

Abstract
Objective
Copy number variation of the C4 complement components, C4A and C4B, has been associated with systemic inflammatory autoimmune diseases. This study was undertaken to investigate whether C4 copy number variation is connected to the autoimmune repertoire in systemic lupus erythematosus (SLE), primary Sjögren's syndrome (SS), or myositis.
Methods
Using targeted DNA sequencing, we determined the copy number and genetic variants of C4 in 2,290 well‐characterized Scandinavian patients with SLE, primary SS, or myositis and 1,251 healthy controls.
Results
A prominent relationship was observed between C4A copy number and the presence of SSA/SSB autoantibodies, which was shared between the 3 diseases. The strongest association was detected in patients with autoantibodies against both SSA and SSB and 0 C4A copies when compared to healthy controls (odds ratio [OR] 18.0 [95% confidence interval (95% CI) 10.2–33.3]), whereas a weaker association was seen in patients without SSA/SSB autoantibodies (OR 3.1 [95% CI 1.7–5.5]). The copy number of C4 correlated positively with C4 plasma levels. Further, a common loss‐of‐function variant in C4A leading to reduced plasma C4 was more prevalent in SLE patients with a low copy number of C4A. Functionally, we showed that absence of C4A reduced the individuals’ capacity to deposit C4b on immune complexes.
Conclusion
We show that a low C4A copy number is more strongly associated with the autoantibody repertoire than with the clinically defined disease entities. These findings may have implications for understanding the etiopathogenetic mechanisms of systemic inflammatory autoimmune diseases and for patient stratification when taking the genetic profile into account.
INTRODUCTION
Systemic inflammatory autoimmune diseases are a group of diseases characterized by inflammation in multiple organs and the presence of antibodies targeting different ubiquitously expressed cytoplasmic or nuclear proteins. Systemic lupus erythematosus (SLE), primary Sjögren's syndrome (SS), and idiopathic inflammatory myopathies (myositis) are all categorized as systemic inflammatory autoimmune diseases. In SLE, multiple tissues and organs such as the skin, joints, cardiovascular system, and kidneys are commonly affected, whereas a more specific involvement is seen with lacrimal and salivary glands affected in primary SS, and muscle as well as connective tissues in myositis. Despite different patterns of tissue and organ involvement, the 3 diseases share important features, including clinical manifestations, presence of autoantibodies to nuclear antigens, and several genetic loci (1, 2, 3, 4, 5, 6).
The complement system plays an important role for clearance of immune complexes and apoptotic cells. Impaired removal of cellular debris may lead to exposure of cellular self‐antigens and loss of tolerance (7, 8). Although mainly studied in SLE, the complement system has also been shown to play a role in the pathogenesis of primary SS and myositis (9, 10). Particularly relevant are the genes in the early classical complement pathway, for which deficiency in any of the genes C1Q, C1R, C1S, C2, or C4 may lead to SLE or lupus‐like disease (11). Further, low plasma levels of C3 and C4 are routinely used in the clinical setting as biomarkers for complement activation associated with disease activity and flares in SLE (12, 13).
C4 is coded by the paralogous genes C4A and C4B located between the HLA class I and class II regions on chromosome 6. A high level of copy number variation exists for the 2 C4 genes, and while most individuals carry 2 C4A copies and 2 C4B copies, the number of genes may range from 0 to 5 copies for C4A and 0–4 copies for C4B (14). By definition, C4A and C4B differ by 4 amino acids (PCPVLD and LSPVIH, respectively) in exon 26 that also affect the biochemical reactivity toward either amino groups or hydroxyl groups, respectively (15, 16, 17).
Previous studies have shown an association between low copy number of C4A and systemic inflammatory autoimmune diseases (14, 18, 19, 20). However, due to the strong linkage disequilibrium (LD) between C4A copy number and the extended C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 risk haplotype, which is associated with SLE, primary SS, and myositis in populations of European origin, it has been difficult to define whether the association is with HLA or with the complement system (21). In a recent study, C4 copy number rather than HLA was suggested to be the main risk factor for SLE (22). This conclusion was based on a parallel analysis of C4 copy number and HLA alleles in patients of European and African American origin, for which the latter population shows low LD between C4A copy number and DRB1*03:01.
Due to a high homology between the genomic reference sequences of C4A and C4B (99.91% identical), the 2 genes are generally excluded from variant calling analysis due to ambiguous mapping of sequencing reads. In addition, a diploid state is assumed in variant calling of human autosomes, which is incompatible with the high level of variation in the C4 copy number ranging from 2 to 8 C4 copies. Therefore, variation in C4 genes at nucleotide level and the association to disease remain relatively unexplored.
The purpose of the current study was to perform a focused analysis of C4 copy number and C4 nucleotide variation in patients with 3 systemic inflammatory autoimmune diseases and in healthy controls. In parallel, we aimed to evaluate the relation between HLA alleles and C4 copy number variation, for which the combined analysis of 3 cohorts of patients with SLE, primary SS, or myositis allowed the comparison of shared and distinct patterns within and between the diseases.
PATIENTS AND METHODS
Study participants
Patients diagnosed as having SLE, primary SS, or myositis at Scandinavian rheumatology clinics, as well as healthy blood donors/population controls have been previously described in detail (23, 24, 25), and basic characteristics are presented in Supplementary Table 1, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42122. For validation of C4 copy number calls and HLA alleles, we included whole‐genome sequencing data from 75 parent/offspring trios, in which offspring were diagnosed as having SLE (26). The individual studies were approved by local ethics committees, and all study participants provided written informed consent.
Targeted sequencing, genotyping, and quality control
The capturing array and the targeted DNA sequencing of patients and healthy controls has previously been described (23, 24, 25, 27). A detailed description of the sequencing workflow, alignment, genotype calling, and quality control at variant‐ and individual‐based levels can be found in the Supplementary Information, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42122.
DNA sequencing and genotyping was performed at the SNP&SEQ Technology Platform in Uppsala, part of the National Genomics Infrastructure Sweden and Science for Life Laboratory. The computations were enabled by resources in projects sens2017142 and sens2020577, provided by the Swedish National Infrastructure for Computing at Uppsala Multidisciplinary Center for Advanced Computational Science.
Estimation of C4 copy number and calling of HLA alleles from targeted sequencing data
The total copy number of C4 was estimated based on read depth using the GermlineCNVCaller (GATK), and the relative read depths of 5 C4A/C4B‐specific single‐nucleotide variants were used for ascertainment of total number of C4 copies into copy number of C4A and C4B. Genetic variants in the C4A/C4B genes were called at bp resolution using the HaplotypeCaller (GATK). The human endogenous retrovirus element present in some C4 genes (28) was not analyzed (Supplementary Information, https://onlinelibrary.wiley.com/doi/10.1002/art.42122).
