

Abstract
Plant metabolomics has been used widely in plant physiology, in particular to analyse metabolic responses to environmental parameters. Derivatization (via trimethylsilylation and methoximation) followed by GC‐MS metabolic profiling is a major technique to quantify low molecular weight, common metabolites of primary carbon, sulphur and nitrogen metabolism. There are now excellent opportunities for new generation analyses, using high resolution, exact mass GC‐MS spectrometers that are progressively becoming relatively cheap. However, exact mass GC‐MS analyses for routine metabolic profiling are not common, since there is no dedicated available database. Also, exact mass GC‐MS is usually dedicated to structural resolution of targeted secondary metabolites. Here, we present a curated database for exact mass metabolic profiling (made of 336 analytes, 1064 characteristic exact mass fragments) focused on molecules of primary metabolism. We show advantages of exact mass analyses, in particular to resolve isotopic patterns, localise S‐containing metabolites, and avoid identification errors when analytes have common nominal mass peaks in their spectrum. We provide a practical example using leaves of different Arabidopsis ecotypes and show how exact mass GC‐MS analysis can be applied to plant samples and identify metabolic profiles.
Keywords: database, high resolution, isotope, mass spectrometry, metabolomics
1. INTRODUCTION
Plant metabolome analysis is now of crucial importance to describe responses to environmental conditions and thus understand associated metabolic mechanisms. Standardised metabolomics protocols have been proposed to characterise crop metabolism (Zheng et al., 2021). The term ‘metabolomics’ is used to refer to techniques exploited to investigate ‘small’ biological molecules (metabolites), that is, extractible molecules with limited molecular weight, usually less than 1200 atomic mass units (a.m.u.) (Roessner & Bowne, 2009). Two mainstream mass spectrometry techniques can be used: gas chromatography coupled to mass spectrometry (GC‐MS) and liquid chromatography coupled to mass spectrometry (LC‐MS) (Allwood et al., 2011; Perez de Souza et al., 2019). The output of metabolomics is a data set of metabolic features (m/z metabolite peaks with retention time in LC‐MS; analytes resulting from metabolite derivatisation in GC‐MS) that can be used for statistics and detect metabolic changes between samples.
GC‐MS metabolomics (also sometimes referred to as metabolic profiling) has been used extensively in plants under many conditions, species, or genetic backgrounds: a simple search with a literature database with query keywords ‘plant’, ‘metabolomics’ and ‘gc‐ms’ returns 43,700 entries. When restricted to Arabidopsis, it returns 13,500 entries. It shows the massive utilisation of GC‐MS for plant physiology and molecular biology. In particular, this technique is very useful to have, in a single sample analysis, a relative quantitation of most important metabolites of plant primary metabolism, such as amino acids, small soluble sugars, polyamines or organic acids. It has thus been used to describe the response of C and N primary metabolism to major environmental cues, for example, herbivores (Jansen et al., 2009), CO2 mole fraction (Högy et al., 2010; Misra & Chen, 2015), drought (Bowne et al., 2012; Sanchez et al., 2012), nutrient conditions (Cui, Abadie et al., 2019; Cui et al., 2021; Cui, Davanture, et al., 2019), or abiotic stress combinations (Ghatak et al., 2018; Nakabayashi & Saito, 2015; Shulaev et al., 2008).
To date, the vast majority of GC‐MS analyses for metabolic profiling utilise nominal mass acquisition (i.e. at a.m.u. resolution). Accordingly, databases associated with GC‐MS metabolomics such as the Golm Metabolomics Database provide spectral data at a.m.u. resolution, and in a recent review of metabolomics resources, only nominal mass databases are discussed for GC‐MS analyses (Vinaixa et al., 2016). In other words, to our knowledge, there is no directly accessible, high resolution (exact mass, i.e. at 0.0001 a.m.u. resolution or lower) and comprehensive GC‐MS resource for plant metabolomics. This lack of curated, accessible and available resource for GC‐MS analyses has three origins: (1) The availability of (affordable) exact mass GC‐MS instruments is relatively recent, since the implementation of the orbitrap technology took place in the 2000s (Makarov, 2000; Makarov et al., 2009; Peterson et al., 2010) and the description of standard practices for high resolution GC‐MS analyses has been proposed in 2021 only (Misra, 2021); (2) Many ordinary applications of GC‐MS metabolomics profiling do not require exact mass resolution since they are targeted on common, well‐known compounds; and (3) Whenever high resolution is required, LC‐MS can be used. The use of high resolution in LC‐MS may be important using full scan analyses, because there is a limited number of fragments (mainly parental ion and adducts) and therefore, identification essentially relies on both exact mass and isotopic pattern (De Vos et al., 2007; Kind & Fiehn, 2006). By contrast, in GC‐MS analyses, the fragmentation pattern along with the retention index are used to identify analytes, with generally good accuracy. Several tools have been recently proposed to automatically annotate ions or fragments in mass spectra, in particular from LC‐MS spectral data, for example in (Doerfler et al., 2014; Gaquerel et al., 2013; Matsuda et al., 2011; Qiu et al., 2016).
However, there are circumstances where high mass resolution may be desirable with GC‐MS, since (1) several compounds with the same retention time could generate fragments with the same nominal mass and (2) it could be useful to distinguish isotopic species (isotopologues) using their mass difference (e.g., there is a mass excess of +1.003355 Da with 13C while it is +1.006277 Da with 2H), and (3) one may desire to perform untargeted GC‐MS analyses with broad chemical coverage. Here, we describe an exact mass GC‐MS method for high resolution routine plant metabolic profiling and provide the associated curated database, checked with authentic standards. This allows us to address both (1) and (2). We also provide the list of current compounds having similar nominal‐mass fragments and similar retention time but can be distinguished easily with exact mass, avoiding quantification errors. We also take advantage of sulphur isotopes at natural abundance to allow the identification of S‐containing fragments in datasets. Finally, we used our protocol and the database using Arabidopsis leaves to show how it can be applied to real samples, allowing facile differentiation of genetic accessions.
2. MATERIALS AND METHODS
2.1. Chemicals
Chemicals were from Sigma Aldrich (Merck; with reference numbers between parentheses): Mass Spectrometry Metabolite Library (MSMLS, IROA Technologies), 2‐Oxoadipic acid (75447), 3‐ Iodo‐l‐Tyrosine (I8250), 5‐Aminolevulinic acid (08339), α‐Tocopherol (T3251), β‐Gentiobiose (G3000), Betain (W422312), Chlorogenic acid (C3878), Choline (C1879), Citraconic acid (C82604), D‐2‐Aminobutyric acid (116122), D‐3 phosphoglyceric acid (P8877), d‐Erythronic acid (75025), d‐Erythronic acid γ‐lactone (374385), d‐Galacturonic acid (48280), d‐Glucuronic acid (G5269), Diaminopimelate (33240), d‐Melibiose (92413), d‐Pinitol (441252), d‐Quinic acid (138622), d‐Threitol (377619), d‐Xylonic acid γ‐lactone (89339), Dulcitol (D0256), Galactinol (79544), Hydrocinnamic acid (135232), L‐α‐Glycerophosphocholine (G5291), Itaconic acid (I29204), l‐carnitine (C0283), l‐Cystathionine (C7505), Levoglucosan (06724), l‐Fucose (F2252), l‐Pyroglutamic acid (83160), Nicotinic acid (N4126), Norleucine (N6877), O‐Acetyl‐Serine (CDS020792), Phosphocholin (P0378), Stigmasterol (S2424), Sinapinic Acid (D7927), Myristic acid (M3128), Tryptamine (193747), Urea (U5378) and Xanthosine (CDS020790).
