

Abstract
While inherited hemizygous variants in PHF6 cause X‐linked recessive Borjeson‐Forssman‐Lehmann syndrome (BFLS) in males, de novo heterozygous variants in females are associated with an overlapping but distinct phenotype, including moderate to severe intellectual disability, characteristic facial dysmorphism, dental, finger and toe anomalies, and linear skin pigmentation. By personal communication with colleagues, we assembled 11 additional females with BFLS due to variants in PHF6. We confirm the distinct phenotype to include variable intellectual disability, recognizable facial dysmorphism and other anomalies. We observed skewed X‐inactivation in blood and streaky skin pigmentation compatible with functional mosaicism. Variants occurred de novo in 10 individuals, of whom one was only mildly affected and transmitted it to her more severely affected daughter. The mutational spectrum comprises a two‐exon deletion, five truncating, one splice‐site and three missense variants, the latter all located in the PHD2 domain and predicted to severely destabilize the domain structure. This observation supports the hypothesis of more severe variants in females contributing to gender‐specific phenotypes in addition to or in combination with effects of X‐inactivation and functional mosaicism. Therefore, our findings further delineate the clinical and mutational spectrum of female BFLS and provide further insights into possible genotype–phenotype correlations between females and males.
Keywords: Borjeson‐Forssman‐Lehmann syndrome, de novo, PHF6, X‐chromosomal

1. INTRODUCTION
X‐linked recessive Borjeson‐Forssman‐Lehmann syndrome (BFLS, OMIM#301900) was first described in 1962. 1 In 2002, variants in the gene encoding PHD finger protein 6 (PHF6) were identified as the underlying cause. 2 PHF6 contains two extended atypical PHD‐like zinc finger domains (PHD1 and PHD2), two nuclear and one nucleolar localization sequences 2 and is assumed to play a role in transcription, ribosomal RNA transcription and neuronal migration. 3 , 4 , 5 Affected males present with developmental delay, moderate to severe intellectual disability (ID), truncal obesity, hypogonadism, tapering fingers, toe anomalies, and a typical facial gestalt with long ears or prominent earlobes and prominent cheekbones. 6 , 7 Some of the female carriers in these families show mild aspects of BFLS such as learning difficulties, mild facial features or toe and finger anomalies. 7 , 8 , 9 Skewing of X‐inactivation (XI) in female carriers in these reports was inconsistent and did not correlate with clinical findings. 9 , 10 , 11
In 2013, a series of seven females with de novo variants in PHF6 was reported. 12 Affected individuals presented with a neurodevelopmental disorder overlapping with BFLS in males, but also displaying additional distinct features. 12 Next to moderate to severe intellectual disability, a characteristic facial gestalt with long shaped ears, bitemporal narrowing, prominent supraorbital ridges, high eyebrows, a short nose, and a bulbous nasal tip was delineated. Furthermore, oligomenorrhea, more prominent finger and toe deformities, dental anomalies, and linear skin hyperpigmentation occurred. In accordance with streaky skin pigmentation, skewed XI in blood samples and random XI in fibroblasts indicated functional mosaicism of the active and inactivated mutant allele. 12 Up to now, a total of twelve female individuals with such de novo germline deletions, duplications or single nucleotide variants in PHF6 were reported. 10 , 12 , 13 , 14 , 15 , 16 , 17
Male individuals with BFLS predominantly harbor missense variants and only a few truncating variants distributed all over the gene/protein, 2 while in females with de novo variants, mostly deletions, (likely) truncating aberrations and only one missense variant located within the second PHD zinc finger domain were identified to date. 12 , 17 , 18 Observing differential cellular localization between “male” and “female” variants in vitro and predicting more severe effects of the single female missense variant compared to male missense variants in the PHD2 zinc finger on domain stability, a possible genotype–phenotype correlation between nature and localization of variants and gender‐specific phenotypic manifestation was recently suggested. 18
We now further delineate the mutational and clinical spectrum of female BFLS by assembling 11 additional cases with aberrations in PHF6. Variants occurred de novo in 10 individuals, of whom one was only mildly affected and transmitted it to her more severely affected daughter. Identification of three further missense variants within the PHD2 domain and subsequent structural modeling support the previously suspected genotype–phenotype correlation.
2. MATERIAL AND METHODS
2.1. Patient material and data
Personal communication with colleagues following the initial reports 12 , 16 enabled us to collect clinical and mutational details on 11 female individuals with BFLS due to variants in PHF6. The study was approved by the ethics committee of the medical faculty of the Friedrich‐Alexander‐University Erlangen‐Nuremberg (approval 142_15B). Testing in the majority of individuals was performed in a diagnostic setting. Individual 10 was analyzed within a study to unravel the diagnosis of patients with developmental disorders. Informed consent for publication of mutational and clinical data and particularly for publication of patient photographs was obtained from the parents or legal guardians.
