

Abstract
Leber hereditary optic neuropathy is a mitochondrial disease mainly due to pathologic mutations in mitochondrial genes related to the respiratory complex I of the oxidative phosphorylation system. Genetic, physiological, and environmental factors modulate the penetrance of these mutations. We report two patients suffering from this disease and harboring a m.15950G > A mutation in the mitochondrial DNA‐encoded gene for the threonine transfer RNA. We also provide evidences supporting the pathogenicity of this mutation.
Keywords: LHON, mtDNA, mutation, tRNA
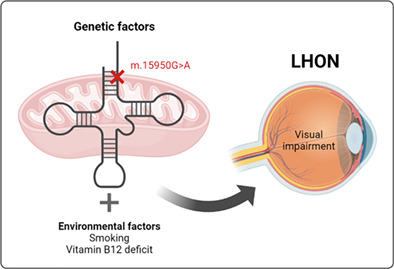
1. INTRODUCTION
Leber hereditary optic neuropathy (LHON) (OMIM#535000) is characterized by bilateral subacute loss of vision due to the preferential death of retinal ganglion cells (RGC) within the inner retina, resulting in optic nerve degeneration. 1 Additional extraocular abnormalities have been described in some LHON pedigrees, including a multiple sclerosis‐like presentation known as Harding disease. 1
About 90% of LHON patients of Northern European descent carry the m.3460G > A, m.11778G > A or m.14484T > C pathogenic variants in genes for mitochondrial DNA (mtDNA)‐encoded respiratory complex I (CI) subunits. Some other rare LHON‐associated mtDNA pathogenic variants are also located in genes for CI subunits. 1 LHON has been recently associated with peculiar combinations of individually non‐pathogenic missense mtDNA variants, affecting genes for mtDNA‐encoded CI subunits. 2 Very interestingly, several genetic variants in nuclear DNA (nDNA) genes, such as NDUFS2 that codes for a CI structural subunit; NDUFAF5 that codes for a CI assembly factor; and DNAJC30 that codes for a chaperone protein needed for CI repair have been also associated with LHON. 3 , 4 Therefore, a dysfunctional CI appears to be the main etiologic factor for LHON.
Interestingly, many of the individuals carrying the CI pathogenic variant do not seem to develop the disease. This is known as incomplete penetrance. Several genetic and physiological factors have been shown to modulate the penetrance in LHON. 5 Evenly, the vulnerability to toxic compounds or nutritional deficiencies may be different in carriers of the CI pathogenic variant. 6 , 7
Here, we report a LHON and a Harding disease patient from different pedigrees and their associated risk factors (Case Reports in Appendix S1). We also show the evidences that support the pathogenicity of the mutation that harbor in the MT‐TT gene, which encodes for the mitochondrial tRNAThr (Material and Methods in Appendix S1).
2. RESULTS
After ruling out the three most frequent LHON pathological mutations, we sequenced the whole mtDNAs and, besides the polymorphisms defining mtDNA haplogroup H3 in both patients, we found two private homoplasmic mutations in patient 1: m.14053A > G in MT‐ND5 and m.15950G > A in MT‐TT (b15950H3‐1, GenBank MW626912) and two private homoplasmic mutations in patient 2: m.4435A > G in MT‐TM and m.15950G > A in MT‐TT (b15950H3‐2, GenBank MZ064555) (Figure 1A,B).
FIGURE 1.
