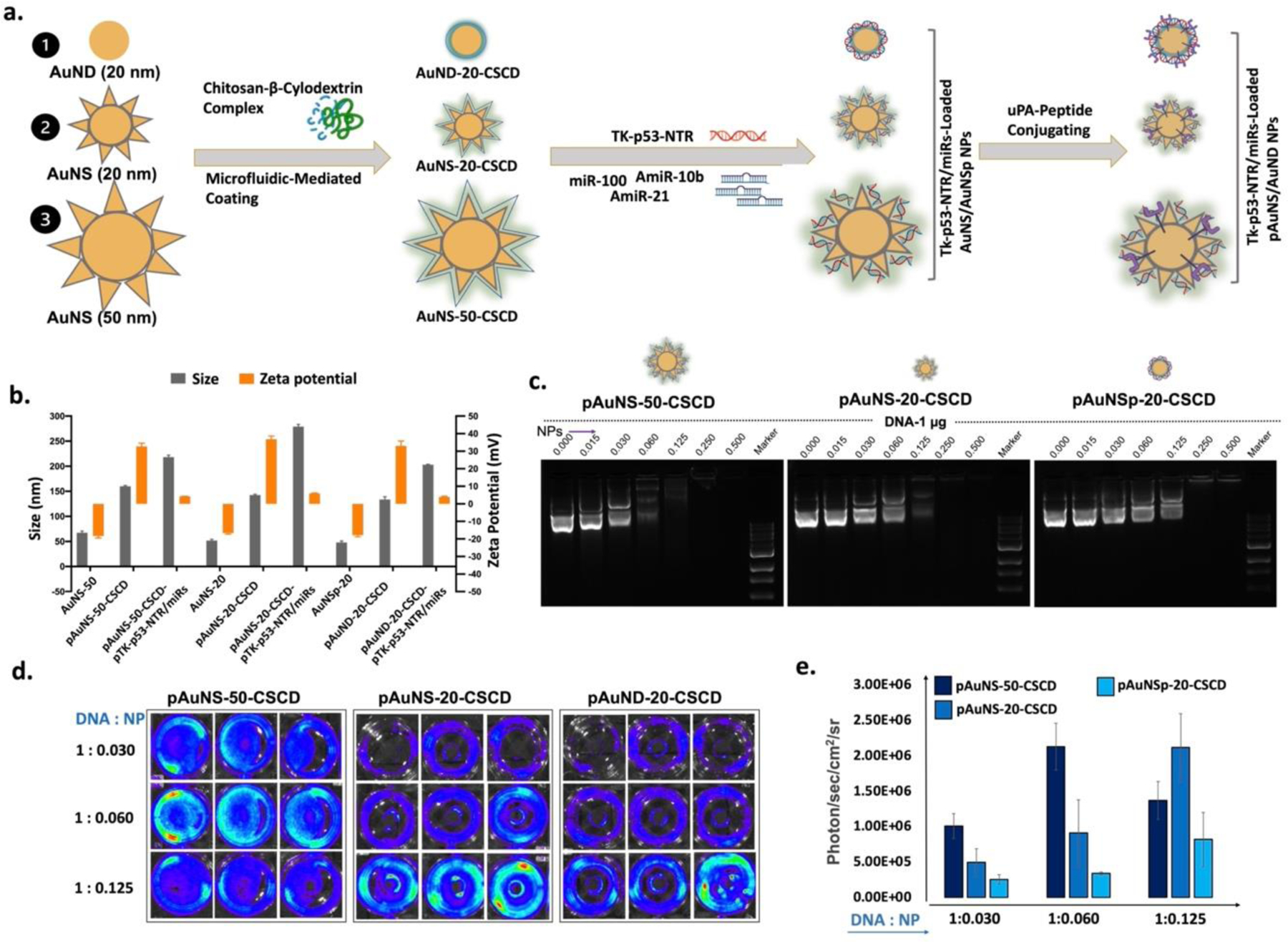
(a) Schematic illustration of uPA-targeted, CS-CD coated gold nanostar (pAuNS), and nanodot (AuND) NPs loaded with therapeutic miRs and a plasmid coding for TK-p53-NTR triple therapeutic gene; (b) Hydrodynamic sizes and surface charges of AuNS and AuND with and without loaded miRs/pTK-p53-NTR by dynamic light scattering (DLS) measurements (n=3, the data are presented as mean ± SD); (c) DNA loading efficiency of AuNS and AuND NPs at multiple concentrations based DNA/NPs ratio measured using the gel retardation assay; (d) Transfection efficiency of three different gold NPs estimated using delivery of Fluc-eGFP-pcDNA reporter plasmid (eGFP-pcDNA) by bioluminescence imaging and its quantitative graph (e), (n=3, the data are presented as mean ± SD).
To further confirm the successful loading of the genes and to determine the optimum polyplex ratio needed for plasmid DNA delivery, we loaded pAuNS-50 nm, pAuNS-20 nm, and pAuND-20 nm with Fluc-eGFP-pcDNA and evaluated them for transfection efficiency in HEK-293T and 4T1 cells using bioluminescence imaging (BLI) (Figure 1d). In accordance with the gel retardation assay, the maximum transfection efficiency was observed with an amalgamation of 1 μg pDNA and 0.06 μL of pAuNS-50 nm, while double the amount (0.125 μL) was required for a smaller-sized pAuNS-20 nm to encapsulate a similar amount of DNA. In contrast, we observed the maximum BLI signals at a ratio of 1:0.25 of pDNA and NPs for the pAuND-20 nm NPs. The quantitative graph shown in Figure 1e reveals the measured BLI signals from the transfected cells. After confirming the gene loading capacity and transfection efficiency of the three different forms of AuNPs, we selected the pAuNS-50 nm for subsequent in vitro and in vivo studies, addressed as pAuNS.
The uniform monolayer of CS-β-CD on the AuNS surface was evident on the transmission electron micrographs (TEM), which showed a smaller size distribution of as-prepared AuNS, with an average size of ~20 nm as the AuNS core, and projecting spikes of ∼20–30 nm (Figure 2a). Unlike DLS, where we measured some increase in particle size upon CS-CD coating, TEM analysis revealed only a 2–5 nm coating on top of the spikes. Further, we estimated the TK-p53-NTR plasmid DNA plus miRs loading efficiency for pAuNS using a gel retardation assay. As shown in Figure 2b, for the loading capacity of AuNS for TK-p53-NTR pDNA indicated that 1 μg of plasmid could be entrapped in 0.015 μL AuNS NPs (0.98 ×105 ± 1.95×104 NPs) (Figure 2b, upper left panel), and, 0.03 μL (1.96 ×105 ± 3.9 ×104 NPs) AuNS could entrap 200 pmol of miRNA (Figure 2b, upper right panel). Whereas, for co-loading of TK-p53-NTR pDNA and miRNA, an ideal formulation demonstrated was 0.06 μL AuNS NPs (3.92 ×105 ± 7.80 ×104 NPs) with 1 μg of TK-p53-NTR pDNA and 200 pmol of miRNA (Figure 2b, lower lane). At the optimized ratio of 0.06 μL, AuNS NPs were able to entrap 1 μg TK-p53-NTR pDNA and 200 pmol of miRNA (66.6 pmols of each) for co-loading experiments.
Figure 2. In vitro characterization of AuNS loaded with DNA.
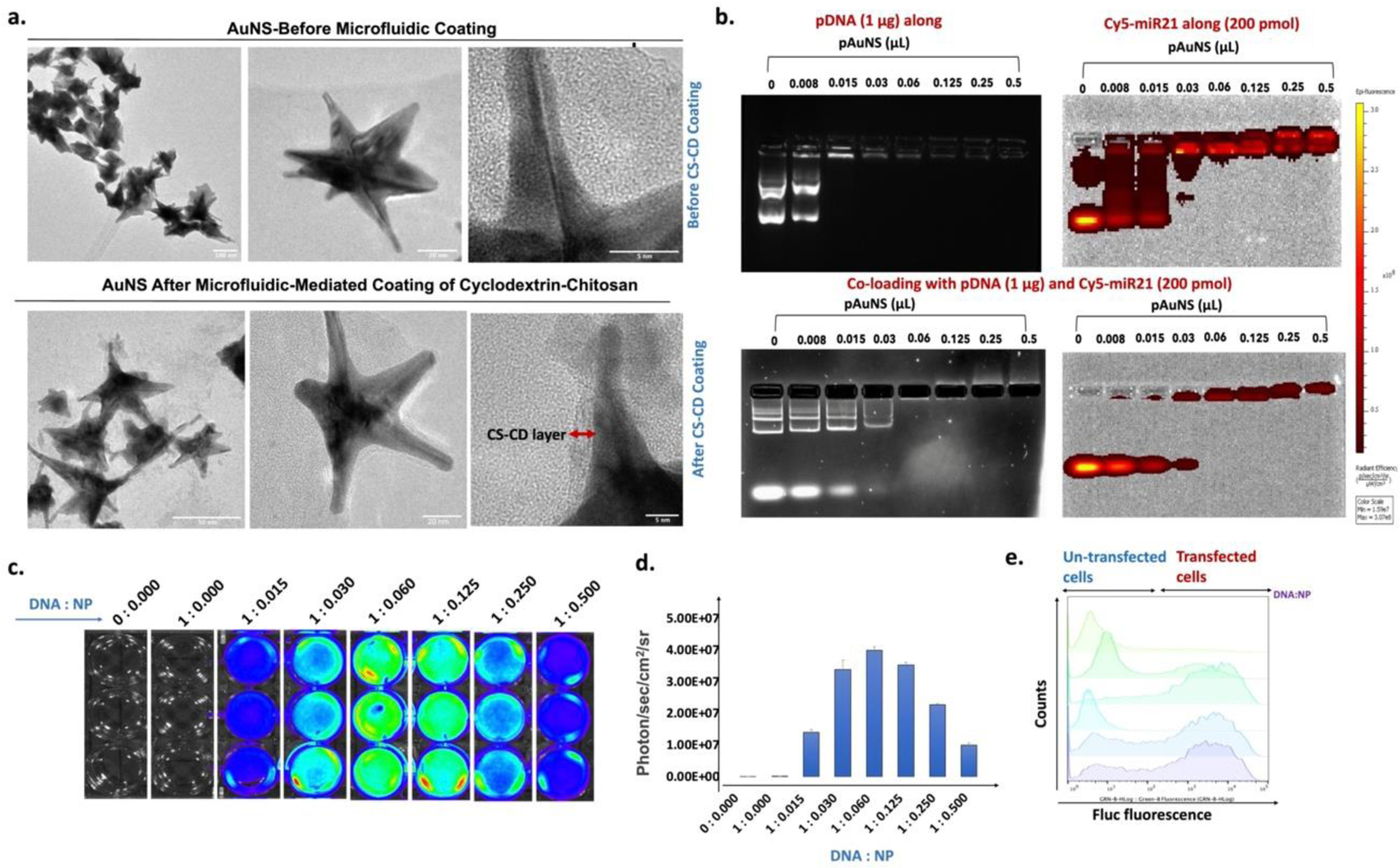
(a) Transmission electron microscopy (TEM) images showing uniform morphology of AuNS before and after microfluidic-mediated coating of CS-CD; Scale bar:100 nm (upper-left panel), 20 nm (upper-middle panel), 5 nm (upper-right panel), 50 nm (lower-left panel), 20 nm (lower-middle panel), and 5 nm (lower-right panel). (b) Optimization of DNA/miR to NPs ratio measured using a gel retardation assay for the co-loading of Cy-5-tagged miR and TK-p53-NTR plasmid DNA; (c) Transfection efficiency of pAuNS- loaded Fluc-eGFP-pcDNA plasmid (pAuNS@eGFG-pcDNA) estimated by bioluminescence imaging; (d) Quantitative graph showing the transfection efficiency measured in “c” (n=3, the data are presented as mean ± SEM); and (e) Flow cytometry analysis measuring the in vitro transfection efficiency of Fluc-eGFP-pcDNA delivered by pAuNS in different N/P ratio by the expressed GFP signal.