2.2. PHF6 analysis and structural modeling
In four individuals, targeted analysis of PHF6 (NM_032458) based on clinical suspicion was performed by Sanger sequencing and/or MLPA, as described previously. 12 Further details on primer and probe sequences and conditions are available on request. Trio‐exome sequencing was performed in one, panel sequencing in two and single exome sequencing in three individuals (Table 1). Segregation analysis in the non‐trio cases was performed by Sanger sequencing or MLPA, respectively. XI analysis in blood samples was performed in seven individuals in the respective centers within routine diagnostics. VIPUR scores 19 of the three novel and one published 20 missense variants were determined as described previously. 18 VIPUR is designed to distinguish between neutral (score <0.5) and deleterious (score >0.5) protein variants by modeling their effect on the three‐dimensional protein structure. Thus, high scores indicate a large effect of the respective variant on the protein structure.
TABLE 1.
Mutational and clinical details of females with BFLS due to variants in PHF6 and comparison to published males and female carriers
| Patient # | 1 | 2.1 (index) | 2.2 (mother) | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | female BFLS n = 27, 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 current study | male BFLS n = 62 2 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 | female carriers in XLR families n = 63 2 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 20 , 21 , 22 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PHF6 variant (NM_032458) | exon 6–7 deletion | c.88C>T, p.(Gln30*) | c.88C>T, p.(Gln30*) | c.129dup, p.(Lys44*) | c.346C>T, p.(Arg116*) | c.374+ 1G>A, r.spl | c.590_593del, p.(Asp197 Glyfs*20) | c.820C>T, p.(Arg274*) | c.845T>C, p.(Leu282 Pro) | c.859G>A, p.(Gly287 Ser) | c.898A>G, p.(Thr300Ala) |
19x truncating 5x missense |
7 families truncating 15 families missense |
NA |
| De novo | Yes | Maternal | Yes | Excl. In the mother | Yes | Yes | Yes | Yes | Yes | Yes | Yes | 23/26 | 1/56 | 1/43 |
| Method | MLPA | Panel | Sanger segr. | Sanger | Panel | Exome | Sanger | Exome | Sanger | Exome | Trio exome | NA | NA | NA |
| skewed XI in blood | NA | XI 97.2 | XI 91.4 | XI 100 | XI 100 | XI 100 | XI 100 | NA | NA | XI 100 | NA | 22/22 (100%) | NA |
23/29 (79.3%) |
| Sex | Female | Female | Female | Female | Female | Female | Female | Female | Female | Female | Female | Female | Male | Female |
| Age | 4 y | 7 y 8 m | 36 y 2 m | 6 y 10 m | 10 y 10 m | 12 m | 12 m | 20 m | 15 y | 3 y | 12 y 7 m | several m to 41 y | 10 m to 62 y | NA |
| Body measurements | ||||||||||||||
| Gestational week | NA | 39 | term | 38 | NA | NA | NA | NA | 38 | 39 | NA | NA | NA | NA |
| Birth weight (g) | 4564 | 2850 | 2800 | 2950 | 3230 | 2300 | 2410 | 2140 | 2739 | 2644 | 3100 | NA | NA | NA |
| Birth length (cm) | NA | 49 | 50 | 49 | 49 | 47.5 | 49 | 44 | NA | 47 | 47 | NA | NA | NA |
| OFC at birth | NA | 36 | 34 | 36.5 | NA | 31 | 32 | 31.5 | NA | NA | 36.5 | NA | NA | NA |
| Weight (kg) / SD | NA | 22 / ‐0.58 | 88 / NA | 24.3 / 0.63 | 57.9 / 2.18 | 6.5 / ‐2.69 | 8.5 / ‐0.42 | 10.8 / 0.12 | NA | 12.1 / ‐1.82 | 67.7 / 2.01 | 2x under‐weight,15x normal, 5x obese | 2x underweight, 3x normal, 6x overweight | 14x normal, 4x obese |
| Height (cm) / SD | NA | 119 / ‐0.99 | 180 / 2.56 | 117.9 / ‐0.36 | 158.6 / 2.22 | 70.5 / ‐1.37 | 76 / 0.77 | 80 / ‐0.9 | NA | 92 / ‐2 | 161.2 / 0.98 | 2x short, 17x normal, 4x tall | 9x short, 12x normal | 2x short, 16x normal |
| OFC (cm) / SD | NA | 51 / ‐0.63 | NA | 51.9 / 0.38 | 53.3 / 0.26 | 43.5 / ‐1.86 | 46 / 0.3 | 45 / ‐2.25 | NA | 49 / ‐0.71 | 58.8 / 3.54 | 4x microcephaly, 14x normocephaly, 2x macrocephaly | 5x microcephaly, 25x normocephaly, 6x macrocephaly | 3x normal |
| Obesity | No | No | No | No | Yes | No | No | No | Yes | No | Yes |
5/25 (20%) |
44/48 (91.7%) |
4/19 (21.1%) |
| Characteristic facial gestalt | Yes | Yes | No (subtle) | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
23/25 (92%) typical female BFLS |
47/47 (100%) typical male BFLS |
16/23 (69.6%) subtle male BFLS |
| Development | ||||||||||||||
| Age at walking | 18 m |
Supported (7 y) |
10 m | 1 y 10 m | 15 m | Unsteady sitting (1 y) | NA |
Not yet (1 y 8 m) |
NA |
Sitting (2 y 2 m) not yet (3 y) |
1 y 1 m |
1 y to not yet at 7 y |
1 y 11 m to 4 y |
NA |
| First words | 2 y | No words | Normal | 3 y | 1 y 6 m | Not yet | Not yet | Not yet | No words | No words | 3 y 6 m | 15 m to not speaking | 4 y to not speaking | NA |
