

Abstract
Activation of the coagulation cascade is a critical, evolutionarily conserved mechanism that maintains hemostasis by rapidly forming blood clots in response to blood‐borne infections and damaged blood vessels. Coagulation is a key component of innate immunity since it prevents bacterial dissemination and can provoke inflammation. The term immunothrombosis describes the process by which the innate immune response drives aberrant coagulation, which can result in a lethal condition termed disseminated intravascular coagulation, often seen in sepsis. In this review, we describe the recently uncovered molecular mechanisms underlying inflammasome‐ and STING‐driven immunothrombosis induced by bacterial and viral infections, culminating in tissue factor (TF) activation and release. Current anticoagulant therapeutics, while effective, are associated with a life‐threatening bleeding risk, requiring the urgent development of new treatments. Targeting immunothrombosis may provide a safer option. Thus, we highlight preclinical tools which target TF and/or block canonical (NLRP3) or noncanonical (caspase‐11) inflammasome activation as well as STING‐driven TF release and discuss clinically approved drugs which block key immunothrombotic processes and, therefore, may be redeployed as safer anticoagulants.
Keywords: coagulation, immunothrombosis, inflammasomes, STING, tissue factor
Detection of pathogens, such as bacteria and viruses, by pattern recognition receptors (PRRs) triggers activation of components of immune cell defense pathways such as the inflammasome, type I interferon, and STING. This culminates in proinflammatory cell death, termed pyroptosis, and release of the coagulation activator, tissue factor (TF). This process of immune cell activation triggering coagulation, termed immunothrombosis, is a hallmark of coagulopathies such as sepsis and disseminated intravascular coagulation.

Introduction
Coagulation is a core component in maintaining physiological hemostasis and the host response to infection. The coagulation cascade is defined by two major pathways—the intrinsic and extrinsic pathways—which culminate in a common pathway which ultimately results in formation of a thrombus and fibrin clot, stopping bleeding. The intrinsic pathway, which primarily contributes to pathological clot formation [1], is initiated via injury to blood vessels by autoactivation of coagulation factor (F)XII upon exposure of plasma to a diverse range of blood‐borne artificial or pathological surfaces, including negatively charged endogenous activating surfaces such as RNA, DNA, polyphosphate, and/or components of atherosclerotic plaques [2]. The extrinsic pathway is initiated by coagulation FIII, also called tissue factor (TF) or CD142, which is expressed at low, basal levels in a complex with FVII on the membrane of circulating immune cells and cells in the blood vessel wall [3, 4, 5]. Blood clotting is controlled by endogenous anticoagulants such as tissue factor pathway inhibitor (TFPI), activated protein C, or antithrombin [6]. However, under pathogenic circumstances, exposure to, and detection of, microbes by innate immune cells amplifies the procoagulant activity of TF up to 100‐fold, resulting in clot formation with the dual role of preventing bleeding but also inhibiting the dissemination of the provoking pathogen [7, 8]. Exposure to bacteria or viruses is detected by pattern recognition receptors (PRRs) on immune cells, such as monocytes, macrophages, endothelial cells (ECs), neutrophils, and platelets, triggering TF production and release. TF is released from macrophages, ECs, and neutrophils via inflammasome‐mediated pyroptosis [9, 10]. This activates the coagulation cascade, restoring, and maintaining hemostasis via rapid development of a thrombus, or blood clot, and subsequent clearance of the pathogen. Thrombin in turn feeds back to drive further inflammation via cleavage of protease‐activated receptors (PARs) and activation of the proinflammatory cytokine IL‐1α [11, 12]. Thus, inflammation and coagulation are innately connected, evolutionarily conserved processes. This interplay has been termed immunothrombosis [13]. Dysregulated immunothrombosis, termed thromboinflammation, characterizes life‐threatening conditions, such as sepsis and disseminated intravascular coagulation (DIC), but also acute respiratory distress syndrome, stroke, myocardial infarction, venous thromboembolism, and coronavirus disease 2019 (COVID‐19) [13, 14, 15, 16, 17, 18].
Coagulopathies, including sepsis and DIC, are conditions of significant microvasculature damage and multiorgan failure, and are the primary cause of death in intensive care units [19]. Appropriately, the World Health Organization has recently recognized sepsis as a global health priority [20], with 48.9 million cases of sepsis and 11 million associated deaths reported in 2017, accounting for just under one‐fifth of all global deaths [21]. In this review, we will describe the critical role of TF in initiating immunothrombosis, and focus on recent developments describing novel mechanisms by which bacterial‐ and viral‐induced immunothrombosis can be triggered via PRRs. In addition, we will discuss new approaches toward targeting these pathways that drive immunothrombosis and thromboinflammation, as a means to treat coagulopathies.
Tissue factor: The initiator of trauma‐induced coagulation
TF is a 47‐kDa membrane glycoprotein and receptor and the key trigger of infection‐ and injury‐induced coagulation [5, 22–24]. TF is critical for survival, as deletion in mice leads to universal embryonic death [25, 26, 27], and defects in TF gene expression are associated with differing clinical outcomes in patients with severe sepsis [28]. TF is expressed by adventitial tissues, such as ECs, and blood‐borne circulating immune cells such as monocytes, macrophages, and neutrophils [5]. During hemostasis, blood vessel injury triggers exposure and release of extravascular TF into the bloodstream, where it forms a complex with FVII and contributes to blood clotting via low‐level activation of the extrinsic pathway of the coagulation cascade, before rapid inhibition by TFPI (Fig. 1) [4]. However, detection of pathogen‐associated molecular patterns (PAMPs), such as LPS by PRRs such as TLR4, triggers immunothrombosis via rapid induction of TF at the mRNA level. This occurs via PAMP‐induced activation of the transcription factor NF‐κB both in vitro and in vivo [29], in monocytes and macrophages [29, 30, 31], neutrophils [32, 33], ECs [34, 35], and epithelial cells [36], the primary sources of TF [37].
Figure 1.

Two major pathways of coagulation converge during hemostasis to form a blood clot. The original “waterfall” model of the coagulation cascade comprises the intrinsic and extrinsic pathways which converge into a common pathway to generate thrombin and form a fibrin clot. The intrinsic pathway primarily contributes to pathological clot formation and is activated when FXII encounters blood‐borne, negatively charged surfaces such as RNA, DNA, and components of atherosclerotic plaques. The extrinsic pathway is activated when subvascular TF is exposed to plasma, or released into the bloodstream via innate immune cell pyroptosis, where TF forms a cell‐surface complex with FVIIa. The intrinsic and extrinsic pathways combine to activate FX, which drives thrombin generation and ultimately blood clot formation. Endogenous inhibitors of the coagulation cascade include TFPI, activated protein C, and antithrombin.
TF is modified in a process termed decryption, which occurs in‐part via changes in the lipid composition in the outer leaflet of the cell membrane [8, 10, 38], increasing the procoagulant activity of TF [7, 8]. Decrypted TF is then released from immune cells through inflammasome‐induced pyroptotic pores, via activation of the NOD‐, LRR‐, and pyrin domain‐containing protein 3 (NLRP3, via caspase‐1) or noncanonical (via caspase‐11) inflammasomes [9, 10]. The molecular mechanisms underlying this process have recently been studied in detail in monocytes and macrophages, as they are the main source of circulating TF [23, 37]. For example, deletion of monocytes and macrophages using clodronate or gadolinium chloride significantly attenuates thrombin generation and septic shock‐induced mortality in mice in vivo [9, 10].
Following its release via pyroptotic pores, decrypted TF is expressed in the circulation on outer membrane vesicles [39, 40, 41, 42] and forms a high‐affinity cell‐surface complex with FVII/VIIa to proteolytically activate factors IX to IXa and X to Xa, resulting in thrombin generation [5, 43]. Thrombin then activates PARs which are critical for the interplay between inflammation and coagulation, boosting proinflammatory cytokine secretion but also activating platelets [44, 45]. Thrombin also cleaves fibrinogen to fibrin which generates a clot by forming a mesh at the site of infection, in conjunction with activated platelets and neutrophils which expel their DNA, histones, and granule‐derived enzymes to form networks of extracellular fibres called neutrophil extracellular traps (NETs), in a process termed NETosis [46, 47, 48, 49, 50]. NETs then propagate thrombosis by capturing TF and TF‐positive extracellular vesicles from the circulation, further driving coagulation [51, 52]. Thus, detection of PAMPs by PRRs triggers induction and decryption of TF, increasing its procoagulant activity, which is the key initiating step in coagulopathy associated with immunothrombosis and thromboinflammation (Fig. 2).
