

Abstract
Chemokine (C–C motif) receptor‐like 2 (CCRL2), is a seven transmembrane receptor closely related to the chemokine receptors CCR1, CCR2, CCR3, and CCR5. Nevertheless, CCRL2 is unable to activate conventional G‐protein dependent signaling and to induce cell directional migration. The only commonly accepted CCRL2 ligand is the nonchemokine chemotactic protein chemerin (RARRES2). The chemerin binding to CCLR2 does induce leukocyte chemotaxis, yet, genetic targeting of CCRL2 was shown to modulate the inflammatory response in different experimental models. This mechanism was shown to be crucial for lung dendritic cell migration, neutrophil recruitment, and Natural Killer cell‐dependent immune surveillance in lung cancer. To gain more insight in the interactions involved in the CCRL2‐chemerin, the binding complexes were generated by protein–protein docking, then submitted to accelerated molecular dynamics. The obtained trajectories were inspected by principal component analyses followed by kernel density estimation to identify the ligand‐receptor regions most frequently involved in the binding. To conclude, the reported analyses led to the identification of the putative hot‐spot residues involved in CCRL2‐chemerin binding.
Keywords: accelerated molecular dynamics, CCRL2, Chemerin, protein–protein docking, protein–protein interaction
1. INTRODUCTION
The chemokine (C‐C motif) receptor‐like 2 (CCRL2) is a seven transmembrane domain receptor that maps within a CC chemokine receptor cluster in human chromosome 9 and mouse chromosome 3 1 and therefore, CCRL2 is most related to chemokine receptors CCR2, CCR3, and CCR5. 2 Since CCRL2 is unable to activate signal transduction through G proteins or β‐arrestin, it is considered to be related to the family of Atypical Chemokine Receptors. 3 , 4
So far, chemerin (encoded by the RARRES2 gene) is the only accepted CCRL2 ligand. Chemerin is produced by mammalian cells as a 163 amino acid (aa) pro‐precursor. The N‐terminal processing of 20 aa leads to the secretion of a precursor form of chemerin. 5 The further C‐terminal cleavage results in both active and deactivated chemerin forms, according to the extent of processing. For example, proteases like plasmin, elastase, and cathepsin G activate chemerin and generate various chemerin isoforms with different affinity to CMKLR1, the active chemerin receptor. Further cleavage of bioactive chemerin by chymase produces an inactive form of chemerin. 6 Thus, the C‐terminal proteolytic processing acts as a regulatory mechanism to control the concentration of active chemerin.
In addition to CCRL2 and CMKLR1, chemerin also binds G protein‐coupled receptor 1 (GPR1); these three receptors have distinct patterns of expression and biological functions.
CMKLR1 is mostly expressed by innate immune cells, such as dendritic cells, macrophages, and Natural Killer (NK) cells 5 , 7 CCRL2 is expressed by a large variety of leukocytes subsets and by barrier cells, such as vascular and lymphatic endothelium and some epithelium. 3 , 8 GPR1 is predominantly expressed in the central nervous system and skin. 5 Among these three receptors, CCRL2 is the only one devoid of the ability to activate an intracellular signaling cascade.
CCRL2 was shown to regulate inflammation‐related diseases such as experimental autoimmune encephalitis, hypersensitivity, and inflammatory arthritis 8 , 9 , 10 , 11 and the recruitment of the NK cells in pathological conditions. 12 , 13 We decided to further characterize CCRL2‐chemerin interaction, by a protein–protein docking followed by accelerated molecular dynamic (aMD) simulation of the proposed binding conformations.
Instead of a classical molecular dynamic approach, which is limited by kinetic trapping effects and limited sampling of the conformational space, it was established a protocol of aMD. 14 This method reduces the energy barrier between different low‐energy states by applying a potential energy boost, increasing the transition probabilities between two different conformations. 15 We obtained a total of 5.5 μM second trajectories, which were analyzed by principal component analyses (PCA) to reduce the complexity of the data and to identify the greatest variance. 16 , 17 Then, the residues more often involved in binding interactions were highlight as hot‐spot residues of the CCRL2 chemerin complex.