| Current speech ability | NA | Only vocalizations | Normal | 2–3 word sentences | Simple sentences | NA | NA | Hoarse voice | Growls | NA | Simple sentences | NA | NA | NA |
| Intellectual disability | Moderate | Severe/profound | No | Moderate | Mild/Moderate | Mild | Moderate | Severe | Severe | Moderate | Moderate |
24/27 (88.9%) |
54/54 (100%) |
6/32 (18.8%) learning disability |
| Neurological | ||||||||||||||
| Seizures | No | No | No | No | No | No | No | Yes | No | No | No | 5/23 | 3x | 2x |
| MRI anomalies | NA | Prominent outer cerebrospinal fluid space | NA | NA | Periventricular white matter lesions, frontal subcortical heterotopia | Small pituitary gland | NA | Delayed myelination, white matter loss, dysplastic pons, 3rd ventriculomegaly | NA | Abnormal position of the cerebellar tonsil | Normal MRI | 10/15 | NR | NR |
| Behavioral anomalies | No | Too impaired to assess | No | No | Earlier physically, now more verbally abusive | No | NA | Too impaired to assess | NA | Happy demeanor | Shy, anxious, friendly demeanor | 6/15 | 13/34 | 1x |
| Extremities | ||||||||||||||
| Finger anomalies | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | 24/25 (96%) |
16/16 (100%) reported as common in the rest |
8/21 (38.1%) |
| Clinodactyly | V | IV | IV | IV + V | No | IV + V | No | V | No | No | No | 16/25 (64.0%) | 5/16 (31.2%) | 0/21 (0%) |
| Brachydactyly | V | No | No | No | No | No | No | V | V | No | V | 13/25 (52.0%) | 3/16 (18.8%) | 0/21 (0%) |
| Camptodactyly | No | No | No | IV + V | Several fingers | No | No | No | No | No | No | 3/25 (12.0%) | 0/16 (0%) | 0/21 (0%) |
| Tapering fingers | No | No | No | No | Yes | No | No | No | No | Yes | Yes |
7/25 (28.0%) |
11/16 (68.8%) |
7/21 (33.3%) |
| Hypoplastic nails | No | No | No | No | No | No | Dysplastic, brittle | Hypoplastic V |
Hypoplastic V |
No | No |
8/25 (32.0%) |
2/16 (12.5%) |
1/21 (4.8%) |
| Toe anomalies | No | Yes, crowded toes | Yes, crowded toes | Yes, broad toes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
21/23 (91.3%) |
21/22 (95.5%) |
9/21 (42.8%) |
| Brachydactyly | No | No | No | No | No | No | No | Yes | No | No | IV + V |
5/23 (21.7%) |
12/22 (54.5%) |
8/21 (36.4%) |
| Camptodactyly/hammer toes | No | No | Yes | III, IV + V | II + III | No | No | No | No | No | No |
6/23 (43.5%) |
common in a sample of 25 22 | 0/21 (0%) |
| Sandal gap | No | No | No | No | No | No | No | No | No | No | Yes | 2/23 (8.7%) | 13/22 (59.1%) | 1/21 (4.8%) |
| Syndactyly | No | No | No | No | No | II‐IV right, II‐III left | Minimal II‐III | No | II‐III | II‐III | II‐III | 10/23 (43.5%) | 4/22 (18.2%) | 0/21 (0%) |
| Hypoplastic nails | No | Dysplastic v | Dysplastic v | No | No | No | Dysplastic, brittle | Hypoplastic | No | No | No | 7/23 (30.4%) | 1/22 (4.5%) | 1/21 (4.8%) |
| Other | ||||||||||||||
| Linear skin hyper‐pigmentation | No | Yes (both thighs) | No | No | Yes (groin + armpits) | No | Yes (legs) | No | Yes (arm + trunk) | Yes (trunk, bottom + legs) | No | 17/25 (68.0%) | NR |
0/1, NR |
| Dental anomalies | No | Small teeth, misalignment | Misalignment | Enamel defect | Hypodontia, large roots | NA | Small teeth | Hypodontia | NA | NA | Yes | 18/20 (90.0%) | 1x small teeth, widely spaced |
0/1, NR |
| Oligoamenorrhea (age at menarche) | NA | NA | yes (13 y) | NA | NA | NA | NA | NA | NA | NA | not yet | 10/10 (100%) | 44/45 (97.8%) hypogonadism or small external genitalia | 1/15 (6.7%) oligomenorrhea |
| Genital anomalies | No | Hypoplastic labia minora | No | No | No | No | No | No | Hypoplastic clitoris | No | NA | |||
| Eye anomalies | No | Strabism, hyperopia, excavation of the papilla | Myopia | Strabism, impaired stereo vision, astigmatism | Progressive retinal depigmentation, maculopathy | No | NA | No | No | NA | Nystagm, hyperopia, retinal depigmentation, maculopathy | Abnormalities in 13/15 | Abnormalities in 3/5 | NR |
| Other | No | Muscular hypotonia | No | Ectopic kidney, initially muscular hypotonia | Unilateral hydro‐nephrosis, congenital umbilical hernia, mild hearing impairment | Renal pilocalyceal dilatation | Hyertrichosis at the back | Cleft hard and soft palate, possible hearing impairment | No | Feeding difficulties | Muscular hypotonia | NA | NA | NA |
Abbreviations: II, III, IV, V, 2nd, 3rd, 4th, 5th finger or toe, respectively; m, months; NA, not available or not applicable; NR: not reported; XLR, x‐linked recessive (obligate female carriers with proven variant either in the female herself or in the family); XLR, X‐linked recessive; y, years, m, months; OFC, occipito‐frontal head circumference.