2.2. Plant material
The height Arabidopsis thaliana accessions were obtained from INRA Versailles Genomic Resource Centre VNAT collection: An‐1 (96AV), Bl‐1 (42AV), Col‐0 (186AV), Cvi‐0 (166AV), Ge‐0 (101AV), Mt‐0 (94AV), Oy‐0 (224AV) and Shahdara. Plants were grown in a growth chamber under short photoperiod (8 h light/16 h dark), PPFD = 150 µmol m−2 s−1, temperature 22°C/18°C (day/night) and relative humidity 60%. Plants were watered every 3 days and once a week with Plant‐Prod (15−10−30, N−P−K, Fertil). Fifty‐five days after sowing, two fully developed leaves were sampled on each rosette and instantly quenched in liquid nitrogen. Frozen samples were then frozen‐dried, dry material was grinded and 5 mg of dry powder used for metabolites extraction by adding 400 µl of methanol/water (90/10) with adonitol (55 µM) as an internal standard.
2.3. GC‐MS analyses
A detailed, step‐by‐step protocol is provided as a Supporting Information Material. This section summarises how exact mass GC‐MS analyses and data extraction with TraceFinder® were performed. GC‐MS analyses were carried out using a GC‐MS‐Orbitrap Q Exactive (Thermo Scientific). Fifty five µl of each standard solution (50 µM), or 4 µl from the plant leaf extract, were poured into a vial (with insert) and spin‐dried at 39°C. Samples were derivatized (automatically with a preparative robot) with 20 µl methoxyamine (20 mg ml−1 in pyridine; 90 min at 37°C) and 30 µl N‐methyl‐N‐(trimethylsilyl)‐trifluoroacetamide (MSTFA) for 30 min at 37°C. Before injection, 5 µl of alkane mix (14 alkanes from C9 to C36, 3 µg µl−1, Connecticut n‐Hydrocarbon Mix, Supelco) were added in each sample to compute the retention index. Analyses were performed by injecting 1 µl in splitless mode at 230°C (injector temperature) in a TG‐5 SILMS column (30 m × 0.25 mm × 0.25 µm; Thermo Scientific) set in a Trace 1300 Series GC (Thermo Scientific). Helium was used as gas carrier with a constant flow of 1 ml min−1. After 1 min at initial GC oven temperature (70°C), temperature was raised to 325°C at 15°C min−1 and finally kept at 325°C for 4 min. MS analyses were operated in positive polarity in full MS scan mode with the following source settings: mass scan range 50−750 m/z, resolution 60 000, AGC target 1E6, MS transfer line 300°C and filament delay 4.12 min. Ionisation by electron impact (70 eV) was performed at 250°C ion source temperature. Analytes were identified automatically using TraceFinder® (Thermo Scientific) using retention time, major characteristic fragment (m/z ion) and a confirmation fragment, with a maximum tolerance of 0.00007 Da via targeted screening.
3. RESULTS
3.1. Overview of GC‐MS analysis
As an example, the chromatogram associated with an Arabidopsis leaf sample derivatised with methoxamine and N‐methyl‐N‐(trimethylsilyl)‐trifluoroacetamide (MSTFA) in pyridine is shown in Figure 1, where the signal shown in panel (a) is total ion current (TIC). Abundant compounds form peaks that can be easily seen on the chromatogram, including some amino acids (e.g., proline), organic acids (fumarate) and sugars (e.g., sucrose), as trimethylsilylated (–Si(CH3)3, abbreviated TMS) derivatives. The TIC signal does not directly reflect the quantity (in moles) in the sample since analytes do not have all the same response coefficient in the mass spectrometer. The internal standard used for semiquantitation (i.e., relative quantification) is adonitol (ribitol), the derivative of which (ribitol 5TMS) elutes at 10.68 min. As expected, there is some overlapping between analytes (i.e., several analytes have very close retention time and coelute). This is visible, for example, at 15.05 (panel (b) of Figure 1) where many m/z features related to sugars are visible together with 6‐phosphogluconate specific m/z features (e.g., at 387.14254 Da). Interestingly, a relatively close mass (387.32912 Da) can be found at a similar retention time (15.25 min) from arachidic acid 1TMS, but such a mass difference is in practice too large (nearly 0.18 Da) to allow confusion with exact mass analysis.
Figure 1.
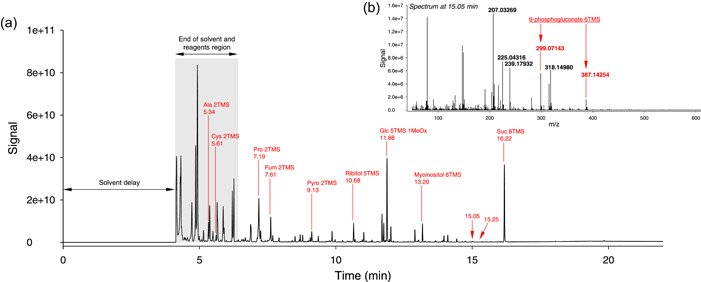
Overview of a GC‐MS chromatogram from an Arabidopsis leaf extract (here, ecotype Cvi‐0). (a) total ion current chromatogram, showing annotation of typical peaks (in red). The observed mass spectrum at 15.05 min is shown in (b), with characteristic ions of 6‐phosphogluconate 3TMS annotated in red. Note the contribution of ion m/z = 387.14254 Da (fragment C13H32O7PSi2 +). Note that a close, but easily distinguishable mass (387.32912 Da, fragment C22H47O3Si+) can be observed at 15.25 min, from arachidic acid 1TMS; nominal mass analysis would have generated the same apparent mass at 387 (or 388) Da. [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Mass resolution and isotopic pattern
Mass resolution across the m/z window (50−750 Da) was not found to be constant, varying from less than 0.0001 Da at low mass (<100 Da) to 0.0017 Da at higher mass (>350 Da), that is, within ≈1 and 5 ppm. In principle, this should allow to solve the natural isotopic pattern and distinguish 13C, 15N and 33S isotopologues (m + 1) and 13C2 and 34S isotopologue (m + 2). However, this depends on whether analytes of interest are silylated or not, because Si has two isotopes (29Si and 30Si), with mass excess values (with respect to the monoisotopic species) very close to that of 33S and 34S (sulphur‐containing species are addressed further below). This is illustrated in Figure 2, using a non‐silylated compounds (naturally volatile and thus not requiring derivatisation), isopropyl‐4‐methylthiazole (IMT). In fact, using the example of the major fragment (molecule minus CH3) (Figure 2a), 15N, 33S, 2H and 13C isotopologues are resolved (Figure 2b,c). With methionine 2TMS (Figure 2d), the presence of Si does not allow full resolution of the isotopic pattern. This is shown using the m + 2 isotopologues (Figure 2e) where some isotopic species are found to be undistinguishable (34S and 30Si). Despite this limitation, the expected isotopologue abundance (predicted from isotope abundance and elemental composition) is very close to observed abundance (Figure 2f). A difference between observed and predicted abundance is visible for the deuterated isotopologue of IMT (red circle), likely due to the very small natural abundance of deuterium (0.015%) and thus the larger imprecision in quantitation. Also, it should be noted that 13C isotopologues are well‐separated (resolved) from other isotopologues, allowing facile monitoring of 13C molecules during plant labelling experiments, for example.
Figure 2.