HLA alleles of 6 HLA genes (HLA–A, HLA–B, HLA–C, HLA–DPB1, HLA–DQB1, and HLA–DRB1) were called at 4‐digit resolution from sequencing data using xHLA (29). A detailed workflow for the analysis of C4 and HLA is available in the Supplementary Information together with method validation using results from previous polymerase chain reaction–based C4 analyses (30, 31, 32).
Plasma C4 concentration and autoantibody status of patients
Measurement of plasma C4 in SLE and primary SS patients was performed at local centers as part of clinical routine (33, 34). Information about autoantibodies was extracted from medical records, and healthy controls were included as a reference cohort under the assumption that autoantibodies are specific to patients. Nevertheless, a small percentage of the individuals in the general population may be positive for autoantibodies (34), but the impact on this study is considered negligible.
C4b deposition on heat‐aggregated human IgG
Serum from healthy individuals carrying only C4A genes or only C4B genes were incubated with heat‐aggregated IgG, and deposition of C4b was detected by enzyme‐linked immunosorbent assay, as described in the Supplementary Information (https://onlinelibrary.wiley.com/doi/10.1002/art.42122).
Statistical analysis
Statistical analyses were performed in R version 4.0.4 (35). Two‐tailed P values less than 0.05 were considered significant. The statistical tests and covariates that were included (e.g., principal components for genetic population structure) are described in the text and figure legends. Odds ratios (ORs) and 95% confidence intervals (95% CIs) were calculated. For analysis of associations in the HLA region, a Bonferroni correction for 5,000 tests was used, corresponding to a statistical significance threshold of 1 × 10−5.
RESULTS
Association of C4A copy number with systemic inflammatory autoimmune diseases
Using targeted sequencing data, we estimated the C4 copy number in 2,290 patients diagnosed as having SLE, primary SS, or myositis, and in 1,251 healthy controls (Figure 1A). The total C4 copy number calls ranged from 2 to 8 C4 copies, but since only 2 individuals had more than 6 C4 copies (Supplementary Information, https://onlinelibrary.wiley.com/doi/10.1002/art.42122), we focused on subjects with 2–6 C4 copies.
Figure 1.
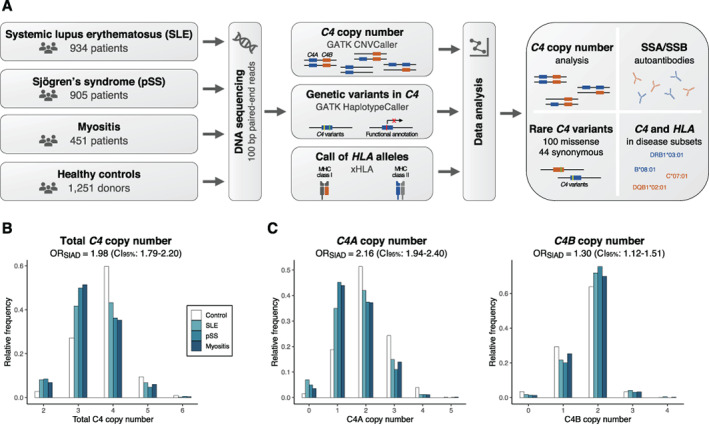
Association between complement C4 copy number and 3 systemic inflammatory autoimmune diseases (SIADs). A Workflow for analysis. Three patient groups with systemic inflammatory autoimmune diseases and 1 reference cohort were analyzed for HLA alleles and copy number of the paralogous C4 genes C4A and C4B, using targeted sequencing data. Association analysis of C4 was performed for clinical subsets of the diseases. Additionally, common and rare variants in the C4 genes were analyzed from the sequencing data. B, Total C4 copy number in healthy controls and patients with systemic lupus erythematous (SLE), primary Sjögren's syndrome (SS), or myositis (n = 3,541). Logistic regression was performed to analyze associations in the combined patient group compared to healthy controls, with adjustment for sex and principal components 1–4 (PC1–PC4). Odds ratios (ORs) represent disease risk in association with each decrease in C4 copy number. C, Copy number of C4A and C4B in each patient group and healthy controls (n = 3,520). Analysis is based on logistic regression with both C4A and C4B included as additive variables and with adjustment for sex and PC1–PC4. ORs represent disease risk in association with each decrease in C4A or C4B copy number. MHC = major histocompatibility complex; CI95% = 95% confidence interval.
Notably, the pattern in C4 copy number was relatively similar among the 3 systemic inflammatory autoimmune diseases, and therefore, we performed both joint and separate analyses of the diseases. As previously observed, a low C4 copy number was associated with an increased risk of all 3 diseases compared to healthy controls (P for C4 = 2 × 10−38) (Figure 1B and Supplementary Figure 1A, https://onlinelibrary.wiley.com/doi/10.1002/art.42122), and this association was almost exclusively explained by the copy number of the C4A gene (P for C4A = 5 × 10−45) (Figure 1C). We noted a negative correlation between C4A copy number and C4B copy number (rs = −0.50) (Supplementary Figure 1B), and although the copy number of C4B was slightly higher in patients compared to controls, each decrease in C4B copy number was modestly associated with an increased risk of systemic inflammatory autoimmune disease when adjusting for the effect of C4A (P for C4B = 6 × 10−4) (Figure 1C and Supplementary Figure 1A). Based on these results, we concluded that low C4A copy number is a strong risk factor for systemic inflammatory autoimmune disease, whereas the effect of C4B is limited.
Overlap of association with autoantibodies against SSA and SSB between systemic inflammatory autoimmune diseases
Autoantibodies against nuclear antigens are a common feature and part of the clinical evaluation of SLE, primary SS, and myositis, and therefore, we analyzed whether C4A copy number was associated with the autoantibody repertoire in patients. A strong and consistent association was detected between low C4A copy number and presence of anti‐SSA and anti‐SSB autoantibodies in all 3 diseases (Supplementary Table 2, https://onlinelibrary.wiley.com/doi/10.1002/art.42122). None of the other autoantibodies investigated for all diseases were consistently associated with C4A copy number (Supplementary Table 2).
The prevalence of anti‐SSA/SSB autoantibodies differed highly among the diseases and was highest in primary SS, with 73% of patients having autoantibodies against SSA and/or SSB (Figure 2A). For myositis, 32% of patients had anti‐SSA/SSB autoantibodies with most being against SSA and only 2.4% of patients being positive for both anti‐SSA and anti‐SSB. In SLE, 41% of patients had autoantibodies against SSA and/or SSB. A few patients (2.4%) had autoantibodies against SSB only, which is in line with the notion that anti‐SSA antibodies appear first and may be followed by anti‐SSB antibodies due to epitope spreading (36).