To further test the in vitro stability of the NPs in different biological solutions, we incubated AuNS-CS-CD (pAuNS) NPs in water, PBS and a complete medium containing 10% FBS for different time periods (0, 1, 3, 5 and 7 days) and evaluated for the charge using DLS, and size and numbers by using a NP tracker analysis (NTA) system. We did not observe any change in the charge of NPs incubated in water (42 ± 4 mV). In contrast, the NPs suspended in PBS showed a significant reduction in overall charge (6 ± 2 mV) compared to original NPs suspended in water (42 ± 4 mV), and this was consistent across all the time points analyzed. The particles suspended in medium with 10% FBS showed negative zeta potential (−8.5 ± 3 mV) independent of analysis time, which is mainly owing to the binding of protein corona of FBS present in the medium on pAuNS particles. The same NPs upon coating with pDNA (pAuNS@TK-p53-NTR/miRs NPs) showed no significant change in the medium upon incubation. The NTA results of NPs suspended in water and PBS showed no significant change in size or numbers analyzed over time. In contrast, the NPs suspended in medium showed the presence of large amounts of protein particles of 50 nm along with the suspended AuNS (Figure S3–S6, Supporting Information).
Meanwhile, we loaded pAuNS plus Fluc-eGFP-pcDNA in multiple ratio combinations and evaluated them for transfection efficiencies in HEK-293T and 4T1 cells using BLI (Figure 2c, d; Figure S9, supporting information). We collected the transfected cells to determine the GFP signal using flow cytometry analysis (Figure 2e). In agreement with BLI outcomes, the highest Fluc-eGFP-pcDNA transfection was observed in pAuNS at an NP to DNA ratio of 1.0 to 0.06.
After selecting the pAuNS with the optimum pDNA loading and transfection efficiency, an increasing amount of Fluc-eGFP-pcDNA with a fixed amount of as-verified NPs-to-DNA ratio was used in a cell transfection experiment to evaluate the optimal dose that can be used in the subsequent in vitro and in vivo therapeutic study without baseline toxicity (Figure S8, and S9, supporting information). We used 4T1 cells and transfected them with increasing amounts of pAuNS@eGFP-pDNA. We observed a dose-dependent increase in Fluc BLI signal with saturation at 3 μg of DNA used for transfection (Figure S7a, c, supporting information). We also performed flow cytometry to verify the GFP signals in treated cells further. The optimized dose of 3 μg pDNA could be used to transfect 10 × 105 4T1 cells with no demonstrable toxicity (Figure S7b, supporting information).
Cellular uptake of uPA-functionalized AuNS NPs co-loaded with TK-p53-NTR gene and miRs in TNBC cells
We evaluated the cellular uptake of AuNS NPs co-loaded with TK-p53-NTR pDNA and Cy5-miR in 4T1 TNBC cells using fluorescence microscopy, flow cytometry, qPCR, and qRT-PCR assays. Confocal fluorescence microscopy images of 4T1 cells treated with the co-loaded AuNS NPs showed clear Cy5 signals from the delivered Cy5-miRNA, and the NTR converted CytoCy5S substrate (Figure 3a). Similarly, flow cytometry analysis further confirmed the presence of intracellular Cy5-miR in cells (Figure 3b). We further analyzed the intracellular delivery of TK-p53-NTR using qPCR and the target primers synthesized against the TK gene and miRs using Taqman qRT-PCR (Figure 3d–f). Flow cytometry analysis of the cells treated with CytoCy5S substrate at 24 h post-transfection further confirmed the successful delivery of Cy5-tagged miR and the expression of NTR gene upon delivery using AuNS NPs co-loaded with TK-p53-NTR and miRs (Figure 3c). The uptake function of uPA-targeted and non-targeted AuNS was also assessed in 4T1 cells at 24 h post-incubation. The AuNS cellular uptake levels of Cy5-miRs and TK-p53-NTR were significantly higher (p<0.05) in cells delivered using uPA functionalized NPs compared to scrambled sc-uPA functionalized NPs. The TK-p53-NTR pDNA and miRs co-loaded AuNS had net positive charges. This allowed passive diffusion of NPs across cell membranes due to the cell membrane’s inherent negative surface potential, and the modification of AuNS with β-CD further facilitated the cellular internalization [42, 43].
Figure 3. Cellular uptake of AuNS loaded with miRs and TK-p53-NTR plasmid DNA in 4T1 TNBC cells.
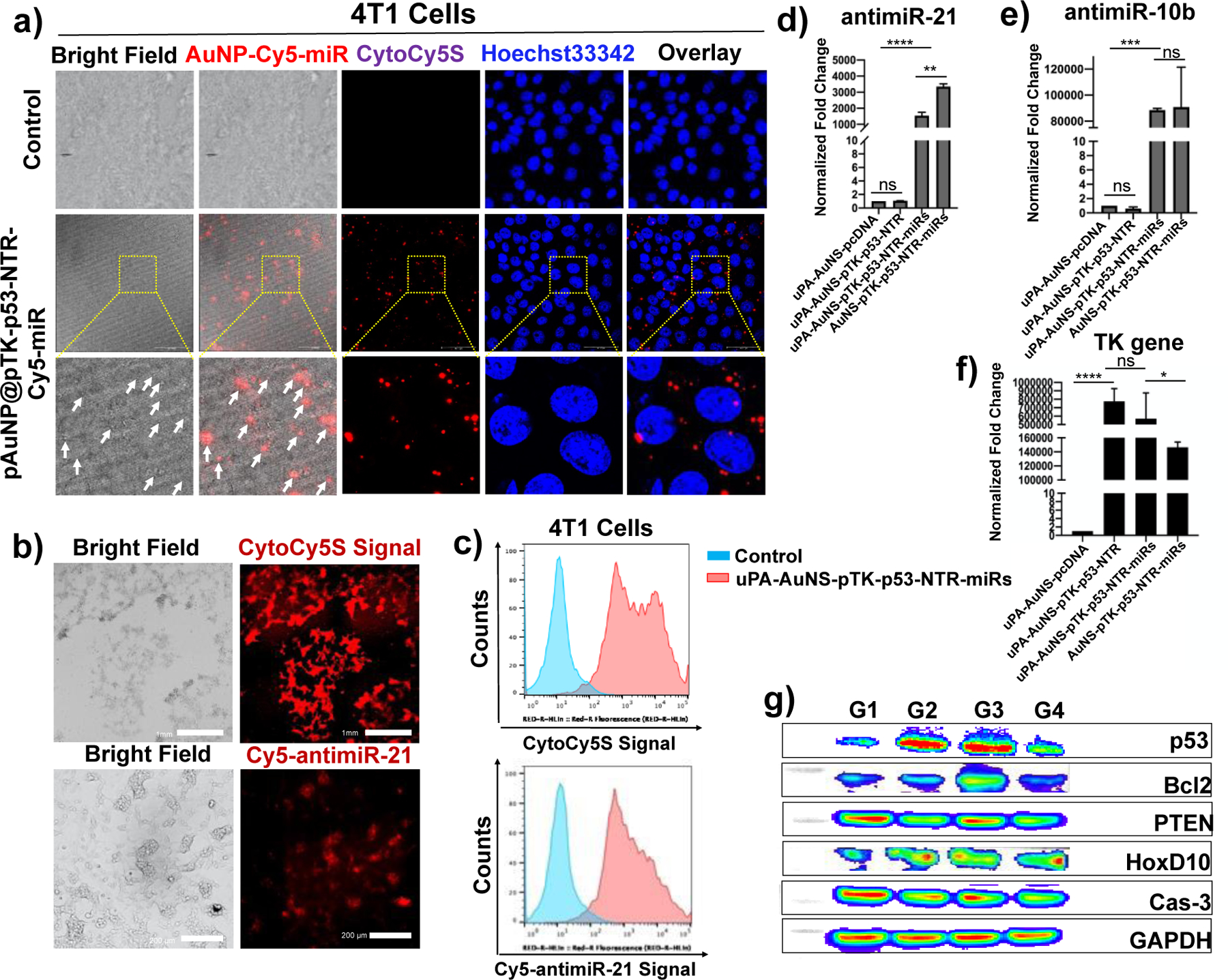
(a) Confocal laser microscopic images showing the internalization of pAuNS coloaded with Cy5-tagged miR and TK-p53-NTR plasmid DNA in 4T1 cells (Red: Cy5-miR, CytoCy5S, and Blue: Hoechst 33342 for nuclear staining); (b) Fluorescent microscopic images indicating the Cy5-signal for miRs delivery and CytoCy5S substrate conversion for TK-p53-NTR expression in 4T1 cells transfected with pAuNS co-loaded with Cy5-miR and TK-p53-NTR plasmid. (c) The same results are shown in ‘b’ measured by flow cytometry analysis. (d-f) Quantitative evaluation of miRs and TK-p53-NTR plasmid transfection in 4T1 cells by AuNS for the intracellular miRs using qRT-PCR, and TK-p53-NTR by using qPCR (n =3; Two-way ANOVA with Tukey T test and confidence interval significance was performed for comparisons; * represents p < 0.05, ** represents p < 0.01, and ***represents p < 0.001; the data were plotted as mean ± SD). (g) Immunoblot analysis of the downstream target proteins of antimiR-21, antimiR-10b, and miR-100 in 4T1 cells transfected with AuNS co-loaded with miRs and TK-p53-NTR plasmid (G1: pAuNS-pcDNA; G2: pAuNS@TK-p53-NTR; G3: pAuNS@TK-p53-NTR-miRs; G4: non-targeted AuNS@pTK-p53-NTR-miRs). All the non-targeted AuNS NPs used in the experiments were conjugated with scrambled-uPA peptide (sc-uPA).
