

Abstract
Objective
We utilized human midbrain‐like organoids (hMLOs) generated from human pluripotent stem cells carrying glucocerebrosidase gene (GBA1) and α‐synuclein (α‐syn; SNCA) perturbations to investigate genotype‐to‐phenotype relationships in Parkinson disease, with the particular aim of recapitulating α‐syn– and Lewy body–related pathologies and the process of neurodegeneration in the hMLO model.
Methods
We generated and characterized hMLOs from GBA1 −/− and SNCA overexpressing isogenic embryonic stem cells and also generated Lewy body–like inclusions in GBA1/SNCA dual perturbation hMLOs and conduritol‐b‐epoxide–treated SNCA triplication hMLOs.
Results
We identified for the first time that the loss of glucocerebrosidase, coupled with wild‐type α‐syn overexpression, results in a substantial accumulation of detergent‐resistant, β‐sheet–rich α‐syn aggregates and Lewy body–like inclusions in hMLOs. These Lewy body–like inclusions exhibit a spherically symmetric morphology with an eosinophilic core, containing α‐syn with ubiquitin, and can also be formed in Parkinson disease patient–derived hMLOs. We also demonstrate that impaired glucocerebrosidase function promotes the formation of Lewy body–like inclusions in hMLOs derived from patients carrying the SNCA triplication.
Interpretation
Taken together, the data indicate that our hMLOs harboring 2 major risk factors (glucocerebrosidase deficiency and wild‐type α‐syn overproduction) of Parkinson disease provide a tractable model to further elucidate the underlying mechanisms for progressive Lewy body formation. ANN NEUROL 2021;90:490–505
Parkinson disease (PD) is a progressive neurodegenerative disorder characterized by the selective loss of midbrain dopaminergic (mDA) neurons in the substantia nigra pars compacta. 1 The key histopathological hallmark of PD is the presence of intraneuronal protein inclusions named Lewy bodies (LBs), which contain α‐synuclein (α‐syn) fibril deposits. 2 , 3 , 4 However, the etiology of LBs in PD is not fully understood. Although LBs are widely observed in both sporadic and familial PD patients, it is not clear whether the formation of LBs, occurring via the amplification of misfolded α‐syn fibrils, causes PD. 5 , 6 , 7 This lack of understanding of LBs and their contribution to disease pathogenesis may be largely due to the absence of in vitro modeling systems that fully recapitulate α‐syn pathologies. Recent advances in human induced pluripotent stem cell (iPSC) derivation, culture, and neuronal differentiation protocols have enabled cellular modeling of brain disorders using patient‐derived iPSCs. 8 However, it is challenging to establish pathological phenotypes of PD, as pathogenesis has been attributed to both mutations in iPSC‐derived mDA neurons and environmental factors. The same challenges have also been reported in animal models, as crucial PD pathologies are largely absent in both transgenic and knockin/knockout mouse genetic models of disease. 9 Several monogenic forms of PD have been identified. Among these, mutations in the glucocerebrosidase (GCase) gene GBA1 10 and triplication of SNCA 11 are among the most significant discoveries.
Here, we utilized human midbrain‐like organoids (hMLOs) 12 generated from human pluripotent stem cells (hPSCs) carrying GBA1 and SNCA perturbations to investigate genotype‐to‐phenotype relationships in PD, with the particular aim of recapitulating α‐syn– and LB‐related pathologies and the process of neurodegeneration in the hMLO model.
Materials and Methods
hPSC Culture and Cell Line Generation
H9 and H1 human embryonic stem cells (hESCs) were obtained from WiCell Research Institute (Madison, WI). GM23338 and ND34391 human iPSCs were purchased from Coriell Institute. PD patient‐derived iPSCs (GBA1‐iPSC‐1 and GBA1‐iPSC‐2; both GBA1 N370S heterozygotes) were obtained as previously described. 13 , 14 The use of the commercial cell lines for in vitro research follows our Institutional (Agency for Science, Technology, and Research) guidelines, which do not require specific institutional review board approval. The hESCs and iPSCs were maintained on Matrigel‐coated plates, in mTeSRTM1 (STEMCELL Technologies) media. GBA1 −/− isogenic hESC lines were generated using a single guide RNA (5′‐CGCTATGAGAGTACACGCAGTGG‐3′) targeting exon 4 of the human GBA1 gene. For doxycycline‐inducible SNCA overexpression, hemagglutinin (HA)‐tagged SNCA cDNA was cloned into a lentiviral backbone (Addgene, Cambridge, MA; #27150; Fig 1H) for cell infection. Cells were selected with appropriate antibiotics from day 3 to 7, and 3 clones with overexpression of SNCA were selected.
FIGURE 1.
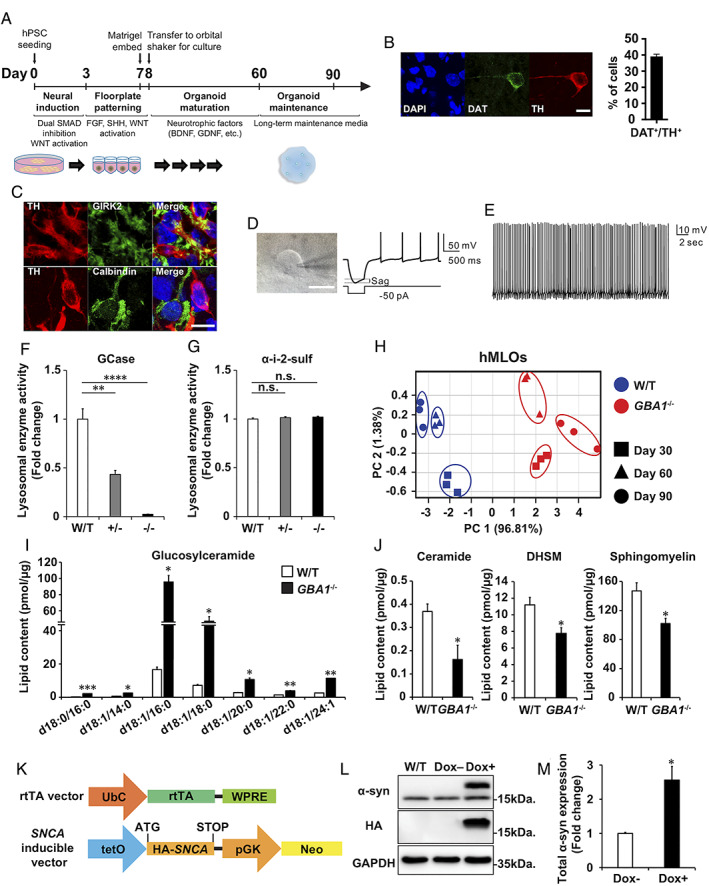
Generation and characterization of human midbrain‐like organoids (hMLOs) and GBA1 +/−, GBA1 −/−, and SNCA overexpressing (O/E) isogenic embryonic stem cells (ESCs). (A) Schematic diagrams of a differentiation protocol to generate hMLOs from starting population of human pluripotent stem cells (hPSCs). (B) Immunostaining of day 60 hMLOs using antibodies against tyrosine hydroxylase (TH) and dopamine transporter (DAT), markers of mature dopaminergic neurons, with quantification (mean ± standard error of the mean [SEM]; n = 3). Scale bar = 10μm. (C) Neurons immunostained with antibodies against TH and calbindin (top panels) or TH and GIRK2 (bottom panels) in day 90 hMLOs. Scale bar = 10μm. (D) A representative voltage trace obtained from TH+ neurons in response to hyperpolarizing current pulse. TH+ neurons displayed rebound depolarization as well as a typical sag. Scale bar = 10μm. (E) Representative traces of spontaneous action potentials from TH+ neurons. (F) Glucocerebrosidase (GCase) activity in day 30 wild‐type (W/T), GBA1 +/−, and GBA1 −/− hMLOs (mean ± SEM; **p = 0.0015, ****p < 0.0001; n = 3). (G) α‐Iduronate‐2‐sulfatase (α‐i‐2‐sulf) activity in day 30 W/T, GBA1 +/−, and GBA1 −/− hMLOs (mean ± SEM; n.s. = no significance; n = 3). (H) Principal component (PC) analysis of lipidomics data from W/T and GBA1 −/− hMLOs at days 30, 60, and 90. (I) Measurement of galactosyl/glucosylceramide in W/T and GBA1 −/− hMLOs demonstrating the specificity of GBA1 loss‐of‐function (mean ± SEM; *p < 0.05, **p < 0.01 ***p < 0.001; n = 3). (J) Quantitative measurements of ceramide, dihydrosphingomyelin (DHSM), and sphingomyelin in W/T and GBA1 −/− hMLOs (mean ± SEM; *p < 0.05; n = 3). (K) Schematic of the use of lentiviral constructs to generate a doxycycline‐inducible SNCA O/E ESC line. (L) Western blot validation of α‐synuclein (α‐syn) overexpression in ESCs with and without doxycycline (Dox) treatment using specific antibodies against α‐syn (17 and 19kDa, indicating endogenous and exogenous α‐syn, respectively) and hemagglutinin (HA; 19kDa). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a loading control. (M) Semiquantitative analysis of the Western blot data for Dox‐induced α‐syn expression (mean ± SEM; *p = 0.04; n = 3). BDNF = brain‐derived neurotrophic factor; DAPI = 4,6‐diamidino‐2‐phenylindole; FGF = fibroblast growth factor; GDNF = glia‐derived neurotrophic factorl pGK = phosphoglycerate kinase; rtTA = reverse tetracycline‐controlled transactivator; SHH = sonic hedgehog; SMAD = a name of a structurally similar protein family. The abbreviation refers to the homologies to the C. elegans SMA ("small" worm phenotype) and MAD family ("Mothers Against Decapentaplegic") of genes in Drosophila (from Wikipedia); tetO = tetracycline operator; UbC = Ubiquitin C; WNT = wingless‐related integration site; WPRE = Woodchuck hepatitis virus posttranslational regulatory element. [Color figure can be viewed at www.annalsofneurology.org]
Generation of hMLOs
hMLOs were generated as described previously. 12 Briefly, dissociated hPSCs were plated into each well of a 96‐well culture plate in neuronal induction media. Three days afterward, this medium was changed to floorplate induction medium. On day 7, organoids were embedded in the reduced growth factor Matrigel (Corning, Corning, NY) and grown in tissue growth medium containing Neurobasal and supplemented with growth factors as described previously. Twenty‐four hours after embedding, hMLOs were cultured on orbital shakers for maintenance.