Figure 2.
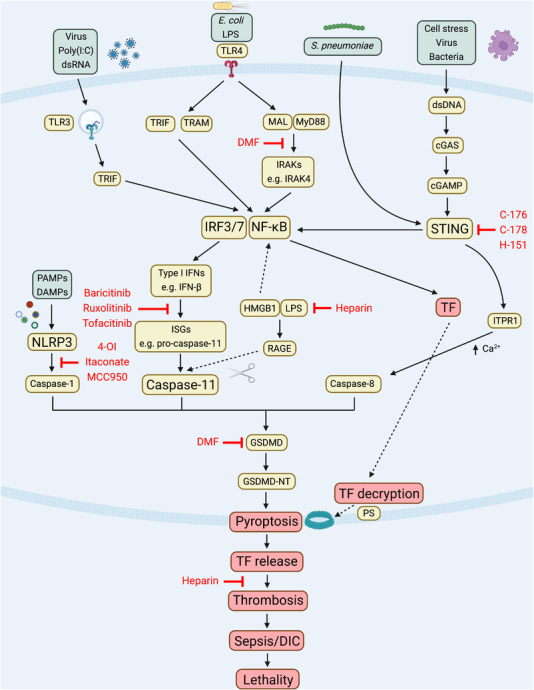
Inflammasome‐ and STING‐mediated TF release drives thrombosis. Detection of a diverse range of microbes (such as viruses and Gram‐negative and Gram‐positive bacteria) by PRRs triggers innate immune signalling cascades which converge to activate IRF3/7 and NF‐κB. IRF3/7 stimulates expression of type I IFNs. This leads to IFN‐β release, which acts via the JAK‐STAT signalling complex to drive transcription of hundreds of ISGs including caspase‐11. Activation of STING can also drive this process. Caspase‐11 is then cleaved and activated upon recognition of cytosolic LPS (which occurs via HMGB1 and RAGE), triggering cleavage and activation of GSDMD, resulting in pyroptosis. GSDMD cleavage can also be triggered by caspase‐1 or caspase‐8 activation. Simultaneously, TF is induced by NF‐κB, before TF is post‐translationally activated, in a process termed decryption. Procoagulant TF is then released through the pyroptotic pores to drive thrombosis, which can result in thromboinflammation, sepsis, and disseminated intravascular coagulation. These signalling cascades have been shown to be blocked by a number of immunomodulatory compounds including DMF, heparin, STING inhibitors (C‐176, C‐178, H‐151), JAK inhibitors (Baricitinib, Ruxolitinib, Tofacitinib), and NLRP3 inflammasome inhibitors (4‐OI, Itaconate, MCC950). Thus, innate immune signalling can trigger TF‐mediated thrombosis via activation of the inflammasome and STING.
Inflammasomes and TF
Caspase‐11 (in mice; caspase‐4/5 in humans) is a member of the evolutionarily conserved family of caspases that mediate cell death [53]. It is induced and activated in response to Gram‐negative bacteria, but not Gram‐positive bacteria [54]. The response of caspase‐11 to Gram‐negative bacteria forms what has been termed as a noncanonical inflammasome. LPS induces transcriptional upregulation of caspase‐11 in a range of immune and nonimmune cells including macrophages, neutrophils, and ECs [14, 53, 55–59]. Activation, and subsequent cleavage, of caspase‐11 occurs upon detection of cytosolic LPS [56, 60, 61], triggering proteolytic cleavage of gasdermin D (GSDMD), a member of the family of gasdermin proteins that cause cell death [57, 62]. The pore‐forming, N‐terminal fragment of GSDMD is released, inserting into the cell membrane to form large oligomeric pores [63]. This leads to a proinflammatory, lytic form of cell death, termed pyroptosis, as first identified by Kayagaki et al. in a seminal paper in 2011 [55]. Pyroptosis, therefore, provides a critical host defense mechanism by killing infected cells and preventing dissemination of a pathogen.
Caspase‐1 forms a canonical inflammasome and processes the proinflammatory cytokines IL‐1β and IL‐18. NLRP3 is a key activator of caspase‐1 and is stimulated upon exposure to a diverse range of pathogens [64] via potassium efflux [65]. Caspase‐1 is then recruited to the complex and autoproteolytically activated where it cleaves GSDMD, forming sublytic pores in the cell membrane [64, 66, 67].
Canonical and noncanonical inflammasome activation has recently been shown to be critical for the release of TF from immune cells. It had previously been reported that caspase‐11 is highly expressed in primary human macrophages in patients with severe sepsis [68], hinting at its importance in immunothrombosis. In 2019, Wu et al. showed that activation of both the canonical (with EprJ type III secretion system rod proteins from Escherichia coli (E. coli)) and noncanonical (with LPS) inflammasomes in macrophages triggers TF release via pyroptosis, leading to severe thrombosis and lethality [9]. Deletion of caspase‐11 and TLR4 (but not caspase‐1) in mice did not affect EprJ‐induced caspase‐1 cleavage and TF release, whereas deletion of both caspase‐11 and ‐1 blocked TF release, highlighting the requirement for caspase‐1 in pyroptosis and TF release [9]. Injection of mice with clodronate‐containing liposomes, which depletes macrophages, significantly reduced EprJ‐induced plasma levels of thrombin‐antithrombin and fibrinogen (which are markers of TF‐mediated thrombosis [15]), as well as lethality [9]. Another 2019 study supported these findings, showing that activation of caspase‐11 and GSDMD is essential for LPS‐induced thrombosis [10]. Notably, GSDMD increased the procoagulant activity of TF via externalization of phosphatidylserine (PS) [10], a cell membrane phospholipid that is mostly expressed on the inner cell membrane during homeostasis [5, 8, 39]. This GSDMD‐mediated increase in TF activity occurs via influx of calcium into the cell [10]. This is consistent with reports from the 1980s and 1990s that PS and calcium are key regulators of TF decryption, and thus, enhance TF‐initiated coagulation [8, 39, 40]. Furthermore, in a mouse model of blood flow restriction‐induced venous thrombosis, deletion of caspase‐1 and GSDMD, but not caspase‐11, protected mice against venous thrombosis [69]. Deletion of macrophages, using gadolinium chloride, also protected against venous thrombosis [69]. These studies directly implicated inflammasome‐mediated macrophage cell death as a trigger of immunothrombosis in response to NLRP3 activation, cytosolic LPS, and in ischemia.
cGAS‐STING and immunothrombosis
Recently, activation of the DNA sensor cyclic GMP–AMP synthase (cGAS)‐STING has been implicated as a driver of sepsis in models of human and mouse coagulopathies. In 2014, mutations in transmembrane protein 173 (TMEM173) (the gene which encodes STING) were found to increase production of IFN‐β in PBMCs and fibroblasts from pediatric patients presenting with recurrent fevers, ulcerative skin lesions, vasculitis, and interstitial lung disease, in addition to systemic inflammation, cutaneous vasculopathy, and pulmonary inflammation [70, 71]. ECs, which express STING, were also found to increase IFN‐β production when stimulated with the second messenger cyclic guanosine monophosphate–adenosine monophosphate [70]. Furthermore, TF expression was upregulated in vascular ECs from patients with a mutation in TMEM173 [70]. These reports, describing a severe autoinflammatory syndrome termed STING‐associated vasculopathy with onset in infancy, were the first to link STING with a coagulopathy.