Figure 2.
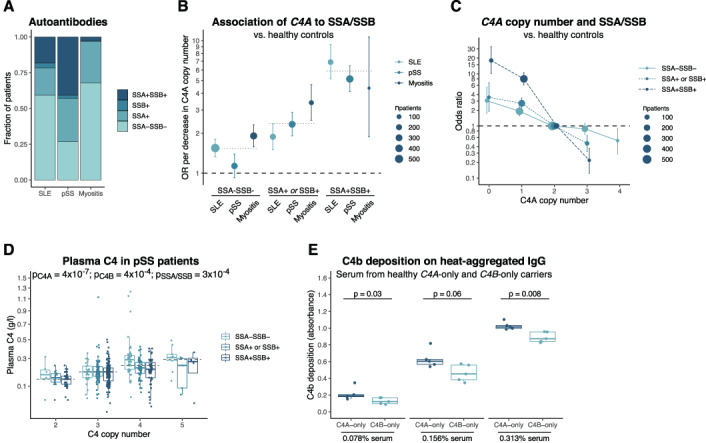
Association between C4 copy number and anti‐SSA/SSB autoantibodies. A, Prevalence of anti‐SSA/anti‐SSB autoantibodies in patients with SLE (n = 919), primary SS (n = 902), or myositis (n = 364). B, Logistic regression analysis of association between each decrease in C4A copy number and anti‐SSA/anti‐SSB autoantibody status among each patient group compared to healthy controls. Dotted lines indicate ORs for association in the combined group of 3 diseases. C, Logistic regression analysis of association between C4A copy number and anti‐SAA/anti‐SSB autoantibody status in the combined patient group compared to healthy controls. In B and C, bars represent 95% confidence intervals, and models have been adjusted for presence of C4B, sex, and PC1–PC4. D, Plasma C4 levels in patients with primary SS. Groups were compared by analysis of variance with square root–transformed values for the C4 concentration, adjusted for sex and cohort (n = 470). C4A and C4B copy number was included in the model; the x‐axis shows total C4 copy number for simplicity. E, Deposition of the complement activation product C4b on heat‐aggregated human IgG, analyzed with varying concentrations of serum from healthy individuals carrying C4A genes only (n = 5) or C4B genes only (n = 5). The samples were analyzed ≥3 times, and the mean absorbance for each sample was evaluated using the Mann‐Whitney U test. In D and E, Data are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median, and whiskers extend to 1.5 times the interquartile range. See Figure 1 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42122/abstract.
Remarkably, we observed a dose–response relationship between C4A copy number and the prevalence of autoantibodies against SSA and SSB in all 3 diseases. Each decrease in C4A copy number was associated with an increased risk of autoantibodies against both SSA and SSB (OR 5.89 [95% CI 4.83–7.23]), whereas the association with the systemic inflammatory autoimmune diseases without any autoantibodies against SSA/SSB was more modest (OR 1.53 [95% CI 1.36–1.73]) (Figure 2B). Patients with autoantibodies against either SSA or SSB showed a similar intermediate association, and therefore, we combined anti‐SSA+SSB− and anti‐SSA−SSB+ patients into 1 group (OR 2.37 [95% CI 2.02–2.77]).
Despite slight differences between the diseases, the associations of C4A copy number were more specific to the presence of anti‐SSA/SSB autoantibodies than to the individual diseases, and thus, we evaluated the prevalence of autoantibodies against number of C4A copies collectively for the 3 diseases. This revealed a strong effect of C4A copy number on the association with autoantibodies against SSA/SSB, in which the risk of disease with anti‐SSA/SSB autoantibodies for individuals with a C4A copy number of 0 was ~80 times higher than for individuals with a C4A copy number of 3 (Figure 2C). Interestingly, each change in C4A copy number was associated with a consistent change in disease risk.
In addition to a genetic association between C4 copy number and autoantibodies against SSA and SSB, we also identified a functional association linking the two parameters. As previously shown (22, 37), plasma C4 levels in patients with primary SS showed higher concentrations with an increasing copy number of C4A and C4B (Figure 2D). Additionally, we detected lower plasma C4 levels in patients with primary SS with autoantibodies against SSA/SSB when compared to patients with primary SS without anti‐SSA/SSB autoantibodies (Figure 2D), suggesting a direct connection between anti‐SSA/SSB autoantibodies and plasma C4. However, a similar association between plasma C4 and anti‐SSA/SSB was not found for SLE patients (P = 0.41; n = 411) (Supplementary Figure 2A, https://onlinelibrary.wiley.com/doi/10.1002/art.42122).
By analyzing serum from healthy individuals carrying C4A genes only or C4B genes only, we detected a higher deposition of the C4 activation product C4b on aggregated human IgG for C4A carriers (Figure 2E). These results are consistent with the proposed role of C4A being more efficient than C4B in clearance of immune complexes and suggest a connection between low C4A copy number and impaired removal of immune complexes in systemic inflammatory autoimmune disease.
In summary, we showed a strong association between C4A copy number and autoantibodies against SSA and SSB, which to a greater extent represented an association with anti‐SSA/SSB autoantibodies rather than with the systemic inflammatory autoimmune diseases themselves.
Higher proportion of SLE patients carrying the C4A loss‐of‐function (LoF) variant rs760602547
We continued by evaluating a common LoF variant (rs760602547) mainly found in C4A. The LoF variant results in a CT insertion in exon 29, which introduces a premature stop codon in C4A (38).
We called the LoF variant rs760602547 for patients and controls and detected the CT insertion in 7.7% of all individuals. Among the LoF carriers, 98% had 1 LoF variant, and the variant was not found among any of those carrying only C4B genes, thus supporting the notion that the CT insertion mainly is found in C4A and rarely in C4B (39). When analyzing the number of LoF variants in patients with SLE, primary SS, or myositis compared to healthy controls, no enrichment of the variant was seen in patients (P > 0.10 by logistic regression). However, logistic regression analysis allowing for interaction between C4A copy number and the LoF variant rs760602547 showed that SLE patients with a low C4A copy number carried the LoF variant to a greater extent than controls (P for LoF = 0.01; P for interaction = 0.03 [n = 2,136]) (Figure 3A), indicating an additional mechanism of impaired C4A function in SLE. The increased frequency of the LoF variant among SLE patients with a low C4A copy number was also seen in a focused analysis of SLE patients and healthy controls with 1–2 C4A copies (P = 0.01 by Fisher's exact test; n = 1,559). For patients with primary SS and those with myositis, no association was found for the LoF variant (P for interaction > 0.10) (Figure 3A). Grouping patients based on anti‐SSA/SSB autoantibody status showed a tendency toward an increased frequency of the LoF variant in SSA/SSB‐negative patients when compared to healthy controls, whereas no association was seen for the patients with SSA/SSB autoantibodies (Figure 3B).