Lysosomal Enzyme Activity Assay
Lysosomal enzyme activity was measured from cell lysates using artificial enzyme substrates: 4MU‐glucopyranoside for GCase and 4MU‐sulfate potassium salt for a‐i‐2‐sulfate (Sigma‐Aldrich, St Louis, MO). hMLOs were harvested in activity assay buffer (0.25% [vol/vol] Triton‐X100, 0.25% [wt/vol] taurocholic acid, and 1mM ethylenediaminetetraacetic acid in citrate/phosphate buffer [pH 5.4]), and 10μg protein was added to 10μl of 10% bovine serum albumin (BSA; in an activity assay buffer) followed by 20μl of 5mM enzyme‐substrate (in an activity assay buffer). The reaction was stopped by adding an equal amount of 1M glycine, pH 12.5. Samples were loaded onto 96‐well plates, and the fluorescence signal was quantified.
Lipidomics Analysis
hMLOs were homogenized in water with 0.6% formic acid using a Precellys 24 beadmill (Bertin Technologies, Montigny‐le‐Bretonneux, France) at 4°C. An equal volume of acetonitrile was added for additional homogenization. For lipid extraction, 20μl of ceramide/sphingoid internal standard mix (LM‐6002; Avanti Polar Lipids, Alabaster, AL) and 10μl of 14:0 phosphatidylcholine were added to 100μl of tissue homogenate. The mixture was incubated with 1.2ml of high‐performance liquid chromatography (HPLC) grade methanol at 50°C for 10 minutes and centrifuged to pellet the precipitated protein. The pellet was dried and reconstituted in 100μl methanol before analysis with a liquid chromatography–mass spectrometer. Lipids were separated using an Agilent 1260 (Agilent Technologies, Santa Clara, CA) liquid chromatography system and a Thermo Fisher Scientific (Waltham, MA) Accucore HILIC column (100 × 2.1mm, particle size = 2.6μm). Mobile phase A consisted of acetonitrile/water (95:5) with 10mM ammonium acetate, pH 8.0, and mobile phase B consisted of acetonitrile/water (50:50) with 10mM ammonium acetate, pH 8.0. For the separation, the column was equilibrated with 100% mobile phase A, increasing to 20% mobile phase B in 5 minutes, then held for 5 minutes. The column was finally equilibrated with 100% mobile phase A for 5 minutes. Mass spectrometry was performed using an Agilent 6430 mass spectrometer. All compounds were ionized in positive mode using electrospray ionization. The chromatograms were analyzed using the Agilent Masshunter Workstation software (vB.06.00).
Immunofluorescence and Immunohistochemistry
For immunofluorescence, hMLOs were fixed in 4% paraformaldehyde (PFA) overnight and incubated in 30% sucrose solution in phosphate‐buffered saline (PBS) at 4°C overnight and were cryosectioned at 20μm thickness. For immunofluorescence, slides were blocked with 3% BSA with 0.5% Triton X‐100 in PBS before primary antibody incubation. Images were taken using an LSM 710 (Zeiss, Oberkochen, Germany) or a TCS SP8 (Leica, Wetzlar, Germany) confocal microscope. For immunohistochemistry, hMLOs were fixed in 4% PFA overnight, embedded in paraffin, and sectioned at 4μm thickness. Deparaffinized and hydrated sections were stained with α‐syn (1:500 dilution; AB5038; Millipore, Billerica, MA) or ubiquitin (1:100 dilution; NB300‐130; Novus Biologicals, Littleton, CO). Primary antibodies were detected with the appropriate horseradish peroxidase (HRP)‐conjugated secondary antibody (VECTASTAIN ABC kit; Vector Laboratories, Burlingame, CA) and developed with 3, 3′‐diaminobenzidine (Sigma‐Aldrich). Nuclei were counterstained with hematoxylin (HHS16‐500ml, Sigma). Images were taken on an Eclipse Ni‐E microscope (Nikon Instruments, Tokyo, Japan).
Transmission Electron Microscopy
hMLOs were fixed in 2% PFA/3% glutaraldehyde in PBS for 4 hours at 4°C and postfixed with 1% OsO4 in PBS (pH 7.4) for 2 hours at room temperature (RT), followed by washing with deionized water twice for 10 minutes. Samples were dehydrated through an ascending ethanol series before staining with 100% acetone. For infiltration, 100 acetone/araldite resin mix (1:1) was added for 30 minutes and 100 acetone/araldite resin mix (1:6) was added to the hMLOs overnight. The pure araldite resin was changed, and hMLOs were then transferred to an oven (40°C for 30 minutes, 45°C for 4 hours, and 50°C for 1 hour). The hMLOs were subsequently embedded in a fresh resin and polymerized at 60°C for 24 hours. Sectioning was performed using an ultramicrotome (Leica EM UC6) at a thickness of 100nm, and sections were collected on copper grids. Before transmission electron microscopy (TEM), the grids were stained with lead citrate. Imaging was performed using JEOL JEM‐1220 and JEOL JEM‐1010 (JEOL, Tokyo, Japan). Processing and imaging of samples were performed in a blinded manner.
Intact Cell Cross‐Linking
hMLOs were dissociated and incubated in 1mM disuccinimidyl glutarate for 30 minutes at 37°C. Samples were quenched with 20mM Tris (pH 7.4) for 15 minutes at RT, and a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) was added before sonication for 15 seconds (1‐second pulse on/off) at 10% amplitude. Cell lysates were then centrifuged at 100,000 × g for 1 hour at 4°C. Supernatants were collected for Western blot analysis.
Thioflavin S Staining
Thioflavin S (ThioS) staining was performed by adding 0.025% ThioS in 50% ethanol to the hMLO sections for 15 minutes of incubation at RT. Cells where ThioS+ signal colocalized with α‐syn were counted. At least 4 fields of view were used, and 100 to 500 cells were counted per condition or cell line. Images were taken using a Leica TCS SP8 confocal microscope.
Flow Cytometry
Flow cytometry was performed as previously described. 12 Briefly, hMLOs were dissociated, suspended in PBS, and subjected to intracellular staining preparation per the manufacturer's instructions (eBioscience, San Diego, CA); 0.15–0.2 × 106 cells were prepared for each 100μl staining reaction. Flow cytometry analysis was carried out with BD LSR Fortessa (Becton, Dickinson, and Co, Franklin Lakes, NJ), and analysis was performed using FlowJo (Ashland, OR). Threshold gates were drawn using cells stained with secondary antibody only.
Cell Viability Assay
Cell viability in hMLOs was assessed using the CellTiter‐Glo 3D viability assay (Promega, Madison, WI) per the manufacturer's instructions.
Purification of Recombinant α‐Syn Protein
The Escherichia coli BL21 (DE3) strain was induced with 0.1mM isopropyl thiogalactoside for 3 hours at 37°C until an absorbance of 0.6 was reached. Cells were pelleted, resuspended with 20mM sodium phosphate buffer (pH 7.4) for sonication, and boiled at 100°C for 20 minutes before centrifugation at 10,000 × g for 10 minutes. The supernatant was subjected to anion exchange chromatography and Superdex‐200 gel filtration column chromatography. Purified α‐syn was dialyzed against distilled water and lyophilized. For monomer preparation, lyophilized α‐syn was reconstituted by PBS, followed by ultrafiltration using a 100,000 molecular weight cutoff centrifugal device (Pall Corporation, Port Washington, NY).
Protein Misfolding Cyclic Amplification Assay and Proteinase K Digestion
hMLOs were sonicated with Vibra‐Cells (Sonics & Materials, Newtown, CT) to 10% (wt/vol) solution with homogenizing buffer (1% Triton X‐100, protease inhibitor cocktail in PBS) and centrifuged at 3,000 × g. The protein concentration of supernatants was determined by bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). Equipment for protein misfolding cyclic amplification (PMCA; Qsonica, Newtown, CT) includes microplate horn, sound enclosure, and thermoelectric chiller. α‐syn monomers were prepared up to a final 5μM concentration in the conversion buffer (1% Triton X‐100, 150mM NaCl), and 50μl was transferred into polymerase chain reaction tubes. Thirty micrograms of hMLO homogenates were used as exogenous seeds. Three Teflon beads were placed before adding the mixture. The samples were subjected to cycles of 20‐second sonication (amplitude = 1%) and 29 minutes 40 seconds of incubation at 37°C.
For proteinase K (PK) digestion, 5μM α‐syn samples were incubated with 100μg/ml PK for 80 minutes at 37°C. Samples were resolved on 16% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels before transfer to nitrocellulose membranes. Membranes were probed with primary antibody overnight at 4°C, washed, and incubated with an HRP‐conjugated secondary antibody. Image detection was performed using an Amersham Imager 600 (GE Healthcare Life Sciences, Marlborough, MA), and Multi‐Gauge (v3.0) software (Fujifilm, Tokyo, Japan).
Seeded Polymerization, Thioflavin T Binding Assay, and TEM Imaging
PMCA products (1% wt/wt) were used as seeds and added to α‐syn monomers (200μM). Samples were incubated at 37°C for 5 days with shaking. Forty microliters of 10μM recombinant α‐syn sample was added to 50μl of 10μM thioflavin T (ThioT) solution in glycine‐NaOH (pH 8.5). Fluorescence was measured with excitation/emission (Ex/Em) of 450/490nm. For TEM imaging of α‐syn fibrils, samples were adsorbed onto 200 mesh grids, followed by negative staining with 2% uranyl acetate for 1 minute. Prepared grids were observed with a transmission electron microscope (JEOL).