STING has been shown to sustain the host procoagulant response at later timepoints by regulating calcium release from macrophages and monocytes to drive GSDMD cleavage, facilitating the release of TF [72]. Notably, however, Zhang et al. found that this occurs in a type I IFN‐independent manner [72]. This occurs in monocytes and macrophages via binding of STING with inositol 1,4,5‐trisphosphate receptor type 1 (ITPR1), the primary calcium release channel from the ER. The authors found that a STING‐ITPR1 complex forms after infection with the Gram‐negative bacterium E. coli, or the Gram‐positive bacterium Streptococcus pneumoniae (S. pneumoniae), which activates caspase‐8. STING‐ITPR1 binding boosts release of calcium from the ER into the cytosol, triggering cleavage of GSDMD via activation of caspase‐1/11 (after E. coli infection) or caspase‐8 (after S. pneumoniae infection). This facilitates pyroptosis and subsequent release of TF, resulting in sepsis and DIC [72]. The authors concluded that this process was type I IFN‐independent as deletion of IFNAR, the type I IFN receptor, did not significantly alter mouse blood coagulation markers, such as platelets, fibrinogen, d‐dimer, and TF, when assayed 48 h after caecal ligation and puncture (CLP)‐induced sepsis [72]. Furthermore, stimulation of human and mouse monocytes and macrophages with IFN‐α and IFN‐β did not induce TF release, whereas stimulation with E. coli and S. pneumoniae both induced TF release [72]. This highlights the specificity of pathways that drive coagulation within certain contexts. Two key signals are required for inflammasome‐mediated coagulation: the first signal is infection‐ or injury‐associated induction of TF at the mRNA and protein levels; the second signal is activation and cleavage of inflammatory caspases to trigger pyroptosis and release of procoagulant TF. After infection with E. coli or S. pneumoniae, TF is induced rapidly at the mRNA level via NF‐κB, in addition to activation of caspase‐1/11/8‐mediated pyroptosis, representing the two key signals of inflammasome‐mediated coagulation. However, when cells are stimulated with IFN‐β, there is no known direct induction of TF mRNA via NF‐κB.
Contrastingly, Yang and Cheng et al. showed a critical role for type I IFN signalling as a driver of coagulation in mouse models of LPS‐ and CLP‐induced septic shock. In this study, the authors assessed coagulation markers between 6 and 16 h after infection, and found that the deletion of IFNAR significantly reduced LPS‐induced plasma levels of thrombin‐antithrombin and d‐dimer, in addition to increasing survival of mice [73]. This was verified using TIR‐domain‐containing adaptor‐inducing interferon‐β (TRIF) KO mice, which were also protected against LPS‐induced septic shock [73]. The different timepoints used in these two studies may explain their differing conclusions, but may also point toward type I IFNs driving a procoagulant phenotype at the onset of infection or injury, while STING may directly trigger coagulation at later timepoints after the type I IFN response has peaked.
HMGB1 and immunothrombosis
The danger‐associated molecular pattern, high‐mobility group box protein 1 (HMGB1), has been linked with coagulation as it is increased in the serum of LPS‐infected mice and septic patients [74]. In addition, HMGB1 expression on circulating platelets is increased in trauma patients [75]. Recent studies have found that HMGB1 derived from platelets, hepatocytes, and myeloid cells mediates LPS‐induced thrombosis in mice in a TLR4‐ and MyD88‐dependent manner [75, 76, 77]. HMGB1 contributes to Gram‐negative sepsis by binding to LPS [78], and it has been shown that hepatocyte‐released HMGB1 transports extracellular LPS into the cytosol of macrophages and ECs [79]. This occurs via endocytosis of HMGB1‐LPS, mediated by the receptor for advanced glycation endproducts (RAGE), and subsequent HMGB1‐induced rupture of the endolysosomal membrane, releasing LPS into the cytosol. Cytosolic LPS is then detected by caspase‐11, triggering noncanonical inflammasome‐induced pyroptosis, releasing TF to drive coagulation [79].
HMGB1 has also been shown to stimulate expression of TF in vitro at the mRNA and protein levels in vascular ECs and macrophages via activation of the transcription factors NF‐κB and Egr‐1 [80]. However, Yang and Cheng et al. did not see an effect on LPS‐induced TF protein levels in vivo after deletion of IFNAR, TRIF, or hepatocyte HMGB1 [73]. Using KO mice, they surmised that type I IFN and extracellular HMGB1 drive procoagulant TF activation and coagulation post‐transcriptionally via caspase‐11‐ and GSDMD‐triggered pyroptosis and subsequent exposure of PS (which decrypts TF to trigger coagulation) [73]. In addition, a recent study assessing the role ninjurin1 (Ninj1) in lytic cell death found that deletion of Ninj1 in macrophages impaired pyroptosis and release of HMGB1, highlighting the importance of cell membrane rupture in driving inflammation and coagulation via release of HMGB1, and likely, TF [81].
Therefore, it is possible that extracellular LPS stimulates caspase‐11‐TF‐induced coagulation initially by activating NF‐κB (and inducing TF at the mRNA level), while simultaneously, extracellular LPS also drives type I IFN‐mediated induction of IFN‐stimulated genes (ISGs) such as caspase‐11. LPS is then delivered to the cytosol via HMGB1, cleaving and activating caspase‐1 (inducing sublytic pores in the cell membrane) and caspase‐11, which triggers lytic pyroptosis and TF release. HMGB1 might then feedback to induce further TF expression, amplifying the available procoagulant TF. Furthermore, as the type I IFN response subsides, STING might then sense bacterial or host‐derived DNA, driving TF release by regulating changes in calcium, activating GSDMD‐induced pyroptosis. Further in vivo studies are required to unravel the differing roles of these key players in immunothrombosis.
Virally‐induced immunothrombosis
Induction and decryption of TF has been shown to occur in vitro and in vivo in response to a range of viruses and the viral ds RNA mimic polyinosinic:polycytidylic acid (poly[I:C]) [82, 83, 84]. TF procoagulant activity is increased in ECs infected with Herpes simplex virus (HSV) [85]. HSV infection in ECs also stimulates increased thrombin generation and platelet activity [86]. Ebola virus infection is also associated with severe hemorrhagic complications, manifesting as DIC which is driven by TF activity [87]. Geisbert et al. showed that TF is increased at the mRNA and protein levels in PBMCs from macaque monkeys infected with Ebola virus, with TF‐positive microvesicles also increased in plasma from infected macaques [87]. A follow‐up study from Geisbert et al. found that inhibition of TF:FVIIa, using recombinant nematode anticoagulant protein c2, following exposure to Ebola virus, significantly reduced coagulation, the cytokine storm, and mortality in rhesus monkeys [88]. Infection of ECs with Dengue virus also induces NF‐κB‐mediated TF expression [89].
Furthermore, HIV is associated with an increased risk of thrombosis. TF expression on the surface of monocytes is increased in humans infected with HIV [90]. Expression of TF in HIV patients correlates with plasma levels of d‐dimer and soluble CD14, the LPS receptor that is released by monocytes after LPS stimulation in vivo [90]. TF expression in human ECs is also increased after infection with Zika virus, boosting thrombin generation [91], which likely contributes to the coagulopathy associated with Zika virus infection [92]. However, further studies are required to decipher the relative roles of immunothrombotic regulators within innate immune cells, such as cGAS‐STING and/or type I IFN, and perhaps as yet unidentified mechanisms, in driving TF induction and release upon viral infection.
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) also drives a profound coagulopathy associated with COVID‐19, which is triggered by the key players of immunothrombosis [18]. SARS‐CoV‐2‐infected ECs release von Willebrand factor [93], promoting inflammation and coagulation by attracting platelets and neutrophils to the site of infection. Neutrophil activation and subsequent release of NETs is increased by SARS‐CoV‐2 infection [94, 95]. NETs then capture TF and TF‐positive microvesicles, triggering activation of the coagulation cascade [51, 52, 96, 97]. TF and TF‐positive microvesicles are also increased in ECs and epithelial cells from patients with severe COVID‐19 [98, 99], propagating the coagulopathy associated with COVID‐19 infection, with TF‐positive microvesicles a clinical marker of severity in patients with COVID‐19 [100, 101]. This may be due to SARS‐CoV‐2‐induced activation of the canonical NLRP3 and noncanonical caspase‐11 inflammasomes [102, 103], resulting in TF release via pyroptosis. Thus, COVID‐19 has been termed a syndrome of dysregulated immunothrombosis [104].
Targeting immunothrombosis to prevent coagulopathies
Current clinically approved anticoagulant therapies, while highly effective, are associated with increased risk of bleeding because blood clotting, platelet aggregation, and fibrin cross‐linking are essential during normal hemostasis [105, 106, 107, 108, 109]. This life‐threatening bleeding risk is significantly increased with treatment of sepsis and DIC [110]. Anticoagulant therapies exert their function by decreasing activity of clotting factors in the common pathway of the coagulation cascade. The widely used anticoagulant heparin exerts its anticoagulant function by activating antithrombin, which in turn inactivates thrombin, FXa, and FIXa [111]. Intriguingly, it has recently been shown that heparin, or a chemically modified form of heparin without anticoagulant function, also blocks HMGB1‐mediated cytosolic delivery of LPS, thus, inhibiting caspase‐11‐driven pyroptosis to prevent aberrant immunothrombosis and subsequent sepsis‐induced lethality in mice [112]. This hints at a potential solution to the bleeding risk associated with existing anticoagulant drugs and an exciting prospect for the development of new anticoagulant therapies: could targeting both PRR‐mediated induction of TF and/or inflammasome activation within immune cells, rather than clotting factors themselves, prevent coagulopathy while also eliminating the associated bleeding risk?