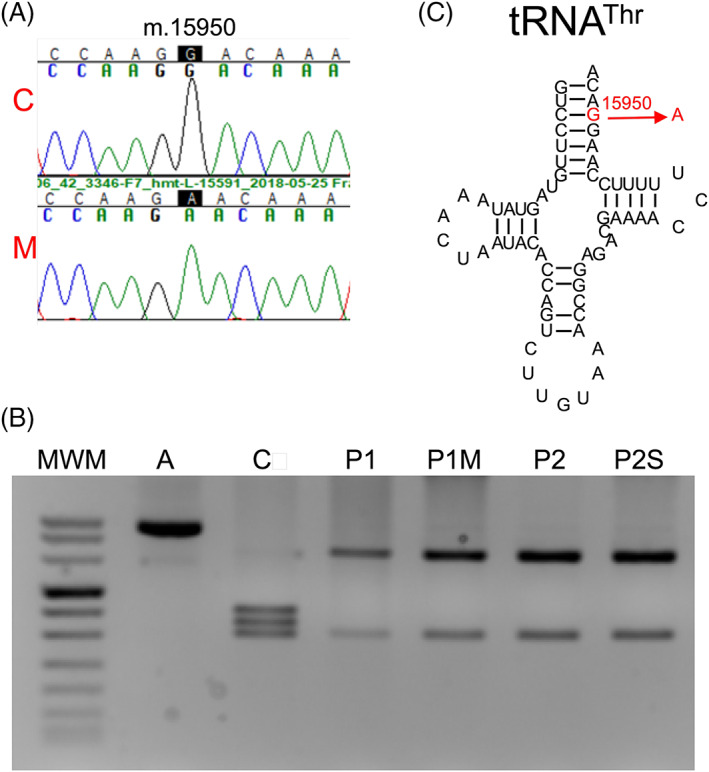
Genetic analysis of the patients. (A) Electropherograms showing the m.15950G wildtype (C) and m.15950A mutated (M, patient 1) alleles. (B) Gel showing the pattern of restriction fragment length polymorphisms from wildtype and m.15950A alleles. A, amplicon; C, control; MWM, molecular weight marker; P1, patient 1; P1M, patient 1's mother; P2, patient 2; P2S, patient 2's sister. (C) Threonine transfer RNA. The m.15950G > A mutation in the acceptor stem is indicated in red color [Colour figure can be viewed at wileyonlinelibrary.com]
The m.14053A > G transition has been found in 1049 out of 304 225 sequences from Mitomap (http://www.mitomap.org. June 22, 2021). This mutation provokes a p.MT‐ND5:T573A substitution. The T573 is conserved in 4% of 5159 p.MT‐ND5 sequences from protists to mammals, 8 and predictors of pathogenicity qualified this change as benign (Mitomap). The m.4435A > G transition has been found in 206 sequences from Mitomap. The MitoTIP predictor considers this mutation as probably benign.
On the other side, the m.15950G > A transition identified in these two independent patients was also found in a patient suffering from Parkinson disease (Mitomap), in a patient with a Tic disorder (Mitomap), in another patient suffering from juvenile myopathy, encephalopathy, lactic acidosis and stroke (MELAS) (gnomAD3.1), and 14 times in Mitomap, a population frequency lower than that of the commonest LHON mutations. The mutation affects the mitochondrial tRNAThr (Figure 1C). The G, at nucleotide position 70 numbered according to conventional rules, is conserved in 130 out of 135 (96.3%) MT‐TT sequences from different organisms (http://mamit-trna.u-strasbg.fr/). This mutation breaks a Watson‐Crick bp in the acceptor stem that is conserved in 133 out of 135 (98.5%) mitochondrial tRNAThr from different organisms. Moreover, it was reported that this mutation provoked a slower migration in native polyacrylamide gel electrophoresis and reduced the tRNA melting temperature, both suggesting a structural alteration. 9 The MitoTIP predictor considers this mutation as possibly pathogenic (Mitomap).
To rule out nuclear genetic variants as ethiologic factors for these phenotypes, we performed whole exome sequencing, but no pathogenic or probably pathogenic variants related to the clinical phenotype were found (Table S1).
Patient's 1 mother and the younger sister of patient 2, with no vision problems, were homoplasmic for the m.15950G > A mutation. The blood mtDNA levels of patient 1's mother were higher than those of her daughter. In the case of patient 2's sister, they were slightly lower than those of her brother (Figure S3).
To determine the pathogenicity of the m.15950G > A genetic variant, we carried out functional studies. For this, we generated osteosarcoma 143B cybrids from a control individual and the patient 1. By genetic fingerprint, we confirmed that these cybrids shared the same nDNA genetic background, the one of osteosarcoma 143B rho0 cell line. We also checked the presence or absence of the m.15950G > A mutation (Figure 2A). The control cybrids harbored a mtDNA sequence from haplogroup H3 (A1H3, GenBank JX081999.1). mtDNA levels were not different between cybrids.
FIGURE 2.