2. MATERIALS AND METHODS
2.1. Homology model of CCRL2 conformational states
CCRL2 primary aminoacidic sequence was derived from Uniprot (UniProtKB: O00421). Since human CCRL2 exist in two isoforms, we modeled the shorter isoform 1 (344 aa, CRAM‐B, O00421‐1). The template choice for CCRL2 active state was performed by aligning all the available chemokine receptor structures, in the active state, resolved by crystallography. The best model was US28 (UniProtKB: P69332; PDB: 5WB1; root mean square deviation (RMSD) 0.776 Å). 18
The resulting CCRL2 structure was obtained with the I‐Tasser server, 19 and as restrain was inserted, the protein topology derived from UniProtKB. Loop refinement and energy minimization was carried out using ModRefiner. 20 The quality of generated model was validated with respect to backbone and side chain geometry. To validate protein backbone quality, the MolProbidity tool was adopted. 21
2.2. Ab initio modeling of chemerin
For chemerin (UniProtKB: Q99969), we lack a proper resolved homologue structure; therefore, we utilized the RAPTOR‐X prediction server 22 to build the model. Loop refinement and energy minimization was carried out using ModRefiner. 20 The quality of generated models was validated with respect to backbone and side chain geometry. To validate protein backbone quality, the MolProbidity tool 21 was adopted.
2.3. Structural comparison of modeled proteins
The optimized CCRL2 and chemerin models were compared to their respective AlphaFold (https://alphafold.ebi.ac.uk/, entry: O00421 CCRL2_HUMAN) conformations using Matchmaker function of UCSF Chimera 23 and RMSD values were obtained.
2.4. Protein–protein docking
Protein–protein docking was performed on HADDOCK server 24 using the 3D models of CCRL2 and chemerin proteins. We set up as “active” residues (residues expected to be involved in the interaction between the two molecules) the N‐terminal residues 1–100 of chemerin and the extracellular loops of the CCRL2 receptor (from UniProtKB, 25–67; 120–128; 190–122; 284–310) following literature data 25 ; the other residues were defined as “passive” (residues accessible to the solvent closed of the active residues). This docking protocol consisted of three stages: rigid body (it0), semi‐flexible refinement (it1), and explicit solvent refinement (water). The docking experiments were carried out by using default parameters. Just MD steps in the TAD and cooling stage were increased to 2000 for it0. 24 HADDOCK produced 1000 models in the first step, then refined to 200 best model in the following steps. The final models were automatically clustered based on the fraction of common contacts that measures the similarity of the intermolecular contacts. At the end, we obtained 12 clusters, and for each of them, it was selected as representative structure the conformation with the best score value.
2.5. Accelerated molecular dynamics
The membrane embedded complexes structures 26 and the Amber parameters 27 were obtained by CHARMM‐GUI server through Membrane builder module. 28
The structural information to have a reliable placement of the bilayer membrane at CCRL2 were obtained by Uniprot (UniProtKB: O00421) at the subcellular localization section. Indeed, in this section were defined both the nonmembrane region (topological domain), and the extent of the membrane‐spanning regions (transmembrane) of CCRL2.
All the CCRL2‐chemerin complexes were inserted in a membrane of 20% cholesterol and 80% POPC (1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine); the system was solvated with TIP3P water model and ionized up to a concentration of 0.15 M NaCl still using the CHARMM‐GUI, additional Cl− ions were added to neutralize the systems. 28 All the system (proteins + membrane + solvent) consists of 67 447 atoms. Each system was then submitted to aMD, carried out on Cineca supercomputer using Amber20. The whole system was minimized (5000 cycle) using restraints for CCRL2 and membrane (10 and 2.5, respectively); then, the CHARMM‐steps equilibration protocol with progressive removal of position restraints was applied to the membrane and protein atoms (http://www.charmm-gui.org/demo/amber_ff/2). This equilibration protocol was carried out by Amber and consists of two NVT (constant number of particles (N), volume (V), and temperature (T)) steps to heat the system to 303.15 K employing as thermostat Langevin dynamics (collision frequency 1 ps) and four NPT (constant number of particles (N), pressure (P), and temperature (T)) steps (125 ps each) with SHAKE algorithm and the particle mesh Ewald (PME) 29 (with a cutoff of 9 Å). The required average dihedral energy and average total potential energy were computed during 5 ns classical molecular dynamics for each studied complex. 30 The aMD production (500 ns) was conducted at 315 K with constant pressure (1 bar) and periodic boundary condition, Shake (ntc = 2) and PME with cut of 10 Å were set, each simulation was repeated three times.