Might inhibition of the transcriptional processes that lead to inflammasome activation and pyroptosis be particularly attractive targets in this context? PAMP‐induced type I IFN and JAK‐STAT signalling is required for expression of ISGs such as caspase‐11. Baricitinib, ruxolitinib, and tofacitinib are clinically approved JAK inhibitors for the treatment of rheumatoid arthritis and myeloproliferative neoplasms [113], and thus, potentially could be redeployed as inhibitors of immunothrombosis. Recently identified STING inhibitors, such as the nitrofurans (C‐176 and C‐178) [114, 115], indole ureas (H‐151) [114], and the acrylamides (BPK‐21 and BPK‐25) [116], which covalently modify STING, might also be useful. Notably, a recent study showed that ex vivo treatment with H‐151 blocked induction of TF mRNA in primary human ECs infected with SARS‐CoV‐2 [99]. In addition, H‐151 reduced lung SARS‐CoV‐2‐induced TF mRNA levels in a mouse model of COVID‐19 [99].
Directly targeting inflammasome activation is another strategy that has been shown to reduce immunothrombosis in several models. MCC950 is a highly selective inhibitor of NLRP3 [117, 118] and attenuates platelet activation and multiorgan injuries in a rat model of CLP‐induced sepsis [119]. Similarly, the endogenous, Krebs cycle‐derived metabolite itaconate, and its potently anti‐inflammatory cell‐permeable derivative, 4‐octyl itaconate (4‐OI), also block NLRP3 activation [120], with 4‐OI attenuating lung injury in a murine model of LPS‐induced coagulopathy [121]. This warrants further testing of these preclinical inhibitors of the canonical (NLRP3) and noncanonical (caspase‐11) inflammasomes as potential treatments for inflammasome‐driven immunothrombosis. Inhibition of GSDMD activation and pyroptosis occurs following treatment with dimethyl fumarate (DMF) [122, 123]. DMF is a clinically approved drug for the treatment of multiple sclerosis and psoriasis, and it exerts its immunomodulatory effects in‐part by blocking induction of type I IFN [124] and inhibiting NLRP3 activation in a murine experimental colitis model via activation of the regulatory transcription factor Nrf2 [125]. Activation of Nrf2 is protective in a model of LPS‐ and NF‐κB‐induced sepsis [126], which would further support the testing of DMF as an anti‐immunothrombotic agent, as TF‐driven thrombosis occurs via activation of NF‐κB. As such, DMF is currently being investigated as a potential broad spectrum anti‐inflammatory therapy for COVID‐19 in the ongoing RECOVERY trial [127].
Clinically approved anti‐inflammatory therapies as potential anticoagulants?
Recent clinical trials have also studied the effects of anti‐inflammatory therapies on thrombosis (discussed in detail in Refs. [18, 109]). The anti‐inflammatory drug, colchicine, utilized for the treatment of gout and pericarditis, significantly lowered the risk of ischemic events in the COLCOT trial when administered to patients after myocardial infarction [128]. Colchicine blocks immunothrombosis by inhibiting NET formation and can also attenuate NLRP3 activation [129, 130]. A follow‐up trial, LoDoCo2, using low‐dose colchicine, found that IL‐18 and myeloperoxidase (an enzyme released during neutrophil activation) were markedly decreased when administered to patients with chronic coronary disease [131, 132], highlighting the importance of drug dosing in anticoagulation treatment. However, a limitation of colchicine is that it is renally excreted, and thus, can be toxic in patients with chronic kidney disease [133], restricting its use as a treatment for cardiovascular diseases.
Concluding remarks
The past decade has seen a flurry of research in the area of immunothrombosis. As targeting mediators of the coagulation cascade downstream of inflammasome activation and pyroptosis has not yielded any new, safer anticoagulant drugs [134], developing therapeutics that inhibit immunothrombosis during activation of the innate immune response to infection, for example, by blocking TF expression and/or inflammasome or STING activation and subsequent pyroptosis, presents an exciting prospect. As this occurs prior to the activation of the coagulation cascade and generation of thrombin, the anti‐inflammatory agents described above may in turn provide a safer method of anticoagulation by preventing any risk of unwanted bleeding, which has been termed the Holy Grail of identifying new treatments for immunothrombosis [135]. In the interim, redeployment of clinically approved anti‐inflammatory drugs for the safer treatment of aberrant coagulation might well be a highly effective way to prevent the coagulopathies associated with immunothrombosis.
Conflict of interest
The authors declare that there is no conflict of interest associated with this manuscript.
Author's contribution
T.A.J.R. wrote the original draft. L.A.J.O'N. critically reviewed and edited the manuscript.
Abbreviations
- 4‐OI
4‐octyl itaconate
- cGAMP
cyclic guanosine monophosphate–adenosine monophosphate
- cGAS
cyclic GMP–AMP synthase
- CKD
chronic kidney disease
- CLP
caecal ligation and puncture
- COVID‐19
coronavirus disease 2019
- DAMP
danger‐associated molecular pattern
- DIC
disseminated intravascular coagulation
- DMF
dimethyl fumarate
- EC
endothelial cell
- E. coli
Escherichia coli
- GSDMD
gasdermin D
- HMGB1
high‐mobility group box protein 1
- HSV
Herpes simplex virus
- IFNAR
IFN‐α/β receptor
- ISG
IFN‐stimulated gene
- ITPR1
inositol 1,4,5‐trisphosphate receptor type 1
- NET
neutrophil extracellular trap
- NINJ1
ninjurin1
- NLRP3
NOD‐, LRR‐ and pyrin domain‐containing protein 3
- PAR
protease activated receptor
- poly(I:C)
polyinosinic:polycytidylic acid
- PS
phosphatidylserine
- RAGE
receptor for advanced glycation endproducts
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- STING
stimulator of interferon genes
- S. pneumoniae
Streptococcus pneumoniae
- TF
tissue factor
- TFPI
tissue factor pathway inhibitor
- TMEM173
transmembrane protein 173
- TRIF
TIR‐domain‐containing adaptor‐inducing interferon‐β
Acknowledgements
The O'Neill laboratory acknowledges grant support from the European Research Council Metabinate (834370), the Wellcome Trust (205455), and Science Foundation Ireland (19/FFP/6507).
Open access funding provided by IReL.
[Correction added on 15 June 2022, after first online publication: IReL funding statement added.]
[Correction added on 15 June 2022, after first online publication: The copyright line was changed.]