Figure 3.
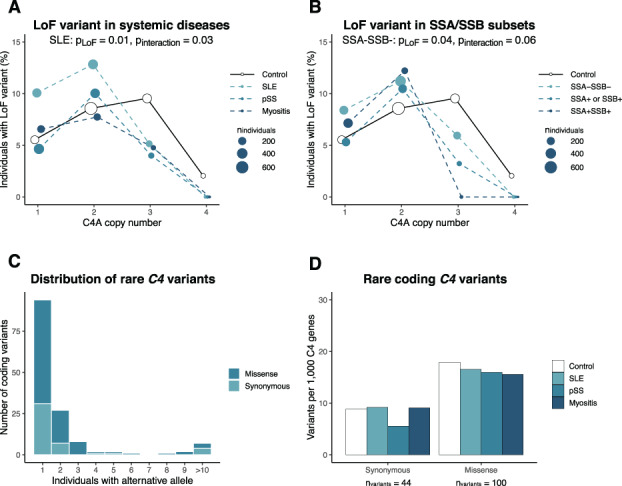
Loss‐of‐function (LoF) variant in C4A and rare variants in C4 genes. A, Proportion of patients and healthy controls carrying the C4A LoF variant rs760602547. B, Proportion of the LoF variant rs760602547 among SSA/SSB autoantibody subgroups combined across the 3 systemic inflammatory autoimmune diseases and healthy controls. Patients (or SSA/SSB subgroups) and controls were grouped based on C4A copy number, and the size of points indicate the total number of individuals in each group with the specific C4A copy number. P values are based on logistic regression with interaction between C4A copy number and rs760602547. The LoF variant was only present among individuals with 1–4 C4A genes. C, Number of individuals carrying rare (present among <0.5% of all individuals) coding variants in ≥1 C4 gene (synonymous, n = 44; missense, n = 100). Variants present among 10–17 individuals have been combined. D, Number of C4 genes carrying a rare coding variant in each disease cohort. The number of variants in each disease group has been adjusted for total C4 copy number in order to account for lower copy number of C4 among patients with systemic inflammatory autoimmune diseases. See Figure 1 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42122/abstract.
Analysis of plasma C4 by linear regression showed lower C4 concentrations for carriers of the LoF variant rs760602547 (SLE: P = 8 × 10−5 [n = 407]; primary SS: P = 5 × 10−4 [n = 471]) (Supplementary Figure 3A, https://onlinelibrary.wiley.com/doi/10.1002/art.42122) after accounting for the copy number of C4A and C4B, demonstrating that the LoF variant directly affects plasma C4 concentration.
To ensure that the enrichment of the LoF variant rs760602547 in SLE patients with a low C4A copy number was not caused by an indirect linkage to DRB1*03:01 or other SLE‐associated HLA alleles, we analyzed the LD between the LoF variant and common variants in C4, HLA alleles, and single‐nucleotide polymorphisms (SNPs) in the HLA region. We detected multiple SNPs and C4 variants in high LD with the LoF variant (Supplementary Figure 3B, Supplementary Data). For HLA alleles, the strongest LD was seen with DQB1*06:04 (R2 = 0.56), whereas C4 copy number or DRB1*03:01 was not in LD with the LoF variant rs760602547 (R2 < 0.10). Therefore, the association of the LoF variant was independent of SLE‐associated HLA alleles.
Overall, we detected lower plasma C4 levels in carriers of the LoF variant rs760602547, and the LoF variant was enriched in SLE patients with a low C4A copy number, thereby adding another level of complexity in the genetic variation of the complement system.
No enrichment of rare C4 variants in systemic inflammatory autoimmune diseases
Due to the common variation in C4 copy number, together with the high sequence homology between C4A and C4B (99.91% identical), nucleotide variants in the C4 genes are generally omitted from sequencing‐based analyses. By using information about the C4 copy number for each individual while analyzing C4 nucleotide variants (Supplementary Information, https://onlinelibrary.wiley.com/doi/10.1002/art.42122), we called common and rare variants for all study participants, focusing on coding variants. As variants could not be assigned unambiguously to C4A or C4B, variants were analyzed relative to the total C4 copy number.
Overall, we detected 144 rare coding variants (present among <0.5% of all study participants). Of these variants, 65% were found in only 1 individual, and 69% of the variants were missense variants (Figure 3C and Supplementary Information). Analysis of rare coding variants in each disease group did not indicate an enrichment of rare synonymous or missense variants in any of the systemic inflammatory autoimmune diseases (Figure 3D). Further, prediction of the effect of missense variants did not show an enrichment of putative benign or deleterious variants in systemic inflammatory autoimmune diseases (Supplementary Figure 3C, https://onlinelibrary.wiley.com/doi/10.1002/art.42122), and no difference was detected for rare C4 variants in patients grouped based on anti‐SSA/SSB autoantibody status (Supplementary Figure 3D).
In summary, we detected 144 rare variants in the coding sequence of C4, but no enrichment was observed in C4 genes of patients with systemic inflammatory autoimmune disease.
Anti‐SSA/SSB autoantibody subgroups and association with HLA and C4
Although the association of C4A copy number was more specific to anti‐SSA/SSB autoantibody status than to the individual disease entity (Figure 2B), we noted some differences between the 3 systemic inflammatory autoimmune diseases. The most striking difference was observed for patients without anti‐SSA/SSB autoantibodies, in which no association was seen between C4A copy number and primary SS (OR 1.13, P = 0.23), while a strong association was seen for a decrease in C4A copy number among myositis patients (OR 1.91, P = 4 × 10−11) and to a lesser extent among SLE patients (OR 1.55, P = 2 × 10−8) (Figure 2B).
In order to evaluate whether other variants in the HLA region could explain these differences, we analyzed the association of SNPs, HLA alleles, C4 copy number, and common variants in the C4 genes in relation to the autoantibody status for each of the individual diseases. Global analysis of the HLA region generally showed an association with the extended C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 haplotype together with C4A copy number in both SLE and myositis patients without anti‐SSA/SSB autoantibodies (Figure 4A). No additional associations were present after conditioning for the HLA allele with the strongest association (Supplementary Figure 4, https://onlinelibrary.wiley.com/doi/10.1002/art.42122). For patients with primary SS without autoantibodies to SSA/SSB, no association was seen with the HLA region (Figure 4A), suggesting at least partially different autoimmune processes among the 3 systemic inflammatory autoimmune diseases for the anti‐SSA/SSB–negative subset of patients.