After this non-specific adsorption and electrostatic interaction-driven transportation, a significantly greater extent of miRs internalization was detected upon loading with AuNS than the controls. The surface modification of the AuNS NPs using uPA peptide further enhanced the cellular internalization of NPs through active targeting of the uPA-receptor-mediated endocytosis in TNBC cells. The uPA receptor is overexpressed in most TNBC cells. Thus, the highest levels of intracellular TK-p53-NTR pDNA and miRs delivery were observed in 4T1 cells treated with uPA-modified AuNS compared to the non-modified counterparts at a 24 h treatment time point (Figure 3d–f). To evaluate the bioactivity of the delivered miRs, the TNBC 4T1 cells were treated with 200 pmols each of antimiR-21, antimiR-10b, and miR-100, either individually or co-loaded with AuNS for 48 h, and cells were collected for their downstream target protein expression analysis. The respective proteins levels were measured using immunoblot analysis. We observed a significant upregulation of PTEN and HoxD10 expression in cells treated with AuNS loaded with antimiR-21, antimiR-10b, and miR-100 combination compared to control. Since p53 plays a vital role in the cellular apoptosis mechanism, the p53 pathway proteins and the pro- and anti- apoptotic proteins (p53, Bcl2, and caspase 3) in 4T1 cells were also evaluated after different treatment conditions. The protein levels significantly changed in cells treated with TK-p53-NTR-pDNA and prodrug combination (Figure 3g). Thus, our findings confirm the efficient loading of the TK-p53-NTR gene and miRs in AuNS and their successful delivery to TNBC cells. This set the stage for further evaluation of our NPs by intranasal delivery in vivo to treat TNBC lung metastases in mice.
Therapeutic evaluation of TK-p53-NTR triple therapeutic gene and miRs co-delivered using pAuNS NPs in TNBC cells
We first investigated the therapeutic potential of the TK-p53-NTR suicide gene with multiple combinations of miRs (antimiR-21, antimiR-10b, and miR-100) against TNBC 4T1 cells in the presence and absence of prodrugs GCV and CB1954 over time for induced cellular apoptosis using flow cytometry analysis. Figure 4a shows the in vitro treatment regimen of the study. First, the 4T1 cells were treated with naked TK-p53-NTR-pDNA and miRs (antimiR-21, antimiR-10b, and miR-100) or single pAuNS without gene loading did not result in any noticeable decline in tumor cell viability as compared to untreated controls. This also indicated the non-toxic and biosafety nature of pAuNS NPs for intranasal delivery applications. To analyze the effects and mechanisms of different miRs or genes on cell apoptosis and to further screen optimized gene therapy combinations, we used multiple gene combination treatments in the presence or absence of prodrugs. Flow cytometry-based cell apoptosis analysis outcomes verified our initial hypothesis: In all miR-alone treated cell groups, even with the prodrug combination treatment, the number of apoptotic cells changed little compared to controls.
Figure 4. In vitro therapeutic efficacy of TK-p53-NTR gene and miRs (antimiR-21, antimiR-10b, and miR-100) co-delivered using pAuNS NPs and treated using prodrugs GCV and CB1954 in TNBC cells.
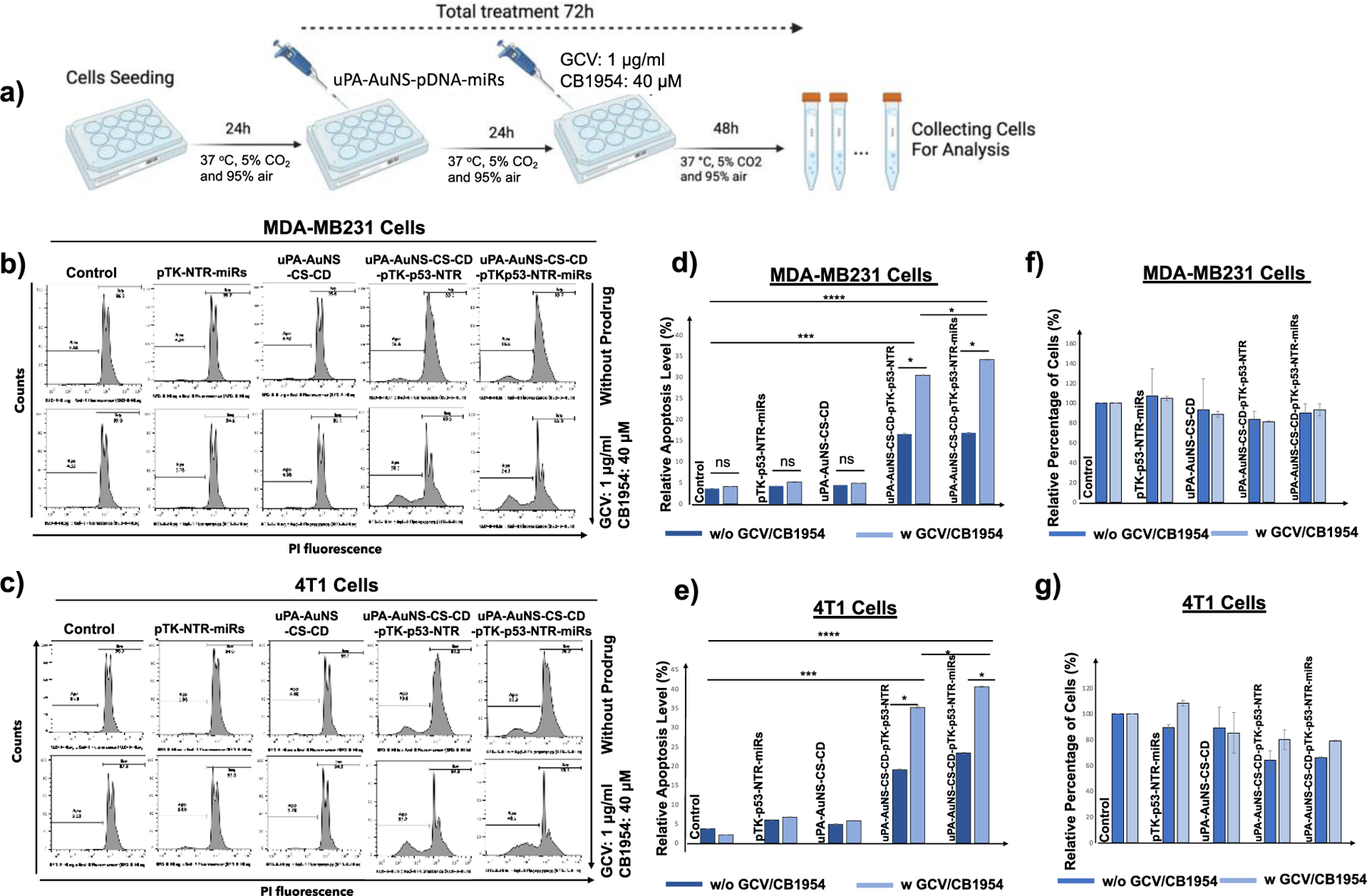
(a) Schematic illustration of TNBC cells treated with pAuNS co-loaded with miRs/TK-p53-NTR and the treatment outcome in the absence and presence of prodrugs GCV and CB1954. (b, c) Propidium Iodide (PI) staining-based FACS analyses measure the apoptotic and live cell populations in human (MDA-MB231) and mouse (4T1) TNBC cells upon treatment using empty pAuNS NPs, pAuNS loaded with TK-p53-NTR alone, miRs alone, and pAuNS co-loaded with TK-p53-NTR/miRs with and without exposure to prodrugs GCV and CB1954, and the quantitative plot of MDA-MB231 (d) and 4T1 cells (e) measured for treatment outcome. (f and g) The measured total cell numbers based on flow cytometry assay used for the quantification of cell proliferation in MDA-MB231 (f) and 4T1 (g) cells (n =3, the data were plotted as mean ± SD; Two-way ANOVA with Tukey T test and confidence interval significance was performed for comparisons; symbols indicating statistical significance were, *p< 0.05, **p< 0.01, and ***p< 0.001).
Moreover, supplementing antimiR-21, antimiR-10b, or miR-100 independently of TK-p53-NTR with prodrugs therapy resulted in limited anti-proliferative effects, as shown by 27.5%, 23.2%, and 21.1% cell apoptosis rates, respectively, as compared to control (5.98%). The highest suicide gene therapeutic efficacy was established in the treatment condition involving the TK-p53-NTR gene and all three miRs (antimiR-21, antimiR-10b, and miR-100) in 4T1 cells (a 42.4% apoptotic rate). The results for different treatment conditions are shown in Figure S10, supporting information.