Data availability statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
- 1. Renné, T. , Pozgajová, M. , Grüner, S. , Schuh, K. , Pauer, H. U. , Burfeind, P. , Gailani, D. et al., Defective thrombus formation in mice lacking coagulation factor XII. J. Exp. Med. 2005. 202: 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Renné, T. , Schmaier, A. H. , Nickel, K. F. , Blombäck, M. and Maas, C. , In vivo roles of factor XII. Blood 2012. 120: 4296–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffman, M. , Colina, C. M. , McDonald, A. G. , Arepally, G. M. , Pedersen, L. and Monroe, D. M. , Tissue factor around dermal vessels has bound factor VII in the absence of injury. J. Thromb. Haemost. 2007. 5: 1403–1408. [DOI] [PubMed] [Google Scholar]
- 4. Mackman, N. , The role of tissue factor and factor VIIa in hemostasis. Anesth. Analg. 2009. 108: 1447–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grover, S. P. and Mackman, N. , Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018. 38: 709–725. [DOI] [PubMed] [Google Scholar]
- 6. Ten Cate, H. and Hackeng, T. M. , García de Frutos, P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost. 2017. 117: 1265–1271. [DOI] [PubMed] [Google Scholar]
- 7. Maynard, J. R. , Heckman, C. A. , Pitlick, F. A and Nemerson, Y. , Association of tissue factor activity with the surface of cultured cells. J. Clin. Invest. 1975. 55: 814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bach, R. and Rifkin, D. B. , Expression of tissue factor procoagulant activity: regulation by cytosolic calcium. Pro. Nat. Acad. Sci USA. 1990. 87: 6995–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu, C. , Lu, W. , Zhang, Y. , Zhang, G. , Shi, X. , Hisada, Y. , Grover, S. P. et al., Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity 2019. 50: 1401–1411.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang, X. , Cheng, X. , Tang, Y. , Qiu, X. , Wang, Y. , Kang, H. , Wu, J. et al., Bacterial endotoxin activates the coagulation cascade through gasdermin d‐dependent phosphatidylserine exposure. Immunity 2019. 51: 983–996.e6. [DOI] [PubMed] [Google Scholar]
- 11. Delvaeye, M. , Conway, E. M. Coagulation and innate immune responses: can we view them separately? Blood 2009. 114: 2367–2374. [DOI] [PubMed] [Google Scholar]
- 12. Burzynski, L. C. , Humphry, M. , Pyrillou, K. , Wiggins, K. A. , Chan, J. N. E. , Figg, N. , Kitt, L. L. et al., The Coagulation and immune systems are directly linked through the activation of interleukin‐1α by thrombin. Immunity 2019. 50: 1033–1042.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Engelmann, B. and Massberg, S. , Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013. 13: 34–45. [DOI] [PubMed] [Google Scholar]
- 14. Cheng, K. T. , Xiong, S. , Ye, Z. , Hong, Z. , Di, A. , Tsang, K. M. , Gao, X. et al., Caspase‐11‐mediated endothelial pyroptosis underlies endotoxemia‐induced lung injury. J. Clin. Invest. 2017. 127: 4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jackson, S. P. , Darbousset, R. and Schoenwaelder, S. M. , Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. 2019. 133: 906–918. [DOI] [PubMed] [Google Scholar]
- 16. Nicolai, L. , Leunig, A. , Brambs, S. , Kaiser, R. , Weinberger, T. , Weigand, M. , Muenchhoff, M. et al., Immunothrombotic dysregulation in COVID‐19 pneumonia is associated with respiratory failure and coagulopathy. Circulation. 2020. 142: 1176–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ackermann, M. , Verleden, S. E. , Kuehnel, M. , Haverich, A. , Welte, T. , Laenger, F. , Vanstapel, A. et al., Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid‐19. N. Engl. J. Med. 2020. 383: 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stark, K. and Massberg, S. , Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seymour, C. W. , Liu, V. X. , Iwashyna, T. J. , Brunkhorst, F. M. , Rea, T. D. , Scherag, A. , Rubenfeld, G. et al., Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (sepsis‐3). JAMA. 2016. 315: 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reinhart, K. , Daniels, R. , Kissoon, N. , Machado, F. R. , Schachter, R. D. and Finfer, S. , Recognizing sepsis as a global health priority–a WHO resolution. N. Engl. J. Med. 2017. 377: 414–417. [DOI] [PubMed] [Google Scholar]
- 21. Rudd, K. E. , Johnson, S. C. , Agesa, K. M. , Shackelford, K. A. , Tsoi, D. , Kievlan, D. R. , Colombara, D. V. et al., Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet (London, England). 2020. 395: 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Josso, F. , Prou‐Wartelle, O. Interaction of tissue factor and factor VII at the earliest phase of coagulation. Thromb. Diath. Haemorrh. Suppl. 1965. 17: 35–44. [PubMed] [Google Scholar]
- 23. Pawlinski, R. , Wang, J. G. , Owens A. P., 3rd , Williams, J. , Antoniak, S. , Tencati, M. , Luther, T. et al., Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood. 2010. 116: 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryan, T. A. J. , Preston, R. J. S. and O'Neill, L. A. J. , Immunothrombosis and the molecular control of tissue factor by pyroptosis: prospects for new anticoagulants. Biochem. J. 2022. 479: 731–750. [DOI] [PubMed] [Google Scholar]
- 25. Bugge, T. H. , Xiao, Q. , Kombrinck, K. W. , Flick, M. J. , Holmbäck, K. , Danton, M. J. , Colbert, M. C. et al., Fatal embryonic bleeding events in mice lacking tissue factor, the cell‐associated initiator of blood coagulation. Proc. Natl. Acad. Sci. USA 1996. 93: 6258–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carmeliet, P. , Mackman, N. , Moons, L. , Luther, T. , Gressens, P. , Van Vlaenderen, I. , Demunck, H. et al., Role of tissue factor in embryonic blood vessel development. Nature 1996. 383: 73–75. [DOI] [PubMed] [Google Scholar]
- 27. Toomey, J. R. , Kratzer, K. E. , Lasky, N. M. , Stanton, J. J. and Broze, G. J., Jr. , Targeted disruption of the murine tissue factor gene results in embryonic lethality. Blood 1996. 88: 1583–1587. [PubMed] [Google Scholar]
- 28. Shi, D. , Song, Z. , Yin, J. , Xue, M. , Yao, C. , Sun, Z. , Shao, M. et al., Genetic variation in the tissue factor gene is associated with clinical outcome in severe sepsis patients. Crit. Care 2014. 18: 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackman, N. , Brand, K. and Edgington, T. S. , Lipopolysaccharide‐mediated transcriptional activation of the human tissue factor gene in THP‐1 monocytic cells requires both activator protein 1 and nuclear factor kappa B binding sites. J. Exp. Med. 1991. 174: 1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gregory, S. A. , Morrissey, J. H. and Edgington, T. S. , Regulation of tissue factor gene expression in the monocyte procoagulant response to endotoxin. Mol. Cell. Biol. 1989. 9: 2752–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Franco, R. F. , de Jonge, E. , Dekkers, P. E. , Timmerman, J. J. , Spek, C. A. , van Deventer, S. J. , van Kerkhoff, L. et al., The in vivo kinetics of tissue factor messenger RNA expression during human endotoxemia: relationship with activation of coagulation. Blood 2000. 96: 554–559. [PubMed] [Google Scholar]
- 32. Ritis, K. , Doumas, M. , Mastellos, D. , Micheli, A. , Giaglis, S. , Magotti, P. , Rafail, S. et al., A novel C5a receptor‐tissue factor cross‐talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 2006. 177: 4794–4802. [DOI] [PubMed] [Google Scholar]
- 33. Kambas, K. , Markiewski, M. M. , Pneumatikos, I. A. , Rafail, S. S. , Theodorou, V. , Konstantonis, D. , Konstantonis, D. et al., C5a and TNF‐alpha up‐regulate the expression of tissue factor in intra‐alveolar neutrophils of patients with the acute respiratory distress syndrome. J. Immunol. 2008. 180: 7368–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parry, G. C. and Mackman, N. , Transcriptional regulation of tissue factor expression in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1995. 15: 612–621. [DOI] [PubMed] [Google Scholar]