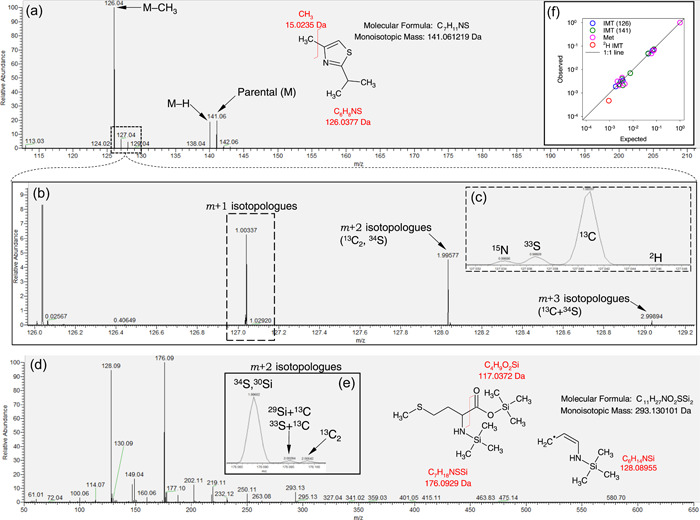
Isotopic pattern in two N‐ and S‐containing molecules, isopropyl‐4‐methylthiazole (IMT) and methionine 2TMS. (a) overall mass spectrum of IMT, showing the prevalence of the demethylated fragment at 126.03770 Da. (b) magnification of the mass spectrum showing the isotopic pattern of the major fragment of IMT and (c) detailed view of m + 1 isotopologues showing the resolution of 15N, 33S, 13C and 2H isotopologues. (d) overall spectrum of methionine 2TMS with two major fragments (128.08955 Da and 176.09290 Da) and (e) magnification showing m + 2 isotopologues. (f) relationship between expected and observed relative abundance of isotopologues in IMT using two fragments (blue and green) and methionine 2TMS (pink), plotted using a log scale. Note the underestimation of deuterium (2H) content (red circle). The black line stands for the 1:1 line. In (a) and (d), fragmentations are shown with chemical formulas to illustrate how the fragments of interest are formed. Note IMT does not require derivatisation while methionine is silylated (two TMS groups). In (e), note that there is a common peak for 34S (+1.995796 Da) and 30Si (+1.996844 Da) and also 33S (+0.999387 Da) and 29Si (+0.999568 Da). [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Differentiation of metabolites with exact mass
Exact mass allows one to distinguish m/z features with the same nominal mass but with very slight differences (well below 0.1 Da), reflecting differences in elemental composition. This is particularly useful when retention time of analytes at the origin of similar m/z features are very close. We took advantage of GC‐MS exact mass analyses of authentic standards used to construct the database (available as a Supporting Information Files), to tackle this problem. Amongst most common plant metabolites, many analytes were found to have both similar retention time and identical nominal mass m/z signals, and they are listed in Table 1. This problem can potentially lead to identification mistakes with nominal mass analyses especially when mass spectra are rather similar. Two examples are further illustrated in Figure 3. Ferulic acid 2TMS and tyramine 2TMS have the same retention time (12.09 min) and share the same nominal mass ion 338 a.m.u. However, there is a clear difference in exact mass (Figure 3a,b). Also, their spectra share little similarity (Figure 3a). Malic acid 3TMS and threitol 4TMS have a retention time difference of less than 4 s and share a feature at a nominal mass of 189. In addition, their mass spectra are partly similar, in particular for features <200 a.m.u (Figure 3c). However, the exact mass of features at 189 a.m.u. is readily distinguishable (189.11309 and 189.076741 Da) (Figure 3d).
Table 1.
Common analytes that share both similar nominal mass signals and retention time but can be distinguished using exact mass
| RT window (min) | Nominal mass (a.m.u.) | Analyte | Fragment and exact mass (Da) | Analyte | Fragment and exact mass (Da) |
|---|---|---|---|---|---|
| 4.40−4.42 | 160 | Pentanoic acid 1TMS | 13C‐C8H19OSi | Glyoxylic acid 1TMS | C5H10NO3Si |
| 160.12380 | 160.04299 | ||||
| 8.00−8.32 | 174 | O‐acetylserine 2TMS | C7H16NO2Si | β‐alanine 3TMS | C7H20NSi2 |
| 174.09503 | 174.11343 | ||||
| 8.80−8.86 | 189 | Malic acid 3TMS | C8H21OSi2 | Threitol 4TMS | C7H17O2Si2 |
| 189.11309 | 189.07671 | ||||
| 5.35−5.55 | 190 | 2‐hydroxybutyric acid 2TMS | C7H18O2Si2 | Alanine 2TMS | C7H20NOSi2 |
| 190.08453 | 190.10834 | ||||
| 13.17−13.20 | 191 | Caffeic acid 2TMS | C10H11O2Si | Myoinositol 6TMS | C7H19O2Si2 |
| 191.05283 | 191.09236 | ||||
| 9.85−10.0 | 192 | Anthranilic acid 2TMS | C10H14NOSi | Phenylalanine 2TMS | C11H18Nsi |
| 192.08446 | 192.12085 | ||||
| 12.21−12.24 | 203 | Glucosamine 6TMS | C8H21NOSi2 | Tryptophan 4TMS | 13C‐C12H16NSi |
| 203.11617 | 203.10855 | ||||
| 7.70−7.92 | 204 | Serine 3TMS | C8H22NOSi2 | N‐formylglycine 2TMS | C7H18NO2Si2 |
| 204.12399 | 204.08760 | ||||
| 9.4−9.64 | 205 | Cinnamic acid 1TMS | C11H13O2Si | Glutathione 4TMS | C10H13N2OSi |
| 205.06848 | 205.07971 | ||||
| 6.91–7.13 | 218 | Hydroxypyruvic acid 2TMS | C9H22O2Si2 | Isoleucine 2TMS | C8H20NO2Si2 |
| 218.11583 | 218.10326 | ||||
| 7.94−8.00 | 218 | Threonine 3TMS | C9H24NOSi2 | O‐acetylserine 2TMS | C8H20NO2Si2 |
| 218.13964 | 218.10326 | ||||
| 9.37−9.39 | 220 | Threonic acid 4TMS | C8H20O3Si2 | Cysteine 3TMS | C8H22NSSi2 |
| 220.09510 | 220.10115 | ||||
| 11.82−11.92 | 226 | Methionine sulfoximine 2TMS 2MeOx* | C5H18N4O2SSi | Histamine 3TMS | C10H22N2Si2 |
| 226.09197 | 226.13215 | ||||
| 9.13−9.16 | 230 | Pyroglutamic acid 2TMS | C9H20NO2Si2 | Hydroxyproline 3TMS | C10H24NOSi2 |
| 230.10326 | 230.13964 | ||||
| 8.34−8.70 | 247 | Erythronolactone 2TMS | C9H19O4Si2 | Citramalic acid 3TMS | C10H23O3Si2 |
| 247.08219 | 247.11857 | ||||
| 10.31‐10.42 | 261 | N‐acetylmethionine 2TMS | C10H23NO3Si2 | N‐methylglutamic acid 3TMS | 13C‐C11H26NO2Si2 |
| 261.12165 | 261.15356 | ||||
| 11.78−12.09 | 264 | Adenine 2TMS | C10H18N5Si2 | Tyramine 3TMS | C13H22NOSi2 |
| 264.11007 | 264.12399 | ||||
| 12.66−12.80 | 267 | Urocanic acid 2TMS | C11H19N2O2Si2 | Octopamine 4TMS | C13H23O2Si2 |
| 267.09851 | 267.12366 | ||||
| 11.93‐12.22 | 280 | Pyridoxine 3TMS | C13H22NO2Si2 | Tyrosine 3TMS | C14H26NOSi2 |
| 280.11891 | 280.15529 | ||||
| 12.09−12.26 | 308 | Ferulic acid 2TMS | C14H20O4Si2 | Coumaric acid 2TMS | C15H24O3Si2 |
| 308.09001 | 308.12640 | ||||
| 11.94−12.09 | 308 | 4‐hydroxyphenyl lactic acid 3TMS | C15H24O3Si2 | Ferulic acid 2TMS | C14H20O4Si2 |
| 308.12640 | 308.09001 | ||||
| 12.13−12.38 | 319 | Mannitol 6TMS | C13H31O3Si3 | 3‐indoleacetic acid 2TMS | C16H25NO2Si2 |
| 319.15810 | 319.14238 | ||||
| 12.09 (same RT) | 338 | Ferulic acid 2TMS | C16H26O4Si2 | Tyramine 3TMS | C16H32NOSi3 |
| 338.13696 | 338.17917 |
Note: For each example, the elemental composition of the fragment and its exact mass is shown. When the observed mass of interest belongs to the isotopic pattern, it is indicated with the isotope in front (13C). See also Figure 4 for a detailed analysis of two examples, with fragment chemical structure. This table shows couple of analytes with a difference in retention time of less than 0.4 min. *Note that m/z ions at ≈226.092 Da can also come from a fragment of allantoin 2TMS (C4H18N4O3Si2, 226.09119 Da). All analytes and fragments in this table have been checked using authentic standards.