3. RESULTS
3.1. Clinical spectrum
For a summary of clinical details, see also Table 1. Age at last investigation of the 11 affected individuals ranged from 10 months to 36 years. Developmental delay was variable. The age of unsupported walking was between 10 months and not yet at 7 years. Age of first words ranged from normal to lack of speech at age 7 or 15 years in two individuals, respectively. Three of the individuals communicated in simple sentences, one with correct grammar.
All but one of the individuals at informative ages presented with intellectual disability, ranging from mild/moderate (7/10) to severe/profound (3/10). Formally tested IQs were not available. Of note, the mother of the familial case had normal motor and speech development and later only learning difficulties at school.
Behavioral anomalies such as verbally and physically abusive behavior were observed in a single individual. A happy and friendly demeanor was described in two other individuals.
MRI of the brain was performed in six individuals and revealed unspecific abnormalities in five of them, such as white matter lesions. Subcortical nodular heterotopia was observed in one individual. Neurological aspects such as muscular hypotonia or seizures only occurred in two or a single individual, respectively. Retinal depigmentation with maculopathy was reported in two individuals. Other ophthalmological anomalies as well as further organ abnormalities or cleft palate, occurred in single cases.
Ten individuals in this study showed a distinctive facial gestalt with long shaped ears with prominent earlobes, bitemporal narrowing, prominent supraorbital ridges, synophrys, a high nasal root, and bulbous nasal tip. The mildly affected mother of family 2 showed rather subtle facial aspects with a bulbous nasal tip. Eight individuals had sparse scalp hair, in combination with fine hair texture during infancy (Figure 1).
FIGURE 1.
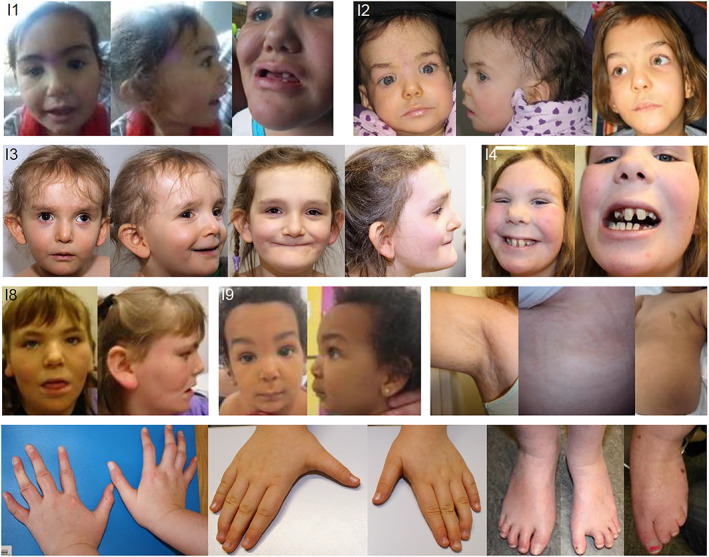
Morphological aspects of females with BFLS due to de novo variants in PHF6. Note the characteristic facial appearance with sparse hair in infancy, long‐shaped ears, bitemporal narrowing, prominent supraorbital ridges, synophrys, and a short nose with bulbous nasal tip. Additionally, irregularly shaped or missing teeth, linear skin hyperpigmentation and finger and/or toe anomalies occur. [Colour figure can be viewed at wileyonlinelibrary.com]
Linear skin hyperpigmentation was present in five individuals affecting different body parts. One individual presented with hypertrichosis. Teeth anomalies were described in six individuals and included hypodontia, enamel defects, rather small teeth or large frontal teeth with long roots, or misalignment. All but one of the individuals showed finger anomalies, including campto‐, brachy‐, clinodactyly or tapering. Nine out of 11 individuals presented with toe anomalies such as syndactyly II/III (5x), brachy‐, clino‐, and camptodactyly, broad or hypoplastic toes. Dysplastic or hypoplastic finger or toe nails were reported in three and four individuals, respectively (Figure 1). Obesity occurred in three individuals.