- 35. Song, D. , Ye, X. , Xu, H. and Liu, S. F. , Activation of endothelial intrinsic NF‐{kappa}B pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood 2009. 114: 2521–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shaver, C. M. , Grove, B. S. , Putz, N. D. , Clune, J. K. , Lawson, W. E. , Carnahan, R. H. , Mackman, N. et al., Regulation of alveolar procoagulant activity and permeability in direct acute lung injury by lung epithelial tissue factor. Am. J. Respir. Cell Mol. Biol. 2015. 53: 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mussbacher, M. , Salzmann, M. , Brostjan, C. , Hoesel, B. , Schoergenhofer, C. , Datler, H. , Hohensinner, P. et al., Cell type‐specific roles of NF‐κB linking inflammation and thrombosis. Front. Immunol. 2019. 10: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang, J. , Pendurthi, U. R. and Rao, L. V. M. , Sphingomyelin encrypts tissue factor: ATP‐induced activation of A‐SMase leads to tissue factor decryption and microvesicle shedding. Blood Adv. 2017. 1: 849–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bach, R. , Gentry, R. and Nemerson, Y. , Factor VII binding to tissue factor in reconstituted phospholipid vesicles: induction of cooperativity by phosphatidylserine. Biochemistry 1986. 25: 4007–4020. [DOI] [PubMed] [Google Scholar]
- 40. Bach, R. R. and Moldow, C. F. , Mechanism of tissue factor activation on HL‐60 cells. Blood 1997. 89: 3270–3276. [PubMed] [Google Scholar]
- 41. Geddings, J. E. , Hisada, Y. , Boulaftali, Y. , Getz, T. M. , Whelihan, M. , Fuentes, R. , Dee, R. et al., Tissue factor‐positive tumor microvesicles activate platelets and enhance thrombosis in mice. J. Thromb. Haemost. 2016. 14: 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peng, Y. , Gao, M. , Liu, Y. , Qiu, X. , Cheng, X. , Yang, X. , Chen, F. et al., Bacterial outer membrane vesicles induce disseminated intravascular coagulation through the caspase‐11‐gasdermin D pathway. Thromb. Res. 2020. 196: 159–166. [DOI] [PubMed] [Google Scholar]
- 43. Li, Y. D. , Ye, B. Q. , Zheng, S. X. , Wang, J. T. , Wang, J. G. , Chen, M. , Liu, J. ‐ G. et al., NF‐kappaB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J. Biol. Chem. 2009. 284: 4473–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Antoniak, S. , Owens A. P., 3rd , Baunacke, M. , Williams, J. C. , Lee, R. D. , Weithäuser, A. , Sheridan, P. A. et al., PAR‐1 contributes to the innate immune response during viral infection. J. Clin. Invest. 2013. 123: 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heuberger, D. M. and Schuepbach, R. A. , Protease‐activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR‐driven inflammatory diseases. Thromb J. 2019. 17: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clark, S. R. , Ma, A. C. , Tavener, S. A. , McDonald, B. , Goodarzi, Z. , Kelly, M. M. , Patel, K. D. et al., Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007. 13: 463–469. [DOI] [PubMed] [Google Scholar]
- 47. McDonald, B. , Urrutia, R. , Yipp, B. G. , Jenne, C. N. and Kubes, P. , Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012. 12: 324–333. [DOI] [PubMed] [Google Scholar]
- 48. Sreeramkumar, V. , Adrover, J. M. , Ballesteros, I. , Cuartero, M. I. , Rossaint, J. , Bilbao, I. , Nácher, M. et al., Neutrophils scan for activated platelets to initiate inflammation. Science (New York, NY) 2014. 346: 1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu, S. , Su, X. , Pan, P. , Zhang, L. , Hu, Y. , Tan, H. , Wu, D. et al., Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Scient. Rep. 2016. 6: 37252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McDonald, B. , Davis, R. P. , Kim, S. J. , Tse, M. , Esmon, C. T. , Kolaczkowska, E. , Jenne, C. N. et al., Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017. 129: 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stakos, D. A. , Kambas, K. , Konstantinidis, T. , Mitroulis, I. , Apostolidou, E. , Arelaki, S. , Arelaki, S. et al., Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015. 36: 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang, H. , Zhou, Y. , Qu, M. , Yu, Y. , Chen, Z. , Zhu, S. , Guo, K. et al., Tissue factor‐enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis‐induced lung injury. Front Cell Infect Microbiol. 2021. 11: 677902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lamkanfi, M. and Dixit, V. M. , Mechanisms and functions of inflammasomes. Cell 2014. 157: 1013–1022. [DOI] [PubMed] [Google Scholar]
- 54. Rathinam, V. A. , Vanaja, S. K. , Waggoner, L. , Sokolovska, A. , Becker, C. , Stuart, L. M. , Leong, J. M. et al., TRIF licenses caspase‐11‐dependent NLRP3 inflammasome activation by gram‐negative bacteria. Cell 2012. 150: 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kayagaki, N. , Warming, S. , Lamkanfi, M. , Vande Walle, L. , Louie, S. , Dong, J. , Newton, K. et al., Non‐canonical inflammasome activation targets caspase‐11. Nature. 2011. 479: 117–121. [DOI] [PubMed] [Google Scholar]
- 56. Shi, J. , Zhao, Y. , Wang, Y. , Gao, W. , Ding, J. , Li, P. , Hu, L. et al., Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014. 514: 187–192. [DOI] [PubMed] [Google Scholar]
- 57. Kayagaki, N. , Stowe, I. B. , Lee, B. L. , O'Rourke, K. , Anderson, K. , Warming, S. , Cuellar, T. et al., Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 2015. 526: 666–671. [DOI] [PubMed] [Google Scholar]
- 58. Kovacs, S. B. , Oh, C. , Maltez, V. I. , McGlaughon, B. D. , Verma, A. , Miao, E. A. , Aachoui, Y. et al., Neutrophil caspase‐11 is essential to defend against a cytosol‐invasive bacterium. Cell Rep. 2020. 32: 107967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schauvliege, R. , Vanrobaeys, J. , Schotte, P. and Beyaert, R. , Caspase‐11 gene expression in response to lipopolysaccharide and interferon‐gamma requires nuclear factor‐kappa B and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem. 2002. 277: 41624–41630. [DOI] [PubMed] [Google Scholar]
- 60. Kayagaki, N. , Wong, M. T. , Stowe, I. B. , Ramani, S. R. , Gonzalez, L. C. , Akashi‐Takamura, S. , Miyake, K. et al., Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (New York, NY) 2013. 341: 1246–1249. [DOI] [PubMed] [Google Scholar]
- 61. Hagar, J. A. , Powell, D. A. , Aachoui, Y. , Ernst, R. K. and Miao, E. A. , Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science (New York, NY). 2013. 341: 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shi, J. , Zhao, Y. , Wang, K. , Shi, X. , Wang, Y. , Huang, H. , Zhuang, Y. et al., Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015. 526: 660–665. [DOI] [PubMed] [Google Scholar]
- 63. Broz, P. , Pelegrín, P. and Shao, F. , The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020. 20: 143–157. [DOI] [PubMed] [Google Scholar]
- 64. Broz, P. and Dixit, V. M. , Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016. 16: 407–420. [DOI] [PubMed] [Google Scholar]
- 65. Muñoz‐Planillo, R. , Kuffa, P. , Martínez‐Colón, G. , Smith, B. L. , Rajendiran, T. M. and Núñez, G. , K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013. 38: 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Evavold, C. L. , Ruan, J. , Tan, Y. , Xia, S. , Wu, H. and Kagan, J. C. , The pore‐forming protein gasdermin d regulates interleukin‐1 secretion from living macrophages. Immunity 2018. 48: 35–44.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Heilig, R. , Dick, M. S. , Sborgi, L. , Meunier, E. , Hiller, S. and Broz, P. , The gasdermin‐D pore acts as a conduit for IL‐1β secretion in mice. Eur. J. Immunol. 2018. 48: 584–592. [DOI] [PubMed] [Google Scholar]
- 68. Napier, B. A. , Brubaker, S. W. , Sweeney, T. E. , Monette, P. , Rothmeier, G. H. , Gertsvolf, N. A. , Puschnik, A. et al., Complement pathway amplifies caspase‐11‐dependent cell death and endotoxin‐induced sepsis severity. J. Exp. Med. 2016. 213: 2365–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang, Y. , Cui, J. , Zhang, G. , Wu, C. , Abdel‐Latif, A. , Smyth, S. S. , Shiroishi, T. et al., Inflammasome activation promotes venous thrombosis through pyroptosis. Blood Adv. 2021. 5: 2619–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu, Y. , Jesus, A. A. , Marrero, B. , Yang, D. , Ramsey, S. E. , Sanchez, G. A. M. , Wittkowski, H. et al., Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014. 371: 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jeremiah, N. , Neven, B. , Gentili, M. , Callebaut, I. , Maschalidi, S. , Stolzenberg, M. C. , Goudin, N. et al., Inherited STING‐activating mutation underlies a familial inflammatory syndrome with lupus‐like manifestations. J. Clin. Invest. 2014. 124: 5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang, H. , Zeng, L. , Xie, M. , Liu, J. , Zhou, B. , Wu, R. , Cao, L. et al., TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe. 2020. 27: 556–570.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang, X. , Cheng, X. , Tang, Y. , Qiu, X. , Wang, Z. , Fu, G. , Wu, J. et al., The role of type 1 interferons in coagulation induced by Gram‐negative bacteria. Blood. 2020. 135: 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang, H. , Bloom, O. , Zhang, M. , Vishnubhakat, J. M. , Ombrellino, M. , Che, J. , Ivanova, S. et al., HMG‐1 as a late mediator of endotoxin lethality in mice. Science (New York, NY) 1999. 285: 248–251. [DOI] [PubMed] [Google Scholar]