Abbreviation: a.m.u., atomic mass units.
Figure 3.
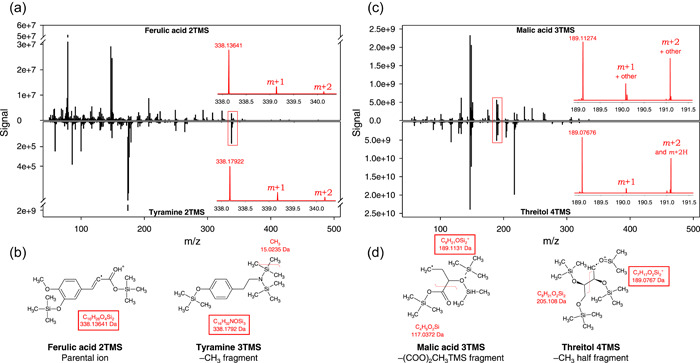
Illustration of some common nominal mass m/z ions between analytes with the same retention time: ferulic acid 2TMS and tyramine 2TMS, both at c. 12.09 min (a, b) and malic acid 3TMS and threitol 4TMS, both at (c) 8.85min (c, d). Comparisons of overall spectra (a, c) with nearly common masses distinguishable with exact mass (insets, in red), and origin of fragments (b, d). In (c), ions ‘other’ refer to C7H18O2Si2 + (190.083983 Da) and its protonated (+1.007825 Da) species (191.091808 Da). Note that tyramine yields a much higher signal (order of magnitude 109) then ferulic acid (107) despite the use of identical concentration in samples (500 ng ml−1). m + 1 and m + 2 refer to +1 a.m.u. (mostly 13C, and 29Si) isotopologues and +2 a.m.u. (mostly 13C2, 30Si and 13C‐29Si) isotopologues, respectively. a.m.u., atomic mass units. [Color figure can be viewed at wileyonlinelibrary.com]
3.4. Arginine derivatives
In terms of biochemical analysis, one of the most problematic metabolites of plant primary N metabolism is arginine, because it can be converted to several by‐products upon derivatisation. It is particularly so during silylation, since the guanidium group of arginine may be cleaved to form ornithine, which in turn can lead to a cyclic form, ornithine lactam. In addition, one N atom of arginine can be lost and form citrulline. Taken as a whole, arginine derivatisation generates ornithine lactam 2TMS, ornithine 3TMS, arginine 3TMS and citrulline 3TMS (Figure 4a). Arginine 3TMS is the minor derivative and has a retention time extremely close to citrulline 3TMS, and therefore it has often been overlooked. Here, arginine 3TMS is visible on the TIC, can also be identified with certainty with exact mass and thus cannot be confused with citrulline 3TMS. Although small, features typical of arginine 3TMS and citrulline 3TMS can be found at 187.108676 Da (guanidium group 2TMS) and 171.08278 Da (decarboxylated citrulline), respectively (Figure 4b−e).
Figure 4.
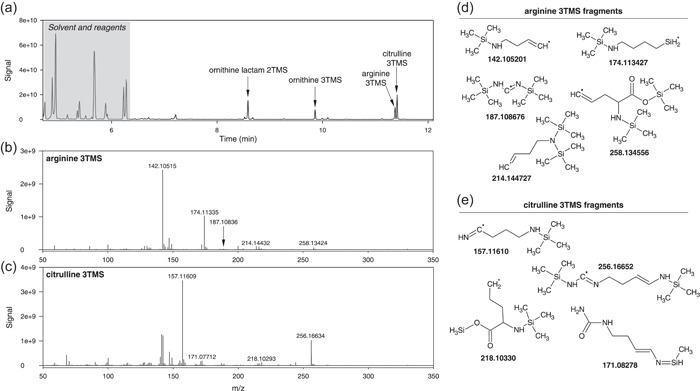
Mass spectrum of arginine derivatives. (a) chromatogram showing the four products of arginine derivatisation: ornithine lactam 2TMS, ornithine 3TMS, arginine 3TMS and citrulline 3TMS. The mass spectrum of arginine 3TMS and citrulline 3TMS are shown in panels (b) and (c). Typical fragments are illustrated in (d) and (e), respectively. Note that arginine 3TMS and citrulline 3TMS have very close retention times (11.37 and 11.41 min) and thus might appear merged in the same peak under low resolutive conditions.
3.5. Identification of s‐containing analytes
The resolution of the isotopic pattern can be exploited to gain information on uncommon (or less common) elements contained by analytes. Nitrogen 15N is not sufficiently abundant (0.36% only) and can be overlapped by 29Si (4.7%) and 33S (0.76%) isotopes. Therefore, the use of the isotopic pattern to detect specific elements in analytes is only suitable for sulphur, provided (1) the signal coming from 34S is high enough (34S abundance is 4.2%), and (2) it can be somehow distinguished from 30Si. The mass excess of the 34S isotopologue is +1.995796 while that of 30Si is +1.996844, meaning a difference of 0.001048 Da (i.e., 1.048‰). If mass resolution during analysis is sufficient, mass excess at nearly exactly +1.995796 can be found. A similar analysis with high resolution LC‐MS (without the issue of Si isotopes) has been undertaken previously (Nakabayashi & Saito, 2017). In Figure 5, automatic searching of mass excess of +1.995796 is shown, with the difference (in ‰) as a function of feature m/z. Using a mixture of authentic standards that includes methionine and cysteine (Figure 5a), only 9 (out of more than 2,000) features were found to fall in the mass excess +1.995796 ± 0.00005 (red shaded area). From these, manual checking allows confirmation of 3 of them, another one (labelled as 4 in Figure 5a) being ambiguous: 1 and 2 are S‐containing fragments of cysteine 2TMS (61.011195 and 131.035071 Da at 5.61 min), while 3 is a fragment of methionine 2TMS (176.092921 at 9.12 min). Four appears at the retention time of methionine 2TMS but in the spectrum, the peak is too wide to ascertain that it is not another fragment with 30Si (i.e., other fragments without S with nearly the same exact mass). Using a plant sample (Arabidopsis leaf extract, Figure 5b), about 10 features fall in the ±0.05‰ window, of which two can be confirmed further: 1 is the same as in Figure 5a (S‐containing fragment of cysteine 2TMS) and 5 is C3H6S (74.018421 Da) and comes from methionine 2TMS, at 9.12 min. The appearance of this feature is further illustrated in Figure 5c, where it is found to be very close to the isotopic pattern of another methionine 2TMS fragment (C2H6OSi). Of course, looking at the mass excess (due to 34S) with a high precision in Figures 5a,b is only possible if resolution is high enough to have a better probability to have an acquired m/z datapoint exactly at, or very close to +1.995796. This is illustrated in Figure 5d−f, where the observed signal (grey) is likely to show a mass signal coming from 34S at medium and high resolution. This can optimally be achieved for relatively small features (m/z < 150 a.m.u.) for which resolution is larger.