Total Accessible Surface Area and Buried Surface Area (BSA) were computed by Pisa server (http://pdbe.org/pisa/). The property maps were calculated by Coco server. 31
2.6. Trajectories analyses
Trajectories analyses were carried out by mdtraj. 32 The PCA analyses was carried out with scikit‐learn using the decomposition module. 33 Scipy library 34 was used to calculate Gaussian Kernel density estimation (KDE). Graphics were done with Matplotlib. 35
3. RESULTS AND DISCUSSION
3.1. Modeling of CCRL2 and chemerin
To identify the putative residues involved in the CCRL2 chemerin binding, it was followed a protocol based on protein–protein docking and aMDs.
Since the structures of CCRL2 and chemerin were not experimentally resolved, it was decided to follow a homology modeling approach to obtain CCRL2, and an ab‐initio computations for chemerin.
CCRL2, similarly to other chemokine receptors had two conformational states: active and inactive. Engagement by the ligands turns GPCRs in the active state; therefore, it was decided to model only the active state of the receptor. In details, the CCRL2 model was based on the most similar chemokine receptor (see methods) and was devoid of N‐terminal tail.
On the other hand, chemerin was modeled ab‐initio due to the lack of highly conserved homologous proteins.
To be mentioned, meanwhile the designed computations were accomplished, both the structures of CCRL2 and chemerin became available at the AlphaFold database (alphafold.ebi.ac.uk). A comparison of the AlphaFold and our models was carried out by measuring the RMSD. For CCRL2, it was calculated a Cα RMSD of 1.02 Å and the great amount of this distance was related with the extracellular loop 2 (ECL2, residues 169–192) and the TM6 helix (Figure S1). TM6 was embedded in the membrane, far from the chemerin binding site. Therefore, we assumed that it would only have a marginal effect on the ligand binding. For the ECL2, it is challenging to reliable predict a long loop (23 residues) 36 and also AlphaFold listed this loop as at low confidence (per‐residue confidence score between 70 and 50). Furthermore, the implementation of aMD instead classical MD reduced the bias related with the different loop conformations. Indeed, aMD offered a great advantage in modeling conformational change and to simulate infrequent events required for protein conformational change without previous knowledge of conformational states. 37
For chemerin, the superimposition of our model and the AlphaFold proposed led to Cα RMSD of 1.12 Å. The less fitting domain was the C terminal helix 2 (Figure S2). This region was reported to be not involved in chemerin binding to the CCRL2. 30 In general, it was observed a good superimposition between the AlphaFold and our in‐house models.
3.2. CCRL2‐chemerin protein–protein docking
The docking computations were carried out by Haddock 24 providing 12 different clusters. A representative binding conformation for each cluster, named complexes 1–12, was selected by Haddock post docking quality assessment tools (Figure S3). Further refinement of the selected models were not carried out. Similarly, to be as much unbiased as possible were not taken into account the docking score and the energy of the complexes.
Each selected complex was embedded into the membrane bilayer and submitted to aMD (500 ns). The obtained trajectories were first analyzed by RMSD to evaluate the system stability. For complex 9, during the simulation time the C‐terminal helix moved up to the binding site. Given that, it is accepted that the Chemerin C‐terminal binds CMKLR 25 this complex was not further considered. Also, for complex 10, it was observed a dramatic change of chemerin conformation, with the C‐terminal that moved far from the N‐terminal and also this complex was not further studied.
3.3. Selection of CCRL2‐chemerin binding models
All the other trajectories were analyzed by PCA and the obtained matrixes were investigated by Gaussian kernel (KDE) to create a probability distribution functions in subspaces spanned by principal components 1 and 2 (PC1 vs. PC2). The approach that combined a dimension reduction step (PCA) with subsequent clustering (KDE) to analyze MD trajectories data were shown to be capable of reducing the noise and to generate more compact and well separated clusters of conformations. 38
Thus for each trajectory, the KDE plots allowed the identification of the higher populated conformational basins, and for each of them, it was extracted the representative conformations. This approach provided 23 highly frequent conformations assumed to be as the most relevant (Tables S1 and S3–S22).