RNA Sequencing and Analysis
Total RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA) and treated with DNase I (Ambion, Austin, TX). For RNA‐sequencing (RNA‐seq) library preparation, total RNA was column‐purified (PureLink RNA Mini Kit, Invitrogen), and 4μg of total RNA was applied for the RNA‐seq run according to the manufacturer's protocol (TruSeq RNA Sample Preparation Kit v2; Illumina, San Diego, CA). Samples were multiplexed and sequenced in 150 base pairs paired‐end format (NovaSeq 6000, Illumina). RNA‐Seq data were mapped against hg19 with STAR‐2.6.1. 15 Reads were counted using featureCounts‐1.6.4. 16 Rlog transformed values of the counts and differential expression were calculated using DESeq2‐1.24.0. 17 We used an adjusted p value threshold of 0.05, and a fold change threshold of ±1.5 for up‐ and downregulated genes, respectively. An MA plot was created using ggplot2, 18 and heatmaps for individual genes of interest were plotted using Morpheus (Broad Institute, Cambridge, MA) using a relative color scheme. Clustering analysis was performed by computing the correlation of normalized gene expression using pheatmap‐1.0.12. Gene Ontology (GO) analysis was performed with DAVID v6.8.
Results
Generation and Characterization of hMLOs from GBA1 −/− and SNCA Overexpressing Isogenic ESCs
We generated hMLOs from hPSCs based on stepwise differentiation protocols that guide hPSCs toward the midbrain floor‐plate that gives rise to substantia nigra dopaminergic neurons and progenitors (see Fig 1). Briefly, hPSCs were directed toward neuroectodermal lineages by dual‐SMAD (a name of a structurally similar protein family) inhibition (neural induction) and patterned to FOXA2‐ and LMX1A‐expressing midbrain floor‐plate progenitors by exogenous treatment of wingless‐related integration site (WNT), fibroblast growth factor (FGF) (resulting in caudalization), and sonic hedgehog morphogens (resulting in ventralization). Next, resulting organoids were cultured in media containing neurotrophic factors such as glia‐derived neurotrophic factor and brain‐derived neurotrophic factor, which promote neuronal growth and maturation. During this time (2–3 months after seeding), resulting hMLOs become enriched in tyrosine hydroxylase (TH)‐positive, mDA neurons expressing the dopamine transporter and/or mDA neuron marker, GIRK2, while being predominantly immunonegative for calbindin. Moreover, these TH‐positive neurons within the hMLOs showed rebound action potentials with significant sag currents and rhythmic spontaneous discharges with an average frequency of 3 to 5Hz, 19 , 20 demonstrating the presence of mDA neurons in hMLOs.
Next, we focused on the GBA1 gene encoding the lysosomal enzyme GCase, whose deficiency contributes to the accumulation of glycosphingolipids (GSLs) and the generation of the toxic, fibrillar α‐syn assemblies seen in PD. 21 A higher rate of LB pathology has been reported in early onset PD patients with GBA1 mutations as compared to those without such mutations. 22 Based on these observations, we hypothesized that GBA1 deficiency could affect the production of toxic α‐syn and trigger a neuronal loss in our hMLOs. 23 To test our hypothesis, we established human ESC lines with GBA1 depletion using the CRISPR/Cas9 system. Homozygous GBA1 knockout (GBA1 −/−) isogenic ESC lines exhibited no detectable GCase activity, with the lysosomal enzyme activity assay showing a drastic reduction in GCase activity in the GBA1 −/− lines but relatively normal activity of another lysosomal enzyme (α‐iduronate‐2‐sulfatase; see Fig 1). 24 , 25 To further validate features associated with the loss of GCase activity, we performed a lipidomic analysis of GBA1 −/− hMLOs to monitor global lipid changes upon the loss of GCase activity. 23 Principal component analysis plots showed clear separation of wild‐type and GBA1 −/− hMLOs, and HPLC–liquid chromatography–mass spectrometry measurements revealed significant accumulation of GSLs and consequent reductions in lipid species related to the galactosyl/glucosylceramide metabolic pathways.
To compare the effect of GCase deficiency, which is known to induce α‐syn accumulation itself, 23 with that of increased wild‐type α‐syn production, which has been observed in cases of familial PD harboring SNCA gene triplication, 11 we established isogenic hESC lines that inducibly express wild‐type α‐syn (SNCA overexpressing [O/E] lines; see Fig 1K). In the presence of doxycycline, we observed a robust expression of HA‐tagged α‐syn (19kDa; see Fig 1L, M).
Progressive Loss of mDA Neurons in PD‐Associated hMLOs
We next sought to characterize the growth rate and the cellular lineage progression of hMLOs derived from these isogenic hESC lines. At day 30, early stage hMLOs derived from wild‐type, GBA1 −/−, and SNCA O/E hESCs exhibited similar growth rates, based on their shapes and diameters (Fig 2). By day 60, however, hMLOs derived from GBA1 −/−, and SNCA O/E ESCs showed reductions in diameter of approximately 100μm. To test whether this size reduction was due to cell loss at a specific stage, we quantified the proportion of cells expressing both FOXA2 and OTX2 (markers that together mark midbrain neuronal progenitors) that are actively proliferating (Ki67+). Interestingly, we did not observe a decrease in the FOXA2+/OTX2+/Ki67+ population in GBA1 −/− or SNCA O/E hMLOs as compared to wild‐type controls at day 30. After 25 days of differentiation, FOXA2+ neuronal progenitors began to express TH, a defining marker of dopaminergic (DA) neurons, with the majority of FOXA2+ cells expressing TH by day 45. Interestingly, at day 60, GBA1 −/− and SNCA O/E hMLOs showed an approximately 8% and 11% reduction in FOXA2+/TH+ double‐positive cells, respectively. We also performed immunostaining for other neuronal subtype markers, such as γ‐aminobutyric acid (GABA; a marker of GABAergic neurons), choline acetyl transferase (ChAT; a marker of cholinergic neurons), and 5‐hydroxytryptamine (5‐HT; a marker of serotonergic neurons). As we previously reported, 12 we did not observe any ChAT‐ or 5‐HT–expressing cells in the hMLOs (data not shown). However, we found that approximately 20% of cells within the hMLOs were positive for GABA and that the percentages of GABA+ cells between groups were not different. Taken together, the results showed that GBA1 −/− and SNCA O/E hMLOs exhibited specific degeneration of mDA neurons by day 60, whereas other cell types, including mDA progenitors and GABAergic neurons, were unaffected.
FIGURE 2.
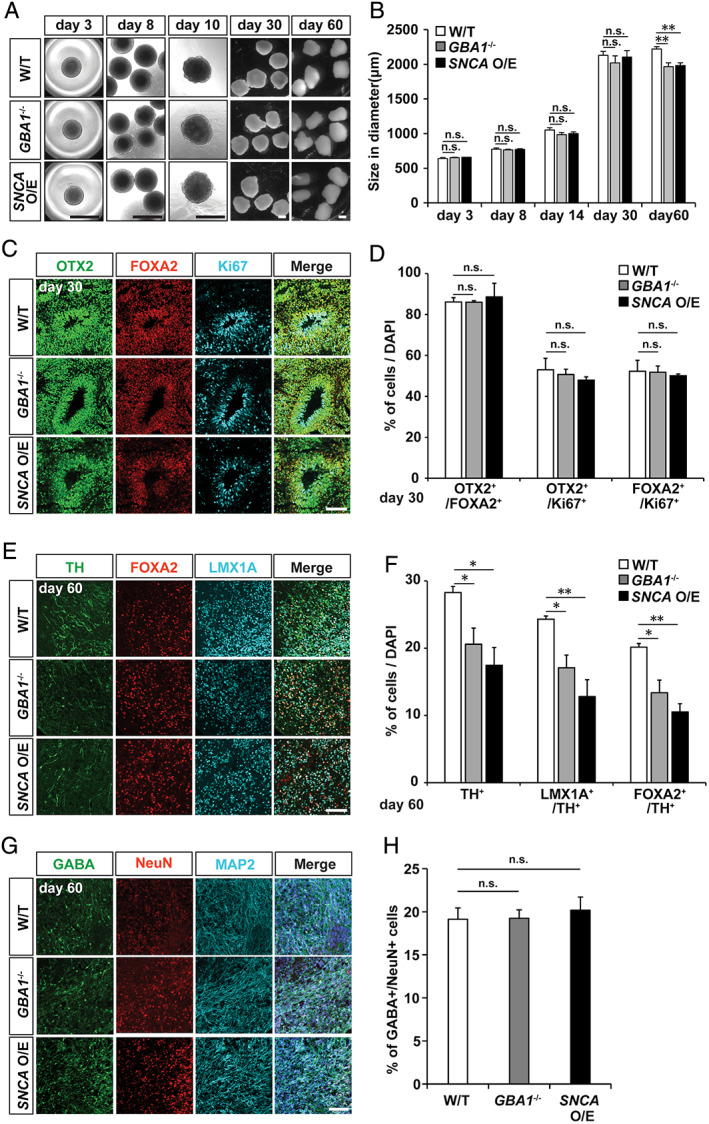
Characterization of midbrain identity in hMLOs. (A) Differential interference contrast images illustrating the typical morphologies of wild‐type (W/T), GBA1 −/−, and SNCA overexpressing (O/E) human midbrain‐like organoids (hMLOs) at the indicated stages. Scale bars = 1mm. (B) Sizes of hMLOs at the indicated stages (mean ± standard error of the mean [SEM]; n.s., no significance, **p < 0.01; n = 5). (C) Cryosections of day 30 hMLOs stained with specific antibodies against OTX2, FOXA2, and Ki67. Scale bar = 100μm. (D) Quantifications for C (mean ± SEM; n.s., no significance; n = 3). (E) Cryosections of day 60 hMLOs stained with specific antibodies against tyrosine hydroxylase (TH), FOXA2, and LMX1A. Scale bar = 100μm. (F) Quantifications for E (mean ± SEM; *p < 0.05, **p < 0.01; n = 3). (G) Cryosections of day 60 hMLOs stained with antibodies against γ‐aminobutyric acid (GABA), NeuN, and MAP2. Scale bar = 50μm. (H) Quantification of the GABA+/NeuN+ cells (mean ± SEM; n = 3). DAPI = 4,6‐diamidino‐2‐phenylindole. [Color figure can be viewed at www.annalsofneurology.org]
Accumulation of α‐Syn Aggregates in PD‐Associated hMLOs
Several recent studies have demonstrated that mice with genetic ablation of Gba1 or PD‐associated point mutations and iPSC‐derived neurons from patients harboring GBA1 mutations exhibit increased levels of neuronal α‐syn aggregation. 24 , 26 Consistent with these findings, we observed increased populations of TH+ mDA neurons with α‐syn aggregates in both GBA1 −/− and SNCA O/E hMLOs as compared to wild‐type controls (52% of mDA neurons in day 60 GBA1 −/− hMLOs and 68% of mDA neurons in day 60 SNCA O/E hMLOs, respectively; Fig 3). Next, we performed sequential detergent‐based protein fractionation and subsequent Western blot analyses using lysates obtained from these hMLOs to assess the levels of detergent‐resistant α‐syn aggregates. 24 We found augmented levels of insoluble α‐syn in lysates from both GBA1 −/− and SNCA O/E hMLOs as compared to wild‐type hMLOs (2.2‐fold and 2.9‐fold increases, respectively). In addition, the expression level of synphilin‐1, an α‐syn–interacting protein that induces the formation of intracellular aggregates, 27 was increased only in insoluble fractions. These data indicated that compared to wild‐type hMLOs, both GBA1 −/− and SNCA O/E hMLOs exhibit enhanced accumulation of insoluble α‐syn at the same developmental stage.