Figure 4.
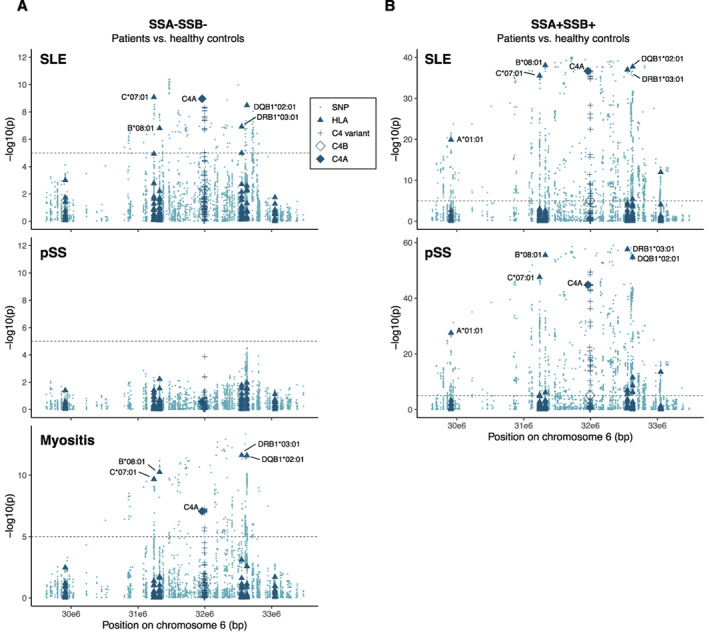
Association of variants in the HLA region with anti‐SSA/SSB–negative and anti‐SSA/SSB–positive patients. A, Association of variants in HLA region in patients with SLE (n = 544), primary SS (n = 241), or myositis (n = 247) who were negative for autoantibodies against SSA/SSB, as compared to healthy controls (n = 1,251). B, Regional association plot of HLA variants in patients with SLE (n = 168) or primary SS (n = 368) who were positive for autoantibodies against both SSA and SSB, as compared to healthy controls (n = 1,251). Few myositis patients (n = 11) had autoantibodies to both SSA and SSB, and therefore the data for these patients were not plotted. HLA alleles for 6 genes (HLA–A, HLA–C, HLA–B, HLA–DRB1, HLA–DQB1, and HLA–DPB1) and variants in C4 present in >1% of individuals were included in the analysis. Groups were analyzed for associations using logistic regression with adjustment for sex and PC1–PC4. Dashed lines represent the Bonferroni‐corrected significance threshold (P = 1 × 10−5). Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42122/abstract.
We continued analyzing genetic variation in the HLA region for patients with autoantibodies against both SSA and SSB. Due to the low number of anti‐SSA/SSB–positive myositis patients (n = 11), we focused on SLE and primary SS patients. Again, the extended C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 haplotype together with C4A copy number generally showed a strong association with SLE and primary SS patients with autoantibodies against both SSA and SSB (Figure 4B). After conditioning for DQB1*02:01 and C4A copy number, no additional association with HLA alleles was found for SLE (Supplementary Figure 5A, https://onlinelibrary.wiley.com/doi/10.1002/art.42122), whereas conditional analysis showed an association with DRB1*03:01, DRB1*15:01, DQB1*04:02, and B*08:01 in anti‐SSA/SSB–positive patients with primary SS patients (Supplementary Figure 5B).
The genomic reference sequence for C4A and C4B differs at 18 nucleotide positions of which 5 variants in exon 26 are used for defining copy number of C4A and C4B (Supplementary Information). In addition to the variants differing between C4A/C4B reference sequences, we detected 78 variants found in ≥1 C4A/C4B gene among more than 1% of the individuals. We included all 96 common variants in the analysis of the HLA region, using the number of C4 genes, with alternative alleles for each variant as variables in the analysis. However, the copy number of C4A generally explained the largest part of the associations with minimal effect of individual variants in the C4 genes (Figure 4 and Supplementary Figure 5).
In addition to the analysis of anti‐SSA/SSB–negative and anti‐SSA/SSB–positive patients against healthy controls, we also performed a pairwise case–case analysis of autoantibody subsets within the individual diseases and for the 3 systemic inflammatory autoimmune diseases combined. Again, the extended C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 haplotype together with C4A copy number explained the main association (Supplementary Figure 6, https://onlinelibrary.wiley.com/doi/10.1002/art.42122).
In summary, no association was seen between the HLA region and primary SS patients without any autoantibodies against SSA and SSB, in contrast with anti‐SSA/SSB–negative SLE and myositis patients. Further, the extended C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 haplotype together with C4A copy number was associated with both anti‐SSA/SSB–positive systemic inflammatory autoimmune diseases as well as SLE and myositis without anti‐SSA/SSB autoantibodies. In conditional analyses, C4A copy number generally followed the extended HLA haplotype, and the copy number of C4A largely explained the association with C4 with a minor effect of individual variants in the C4 genes.
DISCUSSION
In the current study, we demonstrated a strong association between low C4A copy number and the presence of anti‐SSA/SSB autoantibodies in the 3 systemic inflammatory autoimmune diseases: SLE, primary SS, and myositis. The similarities between the disease associations indicated that a low C4A copy number to a higher extent is associated with anti‐SSA/SSB autoantibodies rather than to the individual disease entities.
Autoantibodies, such as anti‐SSA and anti‐SSB, generally appear several years before clinical onset of both SLE (40, 41) and primary SS (42, 43), indicating a slow progression from asymptomatic autoimmunity to clinical manifestations. Here, we demonstrated that a genetic susceptibility with a low C4A copy number is a risk factor for development of anti‐SSA/SSB autoantibodies, and when present, the autoantibodies may contribute to systemic inflammatory autoimmune disease in a subset of individuals. Moreover, these observations may explain the partial overlap in clinical manifestations between diseases for a subgroup of patients (e.g., in SLE, in which 25% of patients are affected by secondary SS [44]). However, the low number of myositis patients with autoantibodies against both SSA and SSB limits the conclusions that can be drawn from this subset of patients.
Although the C4A copy number was associated with anti‐SSA/SSB autoantibodies in a dose‐dependent manner, the association between C4A copy number and systemic inflammatory autoimmune diseases without anti‐SSA/SSB autoantibodies was limited. As described previously, no association with the HLA region was found for anti‐SSA/SSB–negative patients with primary SS (24, 45). In contrast, C4A copy number together with the extended C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 haplotype showed a residual association with anti‐SSA/SSB–negative patients with SLE and myositis, which is consistent with earlier reports indicating DRB1*03:01 as a risk factor in SLE patients without anti‐SSA/SSB autoantibodies (46, 47). This suggests partly different etiopathogenetic mechanisms for anti‐SSA/SSB–negative patients with primary SS compared to patients with SLE or myositis.