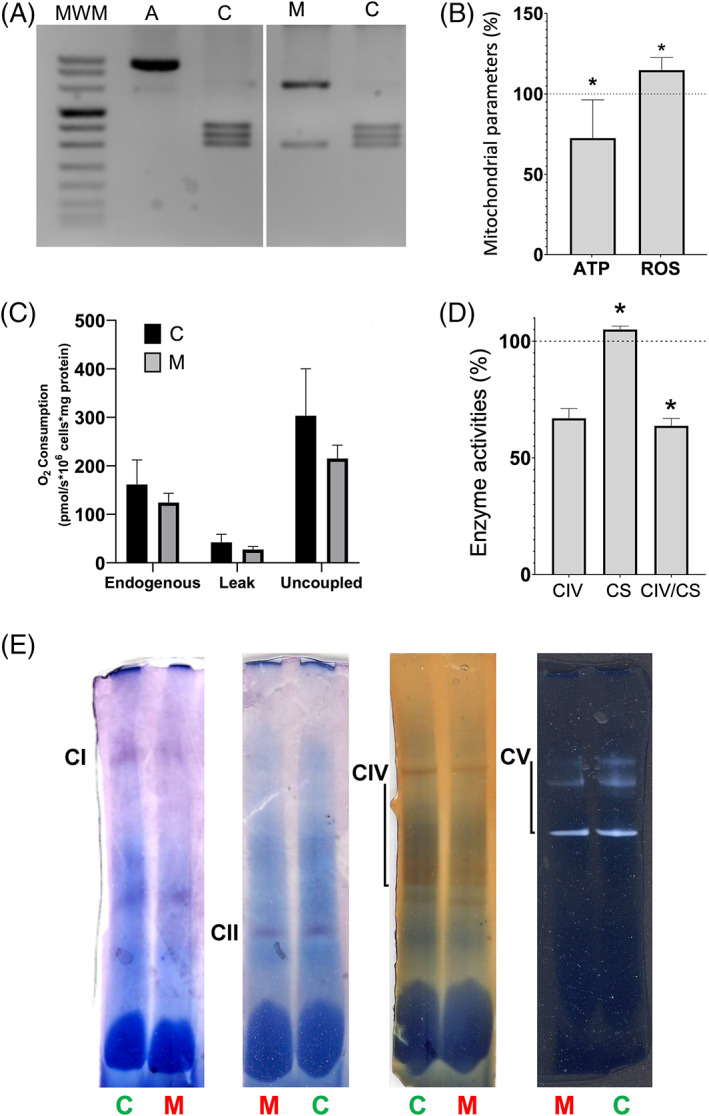
Biochemical variables in control and mutant cybrids. (A) Gel showing the pattern of restriction fragment length polymorphisms from m.15950G and m.15950A alleles in control (C) and mutant (M, patient 1) cybrids, respectively. A, amplicon; MWM, molecular weight marker. (B) Adenosine triphosphate (ATP) amount and reactive oxygen species (ROS) levels. (C) Oxygen consumption. (D) Respiratory complex IV (CIV) and citrate synthase (CS) specific activities and CIV/CS ratio. (E) Respiratory complex I (CI), II (CII), IV (CIV) and ATP synthase (CV) in gel activities. Mean percentages ± standard deviations (SD) are represented. Dotted lines (100%) indicate the mean values in C cybrids. Asterisks, p ≤ 0.0495 [Colour figure can be viewed at wileyonlinelibrary.com]
The adenosine triphosphate (ATP) amount and reactive oxygen species (ROS) levels were significantly decreased and increased, respectively, in mutant cybrids (Figure 2B). Although not statistically significant, basal and uncoupled oxygen consumption was reduced in mutant cybrids (Figure 2C). The citrate synthase (CS) specific activity was higher and respiratory complex IV (CIV)/CS ratio was lower in mutant cybrids (Figure 2D). The CI, CIV and ATP synthase (CV) in gel activities were also decreased in mutant cybrids (Figure 2E). The CIV p.MT‐CO1 subunit levels were reduced in mutant cybrids (Figure 3A). Finally, a mitochondrial protein synthesis assay showed a moderate reduction in the amount of the mtDNA‐encoded polypeptides (Figure 3B,C).
FIGURE 3.