Based on this outcome, we estimated the therapeutic potential of the TK-p53-NTR/miRs (antimiR-21, antimiR-10b, and miR-100) co-delivery regimen, when combined with prodrugs GCV and CB1954 (Figure S11, supporting information) in MDA-MB-231 human TNBC cells and 4T1 mouse TNBC cells under similar conditions, were almost equal in both cell types. The TK and NTR enzymes caused cell cytotoxicity only in the presence of prodrugs, resulting in 30.5% apoptosis in MDA-MB231 cells and 35.2% in 4T1 cells when compared to controls. Further, the concomitant treatment using TK-p53-NTR co-activated by GCV and CB1954, and supplementation of antimiR-21, antimiR-10b, and miR-100, significantly increased the cell apoptosis rates to 34.1% in MDA-MB231 cells (Figure 4b, d) and 40.5% in 4T1 cells (Figure 4c, e), respectively (p<0.001). The relative cell numbers measured in different treatment conditions based on the flow cytometry-based apoptotic analysis are shown in Figure 4f and Figure 4g. These results indicated that the exogenous expression of functional p53 from TK-p53-NTR gene expression could render TNBC cells much more susceptible to the cytotoxic derivates generated by the TK-NTR mediated conversion of GCV and CB1954 prodrug systems. Notably, we observed a significant difference in the various miRs-combination treatment conditions in both 4T1 and MDA-MB231 TNBC cells (p<0.05).
Pulmonary delivery and therapeutic implications of TK-p53-NTR triple therapeutic gene and miRs (antimiR-21, anitmiR-10b, and miR-100) delivered using pAuNS NPs in gene-directed enzyme prodrug therapy of TNBC lung metastases.
We evaluated the delivery, and therapeutic efficacy of TK-p53-NTR and miRs (antimiR-21 anitmiR-10b and miR-100) delivered using AuNS-NPs in vivo, in the presence of prodrugs (GCV and CB1954) (Figure 5a). We used a strategy entailing intranasal/pulmonary delivery of this nanoformulation in an orthotopic TNBC lung metastasis xenograft model of 4T1-Fluc-eGFP cells. For tumor cell implantation, we used the 4T1 cells stably expressing Fluc-eGFP to facilitate in vivo BLI as a marker to visualize the TNBC lung metastatic tumors and track their growth in response to treatment (Figure 5b). We evaluated four treatment groups: 1) pAuNS NPs loaded with control pcDNA, 2) pAuNS@TK-p53-NTR NPs, 3) pAuNS@TK-p53-NTR/miRs NPs, and 4) non-targeted AuNS@TK-p53-NTR/miRs NPs. All mice, including the pcDNA-AuNS control group, periodically received a fixed dose of prodrugs, GCV (40 mg/kg) and CB1954 (40 mg/kg). Acquisition of mouse BLI during the treatments showed a consistent tumor growth pattern.
Figure 5. In vivo estimation of the therapeutic potential of the TK-p53-NTR gene combined with therapeutic miRs in the presence of GCV/CB1954 prodrugs in mice bearing TNBC lung metastatic xenografts and delivered using uPA-targeted AuNS NPs.
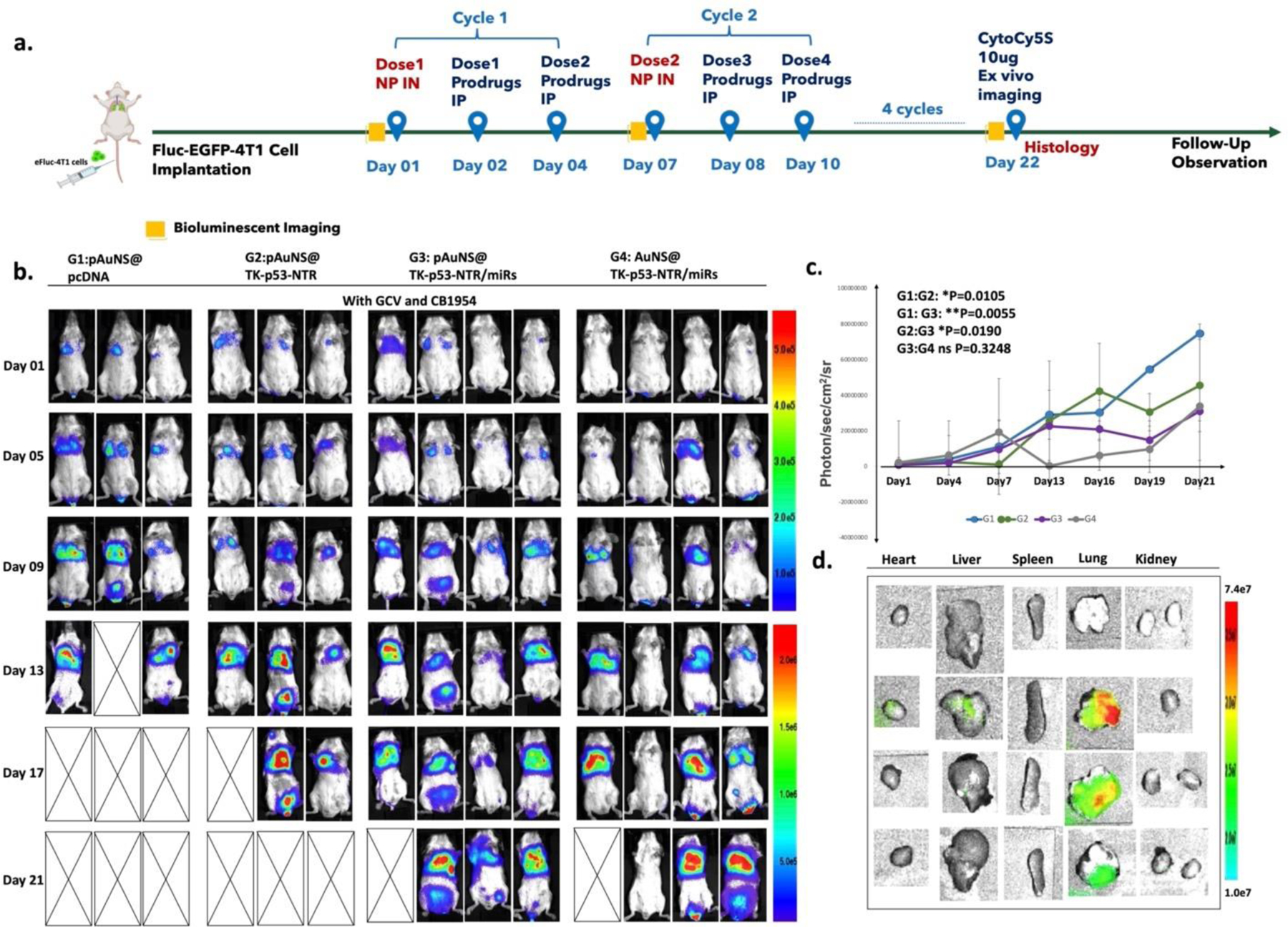
(a) Schematic outline of the in vivo study design for imaging and treatment timelines. (b) Bioluminescence imaging (BLI) of Balb/c mice bearing 4T1 TNBC lung metastasis xenografts expressing Fluc-eGFP reporter gene at multiple time points (G1 denotes pcDNA delivered by pAuNS NPs, G2 denotes TK-p53-NTR gene alone delivered by pAuNS NPs, G3 denotes TK-p53-NTR and miRs (antimiR-21, antimiR-10b and miR-100) delivered by pAuNS NPs, and G4 denotes TK-p53-NTR gene and miRs delivered using non-targeted AuNS NPs. All mice received a combination of prodrugs (GCV and CB1954). (c) Tumor volume estimation by the quantitative plot of the BLI intensity during the treatments. (d) TK-p53-NTR gene delivery evaluation in mice by ex vivo CytoCy5S fluorescence imaging at the termination of treatment (n=3–5/group; data are presented as mean ± SD; the significance of comparisons, as indicated, is drawn using One-way ANOVA with Bonferroni post hoc test. Adjusted p - values were considered statistically significant if p- values were < 0.05. The symbols indicating statistical significance are as follows: ns- represents non-significant difference, * represents p< 0.05, ** represents p< 0.01, *** represents p< 0.001, and **** represents p< 0.0001).
The findings of tumor BLI in mouse lungs and fluorescent images (Cy5 signals from reduced CytoCy5S by the expressed NTR enzyme) of all major organs harvested from mice in each treatment cohort upon completion of the treatment protocol are summarized in Figure 5b and Figure 5d. The CytoCy5S fluorescence signals depicted a predominant accumulation of NTR gene expression in the lungs of mice. Aside from some minor signals detected in the livers, no other significant signals were detected in major organs such as the spleen, kidneys, heart, or brain (Figure 5d). The delivered TK-p53-NTR genes had significantly accumulated in the lungs, indicating that the NPs administered intranasally had successfully trafficked along the airways into the lung peripheries.
We observed that the TK-p53-NTR delivery group achieved ~39.3% tumor growth inhibition compared to the control (p = 0.0105). The tumor growth inhibition remained at 19.4% in the miRNAs supplemented group compared with TK-p53-NTR delivery alone (p = 0.019). Although we found that neither the uPA-targeted co-delivery of TK-p53-NTR plus miRs nor the same delivered by non-targeted AuNS showed no significant difference in tumor growth inhibition (~58.7% and ~55.0%; p = 0.32). However, they were highly effective compared to controls over the 21-day treatment course (Figure 5b, c). Overall, we found that, aside from the comparatively low cellular uptake levels of non-targeted AuNS NPs in 4T1 cells (as evaluated in the in vitro experiments), the in vivo tumor suppression profile of the pAuNS-mediated co-delivery of therapeutic genes plus miRNAs achieved promising levels. This likely reflects some benefits derived from using the direct pulmonary delivery pathway. However, a further precise definition of any such benefit will require further mechanistic analysis in future small and large animal studies. We speculate that the pulmonary delivery of NPs may enhance the overall concentration of NPs in the lungs, and this facilitates direct delivery of genes to rapidly proliferating tumor cells present within the lungs. Hence, there is no specificity observed for the targeted NPs. In addition, it has been clearly shown in the literature that in vivo targeting efficiency is still not an ideal method for nano-based targeted cancer therapy, which is also a major challenge for clinical translation of this approach for various applications [44].