For all these conformations, the BSA was computed. The conformations with BSA lower than 600 Å2 was rejected as consequence of the small size of the interface between CCRL2 and chemerin (Table S2). 39 The 22 remaining conformations were analyzed by visual inspection focusing on the salt bridge interactions. This type of contact has a considerable contribution to the specificity of interaction of proteins with other biomolecules. 40 Indeed, the energetic penalty due of dehydration of polar groups was paid off by favorable energy of salt bridge formation limiting the number of conformations of a molecule or complex, thus playing a crucial role in determining specificity. 41 , 42
By visual inspection, the studied conformations have been grouped into two general chemerin binding modes and it was also possible to identify the CCRL2 and Chemerin regions more often involved in the binding. For CCRL2, the two extracellular loops ECL2 (residues 169–192) and ECL3 (residues 264–270), and the residue lining the entrance of the receptor channel. For Chemerin, the three regions mainly involved in the binding with the cognate receptor were the α1 helix, the β1 sheet, and the loop between β2 and β3 helixes (β2β3‐loop residues 49–73).
The first binding mode (defined BM1) was shared by 12 of the 22 inspected conformations. This binding mode was featured by the contacts between chemerin β2β3‐loop with ECL3 (6 conformations of 12) and with ECL2 (6 conformations of 12). Furthermore, the chemerin α1 helix contacted the entrance of the channel (9 conformations of 12).
For BM2, shared by seven of the studied conformations, the chemerin β2β3‐loop contacted both the CCRL2 ECL2 and ECL3 (seven conformations), the α1 helix interacted with the CCRL2 ECL2 (seven conformations), and the β1 sheet had contacts with both the ECL3 and the residues lining the entrance of the receptor channel (four conformations). The three remaining conformations were featured by the significative involvement of the Chemerin C‐terminal domain in the binding to CCRL2. Since it was reported that the C‐terminal was only involved in the binding of the CMKLR1, 25 these three conformations were rejected.
Worthily, the main differences between the two binding modes, BM1 and BM2, was a 180° rotation of the chemerin conformation. For the BM1, the chemerin α1 helix was located behind the β sheets, in contrast to the BM2 where the α1 helix was located in front of the β sheets (Figure 1).
FIGURE 1.
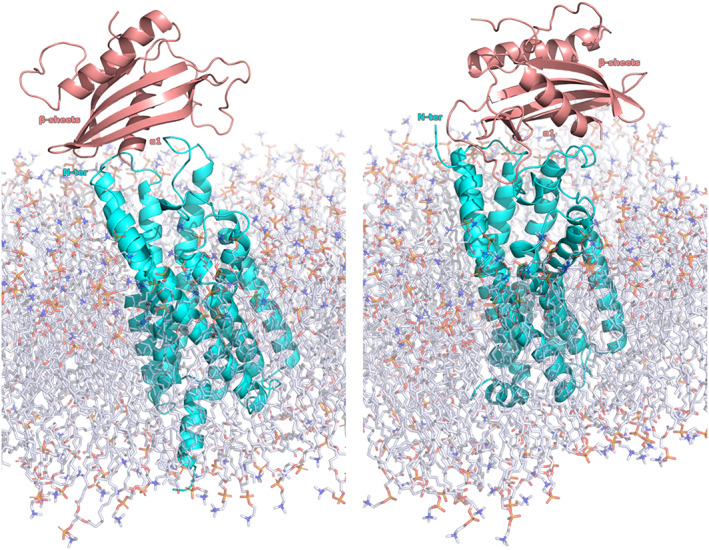
Docking proposed chemerin binding modes
3.4. Proposed interaction models for CCRL2‐chemerin binding
To gain more insight, the residues involved in the binding was analyzed the types and the frequencies of the observed interactions. Actually, for the BM1 it was observed two patterns of interactions. For the first one, we had that the chemerin β2β3 loop established contacts with the residues of CCRL2 ECL2. The residues of the chemerin β2β3 loop were mostly polar and the most frequently observed interactions were salt bridges and H‐bonds. Indeed, we found a conserved array of polar contacts (6 conformation of 12) Lys60chem with Asp271CCRL2, Lys61chem with Glu265CCRL2, Glu63chem with Lys197CCRL2, and Lys72chem with Asp176CCRL2. It was also observed hydrophobic interaction between Val66chem and Phe188CCRL2 (Figure 2 and Figure S4).