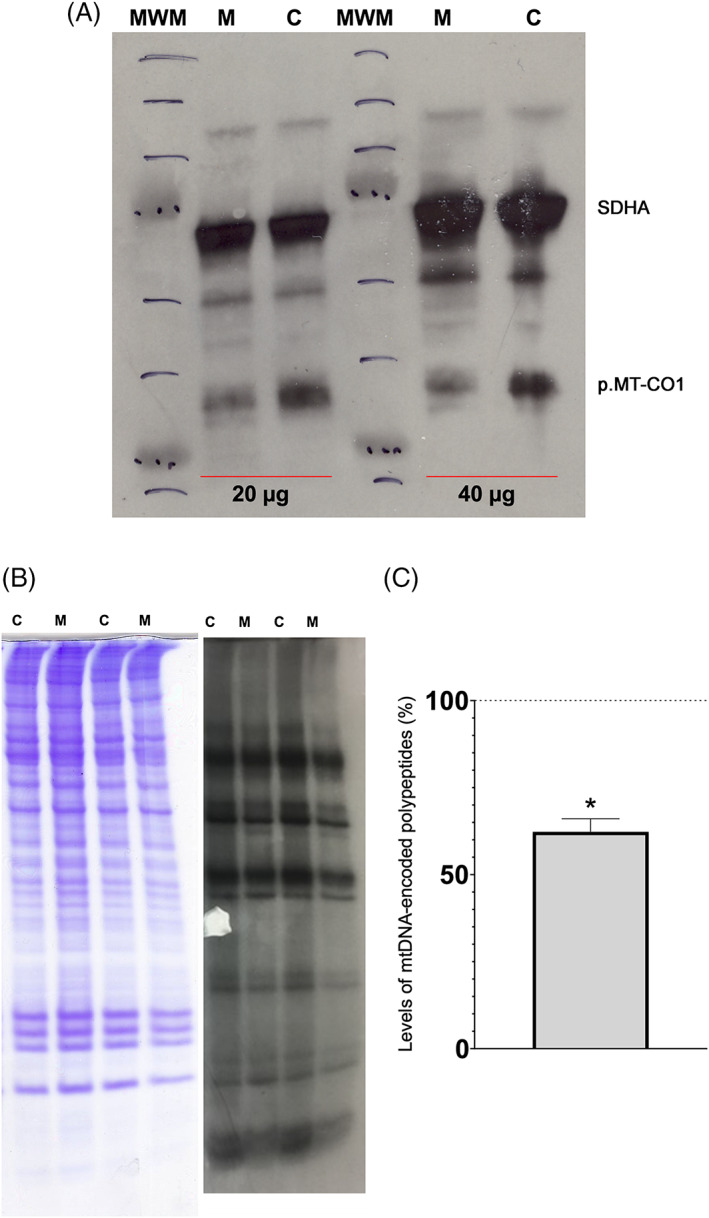
Mitochondrial proteins in control (C) and mutant (M) cybrids. (A) Levels of respiratory complex subunits. Representative image of an immunoblot showing the mitochondrial DNA‐encoded p.MT‐CO1 subunit from respiratory complex IV (CIV). The nuclear DNA‐encoded SDHA subunit from respiratory complex II (CII) is shown as a loading control. The amount of loaded protein is indicated (20 and 40 μg). MWM, molecular weight marker. (B) Mitochondrial protein synthesis. Gel shows loading control (left) and electrophoretic patterns of mitochondrial translation products (right) from control and mutant cybrids. (C) Graphic quantifying the reduction in mitochondrial protein synthesis. Mean percentage ± standard deviation (SD) is represented. Dotted line (100%) indicates the mean value in the C cybrid. Asterisk, p = 0.0104 [Colour figure can be viewed at wileyonlinelibrary.com]
3. DISCUSSION
We report two independent LHON and Harding disease patients harboring a m.15950G > A transition. Both phenotypes slightly vary from the most frequent ones in LHON and Harding disease due to pathological mutations in mtDNA‐encoded CI genes. For example, the fundus at diagnosis was normal in both cases, but this occurs in 20%–40% of LHON cases. 10 Furthermore, LE vision improved in the patient suffering from Harding disease. However, this observation has also been previously described in 30% of Harding disease patients. 11
The patients phenotype (ACMG‐PP4 criterium) 12 ; the extremely low population frequency of this genetic variant (ACMG‐PS4 criterium); the high interspecific conservation of the affected tRNAThr nucleotide; the results of the programs to predict pathogenicity (ACMG‐PP3 criterium); the reported structural alteration provoked by this genetic variant, along with the results of the functional assays in cybrids, in particular those related with the mtDNA‐encoded subunits amount (ACMG‐PM10 criterium) indicated that this genetic variant could be considered as a pathogenic variant and responsible for the LHON disease of these patients.