We observed that the lung metastases grew during all the treatment conditions, but control mice could not survive beyond 13 days. In contrast, the use of AuNS-mediated TK-p53-NTR plus miRs co-delivery (either uPA-targeted or non-targeted) resulted in a significant improvement in the survival rates (p < 0.01). This was also in agreement with the tumor suppression observations depicting the roles of AuNS mediated gene delivery in enhancing prodrugs antitumor efficacy in the TNBC lung metastasis mouse models (Figure 6e). The quantitative analysis of changes in the delivered miRs and TK gene were also ex vivo evaluated in mice lungs and other organs using Taqman qRT-PCR and qPCR, respectively, for both antimiR-21 and TK gene. The quantification of antimiR-21 (which is endogenously absent) revealed a ~4.29×109-fold increase in antimiR-21, and a ~51.35-fold increase in TK gene expression, in the lungs of mice co-treated with TK-p53-NTR gene and miRNAs using pAuNS NPs (Figure 6b, c). This ex vivo quantitative analysis of delivered genes plus miRNAs in organs harvested from mice showed that although a significant amount of administered TK-p53-NTR gene plus miRs homed to TNBC metastasis in mouse lungs, some NPs were also found in other organs. Quantitative analysis of TK gene levels in various organs indicated that small proportions were present in the heart (34%), liver (0.4%), kidneys (17.1%), and brain (13.6%), with respect to the quantities in lung tissues (100%), and accumulation of NPs in livers and kidneys were not significant. To verify the in vivo targeted delivery of pAuNS@TK-p53-NTR/miRs to tumor cells, we quantified the antimiR-21 level in tumor cells (eGFP positive) and normal lung cells (eGFP negative) using qRT-PCR (Figure S12, supporting information). We observed a significant difference in the quantified antimiR-21 level in the tumor cells compared to lung cells (p < 0.01). We also observed some uptake in eGFP negative lung cells. This could be owing to the uptake by the resident macrophages present in the lungs. Thus, the AuNS mediated gene delivery through the pulmonary route could better evade the systemic first-pass effects and deliver the therapeutic cargos selectively to the orthotopic TNBC metastases in the lungs.
Figure 6. Therapeutic efficacy evaluation of pAuNS NPs-mediated TK-p53-NTR/miRs co-delivery in the presence of prodrugs.
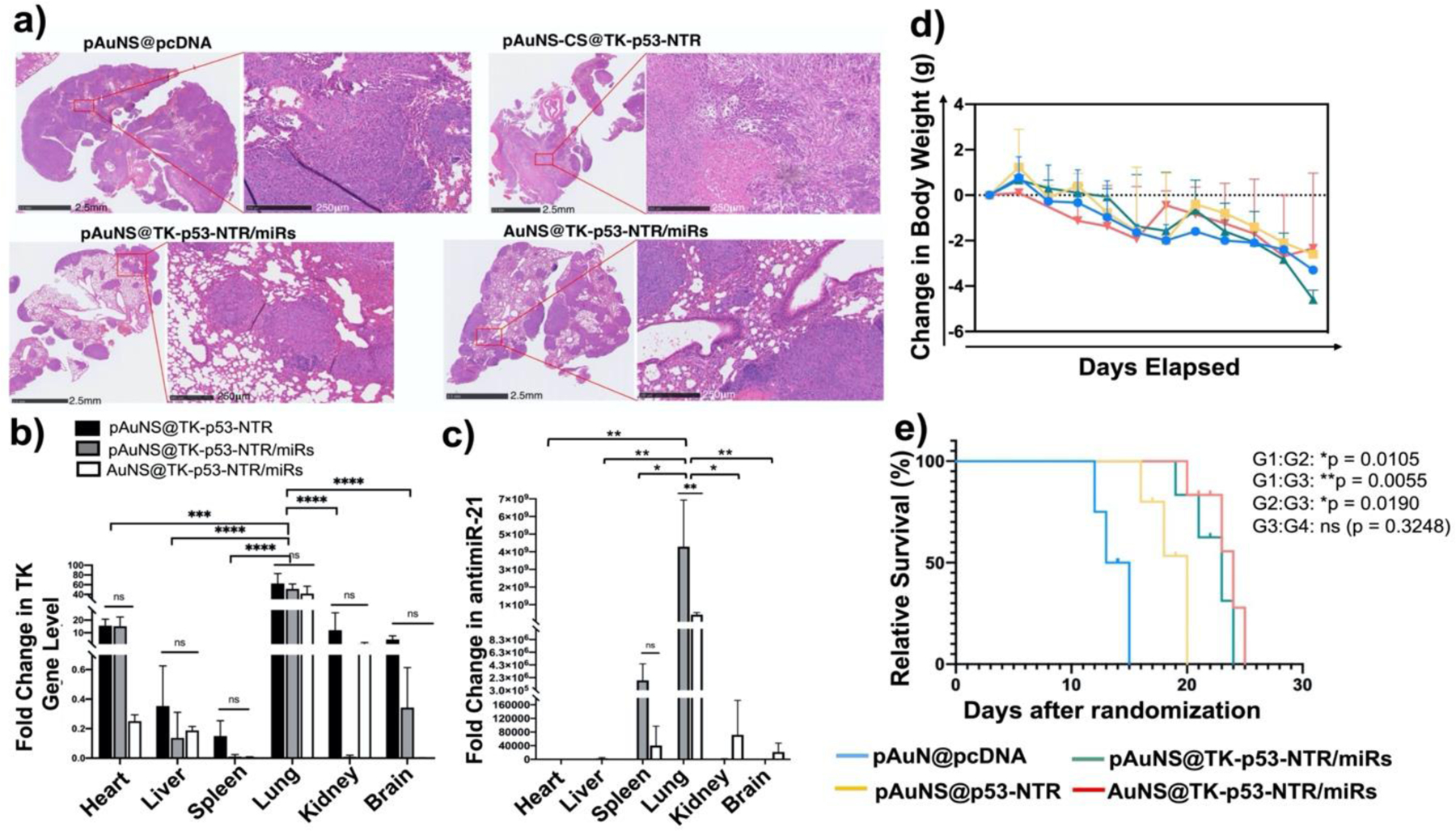
(a) Histologic analysis of lung tissue sections of different treatment groups using H&E staining assays. (b-c) Quantitative estimation of TK-p53-NTR gene and miR delivery to lungs and their biodistribution in other organs using Taqman-qPCR. (d) Bodyweight changes, and (e) survival analysis using the Gehan-Breslow-Wilcoxon method in mice during the treatment period (G1: pAuNS@pcDNA; G2: pAuNS@TK-p53-NTR; G3: pAuNS@TK-p53-NTR/miRs; G4: non-targeted AuNS@TK-p53-NTR/miRs (N=3–5/group, parts of animals were used for histological evaluations). Data are presented as mean ± SD; the significance of comparisons, as indicated, is drawn using One-way ANOVA with Bonferroni post hoc test. Adjusted p- values were considered statistically significant if p- values were < 0.05. The symbols indicating statistical significance are as follows: ns- represents non-significant difference, * represents p< 0.05, ** represents p< 0.01, *** represents p< 0.001, and **** for p< 0.0001.
Histology
The typical H&E staining of lung tissues obtained from control mice and mice treated with AuNS co-loaded with TK-p53-NTR plus miRs and prodrugs are shown in Figure 6a. In control pcDNA treated animals, H&E staining showed large tumor cell populations characterized by dense nuclear distribution with a higher instance of actively dividing atomic morphology. On the other hand, H&E stained sections of treated lungs revealed a significantly decreased tumor cell burden. This confirmed the successful effect of delivered therapeutic genes plus miRNAs using AuNS NPs to TNBC cells upon pulmonary delivery and provided evidence of their therapeutic efficacy in targeted growth arrest or reversal of TNBC lung metastases in mice. These observations were in close agreement with those of the in vivo BLI. Furthermore, although the body weights measured over time during treatment showed a decrease in all conditions, demonstrating tumor growth (Figure 6d), and the AuNS NPs could also partially accumulate in other organs upon intranasal administration (as analyzed by delivered gene quantification), H&E staining also revealed that they did not inflict any tissue damage in those other organs (Figure S13, supporting information).
Conclusion
Nanocarriers for the pulmonary delivery of therapeutics are promising against lung malignancies. NPs can be used to deliver chemotherapeutic drugs, RNAs, and DNAs, and release them in a site-specific and controlled manner. The challenges for pulmonary delivery of NP-based gene therapeutics mainly arise from their low accumulation, and inefficiency in gene transfection. To enhance the pulmonary tumor deposition upon intranasal delivery and minimize the clearance from the lungs, it would be essential to engineer NPs capable of carrying increased gene or drug loads while maintaining NPs at an optimal size. Here, we comprehensively examined and compared three different gold NPs for their gene loading and transfection efficacy, and we optimized the NP formulations using a novel microfluidic-based strategy. The NPs prepared in this manner were more uniform in size and morphology, which is advantageous when aiming to achieve higher drug loading efficiency and reproducible calculation of therapeutic dosage, especially for future clinical applications. This microfluidic-based method is also conducive to scalable mass production for forthcoming clinical translation.