FIGURE 2.
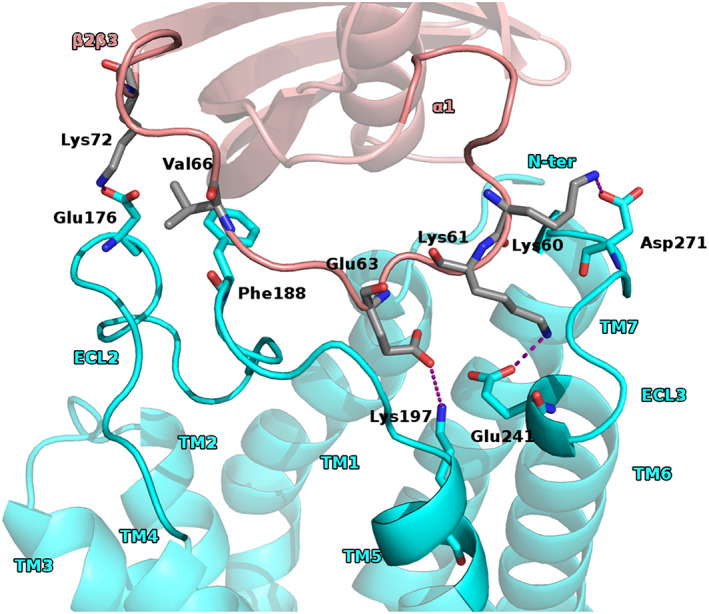
BM1 first proposed pattern of interactions
FIGURE 4.
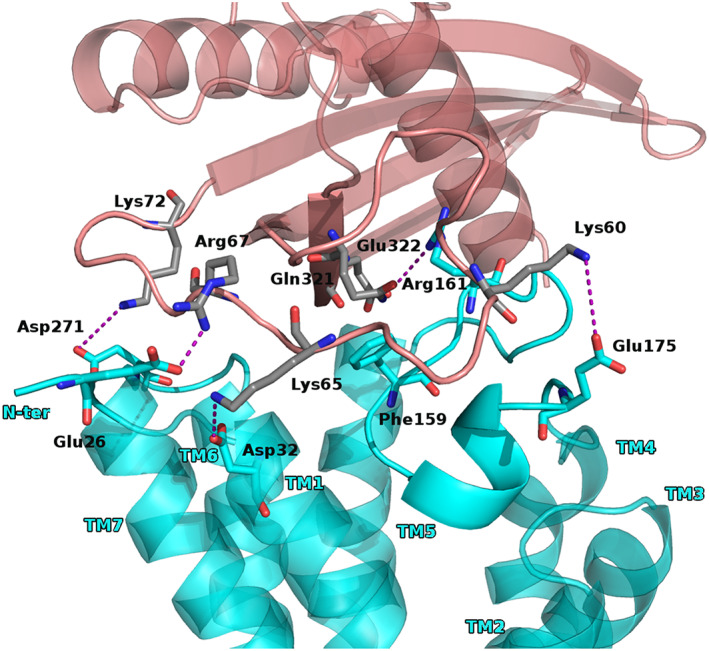
Proposed interactions for BM2
The second pattern of interactions, for the conformation falling within BM1, consisted of the chemerin α1 helix residue Glu1, and Arg4 involved in salt bridge with CCRL2 Lys30 and Glu175, respectively. Also, chemerin Arg5 had polar contact with Glu26 or Asp29 of CCRL2. Worthily, also the residues of the chemerin β1 sheet were involved in interactions with the CCRL2 ECL2 and a polar contact between Glu26chem and Arg185CCRL2 was observed. Another polar interaction was seen between the chemerin β2β3 loop Lys61 and Glu192 of the CCRL2 ECL2 (Figure 3 and Figure S5).