FIGURE 3.
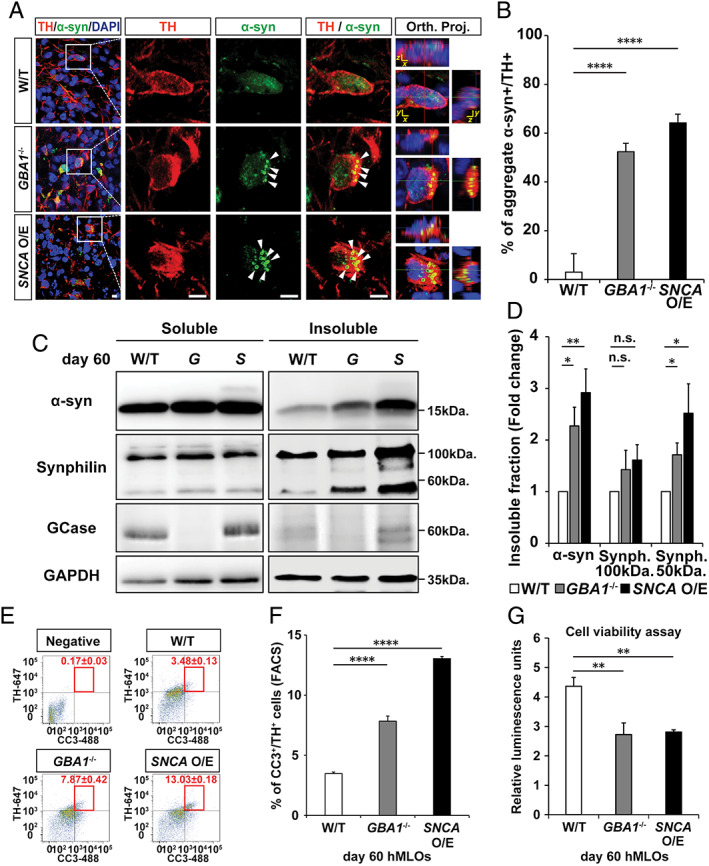
GBA1 −/− and SNCA overexpressing (O/E) human midbrain‐like organoids (hMLOs) show α‐synuclein (α‐syn) aggregation and apoptosis‐driven neurodegeneration. (A) Cryosections of wild‐type (W/T), GBA1 −/−, and SNCA O/E hMLOs at day 60 stained with specific antibodies against tyrosine hydroxylase (TH) and α‐syn. The merged images show orthogonal Z‐stack projections (Orth. Proj.). Scale bars = 10μm. (B) Percentage of TH+ cells that have aggregated α‐syn (mean ± standard error of the mean [SEM]; ****p < 0.0001; n = 3). (C) Western blots of detergent‐soluble and detergent‐insoluble α‐syn, synphilin‐1, and glucocerebrosidase (GCase) proteins in wild‐type, GBA1 −/− (G), and SNCA O/E (S) hMLOs. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a loading control. (D) Quantification of changes in the compositions of insoluble protein fractions (mean ± SEM; n.s. = no significance, *p < 0.05, **p < 0.005; n = 4). (E) Fluorescence‐activated cell sorting (FACS) analysis of cells dissociated from day 60 hMLOs to quantify the percentages of CC3+/TH+ cells (mean ± SEM; n = 3). A threshold gate was drawn based on a secondary antibody‐only negative control. (F) Quantification of CC3+/TH+ cells by FACS revealed a significant increase in apoptotic midbrain dopaminergic neurons in PD‐related hMLOs compared to non–Parkinson disease–related hMLOs (mean ± SEM; ****p < 0.0001; n = 3). (G) Cell viability assay for hMLOs (mean ± SEM; **p = 0.005, 0.004; n = 5). DAPI = 4,6‐diamidino‐2‐phenylindole. [Color figure can be viewed at www.annalsofneurology.org]
Given that mDA neuron numbers were reduced significantly in GBA1 −/− and SNCA O/E hMLOs at day 60, we performed fluorescence‐activated cell sorting analysis and observed significantly higher percentages of TH+/CC3+ cells in the mutant hMLOs (3.48, 7.87, and 13.03% on day 60 wild‐type, GBA1 −/−, and SNCA O/E hMLOs, respectively; see Fig 3E, F). In support of this finding, a cellular viability assay to approximate adenosine triphosphate content 28 showed approximately 50% reduction in the viabilities of GBA1 −/− and SNCA O/E hMLOs (see Fig 3G). Taken together, these data indicated that mDA neurons in both GBA1 −/− and SNCA O/E hMLOs were more prone to apoptosis (which could also explain the size differences between them and wild‐type controls), possibly due to accumulations of insoluble α‐syn aggregates.
Generation of LB‐Like Inclusions in GBA1/SNCA Dual Perturbation hMLOs and Conduritol‐b‐Epoxide–Treated SNCA Triplication hMLOs
Previously, Taguchi et al generated Gba1 mutant (with N370S and L444P mutations) and knockout mice and subsequently crossed these mice with previously characterized human α‐syn A30P transgenic models of PD. 26 These mice exhibited early accumulation of GSLs in the brain, increased fibrillar α‐syn protein levels, and clinical PD symptoms including resting tremor and progressive motor decline but lacked LB pathology. 26 Although mutations in both the GBA1 and SNCA loci have not been reported in PD patients, reductions in GCase activity, as well as increases in pathogenic a‐syn and the appearance of LBs, have been well‐documented in both familial and idiopathic forms of the disease. In particular, heterozygous mutations in GBA1 are significant risk factors for PD. 10 It is therefore possible that the combination of both genetic perturbations would be valuable as an accelerated model for recapitulating hallmarks of PD in vitro and studying disease pathogenesis. To this end, we combined both genetic risk factors (GBA1 −/− in an SNCA O/E background) to test a multi‐hit pathogenic scenario for PD. The resulting GBA1 −/−::SNCA O/E ESC lines lacked the GBA1 protein and GCase enzyme activity and exhibited doxycycline‐dependent α‐syn expression (Fig 4). Intriguingly, we observed α‐syn–containing LB‐like inclusions (LBLIs) in TH+ mDA neurons in the GBA1 −/−::SNCA O/E hMLOs at day 90. Although these LBLIs shared similar spherically symmetric morphology, they were smaller (ranging in size from 1 to 3μm) than LBs reported in PD patients. 9 , 29 These LBLIs were rarely observed in hMLOs derived from either GBA1 −/− or SNCA O/E ESCs alone. In addition, these LBLIs displayed key features of LBs, such as an eosinophilic core as revealed by hematoxylin and eosin staining, a common pathological staining method to detect LBs in postmortem brains. 29 LBs isolated from PD patients have been reported to contain a variety of proteins, including α‐syn and ubiquitin. 30 Immunohistochemical analyses of LBLIs revealed the presence of a peripheral halo that was immunopositive for α‐syn, with a ubiquitin‐immunopositive core. TEM showed concentric lamination in LBLIs; they were ultrastructurally comprised of a granular core and filamentous outer layers. 29 The exclusive presence of LBLIs in GBA1 −/−::SNCA O/E hMLOs was striking and suggested that GCase deficiencies in the context of increased α‐syn could accelerate the formation of LBLIs in TH+ neurons, thus presenting a viable option for studying the mechanisms and role of LB formation in PD.
FIGURE 4.