- 75. Vogel, S. , Bodenstein, R. , Chen, Q. , Feil, S. , Feil, R. , Rheinlaender, J. , Schaffer, T. E. et al., Platelet‐derived HMGB1 is a critical mediator of thrombosis. J. Clin. Invest. 2015. 125: 4638–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tsung, A. , Klune, J. R. , Zhang, X. , Jeyabalan, G. , Cao, Z. , Peng, X. et al., HMGB1 release induced by liver ischemia involves Toll‐like receptor 4 dependent reactive oxygen species production and calcium‐mediated signaling. J. Exp. Med. 2007. 204: 2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lu, B. , Nakamura, T. , Inouye, K. , Li, J. , Tang, Y. , Lundbäck, P. et al., Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012. 488: 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Youn, J. H. , Oh, Y. J. , Kim, E. S. , Choi, J. E. , Shin, J. S. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide‐mediated TNF‐alpha production in human monocytes. J. Immunol. 2008. 180: 5067–5074. [DOI] [PubMed] [Google Scholar]
- 79. Deng, M. , Tang, Y. , Li, W. , Wang, X. , Zhang, R. , Zhang, X. , Zhao, X. et al., The endotoxin delivery protein HMGB1 mediates caspase‐11‐dependent lethality in sepsis. Immunity 2018. 49: 740–753.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lv, B. , Wang, H. , Tang, Y. , Fan, Z. , Xiao, X. and Chen, F. , High‐mobility group box 1 protein induces tissue factor expression in vascular endothelial cells via activation of NF‐kappaB and Egr‐1. Thromb Haemost 2009. 102: 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kayagaki, N. , Kornfeld, O. S. , Lee, B. L. , Stowe, I. B. , O'Rourke, K. , Li, Q. , Sandoval, W. et al., NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021. 591: 131–136. [DOI] [PubMed] [Google Scholar]
- 82. Shibamiya, A. , Hersemeyer, K. , Schmidt Wöll, T. , Sedding, D. , Daniel, J. M. , Bauer, S. , Koyama, T. et al., A key role for Toll‐like receptor‐3 in disrupting the hemostasis balance on endothelial cells. Blood 2009. 113: 714–722. [DOI] [PubMed] [Google Scholar]
- 83. Antoniak, S. and Mackman, N. , Multiple roles of the coagulation protease cascade during virus infection. Blood. 2014. 123: 2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Subramaniam, S. , Ogoti, Y. , Hernandez, I. , Zogg, M. , Botros, F. , Burns, R. , DeRousse, J. T. et al., A thrombin‐PAR1/2 feedback loop amplifies thromboinflammatory endothelial responses to the viral RNA analogue poly(I:C). Blood Adv. 2021. 5: 2760–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Key, N. S. , Vercellotti, G. M. , Winkelmann, J. C. , Moldow, C. F. , Goodman, J. L. , Esmon, N. L. , Esmon, C. T. et al., Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc. Natl. Acad. Sci USA 1990. 87: 7095–7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Visser, M. R. , Tracy, P. B. , Vercellotti, G. M. , Goodman, J. L. , White, J. G. and Jacob, H. S. , Enhanced thrombin generation and platelet binding on herpes simplex virus‐infected endothelium. Proc. Natl. Acad. Sci. USA 1988. 85: 8227–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Geisbert, T. W. , Young, H. A. , Jahrling, P. B. , Davis, K. J. , Kagan, E. and Hensley, L. E. , Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 2003. 188: 1618–1629. [DOI] [PubMed] [Google Scholar]
- 88. Geisbert, T. W. , Hensley, L. E. , Jahrling, P. B. , Larsen, T. , Geisbert, J. B. , Paragas, J. , Young, H. W. et al., Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet (London, England) 2003. 362: 1953–1958. [DOI] [PubMed] [Google Scholar]
- 89. Huerta‐Zepeda, A. , Cabello‐Gutiérrez, C. , Cime‐Castillo, J. , Monroy‐Martínez, V. , Manjarrez‐Zavala, M. E. , Gutiérrez‐Rodríguez, M. , Izaguirre, R. et al., Crosstalk between coagulation and inflammation during Dengue virus infection. Thromb Haemost 2008. 99: 936–943. [DOI] [PubMed] [Google Scholar]
- 90. Funderburg, N. T. , Mayne, E. , Sieg, S. F. , Asaad, R. , Jiang, W. , Kalinowska, M. , Luciano, A. A. et al., Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood 2010. 115: 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Anfasa, F. , Goeijenbier, M. , Widagdo, W. , Siegers, J. Y. , Mumtaz, N. , Okba, N. , Okba, N. et al., Zika virus infection induces elevation of tissue factor production and apoptosis on human umbilical vein endothelial cells. Front Microbiol. 2019. 10: 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wu, Y. , Cui, X. , Wu, N. , Song, R. , Yang, W. , Zhang, W. , Fan, D. et al., A unique case of human Zika virus infection in association with severe liver injury and coagulation disorders. Scient. Rep. 2017. 7: 11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tang, N. , Li, D. , Wang, X. and Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020. 18: 844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Veras, F. P. , Pontelli, M. C. , Silva, C. M. , Toller‐Kawahisa, J. E. , de Lima, M. , Nascimento, D. C. et al., SARS‐CoV‐2‐triggered neutrophil extracellular traps mediate COVID‐19 pathology. J. Exp. Med. 2020. 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Middleton, E. A. , He, X. Y. , Denorme, F. , Campbell, R. A. , Ng, D. , Salvatore, S. P. et al., Neutrophil extracellular traps contribute to immunothrombosis in COVID‐19 acute respiratory distress syndrome. Blood. 2020. 136: 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fuchs, T. A. , Brill, A. , Duerschmied, D. , Schatzberg, D. , Monestier, M. , Myers D. D., Jr. , Myers, D. D. et al., Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010. 107: 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Skendros, P. , Mitsios, A. , Chrysanthopoulou, A. , Mastellos, D. C. , Metallidis, S. , Rafailidis, P. , Ntinopoulou, M. et al., Complement and tissue factor‐enriched neutrophil extracellular traps are key drivers in COVID‐19 immunothrombosis. J. Clin. Invest. 2020. 130: 6151–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Francischetti, I. M. B. , Toomer, K. , Zhang, Y. , Jani, J. , Siddiqui, Z. , Brotman, D. J. , Hooper, J. E. et al., Upregulation of pulmonary tissue factor, loss of thrombomodulin and immunothrombosis in SARS‐CoV‐2 infection. EClinicalMedicine 2021. 39: 101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Di Domizio, J. , Gulen, M. F. , Saidoune, F. , Thacker, V. V. , Yatim, A. , Sharma, K. , Nass, T. et al., The cGAS‐STING pathway drives type I IFN immunopathology in COVID‐19. Nature. 2022. 603:145‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rosell, A. , Havervall, S. , von Meijenfeldt, F. , Hisada, Y. , Aguilera, K. , Grover, S. P. , Lisman, T. et al., Patients with COVID‐19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality‐brief report. Arterioscler. Thromb. Vasc. Biol. 2021. 41: 878–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Balbi, C. , Burrello, J. , Bolis, S. , Lazzarini, E. , Biemmi, V. , Pianezzi, E. , Burrello, A. et al., Circulating extracellular vesicles are endowed with enhanced procoagulant activity in SARS‐CoV‐2 infection. EBioMedicine 2021. 67: 103369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rodrigues, T. S. , de Sá, K. S. G. , Ishimoto, A. Y. , Becerra, A. , Oliveira, S. , Almeida, L. , Gonçalves, A. V. et al., Inflammasomes are activated in response to SARS‐CoV‐2 infection and are associated with COVID‐19 severity in patients. J. Exp. Med. 2021. 218:e20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Eltobgy, M. , Zani, A. , Kenney, A. D. , Estfanous, S. , Kim, E. , Badr, A. , Carafice, C. C. et al., Caspase‐4/11 exacerbates disease severity in SARS‐CoV‐2 infection by promoting inflammation and thrombosis. bioRxiv. 2021:2021.09.24.461743 [DOI] [PMC free article] [PubMed]