FIGURE 3.
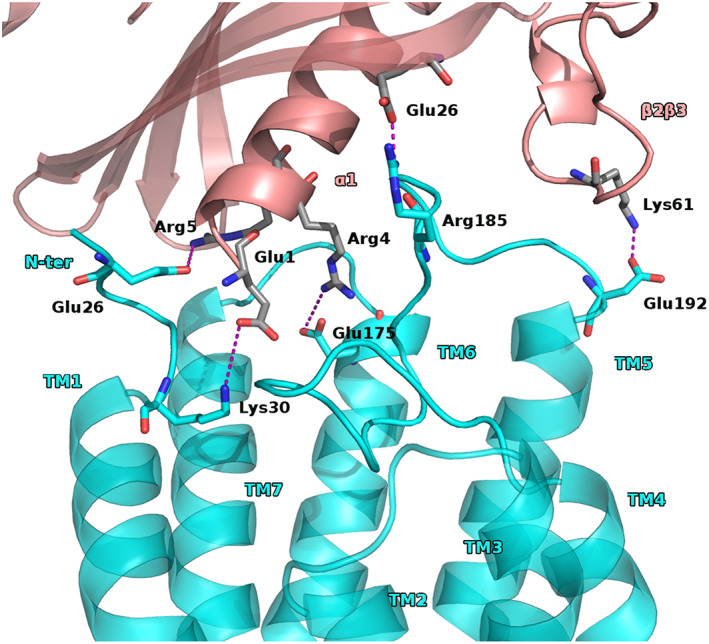
BM1 second proposed pattern of interactions
Thus, the analyses of the BM1 conformations highlighted two main positions named as first and second pattern of interactions. Despite during the simulations time, we did not observe the shifting of one position to the other and we speculated that the chemerin β2β3‐loop might interact with the CCRL2 TM6‐TM7 loop, moving the latter far from the CCRL2 entrance channel enabling the chemerin α1 helix to move toward this channel.
For the BM2, we had that the chemerin β2β3‐loop formed extensive polar interactions and hydrophobic contacts. Indeed, the chemerin residues Lys60, Lys65, Arg67, and Lys72 established salt bridge with Glu175 of ECL2, Asp32 and Glu26 of TM1, and Asp271 of ECL3, respectively (five conformations of seven).
Worthily, it seemed that interactions between chemerin β2β3‐loop and the CCRL2 ECL2, forced the latter farm from the receptor entrance channel creating a space filled by β1 sheet residues (QETSV) doing a salt bridge between Glu322chem and Arg161ECL2 and hydrophobic contact between Gln321chem and Phe159EL2 (Figures 4 and S6).
4. CONCLUSION
The accomplished computations led us to gain more insight in the chemerin binding to CCRL2. A total of 5.5 μs simulations turned back with two binding modes for chemerin, both BMs suggesting a crucial role for the chemerin α1 helix, the β1 sheet and for the β2β3‐loop. It was also postulated that the CCRL2 chemerin complex formation might be dependent by the shift of the CCRL2 ECL2 far from the receptor entrance channel, driven by chemerin approach, lastly facilitating the binding. Furthermore, the analyses of the trajectories produced a short list of hotspot residues that might be crucial in favoring the complex formation and the chemotactic activity. Indeed, we identify for chemerin the α1 helix Glu1, Arg4, and Arg5, at the β2β3‐loop three lysine residues (60, 61, and 65), and for the β1 sheet Gln25 and Glu26. Also, for CCRL2, two regions were highlighted: the ECL2 and the ECL3. For ECL3, a crucial role seemed to be played by Glu175, Asp176, and Asp271 residues. The reported data represent the earliest attempt to shed light to the CCRL2 chemerin interaction. Although these results still need to be experimentally validated, they might help in better clarify CCRL2‐chemerin interaction. Furthermore, the proposed models might pave the way for medicinal chemistry efforts in search for modulators of CCRL2 chemerin interaction and help to better clarify the physio‐pathological role of both the CCRL2 and the chemerin and their potential value as target for therapeutic intervention.