We note that the Scandinavian study population in the current analysis is relatively homogenous, which may limit the generalizability of the study. Still, the variation in C4 copy numbers generally follows the pattern reported in other studies of individuals of European ancestry (14, 18, 22).
While it is difficult to distinguish the association signal from C4A copy number and DRB1*03:01 in populations of European descent, several lines of evidence support the notion that C4A copy number plays a central role in systemic inflammatory autoimmune diseases. Analysis of C4 copy number in SLE patients of East Asian (48) and African American origin (22)—populations in which the LD between C4A copy number and DRB1*03:01 is low—showed a strong risk with a low C4 copy number. The importance of the complement system is further supported by the strong risk of SLE and lupus‐like symptoms often including anti‐SSA autoantibodies in individuals with deficiencies in any of the early classical complement pathway genes C1Q, C1R, C1S, C4, and C2 (10, 11).
The higher frequency of the common deleterious C4A variant among SLE patients with a low C4A copy number adds an additional modulating factor to the variation in C4A copy number and plasma levels of C4 among SLE patients. Previous analyses of the LoF variant did not detect any enrichment in SLE patients (21, 49). However, a larger cohort, along with a slightly increased frequency of the LoF variant, may explain the enrichment detected in the current study. Further, the enrichment was only seen when taking the C4A copy number into account, which was not done in the previous studies. Nevertheless, no enrichment of the LoF variant was seen in patients with primary SS or myositis, suggesting a higher vulnerability among SLE patients.
Despite a high similarity between the C4A and C4B proteins, a low copy number of C4A explained the major risk for systemic inflammatory autoimmune disease with only a minor effect for the C4B copy number. The 4 C4A/C4B‐defining amino acids in exon 26 of C4 alter the reactivity of C4A and C4B toward amino groups and hydroxyl groups, respectively. This is thought to increase the efficiency of C4A in the clearance of immune complexes and apoptotic cells, whereas C4B is more efficient in targeting microbes (9). We found more extensive depositions of the C4 activation product C4b on aggregated human IgG when adding serum from individuals carrying only the C4A gene as compared to serum from individuals carrying only the C4B gene, thus indicating that C4A has enhanced capability to remove immune complexes. The functional differences between C4A and C4B were also investigated in a recent study by Simoni et al (50), in which lupus‐prone mice were genetically modified to express C4 with the human C4A/C4B‐defining amino acids. When compared to mice coding for human C4B, mice coding for C4A showed enhanced clearance of apoptotic cells, less autoreactive B cells and lower titers of anti‐SSA autoantibodies, overall supporting the notion that C4A has a role in prevention of autoantibody generation and autoimmunity.
In conclusion, we demonstrated a strong association between low C4A copy numbers and the presence of anti‐SSA/SSB autoantibodies in SLE, primary SS, and myositis. Similar relationships between C4A copy numbers and autoantibody status were observed in all 3 diseases. Our findings suggest that anti‐SSA/SSB autoantibodies are largely dependent on genetic predisposition, and this subset of autoimmune patients may be considered a specific diagnostic entity.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Lundtoft had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Lundtoft, Pucholt, Martin, Eloranta, Almlöf, Syvänen, Lindblad‐Toh, Nilsson, Blom, Diaz‐Gallo, Svenungsson, Rönnblom.
Acquisition of data
Lundtoft, Pucholt, Martin, Bianchi, Lundström, Eloranta, Sandling, Sjöwall, Jönsen, Gunnarsson, Rantapää‐Dahlqvist, Bengtsson, Leonard, Baecklund, Jonsson, Hammenfors, Forsblad‐d'Elia, Eriksson, Mandl, Bucher, Norheim, Johnsen, Omdal, Kvarnström, Wahren‐Herlenius, Notarnicola, Andersson, Molberg, Diederichsen, Almlöf, Syvänen, Kozyrev, Lindblad‐Toh, Blom, Lundberg, Nordmark, Diaz‐Gallo, Svenungsson, Rönnblom.
Analysis and interpretation of data
Lundtoft, Pucholt, Martin, Bianchi, Lundström, Blom, Diaz‐Gallo, Svenungsson, Rönnblom.
ADDITIONAL DISCLOSURES
Author Pucholt is currently an employee of Olink Proteomics, but was employed by Uppsala University during the time the study was conducted. Author Mandl is an employee of Novartis.
Supporting information
Disclosure Form
Appendix S1: Supporting Information
Appendix S2: Supporting Information
Supplementary Appendix file.
ACKNOWLEDGMENTS
We thank Prof. Chack‐Yung Yu for his support in the laboratory‐based analysis of C4 copy number, and Prof. David Pisetsky for critically reviewing the manuscript.
This study was supported by grants from the Swedish Research Council for Medicine and Health, the Swedish Rheumatism Association, King Gustav V's 80‐Year Foundation, the Swedish Society of Medicine, the Ingegerd Johansson Donation, the Swedish Heart Lung Foundation, ALF funding from Stockholm County, and KID funding from the Karolinska Institutet. Dr. Lindblad‐Toh's work was supported by a Wallenberg Scholarship.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42122&file=art42122‐sup‐0001‐Disclosureform.pdf.
Contributor Information
Lars Rönnblom, Email: lars.ronnblom@medsci.uu.se.