- 104. Bonaventura, A. , Vecchié, A. , Dagna, L. , Martinod, K. , Dixon, D. L. , Van Tassell, B. W. , van Tassell, B. W. et al., Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID‐19. Nat. Rev. Immunol. 2021. 21: 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Holbrook, A. , Schulman, S. , Witt, D. M. , Vandvik, P. O. , Fish, J. , Kovacs, M. J. et al., Evidence‐based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence‐Based Clinical Practice Guidelines. Chest. 2012. 141: e152S–e84S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Doktorova, M. and Motovska, Z. , Clinical review: bleeding— a notable complication of treatment in patients with acute coronary syndromes: incidence, predictors, classification, impact on prognosis, and management. Crit. Care 2013. 17: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lin, L. , Lim, W. S. , Zhou, H. J. , Khoo, A. L. , Tan, K. T. , Chew, A. P. , Foo, D. et al., Clinical and safety outcomes of oral antithrombotics for stroke prevention in atrial fibrillation: a systematic review and network meta‐analysis. J. Am. Med. Director Assoc. 2015. 16: 1103.e1–19. [DOI] [PubMed] [Google Scholar]
- 108. Fan, Y. , Jiang, M. , Gong, D. and Zou, C. , Efficacy and safety of low‐molecular‐weight heparin in patients with sepsis: a meta‐analysis of randomized controlled trials. Scient. Rep. 2016. 6: 25984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mackman, N. , Bergmeier, W. , Stouffer, G. A. and Weitz, J. I. Therapeutic strategies for thrombosis: new targets and approaches. Nat. Reviews Drug Discov. 2020. 19: 333–352. [DOI] [PubMed] [Google Scholar]
- 110. Iba, T. , Connors, J. M. , Nagaoka, I. and Levy, J. H. Recent advances in the research and management of sepsis‐associated DIC. Int. J. Hematol. 2021. 113: 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wardrop, D. and Keeling, D. , The story of the discovery of heparin and warfarin. Br. J. Haematol. 2008. 141: 757–763. [DOI] [PubMed] [Google Scholar]
- 112. Tang, Y. , Wang, X. , Li, Z. , He, Z. , Yang, X. , Cheng, X. , Peng, Y. et al., Heparin prevents caspase‐11‐dependent septic lethality independent of anticoagulant properties. Immunity. 2021. 54: 454–467.e6. [DOI] [PubMed] [Google Scholar]
- 113. Baldini, C. , Moriconi, F. R. , Galimberti, S. , Libby, P. and De Caterina, R. , The JAK‐STAT pathway: an emerging target for cardiovascular disease in rheumatoid arthritis and myeloproliferative neoplasms. Eur. Heart J. 2021. 42: 4389–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Haag, S. M. , Gulen, M. F. , Reymond, L. , Gibelin, A. , Abrami, L. , Decout, A. , Hyemann, M. et al., Targeting STING with covalent small‐molecule inhibitors. Nature 2018. 559: 269–273. [DOI] [PubMed] [Google Scholar]
- 115. Hansen, A. L. , Buchan, G. J. , Rühl, M. , Mukai, K. , Salvatore, S. R. , Ogawa, E. , Anderson, S. D. et al., Nitro‐fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Procs. Natl. Acad. Sci. USA. 2018. 115: E7768–e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Vinogradova, E. V. , Zhang, X. , Remillard, D. , Lazar, D. C. , Suciu, R. M. , Wang, Y. , Bianco, G. et al., An activity‐guided map of electrophile‐cysteine interactions in primary human T cells. Cell. 2020. 182: 1009–1026.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Coll, R. C. , Robertson, A. A. , Chae, J. J. , Higgins, S. C. , Muñoz‐Planillo, R. , Inserra, M. C. , Inserra, M. C. et al., A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015. 21: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Coll, R. C. , Hill, J. R. , Day, C. J. , Zamoshnikova, A. , Boucher, D. , Massey, N. L. , Chitty, J. L. et al., MCC950 directly targets the NLRP3 ATP‐hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019. 15: 556–559. [DOI] [PubMed] [Google Scholar]
- 119. Cornelius, D. C. , Travis, O. K. , Tramel, R. W. , Borges‐Rodriguez, M. , Baik, C. H. , Greer, M. , Giachelli, C. A. et al., NLRP3 inflammasome inhibition attenuates sepsis‐induced platelet activation and prevents multi‐organ injury in cecal‐ligation puncture. PLoS One. 2020. 15: e0234039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hooftman, A. , Angiari, S. , Hester, S. , Corcoran, S. E. , Runtsch, M. C. , Ling, C. , Ruzek, M. C. et al., The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. 2020. 32: 468–478.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Li, Y. , Chen, X. , Zhang, H. , Xiao, J. , Yang, C. , Chen, W. , Wei, Z. et al., 4‐Octyl itaconate alleviates lipopolysaccharide‐induced acute lung injury in mice by inhibiting oxidative stress and inflammation. Drug Des Devel Ther. 2020. 14: 5547–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Miglio, G. , Veglia, E. and Fantozzi, R. , Fumaric acid esters prevent the NLRP3 inflammasome‐mediated and ATP‐triggered pyroptosis of differentiated THP‐1 cells. Int. Immunopharmacol. 2015. 28: 215–219. [DOI] [PubMed] [Google Scholar]
- 123. Humphries, F. , Shmuel‐Galia, L. , Ketelut‐Carneiro, N. , Li, S. , Wang, B. , Nemmara, V. V. , Wilson, R. et al., Succination inactivates gasdermin D and blocks pyroptosis. Science (New York, NY) 2020. 369: 1633–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zaro, B. W. , Vinogradova, E. V. , Lazar, D. C. , Blewett, M. M. , Suciu, R. M. , Takaya, J. , Studer, S. et al., Dimethyl fumarate disrupts human innate immune signaling by targeting the IRAK4‐MyD88 COmplex. J. Immunol. 2019. 202: 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liu, X. , Zhou, W. , Zhang, X. , Lu, P. , Du, Q. , Tao, L. , Ding, Y. et al., Dimethyl fumarate ameliorates dextran sulfate sodium‐induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem. Pharmacol. 2016. 112: 37–49. [DOI] [PubMed] [Google Scholar]
- 126. Thimmulappa, R. K. , Lee, H. , Rangasamy, T. , Reddy, S. P. , Yamamoto, M. , Kensler, T. W. , Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 2006. 116: 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Uo O.. Randomised Evaluation of COVID‐19 Therapy (RECOVERY) 2022 [Available from: https://www.clinicaltrials.gov/ct2/show/NCT04381936?term=recovery+covid‐19+dimethyl+fumarate&draw=2&rank=1.
- 128. Tardif, J. C. , Kouz, S. , Waters, D. D. , Bertrand, O. F. , Diaz, R. , Maggioni, A. P. , Pinto, J. et al., Efficacy and safety of low‐dose colchicine after myocardial infarction. N. Engl. J. Med. 2019. 381: 2497–2505. [DOI] [PubMed] [Google Scholar]
- 129. Vaidya, K. , Tucker, B. , Kurup, R. , Khandkar, C. , Pandzic, E. , Barraclough, J. , Machet, J. et al., Colchicine inhibits neutrophil extracellular trap formation in patients with acute coronary syndrome after percutaneous coronary intervention. J. Am. Heart Assoc. 2021. 10: e018993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Toldo, S. , Mezzaroma, E. , Buckley, L. F. , Potere, N. , Di Nisio, M. , Biondi‐Zoccai, G. , Gullestad, L. et al., Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol. Ther. 2021. 236: 108053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nidorf, S. M. , Fiolet, A. T. L. , Mosterd, A. , Eikelboom, J. W. , Schut, A. , Opstal, T. S. J. , The, S. H. K. et al., Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 2020. 383: 1838–1847. [DOI] [PubMed] [Google Scholar]
- 132. Silvis, M. J. M. , Fiolet, A. T. L. , Opstal, T. S. J. , Dekker, M. , Suquilanda, D. , Zivkovic, M. , Duyvendak, M. et al., Colchicine reduces extracellular vesicle NLRP3 inflammasome protein levels in chronic coronary disease: a LoDoCo2 biomarker substudy. Atherosclerosis. 2021. 334: 93–100. [DOI] [PubMed] [Google Scholar]
- 133. Vargas‐Santos, A. B. and Neogi, T. , Management of gout and hyperuricemia in CKD. Am. J. Kidney Dis. 2017. 70: 422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cavaillon, J. M. , Singer, M. and Skirecki, T. , Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO molecular medicine. 2020. 12: e10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. De Caterina, R. , D'Ugo, E. and Libby, P. , Inflammation and thrombosis: testing the hypothesis with anti‐inflammatory drug trials. Thromb Haemost 2016. 116: 1012–1021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.