3.2. Mutational spectrum
For a summary of identified variants, see Figure 2, Tables 1 and S1.
FIGURE 2.
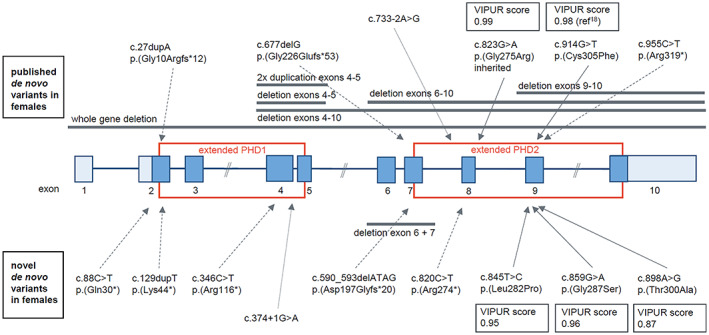
Schematic drawing of PHF6 (NM_032458) with location of variants identified in females. Coding exons are colored in dark blue, non‐coding exons in light blue. Red squares mark exons encoding the extended plant homeodomain 1 and 2 (PHD1/2). Above the gene, published deletions and variants 10 , 12 , 13 , 14 , 15 , 16 , 17 , 20 are indicated, below the scheme novel variants. VIPUR score 19 for all female missense variants in the PHD2 domain is higher than 0.86 indicating a severe deleterious effect of the variants on the protein stability. [Colour figure can be viewed at wileyonlinelibrary.com]
Three missense and six truncating variants, including a splice‐site variant, in PHF6 were detected in the 11 herewith described individuals. The deletion of exons 6 and 7 in one individual was predicted to be frame‐shifting and thus truncating. Nine of the variants were shown to have occurred de novo. In one case, maternal inheritance was excluded, and the father was not available for testing. One individual inherited the variant from her mildly affected mother, in whom the variant was shown to have occurred de novo. Sanger sequencing in blood was not indicative for mosaicism in her (Figure S1).
While the truncating variants were distributed all over the gene/protein, the three missense variants clustered within the PHD2 domain (Figure 2). To our knowledge, none of the variants has been reported as pathogenic before in literature or ClinVar. They are not observed in gnomAD. The missense variants affect highly conserved amino acids and are predicted to be deleterious by at least three of the used in silico prediction programs (Table S1). According to ACMG guidelines, 21 all identified variants were classified as pathogenic or likely pathogenic (Table S1).
XI pattern in blood samples was tested in seven individuals and was skewed (>90%) in all of them. Of note, both the mildly affected mother and the more severely affected daughter of the familial case had a similarly skewed degree of XI of more than 90% (Table 1).
3.3. Structural modeling of the missense variants
We used the VIPUR score, 19 integrating sequence analysis and structural modeling, to assess the effect of the identified missense variants on the three‐dimensional protein structure. All three missense variants in our cohort showed a high VIPUR score of >0.86, thus predicting a strong destabilizing effect on the protein structure (Figure 2, Table S1). Also, the published missense variant c.823G > A, p.(Gly275Arg) within the PHD2 domain 20 was predicted to have a strong effect with a VIPUR score of 0.99.
4. DISCUSSION
PHF6 belongs to the increasing number of X‐chromosomal genes in which both inherited variants in males with an X‐chromosomal recessive neurodevelopmental disorder (NDD) and de novo variants in females with a comparable severe but distinct NDD were identified. Female variant carriers in the X‐linked recessive families are mostly asymptomatic but may display mild and infrequent clinical aspects (Table 1). While the male BFLS phenotype has been known for several decades, 1 , 2 the distinct female phenotype associated with de novo variants in PHF6 was only delineated in 2013. 12 Thus, the available information on the latter is still limited and based on twelve published cases so far. 10 , 12 , 13 , 14 , 15 , 16 , 17 By reporting on eleven further individuals with the female form of BFLS, we further characterize the phenotypic and mutational spectrum.
With this study, we confirm the very distinct phenotype of BFLS in females caused by de novo variants in PHF6 to include variable intellectual disability, a characteristic facial gestalt, acral and dental anomalies and linear skin hyperpigmentation. While the variable degree of intellectual disability is comparable to that of affected male individuals, some of the facial aspects, as well as the presence of dental and pigmentation abnormalities are rather specific for the female phenotype. Finger and toe abnormalities are similarly frequent in both novel and published females and males with BFLS (>90%). However, in males, mainly tapering of fingers has been observed, while finger deformities in females are more prominent and diverse with tapering, campto‐, clino‐, brachydactyly, and hypoplastic nails (Table 1). While sandal gaps have been reported more frequently in males, syndactyly of toes and hypoplastic toe nails occurs more frequently in females (Table 1). The recognizability of the female phenotype is also demonstrated by the fact that targeted testing of PHF6 was performed in four of the individuals based on a specific clinical suspicion.
Furthermore, we confirmed or observed novel or previously under‐recognized aspects of female BFLS in this cohort. Whereas nonspecific ophthalmological abnormalities such as ametropia, nystagmus, or strabismus were frequently reported in about half of the previously published 12 , 16 , 17 and the new cases, more specific, retinal findings such as dystrophy or depigmentation were only observed once previously 12 and now additionally in two of the herewith reported individuals.