the DISSECT Consortium:
Lars Rönnblom, Gunnel Nordmark, Ingrid E. Lundberg, Johanna K. Sandling, Pascal Pucholt, Lina Hultin Rosenberg, Sergey V. Kozyrev, Maija‐Leena Eloranta, Andrei Alexsson, Matteo Bianchi, Christine Bengtsson, Roland Jonsson, Roald Omdal, Øyvind Molberg, Ann‐Christine Syvänen, Andreas Jönsen, Iva Gunnarsson, Elisabet Svenungsson, Solbritt Rantapää‐Dahlqvist, Anders A. Bengtsson, Christopher Sjöwall, Dag Leonard, Kerstin Lindblad‐Toh, Jonas Carlsson Almlöf, Niklas Hagberg, Jennifer R. S. Meadows, Jessika Nordin, Marie Wahren‐Herlenius, Sule Yavuz, Anna Tjärnlund, Antonella Notarnicola, Daniel Hammenfors, Elke Theander, Eva Baecklund, Guðný Ella Thorlacius, Hector Chinoy, Helena Andersson, Helena Enocsson, Helena Forsblad‐d'Elia, Janine Lamb, Johan G. Brun, Jonas Wetterö, Jorge I. Ramírez Sepúlveda, Juliana Imgenberg‐Kreuz, Karin Hjorton, Karl A. Brokstad, Kathrine Skarstein, Katrine Brække Norheim, Lilian Vasaitis, Louise Pyndt Diederichsen, Malin V. Jonsson, Marika Kvarnström, Maryam Dastmalchi, Per Eriksson, Robert G. Cooper, Sara Magnusson Bucher, Silke Appel, Simon Rothwell, Svein Joar Johnsen, Thomas Mandl, Lara Adnan Aqrawi, Janicke Liaaen Jensen, Øyvind Palm, Maria Liden, Thomas Skogh, Balsam Hanna, Christina Ståhl Hallengren, Helena Hellström, Åsa Häggström, Aladdin Mohammad, Tomas Husmark, Anna Svärd, and Awat Jalal
the ImmunoArray Development Consortium:
Kerstin Lindblad‐Toh, Gerli Rosengren Pielberg, Anna Lobell, Åsa Karlsson, Eva Murén, Göran Andersson, Kerstin M. Ahlgren, Lars Rönnblom, Maija‐Leena Eloranta, Nils Landegren, Olle Kämpe, and Peter Söderkvist
Data availability
Raw data for individual figures are available in the Supplementary Information (https://onlinelibrary.wiley.com/doi/10.1002/art.42122). Genotype data at the individual level are not publicly available since they contain information that could compromise research participant privacy and consent. Scripts for calling C4 copy number in GermlineCNVCaller are available upon request. Members of the DISSECT consortium and the ImmunoArray consortium are described in the Supplementary Appendix (https://onlinelibrary.wiley.com/doi/10.1002/art.42122).
REFERENCES
- 1. Bentham J, Morris DL, Graham DS, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 2015;47:1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Langefeld CD, Ainsworth HC, Graham DS, Kelly JA, Comeau ME, Marion MC, et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun 2017;8:16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren's syndrome. Nat Genet 2013;45:1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor KE, Wong Q, Levine DM, McHugh C, Laurie C, Doheny K, et al. Genome‐wide association analysis reveals genetic heterogeneity of Sjögren's syndrome according to ancestry. Arthritis Rheumatol 2017;69:1294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller FW, Chen W, O'Hanlon TP, Cooper RG, Vencovsky J, Rider LG, et al. Genome‐wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun 2015;16:470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rothwell S, Cooper RG, Lundberg IE, Miller FW, Gregersen PK, Bowes J, et al. Dense genotyping of immune‐related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis 2016;75:1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis 2014;73:1601. [DOI] [PubMed] [Google Scholar]
- 8. Thurman JM, Yapa R. Complement therapeutics in autoimmune disease [review]. Front Immunol 2019;10:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sturfelt G, Truedsson L. Complement in the immunopathogenesis of rheumatic disease [review]. Nat Rev Rheumatol 2012;8:458. [DOI] [PubMed] [Google Scholar]
- 10. Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, et al. Early components of the complement classical activation pathway in human systemic autoimmune diseases [review]. Front Immunol 2016;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Truedsson L. Classical pathway deficiencies–a short analytical review. Mol Immunol. 2015;68:14–9. [DOI] [PubMed] [Google Scholar]
- 12. Birmingham DJ, Irshaid F, Nagaraja HN, Zou X, Tsao BP, Wu H, et al. The complex nature of serum C3 and C4 as biomarkers of lupus renal flare. Lupus 2010;19:1272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2002;29:288. [PubMed] [Google Scholar]
- 14. Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene copy‐number variation and associated polymorphisms of complement component c4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet 2007;80:1037–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Law SK, Dodds AW, Porter RR. A comparison of the properties of two classes, C4A and C4B, of the human complement component C4. EMBO J 1984;3:1819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Isenman DE, Young JR. The molecular basis for the difference in immune hemolysis activity of the Chido and Rodgers isotypes of human complement component C4. J Immunol 1984;132:3019. [PubMed] [Google Scholar]
- 17. Yu CY, Belt KT, Giles CM, Campbell RD, Porter RR. Structural basis of the polymorphism of human complement components C4A and C4B: gene size, reactivity and antigenicity. EMBO J 1986;5:2873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lintner KE, Patwardhan A, Rider LG, Abdul‐Aziz R, Wu YL, Lundström E, et al. Gene copy‐number variations (CNVs) of complement C4 and C4A deficiency in genetic risk and pathogenesis of juvenile dermatomyositis. Ann Rheum Dis 2016;75:1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jüptner M, Flachsbart F, Caliebe A, Lieb W, Schreiber S, Zeuner R, et al. Low copy numbers of complement C4 and homozygous deficiency of C4A may predispose to severe disease and earlier disease onset in patients with systemic lupus erythematosus. Lupus 2018;27:600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Savelli SL, Roubey RA, Kitzmiller KJ, Zhou D, Nagaraja HN, Mulvihill E, et al. Opposite profiles of complement in antiphospholipid syndrome (APS) and systemic lupus erythematosus (SLE) among patients with antiphospholipid antibodies (aPL). Front Immunol 2019;10:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boteva L, Morris DL, Cortés‐Hernández J, Martin J, Vyse TJ, Fernando MM. Genetically determined partial complement C4 deficiency states are not independent risk factors for SLE in UK and Spanish populations. Am J Hum Genet 2012;90:445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, et al. Complement genes contribute sex‐biased vulnerability in diverse disorders. Nature 2020;582:577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sandling JK, Pucholt P, Rosenberg LH, Farias FH, Kozyrev SV, Eloranta ML, et al. Molecular pathways in patients with systemic lupus erythematosus revealed by gene‐centred DNA sequencing. Ann Rheum Dis 2021;80:109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thorlacius GE, Hultin‐Rosenberg L, Sandling JK, Bianchi M, Imgenberg‐Kreuz J, Pucholt P, et al. Genetic and clinical basis for two distinct subtypes of primary Sjögren's syndrome. Rheumatology (Oxford) 2021;60:837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bianchi M, Kozyrev SV, Notarnicola A, Rosenberg LH, Karlsson Å, Pucholt P, et al. Contribution of rare genetic variation to disease susceptibility in a large Scandinavian myositis cohort. Arthritis Rheumatol 2022;74:342–52. [DOI] [PubMed] [Google Scholar]