In a TNBC lung metastasis mouse model, we observed a significant increase in overall survival in mice treated with uPA-functionalized AuNS NPs co-loaded with TK-p53-NTR plus miRs and systemic prodrugs GCV and CB1954 compared to other controls. Nevertheless, we found no significant differences in tumor growth inhibition between the uPA-targeted and non-targeted AuNS NPs co-delivered with TK-p53-NTR plus miRs treatment groups along with systemic prodrugs. Although the proportions of delivered genes were quantified using PCR assays at the endpoint of treatments, the genes were generally quantified in whole lung tissues rather than lung metastases alone, making it difficult to precisely evaluate the exact tumor-targeting potential. Hence, it would be necessary to include larger samples from more experimental animal cohorts in future studies to improve our ability to predictability and more precisely quantify the desirable doses of delivered genes that target lung metastases. Overall, our preliminary studies in an animal model suggest that intranasal delivery of therapeutic genes via ligand-anchoring AuNS bears considerable potential for the treatment of metastatic pulmonary cancer. This novel theranostic nanoformulation and its associated inhalation delivery strategy could prove advantageous to patients harboring pulmonary metastases—it could have a strong potential to potentiate the effects of gene-directed prodrug therapy in advanced metastatic malignancies to the lungs, as an addition to current therapeutic options [45–47].
Materials and Methods
Materials
We purchased sodium borohydride (NaBH4), Triton X-100, silver nitrate (AgNO3), tetrachloroauric acid, ascorbic acid (AA), gold (III) chloride trihydrate (HAuCl4·3H2O), L (+)-ascorbic acid (AA), trisodium citrate dihydrate, 1 N hydrochloric acid solution (HCl), chitosan, β- Cyclodextrins and phosphate-buffered saline (PBS) from Sigma-Aldrich (St Louis, MO). We used DEPC water in all NP preparations. We obtained carbon-coated copper TEM grids from VWR (Radnor, PA). We pretreated all glassware used in the study to synthesize gold NPs with aqua regia for 30 min and then washed several times using double-distilled water in an ultrasonication condition (3 min each).
Cells culture
We cultured MDA-MB231, 4T1, and 4T1-eGFP cells in Dulbecco’s Modified Eagle’s Medium with added 10% of FBS, 100 U/mL penicillin, and 0.1% streptomycin in an incubator with 5% CO2 and 95% air environment at 37 °C. We tested all cells for mycoplasma contamination using the MycoAlert kit (Allendale, NJ) and maintained cells at optimal culture conditions according to ATCC guidelines.
Preparation of AuNS
We synthesized the gold nanostars (AuNS) using an optimized seed-assisted growth reaction of the AuNS synthesis process, as reported earlier by [36, 41]. Briefly, the gold nano-seeds were made from HAuCl4− through NaBH4 in Triton X-100 (0.15 M), and the resultant gold nanodot nanoparticles (AuND NPs) were used as seeds for nanostar growth reaction. We added five μL of AuND into 5 mL of a solution containing Triton X-100 (0.1 M) and AgNO3 (0.004 M) in water. We added 5 mL of HAuCl4 (0.001 M) to the resulting solution and stirred at 70 °C for 30 min. After all the components were solubilized into a homogeneous solution, the stirring temperature was lowered to 37 °C, and another 400 μL of ascorbic acid (0.0788 M) was added to the above solution. Then, 600 μL of previously pre- ice cooled NaBH4 (0.001 M) solution was added dropwise into the mixture during stirring. The reacting mixture was stirred for another 30 minutes at 1000 rpm, turning into a brown-red suspension that gradually changed to blue-green color. After the color change, we stopped the gold nanostar growth by decreasing the reaction temperature to ∼4 °C. The resultant AuNS NPs were precipitated by centrifugation at 13,000 rpm for 30 min and washed thrice with double-distilled water before using them in vitro and in vivo experiments.
Synthesis of chitosan-β-cyclodextrin polymer
First, we added β-CD (1 g) to10 mL of NaOH (1% w/v) solution, then placed in an ice-water bath and stirred until completely dissolved. Next, we added 168 mg of p-toluene sulfonyl chloride (dissolved in 500 μl) of acetonitrile to the solution and stirred for 2 h. After that, ammonium chloride was added to quench the reaction and turn it to pH 8.0. The resultant white precipitate was washed with double-distilled water and ethanol twice and vacuum dried for further use.
Synthesis of TK-p53-NTR plasmid, pcDNA or miRs loaded, uPA receptor-targeted peptide (uPA), and scrambled peptide (sc-uPA) conjugated AuNS
We microfluidized the CS-β-CD (5 mg/mL) at 30,000 psi (Microfluidics, Westwood, MA). The CS-β-CD suspension was extracted at a 2.5 mg/mL concentration at the outlet. Then, we mixed 2.5 mg CS-β-CD, 8 mg CS, and the AuNS synthesized earlier (5 mg/mL) and microfluidized these three times. The resultant AuNS-CS-CD complex was centrifuged at 28,000 rpm, 4 °C, for 2 h to pellet the AuNS-CS-CD NPs (addressed as AuNS below) while removing the free, unconjugated CS-β-CD or CS. We pooled together the resultant pellets in 600 μL of DEPC water and used them for further studies.
Formulation of TK-p53-NTR pDNA and miRs co-loaded AuNS nanoparticles
This experiment evaluated TK-p53-NTR pDNA and miRs co-encapsulation on AuNS NP at different NPs/DNA-RNA ratios. Firstly, a stock TK-p53-NTR plasmid (1 μg) was diluted to 200 ng/μL in double-distilled water, complexed with increasing CS-CD capped AuNS and then incubated for 15 min at room temperature. We resolved the compounds in a 0.7% agarose gel by electrophoresing at 40 V for 40 min. We imaged the gel in a BioRad Gel Doc XR+ Gel Documentation system (Bio-Rad, Hercules, CA, USA) to quantify the extent of plasmid DNA encapsulation. Similarly, we diluted a stock of Cy5-antimiR-21 (200 pmol) in 50 μL DEPC water, complexed with increasing AuNS NPs, and incubated for 15 min at room temperature. We resolved the compounds in a 3.0% agarose gel by electrophoresing at 40 V for 60 min. Subsequently, to evaluate the AuNS-CS-CD NPs’ capacity to co-load both TK-p53-NTR pDNA and miRNAs, the Cy5-antimiR-21 complexed AuNS NP prepared earlier at a different NPs/RNA ratio was supplemented with 1 μg of TK-p53-NTR pDNA and then incubated for an additional 15 min at room temperature. We used the optimized TK-p53-NTR plasmid DNA-loaded AuNS NP for subsequent in vitro and in vivo experiments. At the end of the incubation period, the polyplexes were analyzed by gel retardation assay and characterized for size and zeta potential (Malvern Zetasizer Nano Z system, Malvern Instruments Ltd., UK) before use in cell culture and animal experiments.
Gel retardation assay
We performed an agarose gel retardation assay to determine the optimum pAuNS NPs/DNA-miRNAs ratio for miRs and pDNA co-loading on AuNS NPs. As per the aforementioned protocol, different NPs/DNA ratios of pAuNS complexes were prepared in nuclease-free DEPC water and then loaded in 0.7% agarose gel along with the equivalent amount of naked pcDNA as control and electrophoresed in Mini-Sub Cell GT Systems at 40 V for 45 min. After the run, we imaged the gel in the BioRad Gel Doc XR+ Gel Documentation system (Bio-Rad, Hercules, CA, USA) to further quantify and analyze the extent of DNA encapsulation. To visualize the miRNAs migration in gel electrophoresis, we used the Cy5 conjugated antimiR-21 for the encapsulation study. At the end of the run, the gel was imaged in a Lumina III In Vivo Imaging System (IVIS, PerkinElmer) using 2s acquisitions at filters compatible for Cy5 excitation and emission wavelengths.
Size and zeta potential measurements of NP polycomplexes
We determined the NP size distribution using a Malvern Zetasizer 2000 (Malvern Instruments Ltd, Malvern, UK). A volume of 2 μL of each polycomplex or AuNS alone was diluted in 1 mL of deionized water. The size distribution was calculated based on MIE theory with appropriate standard refractive indices of AuNS and the dispersant distilled water. We then analyzed the diluted samples for surface zeta potential using folded capillary zeta cells in a Malvern Zetasizer 2000. To evaluate the in vitro stability of the NPs. AuNS NPs was added into PBS, water, and a complete medium (containing 10% FBS), respectively and assessed for size, and zeta potential for 7-days of incubation at 37 °C using DLS and NTA analysis.
Uptake of antimiR-21, antimiR-10b, miR-100, and TK-p53-NTR co-loaded pAuNS by 4T1 cells
Cellular uptake of miRNAs and TK-p53-NTR co-encapsulated pAuNS was evaluated quantitatively and qualitatively in 4T1 cells using flow cytometry as well as confocal microscopy. We prepared pAuNS NPs with Cy5-antimiR-21 and TK-p53-NTR pDNA according to the procedure outlined earlier and treated 4T1 cells seeded at 30,000 cells per well in a 12-well plate. After 24 h of incubation, we removed the spent media, and briefly washed the cells monolayer twice with PBS to remove the extracellular unbound NPs. We then stained the cells with Hoechst 33342 for 5 min and acquired representative images of each well using a Leica TCS SP8 confocal microscope under Hoechst33342 and Cy5 filters. After acquiring representative images of each treatment, we trypsinized the cells and harvested them for flow cytometry analysis using a Guava easyCyte flow cytometer for quantitative estimation of internalized pDNA, miR, or the co-loaded NPs. Around 10,000 events were acquired for each treatment condition and the results were processed using FlowJo analysis software (Tree Star, Ashland, OR, USA) and represented as scatter plot quadrants for all treatment conditions.
MiRs and TK-p53-NTR GDEPT delivered by pAuNS NPs in 4T1 cells
We evaluated the efficiency of pAuNS NPs mediated GDEPT of TK-p53-NTR therapeutic construct and its therapeutic effects in combination with miRs (antimiR-21 antimiR-10b and miR-100) in 4T1 cells in the presence or absence of one or both prodrugs (GCV and CB1954). We seeded 4T1 cells at a density of 150,000 cells in each well of a 6-well plate and treated them using AuNS NPs loaded with 1 μg of TK-p53-NTR, or 200 pmol of miRs, or a combination of both at an optimized ratio. After 24 h of treatment, we replaced the cells with fresh medium and treated the respective wells with either GCV (1 μg/mL) and/or CB1954 (30 μm). At the end of prodrug treatment time points (i.e., 48 h), we collected the treated cells by trypsinization and fixed them in ice-cold 70% ethanol. We washed and stained the fixed cells with PI (15 nm) in the presence of RNase A (10 μg/mL) and 0.01% Triton X-100 (Life Technologies, Grand Island, NY, USA). We analyzed the stained cells using a Guava easyCyte flow cytometer by collecting 10,000 events for each treatment condition and represented the results as histograms with percentages of live and apoptotic cells. The other set of cells treated with the same conditions were collected for downstream protein analysis using the immunoblotting assay.