In general, neither structural brain abnormalities nor neurological features such as epilepsy seem to be a frequent feature of either male or female BFLS. Two adult females with a similar duplication of exons 4–5 were previously reported with a specific brain phenotype resembling band heterotopia and with adult‐onset epilepsy. 22 Apart from these, MRI data have been only infrequently available for affected individuals with BFLS. In our cohort, MRIs were performed in six individuals, indicating brain anomalies in five of them. These included mainly nonspecific signs such as white matter abnormalities, dysplastic pons, and enlarged ventricles. Interestingly, frontal subcortical heterotopia was observed in a single individual. In accordance with the findings by Kasper et al., 22 and observations in mice, 3 this might support a role of PHF6 in neuronal migration. Epilepsy occurred in only one of the herewith reported individuals. Whether the specific brain and epilepsy phenotype in the two previously reported females 22 reflects a genotype–phenotype correlation regarding the shared exon 4–5 duplication therefore remains elusive and would require further cases with a similar duplication and/or brain phenotype.
While general or truncal obesity was described in more than 90% of males with BFLS, 1 , 6 , 9 this has been only observed in 20% of females with BFLS both in the published and the herewith reported individuals (Table 1). In addition, hypogonadism has been described as one of the prominent features in males with BFLS, 1 , 9 and variable endocrinological abnormalities were observed in individuals carrying PHF6 variants. 23 Oligomenorrhea, frequently observed in females with de novo variants in PHF6, might also reflect hypogonadism. 12 , 14 While hypogonadotropic hypogonadism was confirmed in a single female individual, 14 detailed endocrinological testing was not performed or is not available for other female individuals with de novo variants in PHF6, thus currently limiting the characterization of an endocrinological phenotype in female BFLS. Of note, in the current cohort, a mildly affected female with oligomenorrhea gave birth to a daughter.
Several factors are assumed to contribute to the phenotypic differences between genders and between unaffected and affected female PHF6 variant carriers. Functional mosaicism of the active and inactive mutant PHF6 allele is discussed as a contributing pathomechanistic factor in females with de novo variants in PHF6. 12 , 13 , 18 Streaky skin pigmentation has been observed in the majority of previously reported affected females 12 , 13 , 14 , 15 , 16 , 17 but not in the unaffected carrier females in X‐recessive families. 7 , 8 , 9 Of note, in this study, only half of the females showed skin pigmentation anomalies, and the presence of these was not correlating with the severity of disease manifestation. Random XI might be another indicator of functional mosaicism, supported by a previous report showing skewed XI in blood samples but random XI in fibroblasts. 12 In blood, there is a high frequency of skewed XI both in asymptomatic carriers and symptomatic females. 6 , 9 , 12 , 16 , 23 For two affected females with de novo variants preferential inactivation of the mutant allele in blood was demonstrated (so far unpublished data, Supplementary Figure S2). Thus, the “direction” of XI in blood cells does not provide an explanation for the presence or severity of phenotypes. Furthermore, in the herewith reported and two other published familial cases with the transmission of a PHF6 variant from a mildly affected or asymptomatic mother to a severely affected daughter, 20 , 23 XI pattern in blood was similar in mothers and daughters. However, no data are available if the same allele was preferentially inactivated in both individuals. In summary, previous and new observations demonstrate that determining the degree of XI from blood samples might have only a weak predicting effect and allows no conclusions on the pattern of XI in more relevant tissues such as the brain.
Location and “severity” of the variants in PHF6 were discussed as another factor contributing to phenotypic differences. Although a severe effect by near complete loss of PHF6 protein expression has been demonstrated for the recurrent c.1024C > T, p.(Arg342Ter) variant in males, 24 the frequency of truncating variants is still significantly higher in females with BFLS than in males. 18 Furthermore, “male” and “female” missense variants behave differently in in vitro assays and regarding protein domain stability. 18 While a missense variant in the PHD2 domain identified in a female was predicted to have a strong destabilizing effect, male missense variants in the same domain were predicted to have a milder effect. 18 Missense variants from males in the PHD1 domain were shown to have a strong destabilizing effect 18 and were shown to result in reduced protein expression, 24 but were so far exclusively observed in X‐linked recessive families and not as de novo variants in females with the full clinical picture of BFLS. Missense variants in the PHD1 domain were therefore postulated to be less deleterious than missense variants in the PHD2 domain, and within the PHD2 domain “female” missense variants to be more deleterious than “male” missense variants. 18 By now, we find further evidence for this previously discussed genotype–phenotype correlation. Three additional de novo missense variants identified in females were all located in the PHD2 domain and predicted to result in severe destabilization of the domain, similar to the previously published “female” variant 18 and more severe compared to “male” missense variants in the same domain. 18
This is also supported by another published missense variant detected in a female, c.823G > A, p.(Gly275Arg), 20 which is also located in the PHD2 domain and predicted to have a strong destabilizing effect. Surprisingly, however, this variant was transmitted from an asymptomatic mother to a more severely affected daughter. 20 There is another case of female‐to‐female transmission of a truncating variant in the literature 23 and additionally in the herewith reported family 2 (see also above). Similar degrees of XI skewing in blood in mildly affected mothers and more severely affected daughters 20 , 23 do not provide an explanation for the phenotypic differences. In family 2 and in one of the published cases 20 the variant was shown to be de novo in the mothers, however, without indication of mosaicism for the variant in them. Still, this cannot be excluded as an explanation for the mild presentation. Post‐zygotic mosaicism associated with a milder phenotypic presentation has been reported in a female individual before. 15
In total, the phenotypic manifestation of BFLS in females cannot be attributed to a single factor but seems to result from a complex and variable interplay of different contributing factors including XI, functional mosaicism, as well as localization, nature and severity of variants.