- 26. Almlöf JC, Nystedt S, Leonard D, Eloranta ML, Grosso G, Sjöwall C, et al. Whole‐genome sequencing identifies complex contributions to genetic risk by variants in genes causing monogenic systemic lupus erythematosus. Hum Genet 2019;138:141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eriksson D, Bianchi M, Landegren N, Nordin J, Dalin F, Mathioudaki A, et al. Extended exome sequencing identifies BACH2 as a novel major risk locus for Addison's disease. J Intern Med 2016;280:595–608. [DOI] [PubMed] [Google Scholar]
- 28. Dangel AW, Mendoza AR, Menachery CD, Baker BJ, Daniel CM, Carroll MC, et al. The dichotomous size variation of human complement C4 genes is mediated by a novel family of endogenous retroviruses, which also establishes species‐specific genomic patterns among Old World primates. Immunogenetics 1994;40:425–36. [DOI] [PubMed] [Google Scholar]
- 29. Xie C, Yeo ZX, Wong M, Piper J, Long T, Kirkness EF, et al. Fast and accurate HLA typing from short‐read next‐generation sequence data with xHLA. Proc Natl Acad Sci 2017;114:8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu YL, Savelli SL, Yang Y, Zhou B, Rovin BH, Birmingham DJ, et al. Sensitive and specific real‐time polymerase chain reaction assays to accurately determine copy number variations (CNVs) of human complement C4A, C4B, C4‐long, C4‐short, and RCCX modules: elucidation of C4 CNVs in 50 consanguineous subjects with defined HLA genotypes. J Immunol 2007;179:3012. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y, Lundstrom E, Liu CC, Yang Y, Gunnarsson I, Svenungsson E, et al. Low copy‐number of complement C4A, the presence of HLA‐DR3, and the presence of HLA‐DR2 are independent and additive risk factors for human systemic lupus erythematosus (SLE). J Immunol 2010;184 Suppl:93. [Google Scholar]
- 32. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016;530:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Enocsson H, Wirestam L, Dahle C, Padyukov L, Jönsen A, Urowitz MB, et al. Soluble urokinase plasminogen activator receptor (suPAR) levels predict damage accrual in patients with recent‐onset systemic lupus erythematosus. J Autoimmun 2020;106:102340. [DOI] [PubMed] [Google Scholar]
- 34. Svenungsson E, Gustafsson JT, Grosso G, Rossides M, Gunnarsson I, Jensen‐Urstad K, et al. Complement deposition, C4d, on platelets is associated with vascular events in systemic lupus erythematosus. Rheumatology (Oxford) 2020;59:3264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. R Core Team . R: a language and environment for statistical computing: R Foundation for Statistical Computing; 2018. URL: https://www.R-project.org/.
- 36. Routsias JG, Tzioufas AG. B‐cell epitopes of the intracellular autoantigens Ro/SSA and La/SSB: tools to study the regulation of the autoimmune response. J Autoimmun 2010;35:256–64. [DOI] [PubMed] [Google Scholar]
- 37. Yang Y, Chung EK, Zhou B, Blanchong CA, Yu CY, Füst G, et al. Diversity in intrinsic strengths of the human complement system: serum C4 protein concentrations correlate with C4 gene size and polygenic variations, hemolytic activities, and body mass index. J Immunol 2003;171:2734. [DOI] [PubMed] [Google Scholar]
- 38. Barba G, Rittner C, Schneider PM. Genetic basis of human complement C4A deficiency. Detection of a point mutation leading to nonexpression. J Clin Invest 1993;91:1681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lokki ML, Circolo A, Ahokas P, Rupert KL, Yu CY, Colten HR. Deficiency of human complement protein C4 due to identical frameshift mutations in the C4A and C4B genes. J Immunol 1999;162:3687. [PubMed] [Google Scholar]
- 40. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 2003;349:1526–33. [DOI] [PubMed] [Google Scholar]
- 41. Eriksson C, Kokkonen H, Johansson M, Hallmans G, Wadell G, Rantapää‐Dahlqvist S. Autoantibodies predate the onset of systemic lupus erythematosus in northern Sweden. Arthritis Res Ther 2011;13:R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Theander E, Jonsson R, Sjöström B, Brokstad K, Olsson P, Henriksson G. Prediction of Sjögren's syndrome years before diagnosis and identification of patients with early onset and severe disease course by autoantibody profiling. Arthritis Rheumatol 2015;67:2427–36. [DOI] [PubMed] [Google Scholar]
- 43. Jonsson R, Theander E, Sjöström B, Brokstad K, Henriksson G. Autoantibodies present before symptom onset in primary Sjögren syndrome. JAMA 2013;310:1854–5. [DOI] [PubMed] [Google Scholar]
- 44. Ruacho G, Kvarnström M, Zickert A, Oke V, Rönnelid J, Eketjäll S, et al. Sjögren syndrome in systemic lupus erythematosus: a subset characterized by a systemic inflammatory state. J Rheumatol 2020;47:865. [DOI] [PubMed] [Google Scholar]
- 45. Gottenberg JE, Busson M, Loiseau P, Cohen‐Solal J, Lepage V, Charron D, et al. In primary Sjögren's syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum 2003;48:2240–5. [DOI] [PubMed] [Google Scholar]
- 46. Morris DL, Fernando MM, Taylor KE, Chung SA, Nititham J, Alarcón‐Riquelme ME, et al. MHC associations with clinical and autoantibody manifestations in European SLE. Gene Immun 2014;15:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diaz‐Gallo LM, Oke V, Lundström E, Elvin K, Wu YL, Eketjäll S, et al. Four systemic lupus erythematosus subgroups, defined by autoantibodies status, differ regarding HLA‐DRB1 genotype associations and immunological and clinical manifestations. ACR Open Rheumatol 2022;4:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen JY, Wu YL, Mok MY, Wu YJ, Lintner KE, Wang CM, et al. Effects of complement C4 gene copy number variations, size dichotomy, and C4A deficiency on genetic risk and clinical presentation of systemic lupus erythematosus in East Asian populations. Arthritis Rheumatol 2016;68:1442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tsang‐A‐Sjoe MW, Bultink IE, Korswagen LA, van der Horst A, Rensink I, de Boer M, et al. Comprehensive approach to study complement C4 in systemic lupus erythematosus: gene polymorphisms, protein levels and functional activity. Mol Immunol 2017;92:125–31. [DOI] [PubMed] [Google Scholar]
- 50. Simoni L, Presumey J, van der Poel CE, Castrillon C, Chang SE, Utz PJ, et al. Complement C4A regulates autoreactive B cells in murine lupus. Cell Rep 2020;33:108330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1: Supporting Information
Appendix S2: Supporting Information
Supplementary Appendix file.
Data Availability Statement
Raw data for individual figures are available in the Supplementary Information (https://onlinelibrary.wiley.com/doi/10.1002/art.42122). Genotype data at the individual level are not publicly available since they contain information that could compromise research participant privacy and consent. Scripts for calling C4 copy number in GermlineCNVCaller are available upon request. Members of the DISSECT consortium and the ImmunoArray consortium are described in the Supplementary Appendix (https://onlinelibrary.wiley.com/doi/10.1002/art.42122).