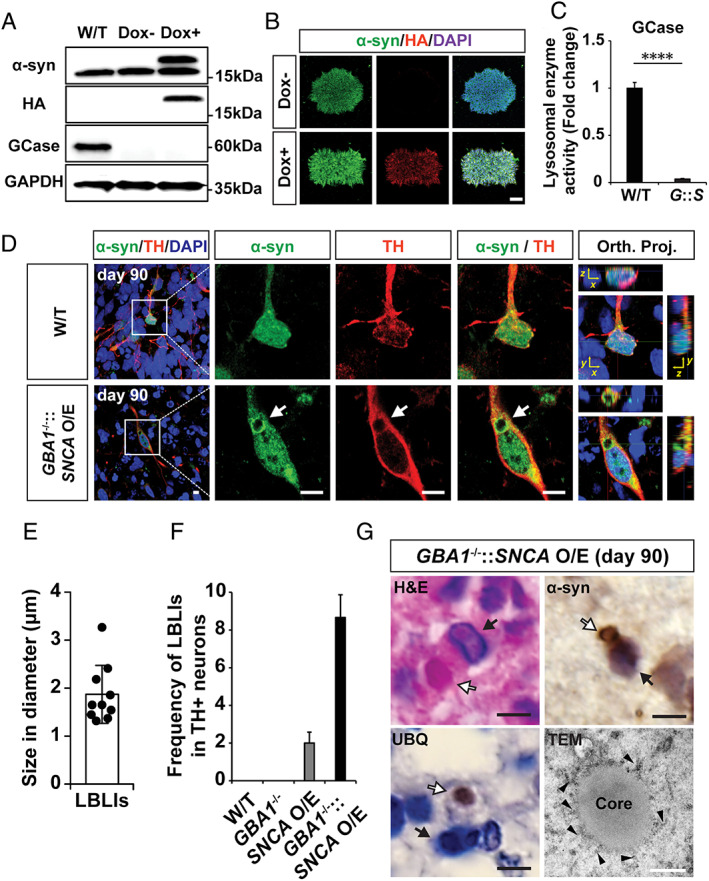
Presence of Lewy body–like inclusions (LBLIs) in human midbrain‐like organoids (hMLOs) derived from GBA −/−::SNCA overexpressing (O/E) embryonic stem cells (ESCs) in the H9 background. (A) Western blot validation of the GBA1 −/−::SNCA O/E ESC lines upon doxycycline (Dox) treatment using specific antibodies against α‐synuclein (α‐syn; 17 and 19kDa, indicating endogenous and exogenous α‐syn, respectively), glucocerebrosidase (GCase; 60kDa), and hemagglutinin (HA; 19kDa). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Immunostaining of GBA1 −/−::SNCA O/E ESC clones upon Dox treatment with specific antibodies against α‐syn and HA. Scale bar = 100μm. (C) Lysosomal enzyme activity in GBA1 −/−::SNCA O/E (mean ± standard error of the mean; ****p < 0.0001; n = 3). (D) Immunostaining of LBLIs exhibiting halolike α‐syn+ immunoreactivity in tyrosine hydroxylase (TH)+ neurons of day 90 hMLOs derived from GBA1 −/−::SNCA O/E ESCs. Scale bars = 10μm. (E) Quantification of the diameters of the identified LBLIs (n = 10). (F) Quantification of LBLIs in cryosections of day 90 hMLOs based on α‐syn immunoreactivity. (G) Hematoxylin and eosin (H&E), immunohistochemistry, and transmission electron microscopy (TEM) images of LBLIs. White and black arrows indicate LBLIs and nuclei, respectively, and the arrowheads indicate α‐syn fibrillar structures. Black scale bars = 5μm. White scale bar = 500nm. DAPI = 4,6‐diamidino‐2‐phenylindole; Orth. Proj. = orthogonal Z‐stack projections (Orth. Proj.); UBQ = ubiquitin; W/T = wild type. [Color figure can be viewed at www.annalsofneurology.org]
To test the robustness of the LBLI phenotype, we established another hESC line with GBA1::SNCA O/E perturbations using the H1 hESC line and confirmed that the cells were deficient for GCase and overexpressed HA‐tagged α‐syn (Fig 5). hMLOs generated from this cell line developed LBLIs by day 90, with LBLI sizes and occurrence frequencies comparable to that of LBLIs from H9 GBA1::SNCA O/E hMLOs. In addition, we tested an alternative pharmacological inhibition of GCase using conduritol‐b‐epoxide (CBE; 50μM) in hMLOs generated from both healthy and SNCA triplication patient‐derived iPSCs. 31 Following 30 days of chronic CBE treatment, we observed a complete loss of GCase activity (data not shown). Importantly, we observed similarly shaped LBLIs in day 90 hMLOs from treated SNCA triplication iPSCs, but not from similarly treated healthy iPSCs. Similar observations were made for hMLOs from patient‐derived iPSCs harboring the GBA1 N370S mutation 10 ; LBLIs were only observed upon induced overexpression of wild‐type α‐syn. Interestingly, compared to hMLOs without LBLIs, all hMLOs with LBLIs showed reduced TH+ mDA neurons with increased expression of apoptotic markers such as CC3 (Fig 6B, C), whereas the midbrain progenitor cells did not seem to be affected (see Fig 6A). Taken together, these results strongly suggest that both impaired GCase function and α‐syn accumulation were crucial to the generation of LBLIs, and underscore the value of the multi‐hit model in interrogating the role of LBLIs and potentially LBs in PD pathogenesis.
FIGURE 5.
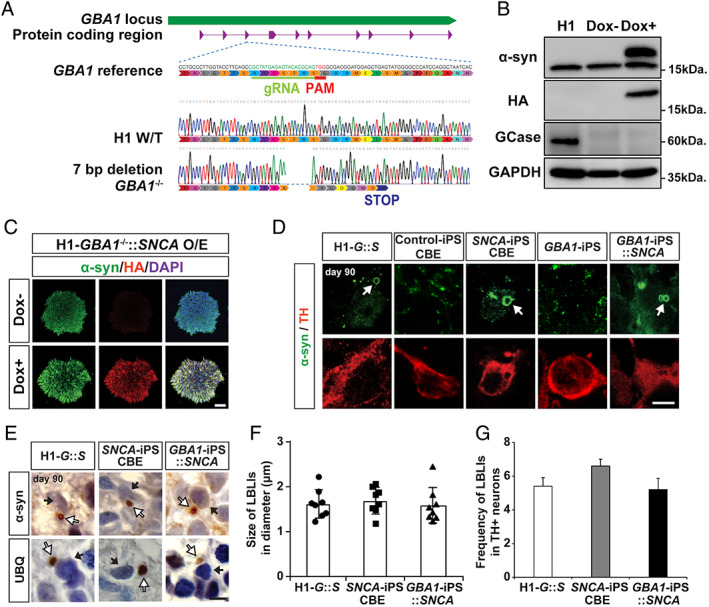
Presence of Lewy body–like inclusions (LBLIs) in human midbrain‐like organoids (hMLOs) from GBA1 −/−::SNCA overexpressing (O/E) embryonic stem cells (ESCs) in the H1 background as well as Parkinson disease patient‐derived induced pluripotent stem cells (iPSCs). (A) Schematic showing CRISPR/Cas9 genome editing to generate H1‐GBA1 −/−::SNCA O/E clones. The gRNA targeting sequence is in green; the protospacer adjacent motif (PAM) sequence is in red. Genomic DNA sequencing of the GBA1 −/− ESC line showed a homozygous 7‐base pair (bp) deletion. (B) Western blot validation of α‐synuclein (α‐syn) protein levels in the H1‐GBA1 −/−::SNCA O/E ESC line upon doxycycline (Dox) treatment using specific antibodies against α‐syn (17 and 19kDa, indicating endogenous and exogenous α‐syn, respectively), glucocerebrosidase (GCase; 60kDa), and hemagglutinin (HA; 19kDa). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a loading control. (C) Immunostaining of H1‐GBA1 −/−::SNCA O/E ESC clones upon Dox treatment with specific antibodies against α‐syn and HA. Scale bar = 100μm. (D) Immunostaining of LBLIs exhibiting halolike α‐syn+ immunoreactivity in tyrosine hydroxylase (TH)+ neurons of day 90 hMLOs derived from H1‐GBA1 −/−::SNCA O/E ESCs (G::S), SNCA triplication iPSCs or control iPSCs with conduritol‐b‐epoxide (CBE) treatment, and GBA1‐iPSCs with and without SNCA overexpression. White arrows indicate LBLIs. Scale bar = 5μm. (E) Immunohistochemical analyses of day 90 hMLOs derived from H1‐GBA1 −/−::SNCA O/E ESCs, SNCA triplication iPSCs with CBE treatment, and GBA1‐iPSCs with SNCA overexpression using specific antibodies against α‐syn and ubiquitin (UBQ). The white and black arrows indicate LBLIs and nuclei, respectively. Scale bar = 5μm. (F) Quantification of the diameters of the identified LBLIs (n = 8). (G) Frequency of LBLI occurrence in cryosections of day 90 hMLOs based on α‐syn immunoreactivity. DAPI = 4,6‐diamidino‐2‐phenylindole. [Color figure can be viewed at www.annalsofneurology.org]
FIGURE 6.