Our study confirms that BFLS caused by de novo variants in PHF6 is a distinct, recognizable neurodevelopmental disorder in females, and we further delineate the clinical and mutational spectrum. By confirming a genotype‐correlation between males and females and between symptomatic and asymptomatic female carriers regarding localization of consequences of missense variants, we support the hypothesis that nature and localization of variants in PHF6 are contributing factors to the female BFLS phenotype.
AUTHOR CONTRIBUTIONS
Céline B. Gerber, Anna Fliedner, Oliver Bartsch, Siren Berland, Malin Dewenter, Marte Haug, Ian Hayes, Purificacion Marin‐Reina, Paul R. Mark, Francisco Martinez‐Castellano, Isabelle Maystadt, Deniz Karadurmus, Katharina Steindl, Antje Wiesener, Markus Zweier, and Christiane Zweier collected mutational and clinical data. Anna Fliedner performed targeted testing of PHF6. Heinrich Sticht performed structural modeling. Céline B. Gerber and Christiane Zweier wrote the manuscript, which was read and revised by all coauthors.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
ETHICS STATEMENT
The study was approved by the ethics committee of the medical faculty of the Friedrich‐Alexander‐University Erlangen‐Nuremberg (approval 142_15B). Testing in all but one individual was performed in a diagnostic setting. Individual 10 was investigated in the frame of a research study approved by the ethical review board of the canton Zurich and respective consent was retrieved from the family. Informed consent for publication of mutational and clinical data and particularly for publication of patient photographs was obtained from the parents or legal guardians.
WEBSITES
https://gnomad.broadinstitute.org
http://www.ncbi.nlm.nih.gov/clinvar/
https://cadd.gs.washington.edu/score
https://sites.google.com/site/revelgenomics/about
http://bejerano.stanford.edu/mcap/
https://sift.bii.a-star.edu.sg/
Supporting information
Appendix S1 Supporting information
ACKNOWLEDGEMENTS
We thank the patients and their families for participating in this study. We furthermore thank Dominique Braun and Anna Kopps for their support. This study was supported by a grant from the “Deutsche Forschungsgemeinschaft (DFG)” to Christiane Zweier (ZW184/3‐1). Open Access Funding provided by Inselspital Universitatsspital Bern.
Gerber CB, Fliedner A, Bartsch O, et al. Further characterization of Borjeson‐Forssman‐Lehmann syndrome in females due to de novo variants in PHF6 . Clinical Genetics. 2022;102(3):182‐190. doi: 10.1111/cge.14173
Funding information Deutsche Forschungsgemeinschaft, Grant/Award Number: ZW184/3‐1
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Borjeson M, Forssman H, Lehmann O. An X‐linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Med Scand. 1962;171:13‐21. doi: 10.1111/j.0954-6820.1962.tb04162.x [DOI] [PubMed] [Google Scholar]
- 2. Lower KM, Turner G, Kerr BA, et al. Mutations in PHF6 are associated with Börjeson‐Forssman‐Lehmann syndrome. Nat Genet. 2002;32(4):661‐665. doi: 10.1038/ng1040 [DOI] [PubMed] [Google Scholar]
- 3. Zhang C, Mejia LA, Huang J, et al. The X‐linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron. 2013;78(6):986‐993. doi: 10.1016/j.neuron.2013.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jahani‐Asl A, Cheng C, Zhang C, Bonni A. Pathogenesis of Börjeson‐Forssman‐Lehmann syndrome: Insights from PHF6 function. Neurobiol Dis. 2016;96:227‐235. doi: 10.1016/j.nbd.2016.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang J, Leung JW, Gong Z, Feng L, Shi X, Chen J. PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis. J Biol Chem. 2013;288(5):3174‐3183. doi: 10.1074/jbc.M112.414839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gécz J, Turner G, Nelson J, Partington M. The Börjeson‐Forssman‐Lehman syndrome (BFLS, MIM #301900). Eur J Hum Genet. 2006;14(12):1233‐1237. doi: 10.1038/sj.ejhg.5201639 [DOI] [PubMed] [Google Scholar]
- 7. Carter MT, Picketts DJ, Hunter AG, Graham GE. Further clinical delineation of the Börjeson‐Forssman‐Lehmann syndrome in patients with PHF6 mutations. Am J Med Genet A. 2009;149a(2):246‐250. doi: 10.1002/ajmg.a.32624 [DOI] [PubMed] [Google Scholar]