It was published that the m.4435A > G transition could increase the penetrance of the m.11778G > A LHON mutation. 13 Similarly, the m.4435A > G transition might contribute to trigger the phenotype in patient 2 harboring the m.15950G > A mutation. However, his healthy sister was also homoplasmic for this mutation. Therefore, other factors must be involved.
Environmental factors can also impact the expression of mtDNA mutations. Patient 1 was treated with hydroxychloroquine (HCQ) and a critical long‐term adverse event of this drug is vision‐threatening toxic retinopathy. 14 In human cell lines, HCQ‐treatment resulted in a marked reduction in the oxygen consumption rate. 15 However, HCQ has not been associated with nerve optic impairment. This woman was a heavy smoker, and cigarette smoking has been implicated as a disease trigger. 16 Therefore, this fact could explain why patient 1, but not her non‐smoker homoplasmic mother, was affected showing visual alterations and also multiple sclerosis signs.
Patient 2 showed a deficiency in vitamin B12 and folate. A vitamin B12 deficiency has been shown to trigger LHON in individuals harboring the m.11778G > A or m.14484T > C primary mutations. Moreover, in a child harboring a m.13816A > G genetic variant of unknown significance in the MT‐ND5 gene, a severe vitamin B12 deficiency sparked an LHON‐like optic neuropathy, and in a young woman harboring a m.14468T > C transition of unknown significance in the MT‐ND6 gene, a vitamin B12 deficiency trigger a LHON phenotype. 17 Vitamin B12 and folate are important to fight some processes intimately link to LHON such as the oxidative damage and the low mtDNA levels. Vitamin B12 is an intracellular superoxide scavenger. 18 Both vitamin B12 and folate are important for thymidine synthesis and DNA replication and the levels of these two vitamins positively correlated with the mtDNA levels. 19 Interestingly, several risk factors for LHON decrease mtDNA content, 5 and mtDNA copy number can differentiate LHON patients from unaffected mutation carriers. 20
The presence of patient 1 m.14053A > G and patient 2 m.4435A > G private mutations in a common genetic background, mtDNA haplogroup H3, suggests that m.15950G > A is not a very recent mtDNA genetic variant. Antiquity is a criterium widely used in mitochondrial medicine to rule out a genetic change as being a pathological mutation. However, these results prevent against simplistic genetic approaches that do not consider the potential effect of environmental conditions. Hence, these results suggest that some relatively ancient genetic variants in mtDNA can be deleterious in association with particular environmental conditions.
Two‐hundred and six out of 348 (59.2%) threonines from mtDNA‐encoded polypeptides are found in CI subunits. Therefore, this m.15950G > A mutation in MT‐TT will affect the tRNAThr and the synthesis of all mtDNA‐encoded polypeptides but very particularly those from CI. This fact could be the link between this genetic variant in the apparatus of mitochondrial protein synthesis and LHON. Similar to our mutant cybrid, an elevated ROS generation is frequently found in osteosarcoma 143B cybrids harboring the commonest LHON mutations. 21 As previously commented, DNAJC30 is involved in the efficient exchange of CI subunits exposed to ROS. 4 Perhaps, an excess of ROS production overpasses the capacity of DNAJC30 to repair CI and this favors the development of LHON in individuals without pathologic mutations in directly CI‐related genes. In this sense, a vitamin B12 deficiency would also increase ROS levels and the risk of developing LHON.
Overall, our work identifies and characterizes a new mutation associated with LHON in a mtDNA‐encoded tRNA gene. This result, along with those of new nDNA mutations associated to LHON, points out that although CI deficiency may be the main etiologic factor for the disease, mutations in genes that act in very different pathways can converge in a CI deficiency leading to LHON.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.14189.
Supporting information
Figure S1 Ophthalmological examination of patient 1. (A) Retinography of the left (on the left) and right eye (on the right). Bilateral optic disc pallor is more evident on the right eye. (B) Automated static perimetry of the left eye (on the left) showed a concentric visual field defect and the right eye (on the right) with a deep generalized defect. (C) Peripapillary retinal nerve fiber layer thickness measured by spectral domain‐optical coherence tomography revealed thinning on superior, inferior and temporal sectors in the right eye and thinning on the temporal sector in the right eye. (D) Automated static perimetry four years after the subacute episode. On the left eye (on the left) showed an important improvement on visual field defect (only an inferior peripheral defect) and on the right eye (on the right) with a slight improvement but persistent deep central defect.