Syngeneic tumor models of TNBC lung metastases in mice
The study was approved by the Institutional Administrative Panel for Laboratory Animal Care, Stanford University, USA (Approval Number: 33144). BALB/c mice (males, bodyweight 18–22 g, 8–11 weeks old) were used to develop 4T1 xenografts. Approximately 1 × 106 4T1 (Fluc-eGFP) cells were suspended in 100 μL saline and then injected intravenously. Tumors were allowed to grow for 1 week after tumor cell injection until the tumor cell-Fluc-eGFP signals were detected before the start of treatment. We assessed tumor growth at different time points after treatment using BLI signal of tumor xenografts following intraperitoneal injection of the substrate D-Luciferin (150 mg/kg). We administered the prodrugs GCV and CB1954 intraperitoneally to all the animals, whereas TK-p53-NTR loaded NPs or TK-p53-NTR and miRs co-loaded NPs were administered intranasally at predetermined time points. Figure 6a details the entire experimental workflow used in this experiment.
Intranasal administration of TK-p53-NTR pDNA/miRs using AuNS NPs
The NPs were administered to mice under deep anesthesia using isoflurane gas with oxygen. The operator held the mouse with one hand by its back and with the mouse head angled upward by 60°. This method (under deep anesthesia and with the head up) ensured that the glottis relaxed, allowing the airway to open and the administered NPs to enter the lower respiratory tract as much as possible. The respiratory rate was monitored until it was slow and stable. Then we applied 20 μL of the DNA/miRs-loaded AuNS NPs (∼1.31× 108 AuNS, loaded with ∼3.4 × 102 μg of TK-p53-NTR pDNA and ~6.68 × 104 pmol of total miRs) as 5 μL/drops into the mouse nares over 10–15 min. After administration of each drop, we waited for 3–5 min to ensure the animal had well-inhaled the drops. The mouse nostrils were monitored for signs of blockage or intolerance. After the full dose was administered, each mouse was closely monitored for complete recovery from anesthesia.
In vivo prodrug administration and imaging for therapy monitoring
Animal treatments and administrations of pDNA- or miRs-loaded AuNS NPs were performed as shown in the schematic workflow in Figure 5a. At every time point, we measured tumor volumes using BLI signals. The prodrugs (GCV (40 mg/kg) and CB1954 (40 mg/kg)) were intraperitoneally injected on Day 2 and Day 4 of each treatment cycle continuously for 22 days. The second cycle of prodrug and NP polyplexes were administered on Day 8, and subsequently followed with the same protocol for four cycles of treatment. On Day 22, we intravenously injected CytoCy5S dye (10 μg) and imaged the mice for CytoCy5S fluorescent signal (640 nm excitation, 690 nm emission) 24 h after injection using a Lago spectral instruments Imaging system (Tucson, Arizona). We sacrificed animals at the endpoint of treatment and collected the organs and tumors for ex vivo quantification of gene delivery and histological analysis.
Quantitative assessment of TK-p53-NTR pDNA and miRs delivery in vitro, and in vivo in mouse models by copy number estimation using qPCR and qRT-PCR
The delivery of therapeutic TK-p53-NTR gene to cells in in vitro experiments and tissues collected from in vivo studies were estimated quantitatively using a modified plasmid extraction procedure and quantitative PCR analysis. We treated the 4T1 cells using AuNS NPs loaded with TK-p53- NTR pDNA and miRs (antimiR-21, antimiR-10b, and miR-100), and at the end of 48 h, we harvested the cells for analysis. Likewise, the stipulated mass of tumor tissues and organs harvested from each treatment group of the in vivo study were used to quantify TK-p53-NTR delivery in tumors and other organs. The pDNA was isolated from treated cells or tissues by a modified plasmid extraction procedure using the QIAGEN plasmid DNA extraction kit. Unlike cells from in vitro experiment, tissue samples were homogenized in cell lysis buffer, all subsequent steps were identical. We probed the isolated DNA for TK-p53-NTR using gene-specific amplification of TK by EVA green-based real-time polymerase chain reaction (qPCR). The forward and reverse primers for targeted amplification of the HSV1-TK segment were TACCCGAGCCGATGACTTAC and CCGATTAGAGGAGCCAGAAC, respectively. A standard graph for both miRNAs and TK-p53-NTR plasmids was generated using RT-PCR and PCR amplification of serially diluted miRs and plasmids of known copy numbers respectively, and the resultant Ct values were plotted to the corresponding log (N0 × 0.1). The linear fit of this plot was used as the standard curve for calculating the copy number of miRNAs, and TK-p53-NTR plasmid delivered into cells for the respective treatments in the cellular uptake studies in living mice. We extracted total RNA from treated 4T1 cells and harvested tissues from different treatment groups of the in vivo study using a mirVana RNA extraction kit (Life Technologies, Grand Island, NY, USA). In brief, treated 4T1 cells and tissues were lysed/homogenized in 300 μL lysis buffer for 3 min at the end of the treatment period. We supplemented the lysis buffer with 30 μL of homogenate additive and incubated at room temperature for 10 min. The RNA was extracted once with phenol-chloroform and purified using a column washed with wash buffer and eluted in 50 μL of sterile distilled water. The total RNA was quantified and was checked for purity using a Nanodrop spectrophotometer (Thermo scientific). After quantification, 15 ng of total RNA was reverse transcribed using RT-primers (Life technologies) using a reverse transcription kit (Life technologies) to produce the corresponding cDNA. We carried out cDNA synthesis in a 15 μL reaction volume, and qRT-PCR was performed using cDNA (5 ng of RNA equivalent) combined with TaqMan-PCR reagents (primer and probe mix). qRT-PCR was performed by 2 min incubation at 50 °C, followed by activation of the DNA polymerase at 95 °C for 10 min, 50 cycles of 95 °C for 15 s, and 60 °C for 60 s in a BioRad CFX real-time PCR system (BioRad, Hercules, USA). The qRT-PCR reaction was carried out in a 20 μL reaction volume. The expression of miRNA was calculated using the 2−ΔΔCT method.
Targeted delivery of pAuNS@TK-p53-NTR/miRs to tumor cells in vivo
At the endpoint of the treatment, whole lung tissues, with metastases, were surgically removed, and single-cell suspension was prepared by collagenase digestion. The tumor cell population were isolated using eGFP signal from the Fluc-eGFP used to engineer the 4T1 cells, and eGFP negative normal lung cells were sorted using FACS. For cell isolation, the lung tissue was grounded using gentleMACS tissue disruptor and incubated in HBSS solution (containing 0.1% collagenase, 0.01% hyaluronidase, and 0.002% deoxyribonuclease) at 37°C for 1 h. The tissue homogenates were sequentially filtered through cell strainers (70 µm followed 40 µm), and collected by centrifugation (800g for 10 min). The eGFP positive tumor cells and eGFP negative lung cells were used for total RNA isolation, and further quantification of the delivered antimiR-21 using Taqman qRT-PCR to evaluate the in vivo targeted delivery of pAuNS@TK-p53-NTR/miRs.
Hematoxylin and eosin staining
After completion of the gene therapy study time points, all animals were euthanized, and tumors and other organs were excised and fixed in 4% paraformaldehyde for H&E staining according to our previously published protocol [36, 37].
Statistical analyses
We pooled data from independent experiments in 5–9 mice and represented results as mean ± standard deviation (SD), or interquartile range between the first range (25th percentile) and third range (75 percentile). All experiments were repeated three times unless otherwise stated. We compared grouped data using a two-tailed Students t-test and calculated multiple comparisons of grouped data using two-way ANOVA with Tukey’s post hoc to determine the significance of differences. One-way ANOVA with Bonferroni post hoc test was used to draw the significance of certain comparisons as indicated. Adjusted p- values were considered statistically significant if p-values were <0.05 and the symbols indicating statistical significance were as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. We used GraphPad Prism 8.0 software (GraphPad Software, Inc., La Jolla, CA, USA) to perform statistical analyses and to plot all the graphs shown in the manuscript.
Supplementary Material
Acknowledgments:
The authors would like to thank the Canary Center at the Department of Radiology, Stanford University, for providing the facility and resources to complete this work. The authors would also like to thank the Stanford Animal Histology Services for the preparation of histology slides and histological analysis of animal tissues. We thank the Small Animal Imaging Facility at Stanford Center for Innovation in In-Vivo Imaging.
Funding:
This work was in part supported by the Focused Ultrasound Foundation and the National Institutes of Health (Grant numbers R01CA209888 and R21EB022298). NIH S10OD023518-01A1 Award for the Celigo S Imaging Cytometer (200-BFFL-S) to RP is acknowledged, as is the Gary Glazer-GE Gift Fund (Department of Radiology, Stanford University).
Abbreviation of relevant nanoparticles:
- AuND
Gold Nanodot
- AuNS
Gold nanostar
- AuNS@TK-p53-NTR/miRs
TK-p53-NTR plasmid DNA and microRNAs-co-loaded gold nanostar
- pAuNS
uPA-peptide conjugated gold nanostar
- pAuNS@TK-p53-NTR/miRs
uPA-peptide conjugated, TK-p53-NTR plasmid DNA and microRNAs-loaded gold nanostar
- pAuNS@pcDNA
uPA-peptide conjugated gold nanostar loaded with control plasmid
Footnotes
Competing interests: The authors do not have any competing interests to declare.
Additional information: Supporting Information, etc.