- 8. Mangelsdorf M, Chevrier E, Mustonen A, Picketts DJ. Börjeson‐Forssman‐Lehmann syndrome due to a novel plant homeodomain zinc finger mutation in the PHF6 gene. J Child Neurol. 2009;24(5):610‐614. doi: 10.1177/0883073808327830 [DOI] [PubMed] [Google Scholar]
- 9. Turner G, Lower KM, White SM, et al. The clinical picture of the Börjeson‐Forssman‐Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin Genet. 2004;65(3):226‐232. doi: 10.1111/j.0009-9163.2004.00215.x [DOI] [PubMed] [Google Scholar]
- 10. Crawford J, Lower KM, Hennekam RC, et al. Mutation screening in Borjeson‐Forssman‐Lehmann syndrome: identification of a novel de novo PHF6 mutation in a female patient. J Med Genet. 2006;43(3):238‐243. doi: 10.1136/jmg.2005.033084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baumstark A, Lower KM, Sinkus A, et al. Novel PHF6 mutation p.D333del causes Börjeson‐Forssman‐Lehmann syndrome. J Med Genet. 2003;40(4):50e‐550e. doi: 10.1136/jmg.40.4.e50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zweier C, Kraus C, Brueton L, et al. A new face of Borjeson‐Forssman‐Lehmann syndrome? De novo mutations in PHF6 in seven females with a distinct phenotype. J Med Genet. 2013;50(12):838‐847. doi: 10.1136/jmedgenet-2013-101918 [DOI] [PubMed] [Google Scholar]
- 13. Garcia‐Melendo C, Roé E, Rodríguez‐Santiago B, et al. A case report of PHF6 mosaicism: beyond the classic Börjeson‐Forssman‐Lehmann syndrome. Pediatr Dermatol. 2021;38(4):919‐925. doi: 10.1111/pde.14636 [DOI] [PubMed] [Google Scholar]
- 14. Berland S, Alme K, Brendehaug A, Houge G, Hovland R. PHF6 deletions may cause Borjeson‐Forssman‐Lehmann syndrome in females. Mol Syndromol. 2011;1(6):294‐300. doi: 10.1159/000330111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Donato N, Isidor B, Lopez Cazaux S, et al. Distinct phenotype of PHF6 deletions in females. Eur J Med Genet. 2014;57(2–3):85‐89. doi: 10.1016/j.ejmg.2013.12.003 [DOI] [PubMed] [Google Scholar]
- 16. Zweier C, Rittinger O, Bader I, et al. Females with de novo aberrations in PHF6: clinical overlap of Borjeson‐Forssman‐Lehmann with Coffin‐Siris syndrome. Am J Med Genet C Semin Med Genet. 2014;166c(3):290‐301. doi: 10.1002/ajmg.c.31408 [DOI] [PubMed] [Google Scholar]
- 17. Wieczorek D, Bögershausen N, Beleggia F, et al. A comprehensive molecular study on coffin‐Siris and Nicolaides‐Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22(25):5121‐5135. doi: 10.1093/hmg/ddt366 [DOI] [PubMed] [Google Scholar]
- 18. Fliedner A, Gregor A, Ferrazzi F, Ekici AB, Sticht H, Zweier C. Loss of PHF6 leads to aberrant development of human neuron‐like cells. Sci Rep. 2020;10(1):19030. doi: 10.1038/s41598-020-75999-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baugh EH, Simmons‐Edler R, Müller CL, et al. Robust classification of protein variation using structural modelling and large‐scale data integration. Nucleic Acids Res. 2016;44(6):2501‐2513. doi: 10.1093/nar/gkw120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daum H, Mor‐Shaked H, Ta‐Shma A, et al. Grandparental genotyping enhances exome variant interpretation. Am J Med Genet A. 2020;182(4):689‐696. doi: 10.1002/ajmg.a.61511 [DOI] [PubMed] [Google Scholar]
- 21. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kasper BS, Dörfler A, Di Donato N, et al. Central nervous system anomalies in two females with Borjeson‐Forssman‐Lehmann syndrome. Epilepsy Behav. 2017;69:104‐109. doi: 10.1016/j.yebeh.2017.01.022 [DOI] [PubMed] [Google Scholar]
- 23. Zhang X, Fan Y, Liu X, et al. A novel nonsense mutation of PHF6 in a female with extended phenotypes of Borjeson‐Forssman‐Lehmann syndrome. J Clin Res Pediatr Endocrinol. 2019;11(4):419‐425. doi: 10.4274/jcrpe.galenos.2019.2018.0220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ahmed R, Sarwar S, Hu J, et al. Transgenic mice with an R342X mutation in Phf6 display clinical features of Börjeson‐Forssman‐Lehmann syndrome. Hum Mol Genet. 2021;30(7):575‐594. doi: 10.1093/hmg/ddab081 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting information
Data Availability Statement
Not applicable.