Figure S2. Ophthalmological examination of patient 2. (A) Peripapillary retinal nerve fiber layer spectral domain optical coherence tomography showing a progressive thinning from the initial visit. (B) Profound loss of retinal ganglion cells in both eyes. (C) Photograph of the patient's right fundus and left fundus showing a diffuse pallor in both optic nerves. (D) Humphrey visual field test revealing a profound defect in both eyes.
Figure S3. Blood mtDNA levels. The mtDNA amount in the index cases was considered 100%. In patient 2, mtDNA levels were studied seven months after vitamin B12 treatment was initiated. P1, patient 1; P1M, patient 1's mother; P2, patient 2; P2S, patient 2's sister. **, P = 0.0079; ***, P < 0.0001.
Table S1. Nuclear variants of unknown significance.
ACKNOWLEDGMENTS
We thank Santiago Morales for his assistance with the figures. This work was supported by grants (FIS‐PI17/00021, PI21/00229) from Instituto de Salud Carlos III and European Regional Development Fund (FEDER); Grant PID2020‐116970GA‐I00 and Grant RYC2020‐029544‐I funded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future”; Gobierno de Aragón (Grupos Consolidados B33_20R) and FEDER 2014‐2020 ‘Construyendo Europa desde Aragón’; and Asociación de Enfermos de Patología Mitocondrial (AEPMI).
Vela‐Sebastián A, López‐Gallardo E, Emperador S, et al. Toxic and nutritional factors trigger Leber hereditary optic neuropathy due to a mitochondrial tRNA mutation. Clinical Genetics. 2022;102(4):339‐344. doi: 10.1111/cge.14189
Ana Vela‐Sebastián and Ester López‐Gallardo contributed equally to this work.
Funding information Asociación de Enfermos de Patología Mitocondrial (AEPMI); Gobierno de Aragón (Grupos Consolidados B33_20R) and FEDER 2014‐2020 'Construyendo Europa desde Aragón'; Instituto de Salud Carlos III (FIs‐PI17/00021; PI21/00229) and European Regional Development Fund (FEDER); MCIN/AEI/10.13039/501100011033 and “ESF investing in your future” (PID2020‐116970GA‐I00; RYC2020‐029544‐I)
Contributor Information
Julio Montoya, Email: jmontoya@unizar.es.
Eduardo Ruiz‐Pesini, Email: eduruiz@unizar.es.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Sundaramurthy S, SelvaKumar A, Ching J, Dharani V, Sarangapani S, Yu‐Wai‐Man P. Leber hereditary optic neuropathy‐new insights and old challenges. Graefes Arch Clin Exp Ophthalmol. 2021;259:2461‐2472. doi: 10.1007/s00417-020-04993-1 [DOI] [PubMed] [Google Scholar]
- 2. Caporali L, Iommarini L, la Morgia C, et al. Peculiar combinations of individually non‐pathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy. PLoS Genet. 2018;14:e1007210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mansukhani SA, Mehta DG, Renaud DL, Whealy MA, Chen JJ, Bhatti MT. Nuclear DNA mutation causing a phenotypic Leber hereditary optic neuropathy plus. Ophthalmology. 2021;128:628‐631. doi: 10.1016/j.ophtha.2020.09.011 [DOI] [PubMed] [Google Scholar]
- 4. Stenton SL, Sheremet NL, Catarino CB, et al. Impaired complex I repair causes recessive Leber's hereditary optic neuropathy. J Clin Invest. 2021;131:e138267. doi: 10.1172/JCI138267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ruiz‐Pesini E, Emperador S, López‐Gallardo E, Hernández‐Ainsa C, Montoya J. Increasing mtDNA levels as therapy for mitochondrial optic neuropathies. Drug Discov Today. 2018;23:493‐498. [DOI] [PubMed] [Google Scholar]
- 6. Oliveira C. Toxic‐metabolic and hereditary optic neuropathies. Continuum. 2019;25:1265‐1288. [DOI] [PubMed] [Google Scholar]
- 7. Roda M, di Geronimo N, Pellegrini M, Schiavi C. Nutritional optic neuropathies: state of the art and emerging evidences. Nutrients. 2020;12:2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martín‐Navarro A, Gaudioso‐Simón A, Álvarez‐Jarreta J, Montoya J, Mayordomo E, Ruiz‐Pesini E. Machine learning classifier for identification of damaging missense mutations exclusive to human mitochondrial DNA‐encoded polypeptides. BMC Bioinf. 2017;18:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y, Zeng QY, Zheng WQ, Ji QQ, Zhou XL, Wang ED. A natural non‐Watson–Crick base pair in human mitochondrial tRNAThr causes structural and functional susceptibility to local mutations. Nucleic Acids Res. 2018;46:4662‐4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu‐Wai‐Man P, Votruba M, Moore AT, Chinnery PF. Treatment strategies for inherited optic neuropathies: past, present and future. Eye. 2014;28:521‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pfeffer G, Burke A, Yu‐Wai‐Man P, Compston DAS, Chinnery PF. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology. 2013;81:2073‐2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong L‐JC, Chen T, Wang J, et al. Interpretation of mitochondrial tRNA variants. Genet Med. 2020;22:917‐926. [DOI] [PubMed] [Google Scholar]