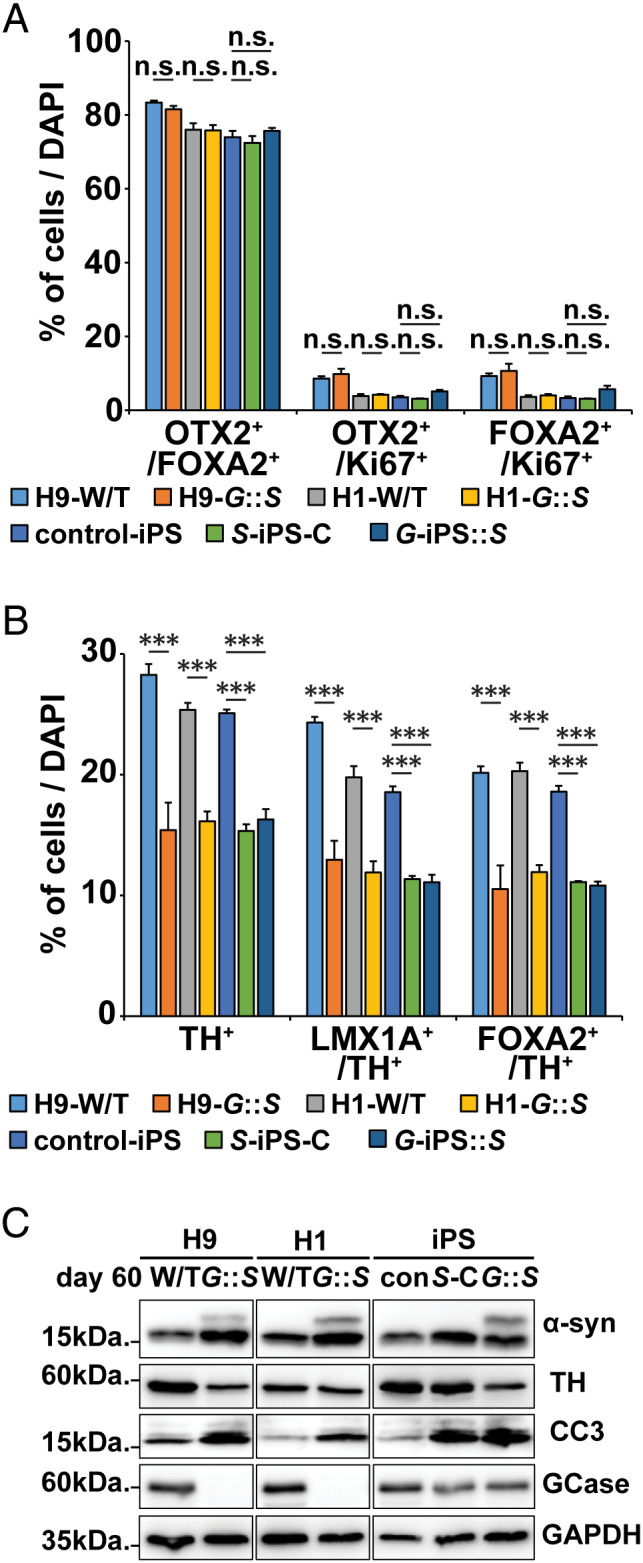
Characterization of H9‐GBA1 −/−::SNCA overexpressing (O/E) and H1‐GBA1 −/−::SNCA O/E human midbrain‐like organoids (hMLOs), SNCA triplication‐induced pluripotent stem cell (iPSC) hMLOs with conduritol‐b‐epoxide (CBE) treatment (S‐iPS‐C), and GBA1‐iPSC hMLOs with SNCA overexpression (G‐iPS::S). (A) Quantification of cells expressing combinations of the midbrain progenitor markers OTX2, FOXA2, and Ki67 in day 60 H9‐ and H1‐GBA1 −/−::SNCA O/E hMLOs (G::S), SNCA triplication iPSC‐derived hMLOs with or without CBE treatment, and GBA1 mutant iPSC‐derived hMLOs (mean ± standard error of the mean [SEM]; n.s. = no significance; n = 3). (B) Quantification of cells expressing combinations of the midbrain progenitor markers FOXA2 and LMX1A with the dopaminergic neuron marker tyrosine hydroxylase (TH) in day 60 H9‐ and H1‐GBA1 −/−::SNCA O/E hMLOs, SNCA triplication iPSC‐derived hMLOs with or without CBE treatment, and GBA1 mutant iPSC‐derived hMLOs (mean ± SEM; ***p < 0.001; n = 3). (C) Western blots for α‐synuclein (α‐syn), TH, CC3, and glucocerebrosidase (GCase) in hMLOs derived from H9‐ and H1‐GBA1 −/−::SNCA O/E hMLOs, SNCA triplication iPSC‐derived hMLOs with or without CBE treatment, and GBA1 mutant iPSC‐derived hMLOs. DAPI = 4,6‐diamidino‐2‐phenylindole; GAPDH = glyceraldehyde‐3‐phosphate dehydrogenase; W/T = wild type. [Color figure can be viewed at www.annalsofneurology.org]
Formation of Distinct Oligomeric and Fibrillar α‐Syn Aggregates in LBLI‐Forming hMLOs
To understand the molecular mechanisms underlying the formation of LBLIs, we analyzed hMLOs at day 60, an earlier time point prior to LBLI formation, and quantified the numbers of α‐syn aggregates, levels of phospho‐Ser129 α‐syn (a marker for pathologic α‐syn), 32 and levels of phosphorylated tau in GBA1 −/−::SNCA O/E organoids (Fig 7). Interestingly, we did not observe any significant differences between SNCA O/E and GBA1 −/−::SNCA O/E hMLOs. Given that prefibrillar oligomeric α‐syn has been proposed to seed and initiate the assembly of fibrillar α‐syn, we assessed the seeding abilities of α‐syn multimeric oligomers from the various hMLO models using an intact cell cross‐linking method. 13 We identified multimeric oligomers (60, 80, and 100kDa) as well as monomeric α‐syn proteins, and observed high levels of high‐molecular weight α‐syn oligomers in GBA1 −/−::SNCA O/E hMLOs as early as day 60.
FIGURE 7.
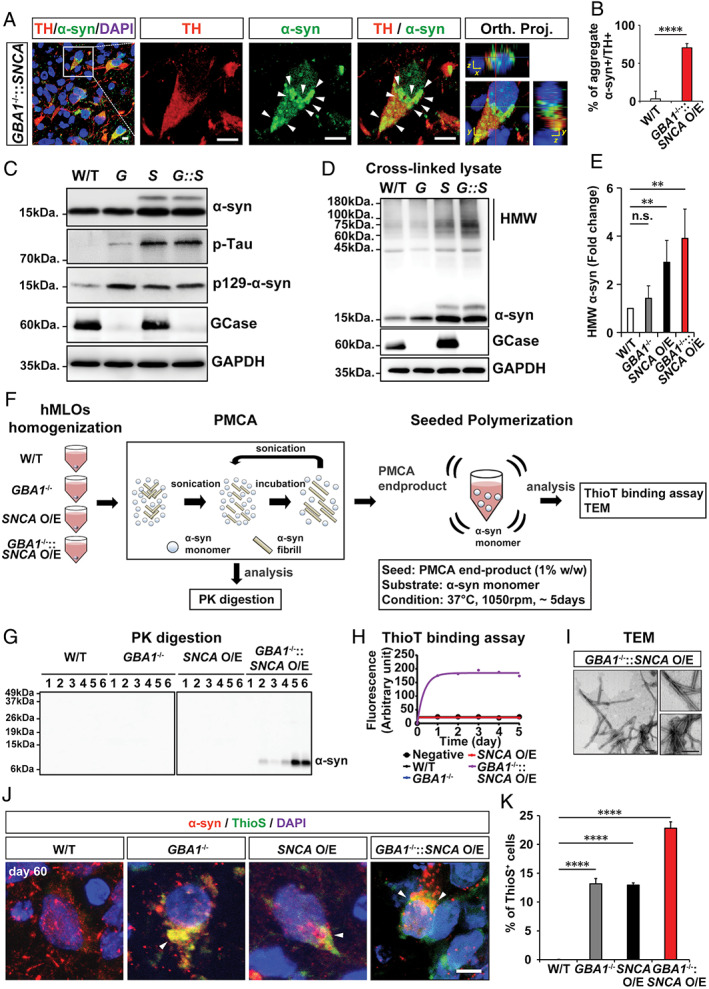
α‐Synuclein (α‐syn) aggregates from GBA1 −/−::SNCA overexpressing (O/E) human midbrain‐like organoids (hMLOs) are pathogenic. (A) Cryosections of GBA1 −/−::SNCA O/E hMLOs at day 60 stained with specific antibodies against tyrosine hydroxylase (TH) and α‐syn. The merged images show orthogonal Z‐stack projections (Orth. Proj.). Scale bars = 10μm. (B) Quantification of the percentages of TH+ cells with α‐syn aggregates. The percentage of TH+ cells containing aggregate α‐syn is shown (mean ± standard error of the mean [SEM]; ****p < 0.0001; n = 3). (C) Western blots of α‐syn, p‐Tau, p129‐α‐syn, and glucocerebrosidase (GCase) proteins in day 60 hMLOs derived from wild‐type (W/T), GBA1 −/− (G), SNCA O/E (S), and GBA1 −/−::SNCA O/E ESCs. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a loading control. (D) Western blotting was performed to detect high‐molecular‐weight (HMW) α‐syn. H9 isogenic hMLOs were subjected to 1mM disuccinimidyl glutarate cross‐linking before whole lysates were extracted. (E) Quantification of HMW α‐syn from the experiment in D (mean ± SEM; n.s. = no significance, **p < 0.005; n = 4). (F) Schematic representation of protein misfolding cyclic amplification (PMCA) and seeded polymerization for the thioflavin T (ThioT) binding assay and transmission electron microscopy (TEM) imaging. (G) Proteinase K (PK) digestion patterns of PMCA products from day 60 hMLOs. (H) ThioT binding kinetics of seeded polymerization using PMCA end products for each type of hMLO. (I) TEM images of seeded aggregates. Scale bars = 200nm. (J) Immunostaining for α‐syn and thioflavin S (ThioS) double‐positive neurons from control, GBA1 −/−, SNCA O/E, and GBA1 −/−::SNCA O/E hMLOs at day 60. The arrowheads point to the colocalization of α‐syn and ThioS. Scale bar = 5μm. (K) Quantification of the immunostaining from J (mean ± SEM; ****p < 0.0001; n = 3). DAPI = 4,6‐diamidino‐2‐phenylindole. [Color figure can be viewed at www.annalsofneurology.org]
Next, we adapted PMCA assays, originally developed for amplification of prion proteins, for α‐syn (see Fig 7). 33 , 34 Briefly, we prepared homogenates from each hMLO as exogenous seeds and amplified prion fibrils by adding recombinant α‐syn monomers as substrates. Multiple cycles of sonication and subsequent incubation resulted in polymerization and fibrillation of α‐syn; the samples were subjected to PK digestion to degrade monomeric α‐syn and were subsequently subjected to Western blotting and TEM. We observed that GBA1 −/−::SNCA O/E hMLOs exhibited accelerated fibril formation, with more β‐sheet oligomers (based on exponential increases in ThioT fluorescence, which emits fluorescence when it binds to β‐sheet oligomers). In addition, the morphology of α‐syn fibrils generated from GBA1 −/−::SNCA O/E hMLOs was filamentous. We further tested whether the multimeric oligomer‐enriched GBA1 −/−::SNCA O/E hMLOs produced more fibrillar α‐syn by performing ThioS staining to label amyloidogenic β‐sheet fibrils. 35 Approximately 13%, 12%, and 23% of cells from GBA1 −/−, SNCA O/E, and GBA1 −/−::SNCA O/E hMLOs, respectively, were double‐positive for α‐syn and ThioS. These data suggest that GCase deficiency with SNCA overexpression accelerated the formation of detergent‐resistant, fibrillar α‐syn, more so than single perturbation mutants, and might account for the formation of LBLIs.