Figure 5.
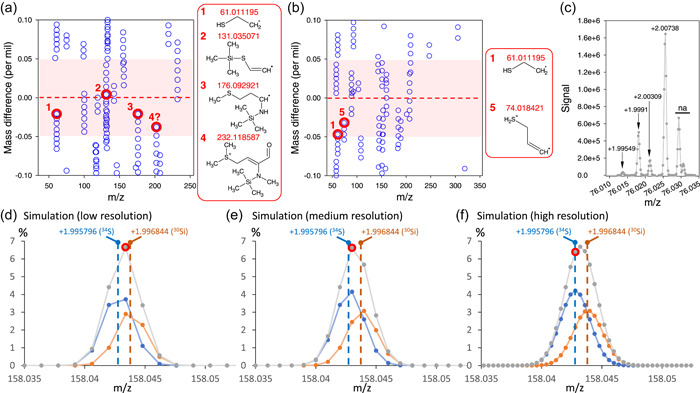
Identification of S‐containing fragments using exact mass data: appearance of m + 2 isotopologues at +1.995796 with a precision of 0.05‰ in a standard mixture containing methionine and cysteine (a) and an extract from Arabidopsis leaves (b). Ions that effectively contain S are circled in red. Other ions do not contain S despite their mass difference value (w.r.t. the isotopologue at +1.995796) close to zero (see main text for calculations). In (a) and (b), red frames illustrate the chemical structure of identified S‐containing fragments. (c) detailed observed isotopic pattern (m + 2 isotopologues) of the fragment ion with a monoisotopic mass of 74.018 Da, showing the 34S isotopologue (observed at +1.99549) of structure 2 in (b), as well as 30Si and 29S + 13C isotopologues of C2H6OSi (74.018791 Da), the monoisotopic form of which also contributes to the observed signal at ≈74.018 Da. The signal at +2.00738 likely corresponds to another fragment rather than the 13C2 isotopologue considering the relatively high signal. Other signals (na) are unrelated to the ion of interest. (d−f) simulation of the peak of m + 2 34S and 30Si isotopologues using the arbitrary example of a monoisotopic mass at 156.04700 Da containing one Si and one S, and three levels of mass resolution. The observed signal (sum) is in grey, while the separate contribution of 34S and 30Si is in blue and orange, respectively. The sampled (i.e., measured by the mass spec) mass that is closest to 34S is circled in red. It shows that there is no sampled mass at exactly +1.995796 (34S mass excess w.r.t. monoisotopic) except at high resolution, and that 34S can be easily confused with 30Si due to their very small mass difference (0.001048 Da). [Color figure can be viewed at wileyonlinelibrary.com]
3.6. Application to plant samples
The compound database used here contains 1,064 target fragments that have all been confirmed with standards, representing 336 compounds with target identification exact mass features, and confirming exact mass features (see Supporting Information: File 1). Using Arabidopsis as a plant model, we found 234 compounds in leaves (266 analytes, some of them generating several derivatives). We assessed the usefulness and repeatability of exact mass analysis by conducting GC‐MS analyses on several Arabidopsis ecotypes (BL1, AN1, Col0, Cvi0, Oy0, Ge0, Mt0 and Sha). Results are shown in Figure 6 as a heatmap of analytes that are significantly different between ecotypes. Hierarchical clustering shows easy discrimination of samples (without any clustering error between ecotypes), suggesting good repeatability. Many metabolites were found to be different between ecotypes, and they are here subdivided into seven groups (Figure 6a). For example, Cvi0 was found to be enriched in various amino acid derivatives, while Shahdara was enriched in sugars. Repeatability (both technical and biological) was assessed by calculating standard errors (SE), expressed in % of mean value, for each genotype. For the vast majority of metabolites, there was a very good reproducibility, with a median value of SE of 9.7% only across all genotypes (Figure 6b). Some metabolites were associated with a high variability, larger than 40%. They were all metabolites present in very low amount, and/or having very poor ionisation, and/or encountering degradation during derivatisation, such that their relative signal was extremely small, 10−5 or less (Figure 6c).
Figure 6.
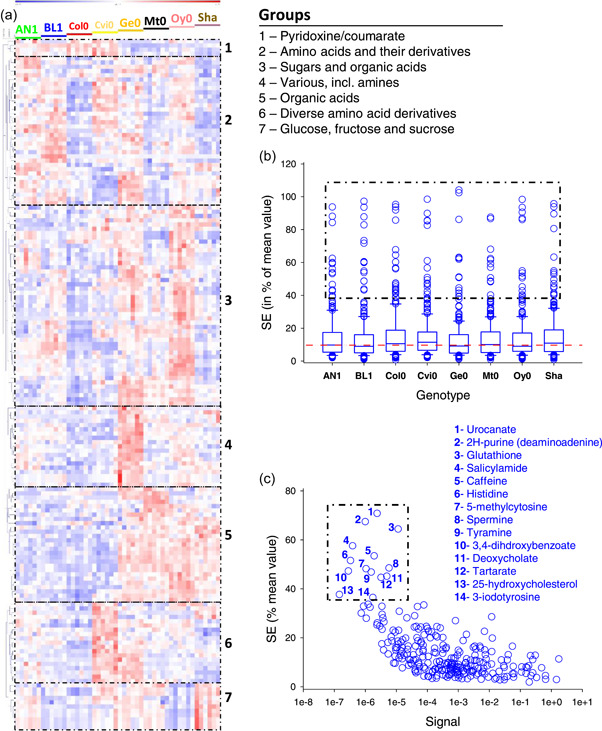
Overview of exact GC‐MS metabolic profiling of Arabidopsis genotypes. (a) heatmap with hierarchical clustering showing groups of metabolites that are significantly different (p < 0.0002, one‐way ANOVA) between genotypes. Numbers refers to groups named on top left. (b) Standard error between 6 biological replicates, in % of average value, in each genotype. The red dotted line stands for the median value(9.7%). The dotted frame comprises metabolites that are also framed and named in panel (c). Data are shown as whisker plots with median, 25% and 75% quartiles, and interquartile range. (c) Relationship between average standard error (in % of overall mean) and relative signal (relative to sucrose set at 1) obtained on the GC‐MS instrument (quantifying target mass). Signals were normalised to the internal standard (ribitol) and dry weight. Metabolites that appear highly variable are framed (and are the same as metabolites framed in panel [b]). Note that metabolite 14 is labelled ‘3‐iodotyrosine’ because it is identified with high certainty by Tracefinder® using the database although it is not expected in plants (see main text). ANOVA, analysis of variance. [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
4.1. Exact mass GC‐MS database for routine analysis
The present curated database contains 336 compounds, 234 of them being identified and quantified in Arabidopsis leaves. This might appear relatively small compared with the total estimated number of small metabolites (several thousands) in plants. However, this compares well with most targeted routine GC‐MS analyses for metabolic profiling, which yield a list of about 80−100 metabolites in the vast majority of cases (e.g., there are 162 metabolites in (Cui, Davanture, et al., 2019), and 178 in (Cui et al., 2021) found in leaves). Of course, the ability of instruments and softwares to extract a proper data set from raw data using the database depends on the quality of analyses. In effect, despite the considerable dynamic range of modern instruments (here, six orders of magnitude in peak height), precise quantification can only be achieved when analytes are not too concentrated (inadequate peak shape does not allow peak extraction by softwares like Tracefinder®) (Kaufmann & Walker, 2017). This can be challenging when some metabolites are present in high amounts (e.g., sucrose or proline) while others are present in trace amounts or generate a weak signal (e.g., salicylamide) (Figure 6). It should be noted that data extraction from raw data can also be processed via untargeted peak searching, providing a much more powerful way to appreciate the diversity of molecules present in extracts (Perez de Souza et al., 2019). However, this has two drawbacks: (1) processing time is very long, at least 20 times slower with Tracefinder®. Note however that other publicly available softwares such as MS‐DIAL (Tsugawa et al., 2015) or MZmine (Pluskal et al., 2010) can be used for high‐resolution data. (2) many peaks would appear as unidentified, with only the m/z value and retention time (and thus post‐hoc identification is required using exact mass and potentially, co‐occurring fragments). Therefore, for routine analyses, it is probably more convenient to rely on targeted analyses with the database we propose here.