CONFLICT OF INTEREST
The authors declare no competing interests.
Supporting information
Appendix S1 Supporting Information.
ACKNOWLEDGMENTS
Antonio Coluccia would like to thank Cineca for supercomputing resources: ISCRA C project HP10CKWI8K. This research was funded by the Italian Ministry of Health (Bando Ricerca COVID‐2020‐12371735 and by AIRC IG‐20776 2017 to SS). ML was the recipient of a fellowship from AIRC (code 25307). Open Access Funding provided by Universita degli Studi di Roma La Sapienza within the CRUI‐CARE Agreement.
Bufano M, Laffranchi M, Sozzani S, Raimondo D, Silvestri R, Coluccia A. Exploring CCRL2 chemerin binding using accelerated molecular dynamics. Proteins. 2022;90(9):1714‐1720. doi: 10.1002/prot.26348
Funding information Associazione Italiana per la Ricerca sul Cancro, Grant/Award Number: COVID‐2020‐12371735; Cineca; Italian Ministry of Health, Grant/Award Number: AIRC IG‐20776 2017
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7(12):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fan P, Kyaw H, Su K, et al. Cloning and characterization of a novel human chemokine receptor 4. Bioochem Biophys Res Commun. 1998;243(1):264‐268. [DOI] [PubMed] [Google Scholar]
- 3. Schioppa T, Sozio F, Barbazza I, et al. Molecular basis for CCRL2 regulation of leukocyte migration. Front Cell Dev Biol. 2020;8:615031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mazzotti C, Gagliostro V, Bosisio D, et al. The atypical receptor CCRL2 (C‐C chemokine receptor‐like 2) does not act as a decoy receptor in endothelial cells. Front Immunol. 2017;8:1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bondue B, Wittamer V, Parmentier M. Chemerin and its receptors in leukocyte trafficking, inflammation and metabolism. Cytokine Growth Factor Rev. 2011;22(5–6):331‐338. [DOI] [PubMed] [Google Scholar]
- 6. Mattern A, Zellmann T, Beck‐Sickinger AG. Processing, signaling, and physiological function of chemerin. IUBMB Life. 2014;66(1):19‐26. [DOI] [PubMed] [Google Scholar]
- 7. Sozzani S, Vermi W, Del Prete A, Facchetti F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010;31(7):270‐277. [DOI] [PubMed] [Google Scholar]
- 8. Del Prete A, Bonecchi R, Vecchi A, Mantovani A, Sozzani S. CCRL2, a fringe member of the atypical chemoattractant receptor family. Eur J Immunol. 2013;43:1418‐1422. [DOI] [PubMed] [Google Scholar]
- 9. Mazzon C, Zanotti L, Li W, et al. CCRL2 regulates M1/M2 polarization during EAE recovery phase. J Leukoc Biol. 2016;99(6):1027‐1033. [DOI] [PubMed] [Google Scholar]
- 10. Del Prete A, Martínez‐Muñoz L, Mazzon C, et al. The atypical receptor CCRL2 is required for CXCR2‐dependent neutrophil recruitment and tissue damage. Blood. 2017;130(10):1223‐1234. [DOI] [PubMed] [Google Scholar]
- 11. Salvi V, Sozio F, Sozzani S, Del Prete A. Role of atypical chemokine receptors in microglial activation and polarization. Front Aging Neurosci. 2017;9:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parolini S, Santoro A, Marcenaro E, et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood. 2007;109(9):3625‐3632. [DOI] [PubMed] [Google Scholar]
- 13. Del Prete A, Sozio F, Schioppa T, et al. The atypical receptor CCRL2 is essential for lung cancer immune surveillance. Cancer Immunol Res. 2019;7(11):1775‐1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys. 2004;120:11919‐11929. [DOI] [PubMed] [Google Scholar]
- 15. Bucher D, Pierce LC, McCammon JA, Markwick PR. On the use of accelerated molecular dynamics to enhance configurational sampling in ab initio simulations. J Chem Theory Comput. 2011;7(4):890‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maisuradze GG, Liwo A, Scheraga HA. Principal component analysis for protein folding dynamics. J Mol Biol. 2009;385(1):312‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wold S, Esbensen K, Geladi P. Principal component analysis. Chemom Intel Lab Syst. 1987;2(1–3):37‐52. [Google Scholar]