Discussion
We have previously developed a method to derive hMLOs from hPSCs; the resulting hMLOs exhibit distinct layers of neuronal cells with functionally mature mDA neurons and produce neuromelanin‐like granules that are in vitro replicas of those isolated from human substantia nigra tissue. 12 However, given the transcriptional resemblance of the hMLOs to human fetal midbrains, it was not clear whether the hMLO model could be utilized to study late onset human midbrain disorders such as PD.
Here, we report that the genetic introduction of two PD risk factors (loss of GCase and α‐syn overexpression) in hMLOs resulted in the recapitulation of many hallmark features of PD, most significantly the formation of LBLIs and the loss of mDA neurons, two key pathophysiological hallmarks of PD. 36 LBLIs were structurally and molecularly similar to PD‐associated LBs. For decades, multiple animal models of PD (pathogenic models established by neurotoxin treatment and in vivo injection of preformed α‐syn fibrils as well as gene‐based, knockin/knockout transgenic mouse models harboring familial PD mutations) have been used as standard PD models for basic mechanistic studies and drug discovery. However, these models do not fully recapitulate human PD, 37 resulting in limited implications for disease pathogenesis. In contrast to these models, our 3‐dimensional hMLO system recapitulated pathogenic cascades of PD in a more physiological environment, which has not been demonstrated in PD animal models or other 2‐dimensional cultures of human mDA neurons.
One of the most interesting aspects of our hMLO PD model is the generation of LBLIs without expression of mutant α‐syn. To date, 6 different autosomal‐dominant missense mutations in SNCA (A53T, A30P, E64K, H50Q, G51D, and A53E) have been identified; these mutations result in α‐syn adopting an oligomeric and/or fibrillar conformation 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 or interacting directly with membrane phospholipids—particularly highly curved, endogenous vesicles and organelles such as mitochondria, the endoplasmic reticulum, and the Golgi. 47 Intriguingly, although complete GBA1 knockout alone could augment the levels of both soluble and insoluble α‐syn in our system and induce levels of cellular α‐syn aggregates comparable to those in SNCA O/E hMLOs (see Fig 3C, D), they failed to form LBLIs (see Fig 4F). This may have been due to qualitative differences in the α‐syn oligomers derived from GBA1 knockout hMLOs as compared to SNCA O/E organoids (see Fig 7G, H). In support of this notion, transcriptome profiling of hMLOs using RNA‐seq also indicated that SNCA overexpression remodeled the transcriptomic landscape much more dramatically than GBA1 loss (differentially expressed genes [DEGs]: SNCA O/E hMLOs: 4,821 upregulated, 2,815 downregulated; GBA1 −/− hMLOs: 286 upregulated, 305 downregulated; false discovery rate ≤ 0.05 and |fold change| ≥ 1.5; Fig 8, Table S1). More importantly, GO analysis indicated that the DEGs in SNCA O/E hMLOs were enriched for biological processes such as protein catabolism, endoplasmic reticulum stress, ion transmembrane transport, and lipid transport (Table S2). In contrast, the DEGs in GBA1 −/− hMLOs were implicated in synaptic transmission, ion channel function and cell division (see Table S2). These findings support the notion that wild‐type SNCA overexpression resulted in substantial transcriptome changes that corroborate well with the results of previous transcriptome analyses of PD patient brains 48 ; in contrast, transcriptome analysis of GBA1 −/− hMLOs showed less pronounced changes in the PD‐related transcriptome, potentially due to the biological function of GBA1, a lysosomal enzyme that cleaves glucosylceramide into glucose and ceramide during GSL metabolism and lies downstream of gene transcription and modification. 49
FIGURE 8.
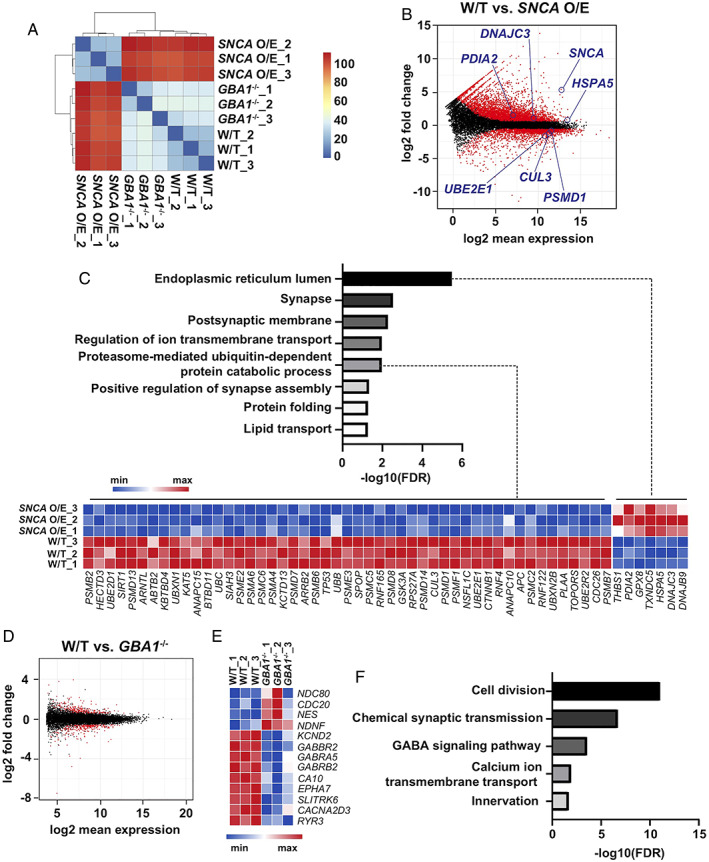
Transcriptomic characterization of GBA1 −/− and SNCA overexpressing (O/E) human midbrain‐like organoids (hMLOs). (A) Clustering analysis of the RNA‐sequencing (RNA‐seq) data. The Euclidean distance of the normalized gene expression among wild‐type (W/T), GBA1 −/−, and SNCA O/E hMLOs was used for sample clustering. (B) MA plot showing the distinct genes differentially expressed in SNCA O/E hMLOs in the RNA‐seq data set. Statistically significant differentially expressed genes (DEGs; |fold change| ≥ 1.5, p‐adj ≤ 0.05) are highlighted in red (related to Table S1). (C) Gene Ontology (GO) enrichment analysis of DEGs dysregulated in SNCA O/E hMLOs. A heatmap representation of the DEGs (|fold change| ≥ 1.5, p‐adj ≤ 0.05) known to be associated with protein catabolism and endoplasmic reticulum stress is shown in the bottom panel (related to Table S1). (D) MA plot showing the DEGs in GBA1 −/− hMLOs. Statistically significant DEGs (|fold change| ≥ 1.5, p‐adj ≤ 0.05) are highlighted in red (related to Table S1). (E) Heatmap representation of selected DEGs between W/T and GBA1 −/− hMLOs (related to Table S1). (F) GO enrichment analysis of selected DEGs in GBA1 −/− hMLOs (select GO terms are ranked by the adjusted p values; related to Table S2). FDR = false discovery rate; GABA =γ‐aminobutyric acid. [Color figure can be viewed at www.annalsofneurology.org]
Another interesting aspect of our results is that the genetic introduction of 2 PD risk factors (loss of GCase activity and wild‐type α‐syn overexpression) resulted in the formation of LBLIs, possibly via specific oligomeric and fibrillar α‐syn formation. Although it is unclear why these 2 factors synergistically influence LBLI formation in hMLOs, further proteomic/lipidomic investigation of isolated α‐syn oligomers and fibrils from LBLI‐forming hMLOs will help us to elucidate the cellular and molecular underpinnings of LB biogenesis in PD and eventually address the physiological function(s) of LB formation in neurons, which in turn will guide therapeutic strategies. 2 In particular, it would be exciting and illuminating to test whether rescue of GCase activity via several avenues, for example, by activation of the autophagic–lysosomal pathway through treatment with chemical chaperones such as ambroxol (currently under phase 2 clinical trials), GCase overexpression, or reduction of GCase substrates such as glucosylceramides with compounds such as Genz‐682452 (currently in phase 2 clinical trials), 50 can reverse or prevent the formation of LBLIs. Some success for such strategies has been reported in human iPSC‐derived mDA neurons, where GBA1 overexpression decreases overall cellular α‐syn levels. 51 Similar strategies in the dual perturbation genetic model presented here could be key in identifying therapies for the rescue of LB formation phenotypes in PD.
In summary, we have successfully recapitulated α‐syn and LB pathologies in a human brain organoid culture system, generated from isogenic hESCs, SNCA triplication, and GBA1 loss‐of‐function iPSCs. Our model can be used to further elucidate the underlying mechanisms of progressive LB formation as well as the selective vulnerability of mDA neurons and to screen potential therapeutic agents for PD.
Author Contributions
J.J., L.Y., K.‐L.L., E.‐K.T., H.‐H.N., and H.S.J. contributed to the conception and design of the study; J.J., L.Y., H.‐D.T., W.Y., A.X.S., Y.Y.C., B.C.J., S.‐J.L., T.Y.S., B.X., A.T.T.K., L.‐P.Y., J.J.X., H.L., W.‐Y.O., and G.G.Y.L. contributed to the acquisition and analyses of data; J.J., L.Y., and H.S.J. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
TABLE S1. Differential expression analysis between wild‐type and SNCA overexpressing or GBA1 −/− human midbrainlike organoids. The table contains gene annotation, fold change, and adjusted p value.