4.2. Advantages of exact mass analysis
Nominal mass GC‐MS analyses rely on two main criteria: retention index and mass spectrum (Garcia & Barbas, 2011). A well‐recognised difficulty with mass spectrum analysis and comparisons in biological samples is coelution of analytes, leading to mixed mass spectra. To address this problem, deconvolution algorithms can be used (e.g., in ChromaTOF® or the deconvolution plugin in TraceFinder®) (Du & Zeisel, 2013; H. Lu et al., 2008) or alternatively, data extraction can focus on individual target m/z, for example, in MetabolomeExpress (Carroll et al., 2010). However, there remains a risk that coelution leads to erroneous identification and quantitation when common m/z nominal mass features are present. Here, we have covered this aspect by searching systematically coeluting nominal masses m/z (Table 1, Figure 3). Rather common plant metabolites are concerned by this problem (e.g., malate, serine, etc.). Exact mass offers an efficient way to avoid it since exact mass values of targeted m/z features are clearly different. In addition, the isotopic pattern reflects elemental composition (Figure 2) and can be used as a post‐hoc verification. Erroneous identification is therefore much less probable. Here, using Arabidopsis samples, there was only one case of misidentification: iodotyrosine 3TMS has been identified by TraceFinder® at 16.12 min (Figure 6c). This molecule is very unlikely since enzymes of iodotyrosine metabolism (thyroperoxidase and iodotyrosine deiodinase) are absent in plants (Phatarphekar et al., 2014; Taurog, 1999). Our Arabidopsis analyte list available in Supporting Information Material has been curated accordingly. Coincidentally, several unusual sugar‐containing molecules with similar retention time can generate fragments (m/z features) with the same exact mass. For example, rutinose 6TMS 1Meox elutes just before sucrose 8TMS (16.17 min here) and can form a target fragment at 218.1033 like iodotyrosine (Supporting Information: Figure S1). The observed signal is very low (10−6 that of sucrose 8TMS), suggesting it is a minor compound. Further work is needed (e.g., using MS2 on a purified fraction where this compound is more concentrated and thus generates well‐visible MS² fragments) to determine what compound generates a signal similar to iodotyrosine in Arabidopsis samples.
Another advantage of exact mass analyses is the resolution of isotopologues. This is of particular interest to identify 13C‐molecules upon double‐labelling (typically 13C‐15N or 13C‐33S) since the signal associated with 13C mass excess (+1.003355 Da) is readily visible. This allows direct quantitation of 13C isotopologues unlike in nominal mass analyses where all m + 1 isotopologues are under the same peak. In a recent study, the systematic, nontargeted analysis of 13C isotopologues (+1.003355 Da, +2.006710 Da, +3.010065 Da, etc.) has been used with high resolution LC‐MS to look at variations in leaf metabolites with CO2 and O2 gaseous conditions (Abadie et al., 2021). For 34S‐isotopologues, there is an interference with 30Si (silicium being carried by trimethylsilyl groups). Despite this limitation, it is worth noting that whenever mass resolution is sufficiently high (in particular for low m/z values), it is possible to identify S‐containing fragments (Figure 5). Resolution is less of an issue upon labelling since 34S would prevail over 30Si and thus the peak apex would be closer to the mass excess of 34S (+1.995796 Da).
4.3. Application to plant samples
Here, we took advantage of Arabidopsis samples to show how exact mass GC‐MS analysis can be performed routinely on plant samples, with a rather good outcome, that is, identification and quantification of more than 200 analytes. Another attempt to carry out high‐resolution GC‐MS for primary metabolites and pesticides analysis in Arabidopsis can be found in, for example, (Peterson et al., 2010). In terms of methodology and instrumentation, high resolution exact mass GC‐MS is similar to nominal mass GC‐MS analysis in that it requires standardised derivatisation (optimally with a robotic facility) ensuring that all samples are treated evenly (including the same time lag between the end of derivatisation and injection), and quality controls to assess reproducibility and quantitativity (for a specific discussion about automation of derivatisation for GC‐MS, see (Zarate et al., 2016)). Of course, a specific feature of exact mass analysis is that it requires checking mass accuracy (W. Lu et al., 2017), and here this was performed on a day‐to‐day basis with calibration gas FC43 containing perfluorotributylamine PFTBA (from ThermoFisher Scientific). Also, it is extremely useful to have specific samples to appreciate mass accuracy, for example with a compound that does not require derivatisation like IMT (Figure 2a−b). This allows one to check not only mass accuracy but also precision of mass excess values associated with all relevant isotopes (C, N, S isotopes; Figure 2c).
It should be noted that exact mass GC‐MS, however, does not solve the issue of multiple derivatives for some specific metabolites. In particular, amino acids can form by‐products upon silylation. So is the case of glutamic acid and glutamine, that can both yield pyroglutamate (for a recent discussion on this issue, see (Miyagawa & Bamba, 2019)). It is also the case of arginine, which is known to yield several products (Molnár‐Perl & Katona, 2000), and here it formed four products (Figure 4). High resolution analysis nevertheless provides a method to have a better picture of derivatives and here, we identified four main products of arginine derivatisation: ornithine lactam 2TMS, ornithine 3TMS, arginine 3TMS and citrulline 3TMS. Since these different compounds do not have the same response coefficient in the mass spectrometer (i.e., distinct observed signal response curve to concentration), arginine cannot be quantified very precisely by this method.