- 13. Qu J, Li R, Zhou X, et al. The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON‐associated ND4 G11778A mutation. Invest Ophthalmol Vis Sci. 2006;47:475‐483. [DOI] [PubMed] [Google Scholar]
- 14. Jorge A, Ung C, Young LH, Melles RB, Choi HK. Hydroxychloroquine retinopathy—implications of research advances for rheumatology care. Nat Rev Rheumatol. 2018;14:693‐703. [DOI] [PubMed] [Google Scholar]
- 15. Lee H‐O, Mustafa A, Hudes GR, Kruger WD. Hydroxychloroquine destabilizes phospho‐S6 in human renal carcinoma cells. PLoS One. 2015;10:e0131464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giordano L, Deceglie S, d'Adamo P, et al. Cigarette toxicity triggers Leber's hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis. 2015;6:e2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bekerman VP, Berman E, You B, Turbin R, Frohman L. A novel mitochondrial mutation for Lebers hereditary optic neuropathy presenting with vitamin B12 deficiency. J Neuroophthalmol. 2021. doi: 10.1097/WNO.0000000000001391 [DOI] [PubMed] [Google Scholar]
- 18. Chan W, Almasieh M, Catrinescu MM, Levin LA. Cobalamin‐associated superoxide scavenging in neuronal cells is a potential mechanism for vitamin B12‐deprivation optic neuropathy. Am J Pathol. 2018;188:160‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Praveen G, Shalini T, Sivaprasad M, Reddy GB. Relative telomere length and mitochondrial DNA copy number variation with age: association with plasma folate and vitamin B12. Mitochondrion. 2020;51:79‐87. [DOI] [PubMed] [Google Scholar]
- 20. Bianco A, Martínez‐Romero I, Bisceglia L, et al. Mitochondrial DNA copy number differentiates the Leber's hereditary optic neuropathy affected individuals from the unaffected mutation carriers. Brain. 2016;139:e1. [DOI] [PubMed] [Google Scholar]
- 21. Beretta S, Mattavelli L, Sala G, et al. Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines. Brain. 2004;127:2183‐2192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Ophthalmological examination of patient 1. (A) Retinography of the left (on the left) and right eye (on the right). Bilateral optic disc pallor is more evident on the right eye. (B) Automated static perimetry of the left eye (on the left) showed a concentric visual field defect and the right eye (on the right) with a deep generalized defect. (C) Peripapillary retinal nerve fiber layer thickness measured by spectral domain‐optical coherence tomography revealed thinning on superior, inferior and temporal sectors in the right eye and thinning on the temporal sector in the right eye. (D) Automated static perimetry four years after the subacute episode. On the left eye (on the left) showed an important improvement on visual field defect (only an inferior peripheral defect) and on the right eye (on the right) with a slight improvement but persistent deep central defect.
Figure S2. Ophthalmological examination of patient 2. (A) Peripapillary retinal nerve fiber layer spectral domain optical coherence tomography showing a progressive thinning from the initial visit. (B) Profound loss of retinal ganglion cells in both eyes. (C) Photograph of the patient's right fundus and left fundus showing a diffuse pallor in both optic nerves. (D) Humphrey visual field test revealing a profound defect in both eyes.
Figure S3. Blood mtDNA levels. The mtDNA amount in the index cases was considered 100%. In patient 2, mtDNA levels were studied seven months after vitamin B12 treatment was initiated. P1, patient 1; P1M, patient 1's mother; P2, patient 2; P2S, patient 2's sister. **, P = 0.0079; ***, P < 0.0001.
Table S1. Nuclear variants of unknown significance.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.