TABLE S2. Gene ontology enrichment analysis (obtained from DAVID v6.8) of dysregulated genes in either SNCA overexpressing or GBA1 −/− human midbrainlike organoids (hMLOs), compared to wild‐type hMLOs.
Acknowledgments
This work was supported by the Singapore Ministry of Education Academic Research Fund (MOE2014‐T2‐2‐071), National Medical Research Council Open‐Fund Individual Research Grant (NMRC/OFIRG/0050/2017), National Research Foundation Competitive Research Program (NRF‐CRP17‐2017‐04), Duke‐NUS Signature Research Program Block Grant (H.S.J.), National Medical Research Council Translational and Clinical Flagship Grant (NMRC/TCR/013‐NNI/2014; E.‐K.T., K.‐L.L, and H.‐H.N.), and Agency for Science, Technology, and Research (H.‐H.N).
We thank Dr H. S. Ko and Dr X. Zeng for kindly sharing GBA1‐N370S iPSC lines; Dr S. Engelender for generously sharing synphilin‐1 antibody; and Dr E. Leman for constructive comments and suggestions.
Contributor Information
Kah‐Leong Lim, Email: kahleong.lim@ntu.edu.sg.
Eng‐King Tan, Email: gnrtek@sgh.com.sg.
Huck‐Hui Ng, Email: nghh@gis.a-star.edu.sg.
Hyunsoo Shawn Je, Email: shawn.je@duke-nus.edu.sg.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Tables. The RNA‐seq data have been deposited in the ArrayExpress European Bioinformatics Institute database under accession number E‐MTAB‐7302. Additional data related to this paper may be requested from the authors.
References
- 1. Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine‐containing neurons in Parkinson's disease. Brain 1999;122:1437–1448. [DOI] [PubMed] [Google Scholar]
- 2. Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 3. Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol 1996;55:259–272. [DOI] [PubMed] [Google Scholar]
- 4. Kalia LV, Lang AE. Parkinson's disease. Lancet 2015;386:896–912. [DOI] [PubMed] [Google Scholar]
- 5. Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson's disease: molecules implicated in the formation and degradation of alpha‐synuclein aggregates. Neuropathology 2007;27:494–506. [DOI] [PubMed] [Google Scholar]
- 6. Milber JM, Noorigian JV, Morley JF, et al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology 2012;79:2307–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tompkins MM, Hill WD. Contribution of somal Lewy bodies to neuronal death. Brain Res 1997;775:24–29. [DOI] [PubMed] [Google Scholar]
- 8. Imaizumi Y, Okano H. Modeling human neurological disorders with induced pluripotent stem cells. J Neurochem 2014;129:388–399. [DOI] [PubMed] [Google Scholar]
- 9. Chesselet MF, Fleming S, Mortazavi F, Meurers B. Strengths and limitations of genetic mouse models of Parkinson's disease. Parkinsonism Relat Disord 2008;14:S84–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singleton AB, Farrer M, Johnson J, et al. Alpha‐synuclein locus triplication causes Parkinson's disease. Science 2003;302:841. [DOI] [PubMed] [Google Scholar]
- 12. Jo J, Xiao Y, Sun AX, et al. Midbrain‐like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin‐producing neurons. Cell Stem Cell 2016;19:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim S, Yun SP, Lee S, et al. GBA1 deficiency negatively affects physiological alpha‐synuclein tetramers and related multimers. Proc Natl Acad Sci U S A 2018;115:798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Momcilovic O, Sivapatham R, Oron TR, et al. Derivation, characterization, and neural differentiation of integration‐free induced pluripotent stem cell lines from Parkinson's disease patients carrying SNCA, LRRK2, PARK2, and GBA mutations. PLoS One 2016;11:e0154890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- 17. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wickham H. ggplot2: elegant graphics for data analysis. New York, NY: Springer, 2009. [Google Scholar]
- 19. Neuhoff H, Neu A, Liss B, Roeper J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci 2002;22:1290–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pfisterer U, Kirkeby A, Torper O, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A 2011;108:10343–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med 2011;17:485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nalls MA, Duran R, Lopez G, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 2013;70:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zunke F, Moise AC, Belur NR, et al. Reversible conformational conversion of alpha‐synuclein into toxic assemblies by glucosylceramide. Neuron 2018;97:92 107.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burbulla LF, Song P, Mazzulli JR, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science 2017;357:1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taguchi YV, Liu J, Ruan J, et al. Glucosylsphingosine promotes alpha‐synuclein pathology in mutant GBA‐associated Parkinson's disease. J Neurosci 2017;37:9617–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Engelender S, Kaminsky Z, Guo X, et al. Synphilin‐1 associates with alpha‐synuclein and promotes the formation of cytosolic inclusions. Nat Genet 1999;22:110–114. [DOI] [PubMed] [Google Scholar]
- 28. Janda CY, Dang LT, You C, et al. Surrogate Wnt agonists that phenocopy canonical Wnt and β‐catenin signalling. Nature 2017;545:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olanow CW, Perl DP, DeMartino GN, McNaught KS. Lewy‐body formation is an aggresome‐related process: a hypothesis. Lancet Neurol 2004;3:496–503. [DOI] [PubMed] [Google Scholar]
- 30. Chung KK, Zhang Y, Lim KL, et al. Parkin ubiquitinates the alpha‐synuclein‐interacting protein, synphilin‐1: implications for Lewy‐body formation in Parkinson disease. Nat Med 2001;7:1144–1150. [DOI] [PubMed] [Google Scholar]
- 31. Ogawa S, Uetsuki S, Tezuka Y, et al. Synthesis and evaluation of glucocerebrosidase inhibitory activity of anhydro deoxyinositols from (+)‐epi‐ and (−)‐vibo‐quercitols. Bioorg Med Chem Lett 1999;9:1493–1498. [DOI] [PubMed] [Google Scholar]
- 32. Fujiwara H, Hasegawa M, Dohmae N, et al. α‐Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 2002;4:160–164. [DOI] [PubMed] [Google Scholar]
- 33. Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001;411:810–813. [DOI] [PubMed] [Google Scholar]
- 34. Jung BC, Lim YJ, Bae EJ, et al. Amplification of distinct alpha‐synuclein fibril conformers through protein misfolding cyclic amplification. Exp Mol Med 2017;49:e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. LeVine H 3rd. Quantification of beta‐sheet amyloid fibril structures with thioflavin T. Methods Enzymol 1999;309:274–284. [DOI] [PubMed] [Google Scholar]
- 36. Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science 2003;302:819–822. [DOI] [PubMed] [Google Scholar]
- 37. Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron 2010;66:646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Puschmann A, Ross OA, Vilariño‐Güell C, et al. A Swedish family with de novo α‐synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat Disord 2009;15:627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Appel‐Cresswell S, Vilarino‐Guell C, Encarnacion M, et al. Alpha‐synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord 2013;28:811–813. [DOI] [PubMed] [Google Scholar]
- 40. Lesage S, Anheim M, Letournel F, et al. G51D α‐synuclein mutation causes a novel Parkinsonian–pyramidal syndrome. Ann Neurol 2013;73:459–471. [DOI] [PubMed] [Google Scholar]
- 41. Choi JM, Woo MS, Ma H‐I, et al. Analysis of PARK genes in a Korean cohort of early‐onset Parkinson disease. Neurogenetics 2008;9:263–269. [DOI] [PubMed] [Google Scholar]
- 42. Ki CS, Stavrou EF, Davanos N, et al. The Ala53Thr mutation in the α‐synuclein gene in a Korean family with Parkinson disease. Clin Genet 2007;71:471–473. [DOI] [PubMed] [Google Scholar]
- 43. Zarranz JJ, Alegre J, Gómez‐Esteban JC, et al. The new mutation, E46K, of α‐synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164–173. [DOI] [PubMed] [Google Scholar]
- 44. Athanassiadou A, Voutsinas G, Psiouri L, et al. Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding α‐synuclein. Am J Hum Genet 1999;65:555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krüger R, Kuhn W, Müller T, et al. AlaSOPro mutation in the gene encoding α‐synuclein in Parkinson's disease. Nat Genet 1998;18:106–108. [DOI] [PubMed] [Google Scholar]
- 46. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science 1997;276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 47. Auluck PK, Caraveo G, Lindquist S. Alpha‐synuclein: membrane interactions and toxicity in Parkinson's disease. Annu Rev Cell Dev Biol 2010;26:211–233. [DOI] [PubMed] [Google Scholar]
- 48. Borrageiro G, Haylett W, Seedat S, et al. A review of genome‐wide transcriptomics studies in Parkinson's disease. Eur J Neurosci 2018;47:1–16. [DOI] [PubMed] [Google Scholar]
- 49. Brady RO, Kanfer J, Shapiro D. The metabolism of glucocerebrosides. I. Purification and properties of a glucocerebroside‐cleaving enzyme from spleen tissue. J Biol Chem 1965;240:39–43. [PubMed] [Google Scholar]
- 50. Marshall J, Sun Y, Bangari DS, et al. CNS‐accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic Gaucher disease. Mol Ther 2016;24:1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Woodard CM, Campos BA, Kuo SH. iPSC‐derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson's disease. Cell Rep et al., 2014;9:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Differential expression analysis between wild‐type and SNCA overexpressing or GBA1 −/− human midbrainlike organoids. The table contains gene annotation, fold change, and adjusted p value.
TABLE S2. Gene ontology enrichment analysis (obtained from DAVID v6.8) of dysregulated genes in either SNCA overexpressing or GBA1 −/− human midbrainlike organoids (hMLOs), compared to wild‐type hMLOs.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Tables. The RNA‐seq data have been deposited in the ArrayExpress European Bioinformatics Institute database under accession number E‐MTAB‐7302. Additional data related to this paper may be requested from the authors.