- 18. Miles TF, Spiess K, Jude KM, et al. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. Elife. 2018;7:e35850. doi: 10.7554/eLife.35850.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I‐TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two‐step atomic‐level energy minimization. Biophys J. 2011;101(10):2525‐2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Williams CJ, Headd JJ, Moriarty NW, et al. MolProbity: more and better reference data for improved all‐atom structure validation. Protein Sci. 2018;27(1):293‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu J, Mcpartlon M, Li J. Improved protein structure prediction by deep learning irrespective of co‐evolution information. Nat Mach Intell. 2021;3:601‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera‐‐a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605‐1612. [DOI] [PubMed] [Google Scholar]
- 24. Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: a protein−protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125(7):1731‐1737. [DOI] [PubMed] [Google Scholar]
- 25. De Henau O, Degroot GN, Imbault V, et al. Signaling properties of Chemerin receptors CMKLR1, GPR1 and CCRL2. PLoS One. 2016;11(10):e0164179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jo S, Kim T, Iyer VG, Im W. CHARMM‐GUI: a web‐based graphical user Interface for CHARMM. J Comput Chem. 2008;29(11):1859‐1865. [DOI] [PubMed] [Google Scholar]
- 27. Lee J, Hitzenberger M, Rieger M, Kern NR, Zacharias M, Im W. CHARMM‐GUI supports the Amber force fields. J Chem Phys. 2020;153(3):035103. [DOI] [PubMed] [Google Scholar]
- 28. Lee J, Patel DS, Ståhle J, et al. CHARMM‐GUI membrane builder for complex biological membrane simulations with glycolipids and Lipoglycans. J Chem Theory Comput. 2019;15(1):775‐786. [DOI] [PubMed] [Google Scholar]
- 29. Darden T, York D, Pedersen L. Particle mesh Ewald: an N‐log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089‐10092. [Google Scholar]
- 30. Gedeon PC, Thomas JR, Madura JD. Accelerated molecular dynamics and protein conformational change: a theoretical and practical guide using a membrane embedded model neurotransmitter transporter. Methods Mol Biol. 2015;1215:253‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vangone A, Spinelli R, Scarano V, Cavallo L, Oliva R. COCOMAPS: a web application to analyse and visualize contacts at the interface of biomolecular complexes. Bioinformatics. 2011;27(20):2915‐2916. [DOI] [PubMed] [Google Scholar]
- 32. McGibbon RT, Beauchamp KA, Harrigan MP, et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J. 2015;109(8):1528‐1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit‐learn: machine learning in python. J Machine Learn Res. 2011;12:2825‐2830. [Google Scholar]
- 34. Virtanen P, Gommers R, Oliphant TE, et al. SciPy 1.0: fundamental algorithms for scientific computing in python. Nat Methods. 2020;17(3):261‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9(3):90‐95. [Google Scholar]
- 36. Marks C, Deane CM. Increasing the accuracy of protein loop structure prediction with evolutionary constraints. Bioinformatics. 2019;35(15):2585‐2592. [DOI] [PubMed] [Google Scholar]
- 37. Allison JR. Computational methods for exploring protein conformations. Biochem Soc Trans. 2020;48(4):1707‐1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wolf A, Kirschner KN. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11·23S subdomain. J Mol Model. 2013;19(2):539‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J, Sawyer N, Regan L. Protein‐protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 2013;22(4):510‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bosshard HR, Marti DN, Jelesarov I. Protein stabilization by salt bridges: concepts, experimental approaches and clarification of some misunderstandings. J Mol Recognit. 2004;17(1):1‐16. [DOI] [PubMed] [Google Scholar]
- 41. Bush J, Makhatadze GI. Statistical analysis of protein structures suggests that buried ionizable residues in proteins are hydrogen bonded or form salt bridges. Proteins. 2011;79(7):2027‐2032. [DOI] [PubMed] [Google Scholar]
- 42. Hendsch ZS, Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 1994;3(2):211‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.