Basically, GC‐MS analysis provides semiquantitative information on plant samples. A good method to have information on effective quantity (i.e., absolute quantity) of analytes is GC‐C‐IRMS (GC coupled to combustion and isotope ratio mass spectrometry). In effect, the same sample (same derivatisation) can be injected and the GC‐C‐IRMS converts quantitatively each peak to CO2 and N2 and thus can provide direct information on the amount of carbon (via mass 44 monitoring) or nitrogen (via mass 28 monitoring) in each peak of interest. This method has been used recently (using alfalfa seed protein extracts) and shown to provide satisfactory estimates of absolute amino acid contents (Domergue et al., 2022). More classical method can also be used, such as external calibration curves, or deuterated standards (internal references). The use of deuterated standards has three drawbacks: Firstly, it complicates mass spectra, with some probability to coincide with m/z features of interest. An example with plant samples is cysteine 3TMS where the target peak (C8H22NSSi2) at 220.1009 Da might coincide with a deuterated fragment ([2H]‐C7H21NOSi3 and 220.0995 Da). Secondly, having all deuterated standards is tedious and expensive. Thirdly, deuteration can cause an isotope effect in either chromatography or ionisation efficiency so that the GC‐MS signal differs between deuterated and protiated forms (Alzweiri et al., 2015; Caban & Stepnowski, 2020; Matucha et al., 1991; Ripszam et al., 2013).
4.4. Perspectives
Our results show that high resolution exact mass GC‐MS can be used in plants, as an efficient way to carry out metabolic profiling. This technique shows several advantages over classical, nominal mass GC‐MS, such as identification less prone to errors thanks to exact mass m/z features. The database we propose here to perform targeted analyses covers more than 200 metabolites of different chemical families. This database is evolutive, in that it will be enriched over the coming years and updated versions kept accessible via the journal website. We recognise that plants contain a lot of sugars, with many isomers. Some of them have identical retention times, and thus cannot be resolved with exact mass since m/z features have the same elemental composition. To solve this problem, specific devices like ion mobility may be required (Morrison & Clowers, 2018; Mu et al., 2018; Przybylski & Bonnet, 2021). In terms of isotopic analysis via exact mass GC‐MS, Si isotopes represent a limitation to distinguish isotopologues. We showed that high mass precision allows reasonable access to 34S isotopologues, but less so for 15N or 33S that are much less abundant. When isotopic enrichments are modest, GC‐C‐IRMS analysis is the best alternative, but this technology is presently not able to perform 34S analysis. Therefore, exact mass LC‐MS seems to be better suited to isotopic analysis of sulphur‐containing compounds.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
Supplementary information.
Supplementary information.
ACKNOWLEDGMENTS
Authors acknowledge the support of the Région Pays de la Loire and Angers Loire Métropole via the grant Isoseed, awarded to G. T. Authors also thank Thermo Scientific for helping us setting up the robotic facility and modifying programs to fix the sample preparation sequence.
Abadie, C. , Lalande, J. & Tcherkez, G. (2022) Exact mass GC‐MS analysis: protocol, database, advantages and application to plant metabolic profiling. Plant, Cell & Environment, 45, 3171–3183. 10.1111/pce.14407
REFERENCES
- Abadie, C. , Lalande, J. , Limami, A.M. & Tcherkez, G. (2021) Non‐targeted 13C metabolite analysis demonstrates broad re‐orchestration of leaf metabolism when gas exchange conditions vary. Plant, Cell & Environment, 44(2), 445–457. [DOI] [PubMed] [Google Scholar]
- Allwood, J.W. , De Vos, R.C. , Moing, A. , Deborde, C. , Erban, A. , Kopka, J. et al. (2011) Plant metabolomics and its potential for systems biology research: background concepts, technology, and methodology. Methods in Enzymology, 500, 299–336. [DOI] [PubMed] [Google Scholar]
- Alzweiri, M. , Khanfar, M. & Al‐Hiari, Y. (2015) Variations in GC–MS response between analytes and deuterated analogs. Chromatographia, 78(3), 251–258. [Google Scholar]
- Bowne, J.B. , Erwin, T.A. , Juttner, J. , Schnurbusch, T. , Langridge, P. , Bacic, A. et al. (2012) Drought responses of leaf tissues from wheat cultivars of differing drought tolerance at the metabolite level. Molecular Plant, 5(2), 418–429. [DOI] [PubMed] [Google Scholar]
- Caban, M. & Stepnowski, P. (2020) The application of isotopically labeled analogues for the determination of small organic compounds by GC/MS with selected ion monitoring. Analytical Methods, 12(30), 3854–3864. [DOI] [PubMed] [Google Scholar]
- Carroll, A.J. , Badger, M.R. & Harvey Millar, A. (2010) The MetabolomeExpress Project: enabling web‐based processing, analysis and transparent dissemination of GC/MS metabolomics datasets. BMC Bioinformatics, 11(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, J. , Abadie, C. , Carroll, A. , Lamade, E. & Tcherkez, G. (2019) Responses to K deficiency and waterlogging interact via respiratory and nitrogen metabolism. Plant, Cell & Environment, 42(2), 647–658. [DOI] [PubMed] [Google Scholar]
- Cui, J. , Davanture, M. , Lamade, E. , Zivy, M. & Tcherkez, G. (2021) Plant low‐K responses are partly due to Ca prevalence and the low‐K biomarker putrescine does not protect from Ca side effects but acts as a metabolic regulator. Plant, Cell & Environment, 44(5), 1565–1579. [DOI] [PubMed] [Google Scholar]
- Cui, J. , Davanture, M. , Zivy, M. , Lamade, E. & Tcherkez, G. (2019) Metabolic responses to potassium availability and waterlogging reshape respiration and carbon use efficiency in oil palm. New Phytologist, 223(1), 310–322. [DOI] [PubMed] [Google Scholar]
- De Vos, R.C.H. , Moco, S. , Lommen, A. , Keurentjes, J.J.B. , Bino, R.J. & Hall, R.D. (2007) Untargeted large‐scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nature Protocols, 2(4), 778–791. [DOI] [PubMed] [Google Scholar]
- Doerfler, H. , Sun, X. , Wang, L. , Engelmeier, D. , Lyon, D. & Weckwerth, W. (2014) mzGroupAnalyzer‐predicting pathways and novel chemical structures from untargeted high‐throughput metabolomics data. PLoS One, 9(5), e96188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domergue, J.‐B. , Lalande, J. , Abadie, C. & Tcherkez, G. (2022) Compound‐Specific 14N/15N analysis of amino acid trimethylsilylated derivatives from plant seed proteins. International Journal of Molecular Sciences, 23(9), 4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X. & Zeisel, S.H. (2013) Spectral deconvolution for gas chromatography mass spectrometry‐based metabolomics: current status and future perspectives. Computational and Structural Biotechnology Journal, 4(5), e201301013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaquerel, E. , Kuhl, C. & Neumann, S. (2013) Computational annotation of plant metabolomics profiles via a novel network‐assisted approach. Metabolomics, 9(4), 904–918. [Google Scholar]
- Garcia, A. & Barbas, C. (2011) Gas chromatography‐mass spectrometry (GC‐MS)‐based metabolomics, Metabolic Profiling. Springer, pp. 191–204. [DOI] [PubMed] [Google Scholar]
- Ghatak, A. , Chaturvedi, P. & Weckwerth, W. (2018) Metabolomics in plant stress physiology. Plant Genetics and Molecular Biology, 164, 187–236. [DOI] [PubMed] [Google Scholar]