References
- 1.Gerull WD, Puri V, and Kozower BD, The epidemiology and biology of pulmonary metastases. J Thorac Dis, 2021. 13(4): p. 2585–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crow J, Slavin G, and Kreel L, Pulmonary metastasis: a pathologic and radiologic study. Cancer, 1981. 47(11): p. 2595–602. [DOI] [PubMed] [Google Scholar]
- 3.Sundquist M, Brudin L, and Tejler G, Improved survival in metastatic breast cancer 1985–2016. Breast, 2017. 31: p. 46–50. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, et al. , NIR-II Absorbing Semiconducting Polymer-Triggered Gene-Directed Enzyme Prodrug Therapy for Cancer Treatment. Small, 2021. 17(23): p. e2100501. [DOI] [PubMed] [Google Scholar]
- 5.Gao Y, et al. , Functionalized DMP-039 Hybrid Nanoparticle as a Novel mRNA Vector for Efficient Cancer Suicide Gene Therapy. Int J Nanomedicine, 2021. 16: p. 5211–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassanzadeh A, et al. , Genetically-Modified Stem Cell in Regenerative Medicine and Cancer Therapy; A New Era. Curr Gene Ther, 2021. [DOI] [PubMed] [Google Scholar]
- 7.Davis ME, et al. , Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature, 2010. 464(7291): p. 1067–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sukumar UK, et al. , SP94-Targeted Triblock Copolymer Nanoparticle Delivers Thymidine Kinase-p53-Nitroreductase Triple Therapeutic Gene and Restores Anticancer Function against Hepatocellular Carcinoma in Vivo. ACS Appl Mater Interfaces, 2020. 12(10): p. 11307–11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sekar TV, et al. , Dual-therapeutic reporter genes fusion for enhanced cancer gene therapy and imaging. Gene Ther, 2013. 20(5): p. 529–37. [DOI] [PubMed] [Google Scholar]
- 10.Kanada M, et al. , Microvesicle-Mediated Delivery of Minicircle DNA Results in Effective Gene-Directed Enzyme Prodrug Cancer Therapy. Mol Cancer Ther, 2019. 18(12): p. 2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahraman M, et al. , MicroRNA in diagnosis and therapy monitoring of early-stage triple-negative breast cancer. Sci Rep, 2018. 8(1): p. 11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maryam M, Naemi M, and Hasani SS, A comprehensive review on oncogenic miRNAs in breast cancer. J Genet, 2021. 100. [PubMed] [Google Scholar]
- 13.McGuire A, Brown JA, and Kerin MJ, Metastatic breast cancer: the potential of miRNA for diagnosis and treatment monitoring. Cancer Metastasis Rev, 2015. 34(1): p. 145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savari B, et al. , Overexpression of microRNA-21 in the Serum of Breast Cancer Patients. Microrna, 2020. 9(1): p. 58–63. [DOI] [PubMed] [Google Scholar]
- 15.Wu G, et al. , miR-100 Reverses Cisplatin Resistance in Breast Cancer by Suppressing HAX-1. Cell Physiol Biochem, 2018. 47(5): p. 2077–2087. [DOI] [PubMed] [Google Scholar]
- 16.Bose RJC, et al. , Tumor Cell-Derived Extracellular Vesicle-Coated Nanocarriers: An Efficient Theranostic Platform for the Cancer-Specific Delivery of Anti-miR-21 and Imaging Agents. ACS Nano, 2018. 12(11): p. 10817–10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devulapally R, et al. , Polymer nanoparticles mediated codelivery of antimiR-10b and antimiR-21 for achieving triple negative breast cancer therapy. ACS Nano, 2015. 9(3): p. 2290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devulapally R, Sekar TV, and Paulmurugan R, Formulation of Anti-miR-21 and 4-Hydroxytamoxifen Co-loaded Biodegradable Polymer Nanoparticles and Their Antiproliferative Effect on Breast Cancer Cells. Mol Pharm, 2015. 12(6): p. 2080–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, et al. , Camouflaged Hybrid Cancer Cell-Platelet Fusion Membrane Nanovesicles Deliver Therapeutic MicroRNAs to Presensitize Triple-Negative Breast Cancer to Doxorubicin. Adv Funct Mater, 2021. 31(41). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangal S, et al. , Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: challenges and opportunities. Acta Pharmacol Sin, 2017. 38(6): p. 782–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patel AK, et al. , Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv Mater, 2019. 31(8): p. e1805116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cuong NV and Hsieh MF, Molecular targeting of liposomal nano-particles to lymphatic system. Curr Cancer Drug Targets, 2011. 11(2): p. 147–55. [DOI] [PubMed] [Google Scholar]
- 23.Seo JB, et al. , Atypical pulmonary metastases: spectrum of radiologic findings. Radiographics, 2001. 21(2): p. 403–17. [DOI] [PubMed] [Google Scholar]
- 24.Khanna C, et al. , Interleukin-2 liposome inhalation therapy is safe and effective for dogs with spontaneous pulmonary metastases. Cancer, 1997. 79(7): p. 1409–21. [DOI] [PubMed] [Google Scholar]
- 25.Moller W, et al. , Mucociliary and long-term particle clearance in the airways of healthy nonsmoker subjects. J Appl Physiol (1985), 2004. 97(6): p. 2200–6. [DOI] [PubMed] [Google Scholar]
- 26.Schmid O, et al. , Dosimetry and toxicology of inhaled ultrafine particles. Biomarkers, 2009. 14 Suppl 1: p. 67–73. [DOI] [PubMed] [Google Scholar]
- 27.TM MW, Lau WM, and Khutoryanskiy VV, Chitosan and Its Derivatives for Application in Mucoadhesive Drug Delivery Systems. Polymers (Basel), 2018. 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernkop-Schnurch A and Dunnhaupt S, Chitosan-based drug delivery systems. Eur J Pharm Biopharm, 2012. 81(3): p. 463–9. [DOI] [PubMed] [Google Scholar]
- 29.Bravo-Osuna I, et al. , Mucoadhesion mechanism of chitosan and thiolated chitosan-poly(isobutyl cyanoacrylate) core-shell nanoparticles. Biomaterials, 2007. 28(13): p. 2233–43. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto H, et al. , Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions. J Control Release, 2005. 102(2): p. 373–81. [DOI] [PubMed] [Google Scholar]
- 31.Palecanda A and Kobzik L, Receptors for unopsonized particles: the role of alveolar macrophage scavenger receptors. Curr Mol Med, 2001. 1(5): p. 589–95. [DOI] [PubMed] [Google Scholar]
- 32.Tabata Y and Ikada Y, Macrophage phagocytosis of biodegradable microspheres composed of L-lactic acid/glycolic acid homo- and copolymers. J Biomed Mater Res, 1988. 22(10): p. 837–58. [DOI] [PubMed] [Google Scholar]
- 33.Wanakule P, et al. , Nano-inside-micro: Disease-responsive microgels with encapsulated nanoparticles for intracellular drug delivery to the deep lung. J Control Release, 2012. 162(2): p. 429–37. [DOI] [PubMed] [Google Scholar]
- 34.Aantaa R, et al. , Dexmedetomidine, an alpha 2-adrenoceptor agonist, reduces anesthetic requirements for patients undergoing minor gynecologic surgery. Anesthesiology, 1990. 73(2): p. 230–5. [DOI] [PubMed] [Google Scholar]
- 35.Borm P, et al. , Research strategies for safety evaluation of nanomaterials, part V: role of dissolution in biological fate and effects of nanoscale particles. Toxicol Sci, 2006. 90(1): p. 23–32. [DOI] [PubMed] [Google Scholar]
- 36.Sukumar UK, et al. , Intranasal delivery of targeted polyfunctional gold-iron oxide nanoparticles loaded with therapeutic microRNAs for combined theranostic multimodality imaging and presensitization of glioblastoma to temozolomide. Biomaterials, 2019. 218: p. 119342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang K, et al. , A Microfluidics-Based Scalable Approach to Generate Extracellular Vesicles with Enhanced Therapeutic MicroRNA Loading for Intranasal Delivery to Mouse Glioblastomas. ACS Nano, 2021. [DOI] [PubMed] [Google Scholar]
- 38.Banys-Paluchowski M, et al. , The prognostic relevance of urokinase-type plasminogen activator (uPA) in the blood of patients with metastatic breast cancer. Sci Rep, 2019. 9(1): p. 2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu W, et al. , Actively targeted nanomedicines for precision cancer therapy: Concept, construction, challenges and clinical translation. J Control Release, 2021. 329: p. 676–695. [DOI] [PubMed] [Google Scholar]
- 40.Gao P, et al. , Synergistic integration of metal nanoclusters and biomolecules as hybrid systems for therapeutic applications. Acta Pharm Sin B, 2021. 11(5): p. 1175–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar US, et al. , Gold-Nanostar-Chitosan-Mediated Delivery of SARS-CoV-2 DNA Vaccine for Respiratory Mucosal Immunization: Development and Proof-of-Principle. ACS Nano, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MJ ON, et al. , Mechanistic studies on the uptake and intracellular trafficking of novel cyclodextrin transfection complexes by intestinal epithelial cells. Int J Pharm, 2011. 413(1–2): p. 174–83. [DOI] [PubMed] [Google Scholar]
- 43.Lakk M, et al. , Dyslipidemia modulates Muller glial sensing and transduction of ambient information. Neural Regen Res, 2018. 13(2): p. 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenblum D, et al. , Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun, 2018. 9(1): p. 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi L, et al. , Transforming a toxic drug into an efficacious nanomedicine using a lipoprodrug strategy for the treatment of patient-derived melanoma xenografts. J Control Release, 2020. 324: p. 289–302. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, et al. , Tuning the efficacy of esterase-activatable prodrug nanoparticles for the treatment of colorectal malignancies. Biomaterials, 2021. 270: p. 120705. [DOI] [PubMed] [Google Scholar]
- 47.Xie B, et al. , Preclinical Evaluation of a Cabazitaxel Prodrug Using Nanoparticle Delivery for the Treatment of Taxane-Resistant Malignancies. Mol Cancer Ther, 2020. 19(3): p. 822–834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.