- Högy, P. , Keck, M. , Niehaus, K. , Franzaring, J. & Fangmeier, A. (2010) Effects of atmospheric CO2 enrichment on biomass, yield and low molecular weight metabolites in wheat grain. Journal of Cereal Science, 52(2), 215–220. [Google Scholar]
- Jansen, J.J. , Allwood, J.W. , Marsden‐Edwards, E. , van der Putten, W.H. , Goodacre, R. & van Dam, N.M. (2009) Metabolomic analysis of the interaction between plants and herbivores. Metabolomics, 5(1), 150–161. [Google Scholar]
- Kaufmann, A. & Walker, S. (2017) Comparison of linear intrascan and interscan dynamic ranges of Orbitrap and ion‐mobility time‐of‐flight mass spectrometers. Rapid Communications in Mass Spectrometry, 31(22), 1915–1926. [DOI] [PubMed] [Google Scholar]
- Kind, T. & Fiehn, O. (2006) Metabolomic database annotations via query of elemental compositions: mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics, 7(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H. , Liang, Y. , Dunn, W.B. , Shen, H. & Kell, D.B. (2008) Comparative evaluation of software for deconvolution of metabolomics data based on GC‐TOF‐MS. TrAC, Trends in Analytical Chemistry, 27(3), 215–227. [Google Scholar]
- Lu, W. , Su, X. , Klein, M.S. , Lewis, I.A. , Fiehn, O. & Rabinowitz, J.D. (2017) Metabolite measurement: pitfalls to avoid and practices to follow. Annual Review of Biochemistry, 86, 277–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov, A. (2000) Electrostatic axially harmonic orbital trapping: a high‐performance technique of mass analysis. Analytical Chemistry, 72(6), 1156–1162. [DOI] [PubMed] [Google Scholar]
- Makarov, A. , Denisov, E. & Lange, O. (2009) Performance evaluation of a high‐field Orbitrap mass analyzer. Journal of the American Society for Mass Spectrometry, 20(8), 1391–1396. [DOI] [PubMed] [Google Scholar]
- Matsuda, F. , Nakabayashi, R. , Sawada, Y. , Suzuki, M. , Hirai, M.Y. & Kanaya, S. et al. (2011) Mass spectra‐based framework for automated structural elucidation of metabolome data to explore phytochemical diversity. Frontiers in plant science, 2, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matucha, M. , Jockisch, W. , Verner, P. & Anders, G. (1991) Isotope effect in gas—liquid chromatography of labelled compounds. Journal of Chromatography A, 588(1), 251–258. [Google Scholar]
- Misra, B.B. (2021) Advances in high resolution GC‐MS technology: a focus on the application of GC‐Orbitrap‐MS in metabolomics and exposomics for FAIR practices. Analytical Methods, 13(20), 2265–2282. [DOI] [PubMed] [Google Scholar]
- Misra, B.B. & Chen, S. (2015) Advances in understanding CO2 responsive plant metabolomes in the era of climate change. Metabolomics, 11(6), 1478–1491. [Google Scholar]
- Miyagawa, H. & Bamba, T. (2019) Comparison of sequential derivatization with concurrent methods for GC/MS‐based metabolomics. Journal of Bioscience and Bioengineering, 127(2), 160–168. [DOI] [PubMed] [Google Scholar]
- Molnár‐Perl, I. & Katona, Z.F. (2000) GC‐MS of amino acids as their trimethylsilyl/t‐butyldimethylsilyl Derivatives: in model solutions III. Chromatographia, 51(1), S228–S236. [Google Scholar]
- Morrison, K.A. & Clowers, B.H. (2018) Contemporary glycomic approaches using ion mobility–mass spectrometry. Current Opinion in Chemical Biology, 42, 119–129. [DOI] [PubMed] [Google Scholar]
- Mu, Y. , Schulz, B.L. & Ferro, V. (2018) Applications of ion mobility‐mass spectrometry in carbohydrate chemistry and glycobiology. Molecules (Basel, Switzerland), 23(10), 2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi, R. & Saito, K. (2015) Integrated metabolomics for abiotic stress responses in plants. Current Opinion in Plant Biology, 24, 10–16. [DOI] [PubMed] [Google Scholar]
- Nakabayashi, R. & Saito, K. (2017) Ultrahigh resolution metabolomics for S‐containing metabolites. Current Opinion in Biotechnology, 43, 8–16. [DOI] [PubMed] [Google Scholar]
- Perez de Souza, L. , Alseekh, S. , Naake, T. & Fernie, A. (2019) Mass spectrometry‐based untargeted plant metabolomics. Current Protocols in Plant Biology, 4(4), e20100. [DOI] [PubMed] [Google Scholar]
- Peterson, A.C. , McAlister, G.C. , Quarmby, S.T. , Griep‐Raming, J. & Coon, J.J. (2010) Development and characterization of a GC‐enabled QLT‐Orbitrap for high‐resolution and high‐mass accuracy GC/MS. Analytical Chemistry, 82(20), 8618–8628. [DOI] [PubMed] [Google Scholar]
- Phatarphekar, A. , Buss, J.M. & Rokita, S.E. (2014) Iodotyrosine deiodinase: a unique flavoprotein present in organisms of diverse phyla. Molecular BioSystems, 10(1), 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskal, T. , Castillo, S. , Villar‐Briones, A. & Oresic, M. (2010) MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry‐based molecular profile data. BMC Bioinformatics, 11, Article no. 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybylski, C. & Bonnet, V. (2021) Discrimination of isomeric trisaccharides and their relative quantification in honeys using trapped ion mobility spectrometry. Food Chemistry, 341, Article no. 128182. [DOI] [PubMed] [Google Scholar]
- Qiu, F. , Fine, D.D. , Wherritt, D.J. , Lei, Z. & Sumner, L.W. (2016) PlantMAT: a metabolomics tool for predicting the specialized metabolic potential of a system and for Large‐Scale metabolite identifications. Analytical Chemistry, 88(23), 11373–11383. [DOI] [PubMed] [Google Scholar]
- Ripszam, M. , Grabic, R. & Haglund, P. (2013) Elimination of interferences caused by simultaneous use of deuterated and carbon‐13 standards in GC‐MS analysis of polycyclic aromatic hydrocarbons (PAHs) in extracts from passive sampling devices. Analytical Methods, 5(12), 2925–2928. [Google Scholar]
- Roessner, U. & Bowne, J. (2009) What is metabolomics all about? Biotechniques, 46(5), 363–365. [DOI] [PubMed] [Google Scholar]
- Sanchez, D.H. , Schwabe, F. , Erban, A. , Udvardi, M.K. & Kopka, J. (2012) Comparative metabolomics of drought acclimation in model and forage legumes. Plant, Cell & Environment, 35(1), 136–149. [DOI] [PubMed] [Google Scholar]
- Shulaev, V. , Cortes, D. , Miller, G. & Mittler, R. (2008) Metabolomics for plant stress response. Physiologia Plantarum, 132(2), 199–208. [DOI] [PubMed] [Google Scholar]
- Taurog, A. (1999) Molecular evolution of thyroid peroxidase. Biochimie, 81(5), 557–562. [DOI] [PubMed] [Google Scholar]
- Tsugawa, H. , Cajka, T. , Kind, T. , Ma, Y. , Higgins, B. , Ikeda, K. et al. (2015) MS‐DIAL: data‐independent MS/MS deconvolution for comprehensive metabolome analysis. Nature Methods, 12(6), 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinaixa, M. , Schymanski, E.L. , Neumann, S. , Navarro, M. , Salek, R.M. & Yanes, O. (2016) Mass spectral databases for LC/MS‐ and GC/MS‐based metabolomics: state of the field and future prospects. TrAC, Trends in Analytical Chemistry, 78, 23–35. [Google Scholar]
- Zarate, E. , Boyle, V. , Rupprecht, U. , Green, S. , Villas‐Boas, S.G. & Baker, P. et al. (2016) Fully automated trimethylsilyl (TMS) derivatisation protocol for metabolite profiling by GC‐MS. Metabolites, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J. , Johnson, M. , Mandal, R. & Wishart, D.S. (2021) A comprehensive targeted metabolomics assay for crop plant sample analysis. Metabolites, 11(5), 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Supplementary information.